
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic deoxygenation of N–O bonds with rhenium complexes: from the reduction of nitrous oxide to pyridine N-oxides†
Marianne
Kjellberg
,
Alexia
Ohleier
,
Pierre
Thuéry
 ,
Emmanuel
Nicolas
,
Lucile
Anthore-Dalion
* and
Thibault
Cantat
*
,
Emmanuel
Nicolas
,
Lucile
Anthore-Dalion
* and
Thibault
Cantat
*
Université Paris-Saclay, CEA, CNRS, NIMBE, 91191 Gif-sur-Yvette CEDEX, France
First published on 30th June 2021
Abstract
The accumulation of nitrogen oxides in the environment calls for new pathways to interconvert the various oxidation states of nitrogen, and especially their reduction. However, the large spectrum of reduction potentials covered by nitrogen oxides makes it difficult to find general systems capable of efficiently reducing various N-oxides. Here, photocatalysis unlocks high energy species able both to circumvent the inherent low reactivity of the greenhouse gas and oxidant N2O (E0(N2O/N2) = +1.77 V vs. SHE), and to reduce pyridine N-oxides (E1/2(pyridine N-oxide/pyridine) = −1.04 V vs. SHE). The rhenium complex [Re(4,4′-tBu-bpy)(CO)3Cl] proved to be efficient in performing both reactions under ambient conditions, enabling the deoxygenation of N2O as well as synthetically relevant and functionalized pyridine N-oxides.
Introduction
The modern development of intensive soil exploitation and – in corollary – the massive production of nitrogen-based fertilizers from atmospheric N2 has enabled us to feed an ever-growing population.1 Collateral damage, however, includes increased production and accumulation of noxious nitrogen oxide wastes such as nitrates NO3−, nitrites NO2− or nitrous oxide N2O, their reduction back to N2 being mostly ensured by the natural nitrogen cycle.2 As defined by environmental scientists, planetary boundaries for the biogeochemical nitrogen cycle have indeed already been crossed, the current nitrogen anthropogenic fixation representing more than twice the estimated boundary (150 Tg N per year vs. 62 Tg N per year).3 Since breaking the highly stable N2 bond via the Haber–Bosch process consumes yearly 2% of worldwide energy, leakage of nitrogen oxides in the environment also represents a loss of energy.4 In the search for a more sustainable economy, there is hence a need for new pathways able to perform efficient interconversion between oxidation states of nitrogen, and especially reduction of nitrogen oxides to less oxidized molecules.5Chemically speaking, this raises the issue of deoxygenating the N–O bond of both inorganic and organic N-oxides. With the aim to explore new pathways, we decided to harness the potential of homogeneous photoredox catalysis. One of its most important applications is the deoxygenation of C–O bonds of C1-molecules, which covers a narrow spectrum of reduction potentials (0.4 V, Fig. 1a).6 The main strategy in optimizing photo- or electro-catalysts has thus been focused on the selectivity between different products, especially between CO and HCOOH.7 Nitrogen oxides however cover a wider spectrum of reduction potentials (over 2.8 V, Fig. 1a) shifting the question from a search for selective catalysts, to the discovery of versatile catalytic systems.8
 | ||
| Fig. 1 Development of a photocatalytic pathway to deoxygenate N–O bonds. (a) Range of potentials involved for the reduction of C–O and N–O bonds. (b) Existing strategies for N–O bond deoxygenation. (c) Proposed strategy for the deoxygenation of N2O and pyridine N-oxides. DIPEA, diisopropylethylamine. | ||
A simple model that can be used to showcase this deoxygenation is the ozone-depleting nitrous oxide N2O, since it leads directly to N2. Although N2O is a strong oxidant (E0(N2O/N2) = +1.77 V vs. SHE),8 homogeneous deoxygenation reactions are scarcely reported, for it is highly inert and a poorly coordinating molecule.9 Among the few thermocatalytic deoxygenating methods, Milstein group disclosed the hydrogenation of N2O to N2 and H2O catalyzed by a PNP pincer ruthenium complex at 65 °C, under a total pressure of 7 bar (Fig. 1b).10 More recently, our laboratory proposed the deoxygenation of N2O using disilanes and a catalytic amount of fluoride source under ambient conditions (Fig. 1b).11 To the best of our knowledge, only heterogeneous photodeoxygenation of N2O has been so far reported.12 A very recent report by the group of Costentin and Chardon-Noblat demonstrated the use of ReI complexes able to catalyse the electroreduction of N2O to N2.13
At the opposite end of the redox scale, pyridine N-oxides have, in contrast, considerably lower reduction potentials (E1/2(pyridine N-oxide (1a)/pyridine (2a)) = −1.04 V vs. SHE, Fig. 1b) and they represent a much greater challenge.14 These compounds are useful intermediates in the synthesis of N-heteroaromatic-containing pharmaceutical or agrochemical products, where N-oxides are used for instance as directing groups for C–H bond functionalization.15 In that context, there is a need for mild methods to post-reduce the N-oxide group and afford the N-heterocycle. The main reported methods involve sacrificial oxophilic reagents or high energy reductant based on phosphines,16 silane17 or borane18 derivatives, with thermochemical or (photo)electrochemical activation.19 Nevertheless, all these methods imply the use of either heating systems or high energy reductants (with E0 < 0 V), leading to a loss of energy. There is moreover no report of systems capable of addressing the wide range of potentials (>2.5 V) required to reduce both nitrous oxide and pyridine N-oxides.
Herein, we demonstrate that a versatile photocatalytic system based on [Re(bpy)(CO)3Cl]-type complexes20 is able to efficiently deoxygenate both N2O and pyridine N-oxides under ambient conditions (20 °C, 1 bar, Fig. 1c).
Results and discussion
Design of the system for the photodeoxygenation of N2O
The potential of [Re(bpy)(CO)3Cl] (Re-1) to photocatalyze the conversion of N2O to N2 was tested, using Re-1 (5 mol%) as catalyst in acetonitrile in the presence of triethanolamine (TEOA) and 1 bar of N2O. After 2 h of irradiation with white LEDs at 20 °C, N2 was formed to our delight in 66% yield according to GC analysis of the gaseous fraction. This result represents the first homogeneous photocatalytic deoxygenation of N2O to N2 (Table 1, entry 1).|
|
||||
|---|---|---|---|---|
| Entry | Deviation from standard conditionsa | % H2b | % N2b | % N2Ob |
a Standard reaction conditions: N2O (1 bar, 100 µmol), Re-1 (5 µmol, 5 mol%), donor (570 µmol, 5.7 equiv.), CD3CN (0.5 mL), 2 h, 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C.
b Determined by GC analysis of the gaseous phase. Percentages correspond to the fraction of named gas in the overall analyzed gaseous phase (corrected with response factors).
c Under argon (1 bar). TEOA: triethanolamine; TEA: trimethylamine; DIPEA: diisopropylethylamine. °C.
b Determined by GC analysis of the gaseous phase. Percentages correspond to the fraction of named gas in the overall analyzed gaseous phase (corrected with response factors).
c Under argon (1 bar). TEOA: triethanolamine; TEA: trimethylamine; DIPEA: diisopropylethylamine.
|
||||
| 1 | None | 1.5 | 66 | 32 |
| 2 | No photocatalyst | 0 | 2 | 98 |
| 3 | No light | 0 | 4 | 96 |
| 4 | No N2Oc | 91 | 9 | 0 |
| 5 | No TEOA | 0 | 8 | 92 |
| 6 | TEA instead of TEOA | 0.2 | 36 | 63 |
| 7 | DIPEA instead of TEOA | 0 | 76 | 24 |
In the absence of Re-1, no conversion of N2O was observed: cleavage of the N–O bond does not occur spontaneously under irradiation (Table 1, entry 2). Other blank experiments confirmed that no N2 is formed in the absence of light, N2O, or electron donor (Table 1, entries 3–5).
Interestingly, the reaction performed without N2O afforded H2 as sole product, as a result of TEOA decomposition in the presence of the catalyst under irradiation (Table 1, entry 4). The detection of traces of H2 at the end of the reaction (1.5% of the gaseous phase, Table 1, entry 1) was thus ascribed to the use of TEOA. Sacrificial amines in photocatalysis are indeed known to supply the catalytic cycle with electrons and may serve as a proton source, which can in turn be reduced by the photocatalyst to H2.6a,21 This prompted us to investigate the influence of the sacrificial electron donor on the outcome of N2O photocatalytic reduction. Another tertiary amine, triethylamine (TEA), was used, inducing a drop of the N2O conversion, the gaseous fraction of N2 after 2 h decreasing from 66% to 36% without suppressing the generation of H2 (Table 1, entry 6). Interestingly, when a bulkier tertiary amine, diisopropylethylamine (DIPEA, Hünig's base), was used as electron donor, H2 could not be detected in GC after 2 hours of irradiation, and the N2 yield increased up to 76% (Table 1, entry 7). We therefore selected DIPEA as the sacrificial electron donor for the rest of our study.
Catalyst optimization
When the reaction was scaled up from 0.1 to 1.2 mmol N2O, the yield with Re-1 as catalyst dropped to 50%, affording a TON of 10 and a TOF0 of 1.9 h−1 (Table 2, entry 1). GC-monitoring showed no evolution after 22 h of reaction, indicating potential deactivation of the catalyst. We hence explored the effects of substitution on the bipyridine ligand. We synthesized a series of [Re(bpy)(CO)3Cl]-type catalysts with bipyridines substituted either on the 4,4′ or the 6,6′-position, and we studied their ability to photocatalyze the deoxygenation of N2O (Fig. 2a).|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | TOF0 (h−1) | % N2 (TON) | Time (h) |
| a Reaction conditions: N2O (1.0 bar, 1.2 mmol), catalyst (60 µmol, 5 mol%), DIPEA (6.9. mmol, 5.7 equiv.), CH3CN (6 mL), 20 °C. | ||||
| 1 | Re-1 | 1.9 | 50 (10) | 22 |
| 2 | Re-2 | 3.7 | 70 (14) | 50 |
| 3 | Re-3 | 4.3 | 86 (17) | 115 |
| 4 | Re-4 | 2.7 | 55 (11) | 100 |
| 5 | Re-5 | 1.1 | 61 (12) | 150 |
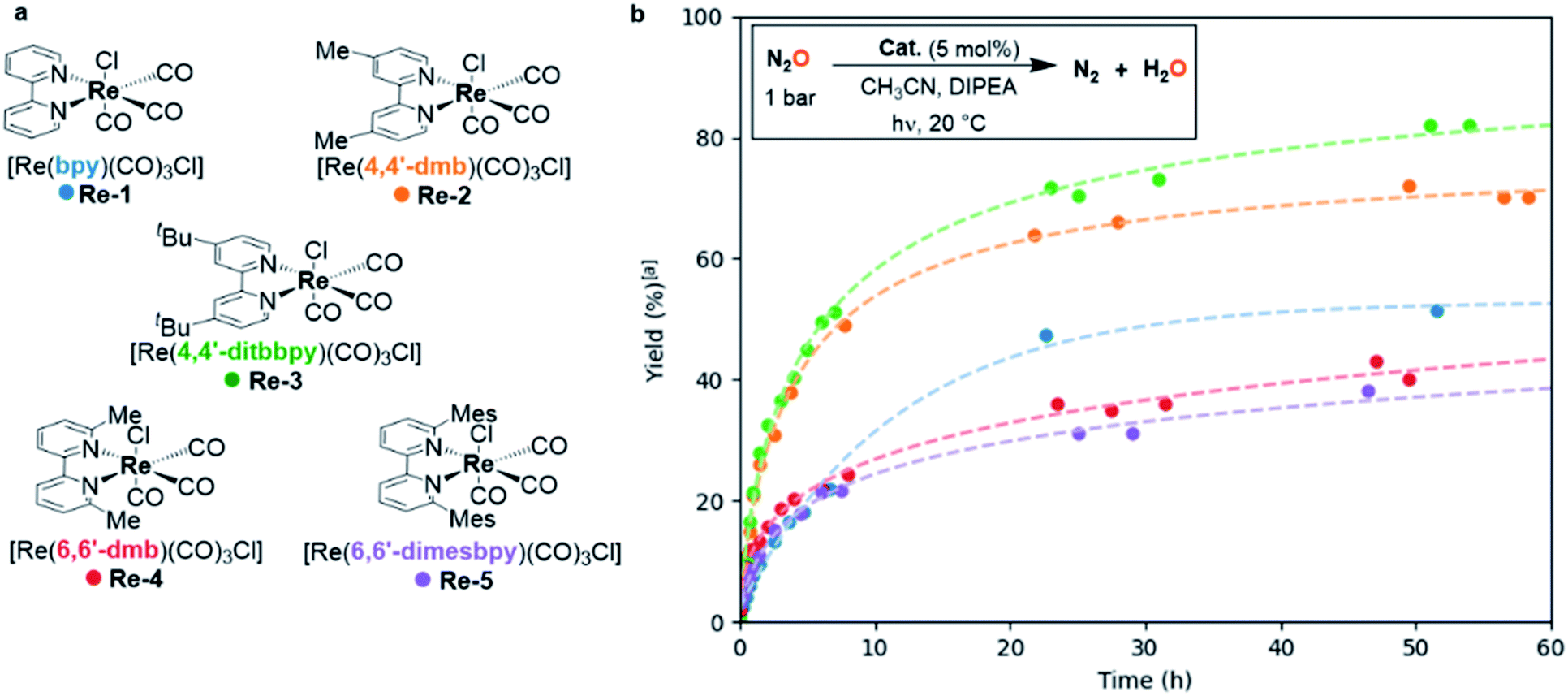 | ||
| Fig. 2 Photodeoxygenation of N2O: screening of catalysts. (a) Photocatalysts Re-1–5 used in the study. (b) Kinetic profiles on 1.2 mmol scale. Reaction conditions: N2O (1.0 bar, 1.2 mmol), catalyst (60 µmol, 5 mol%), DIPEA (6.9 mmol, 5.7 equiv.), CH3CN (6 mL), 20 °C. | ||
Complexes Re-2 (ref. 22) and Re-3,23 possessing bipyridines substituted in the 4,4′-position with methyl or t-butyl groups, respectively, were also active in the deoxygenation of N2O, and led to higher yields in N2 than unsubstituted Re-1, 70% and 86% respectively, corresponding to TONs of 14 and 17. They also showed higher activity than Re-1: TOF0 reached 3.7 h−1 and 4.3 h−1 for Re-2 and Re-3 respectively vs. 0.7 h−1 for Re-1 (Table 2, entries 1–3). On the other hand, Re-4 (ref. 23) and the novel Re-5 complex, featuring methyl and bulky mesityl substituents in the 6 and 6′ positions of bipyridines, were less efficient on a 24 h timescale, leading to moderate yields in N2 of 55% and 61% respectively (Table 2, entries 4 and 5). This behavior is also present in their initial activities: lower TOF0 (2.7 and 1.1 h−1 for Re-4 and Re-5 respectively) were measured compared to Re-2 and Re-3.
A more precise monitoring of kinetic profiles for each catalyst was also performed (Fig. 2b). The evolution of the N2 yield over 60 h showed that Re-1 deactivated after 22 h, sooner than all the other substituted catalysts, which accounts for the low yields obtained with Re-1. In contrast, Re-2 and Re-3, presenting para-substituted bipyridines, showed both high catalytic activity and increased stability. A possible explanation of this phenomenon has been studied in the case of CO2 electroreduction by Kubiak et al.: they showed that substituting the bipyridine ligand in the 4,4′-positions provided sufficient steric hindrance to inhibit the dimerization of a Re(0) reactive intermediate to an inactive bimetallic complex.24,25 The stability increased with the steric bulk of the substituent. The same tendency is observed here in the case of the photoreduction of N2O, and hints towards a similar mechanistic pathway. Complexes Re-4 and Re-5, substituted in the ortho position, demonstrated an increased stability compared to Re-1 but, interestingly enough, the increase in stability brought by substitution on the bipyridine did not come along with an increase in activity. These catalysts indeed required respectively 100 h and 150 h to yield 55% and 61% of N2. A structural explanation could arise from the steric strain between the substituents on the bipyridine and the equatorial carbonyls in Re-4 and Re-5.26
Rhenium photocatalysts of type [Re(bpy)(CO)3Cl] are thus able to catalyze the deoxygenation of N2O, suggesting that other N–O bonds could be deoxygenated under similar reaction conditions.
Photodeoxygenation of pyridine N-oxides
Encouraged by the success of N2O deoxygenation, and to probe the versatility of our catalytic system, we considered the possibility of applying this methodology to the deoxygenation of more challenging N–O bonds across the scale of redox potentials, namely of pyridine N-oxides (E1/2(pyridine N-oxide (1a)/pyridine (2a)) = −1.04 V vs. SHE).The reaction was monitored by NMR using rhenium-based Re-1–5 photocatalysts. To our delight, a first attempt using the tBu-substituted catalyst Re-3 under the reaction conditions developed for N2O enabled deoxygenation of pyridine N-oxide (1a) to pyridine (2a) in 82% yield after 34 h (Table 3, entry 1). The blank experiments confirmed that all components (light, photocatalyst, electron donor) were necessary to perform the reduction (see ESI Table S1†). Remarkably, the other catalysts also afforded full conversion of compound 1a, albeit with longer reaction times (44–87 h), leading to pyridine (2a) in 85–99% yield (Table 3, entries 2–5). This stands in contrast with our observations with N2O: here the yields showed steady increase until full conversion with all catalysts. Two explanations may account for this phenomenon: either the pyridine produced during the reaction delays catalyst deactivation through the stabilization of reactive intermediates,27 or the higher oxidative character of N2O has a detrimental effect on the catalyst stability. This effect was particularly beneficial in the case of the 6,6′-dimethylbipyridine rhenium complex Re-4, which led only to 55% yield of N2 after 100 h and gave full conversion of pyridine N-oxide (1a) within 55 h. Again, 4,4′-substituted catalysts Re-2 and Re-3 displayed higher catalytic activity (TOF0 = 11 and 16 h−1) than unsubstituted Re-1 and 6,6′-substituted Re-4 and Re-5 (TOF0 = 5.5, 7.5 and 2.6 h−1, respectively), suggesting similar mechanistic patterns for the deoxygenation of both N2O and pyridine N-oxide (1a) (Table 3). To study the effect of irradiation, a light on/light off experiment was also performed (Fig. 3a).28 The substrate was only converted when the system was exposed to light, and the reaction stopped once the system was in the dark. Continuous irradiation is thus required to perform the reaction.
| Entry | Cat. | TOF0 (h−1) | Yield (%) | Time to full conversion (h) |
|---|---|---|---|---|
|
a Reaction conditions: pyridine N-oxide (100 µmol), catalyst (5 µmol, 5 mol%), DIPEA (570 µmol, 5.7 equiv.), CD3CN (0.5 mL), 20°C.
|
||||
| 1 | Re-3 | 16 | 82 | 34 |
| 2 | Re-1 | 5.5 | 99 | 63 |
| 3 | Re-2 | 11 | 85 | 87 |
| 4 | Re-4 | 7.5 | 96 | 55 |
| 5 | Re-5 | 2.6 | 87 | 44 |
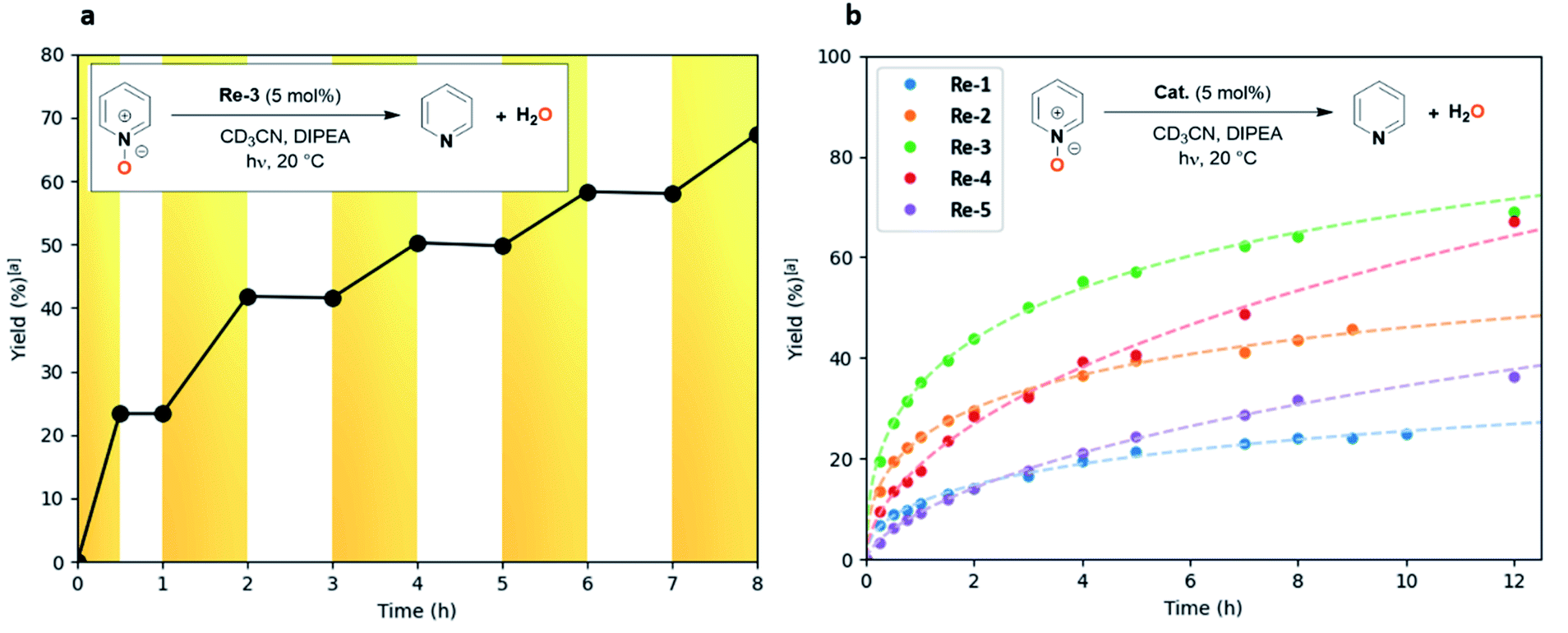 | ||
| Fig. 3 Photoreduction of pyridine N-oxide. (a) Light and dark experiment with Re-3. Yellow: light on; white: light off. (b) Kinetic profiles of pyridine N-oxide photoreduction with different catalysts. Reaction conditions: pyridine N-oxide (100 µmol), catalyst (5 µmol, 5 mol%), DIPEA (570 µmol, 5.7 equiv.), CD3CN (0.5 mL), 20 °C. aDetermined by 1H NMR analysis of the crude reaction mixture. | ||
From a kinetic standpoint, the possible improvement of catalytic performances as pyridine was released was particularly noticeable in the case of 6,6′-substituted catalysts Re-4 and Re-5. If their initial catalytic activities were low (TOF0 = 7.5 and 2.6 h−1), they showed steady catalytic activity past 2 hours of irradiation, while the conversion rates for the other catalysts were gently decreasing (Fig. 3b). This resulted in Re-4 and Re-5 reaching full conversion before Re-1 and Re-2.
Several para-substituted pyridine N-oxides 1b–f could be deoxygenated in 77–99% yield using photocatalyst Re-3 (Fig. 4). The electronic parameters of the substituents affected the initial reaction rate: the yields after 30 minutes of irradiation evolved in line with the electron-withdrawing ability of the substituents as given by their Hammett sigma constants (Fig. 4b).29 Indeed, compounds bearing electron-donating substituents were only poorly converted after 30 minutes: 1b–d bearing dimethylamino, methoxy or methyl groups (σH = −0.83, −0.27 and −0.17 respectively) reached only 3 to 25% yield, while 1a, 1e and 1f, bearing –H, chloro or cyano groups (σH = 0, 0.23 and 0.66 respectively) yielded after 30 min. 26, 30 and 43% of the corresponding pyridines, respectively. This was also reflected in the time to reach full conversion: pyridine N-oxides 1b-d bearing electron-donating substituents (–NMe2, –OMe, –Me) in the para position required longer reaction times (14–126 h) than 1e-f with electron-withdrawing substituents (8 and 0.5 h respectively for the chloro and cyano derivatives). In particular the conversion of the nitrile derivative 1f was extremely fast, maximal (43%) after only 30 minutes of irradiation. Further irradiation however led to over-reduction of 2f to pyridine 2a (for details see ESI Fig. S1†).
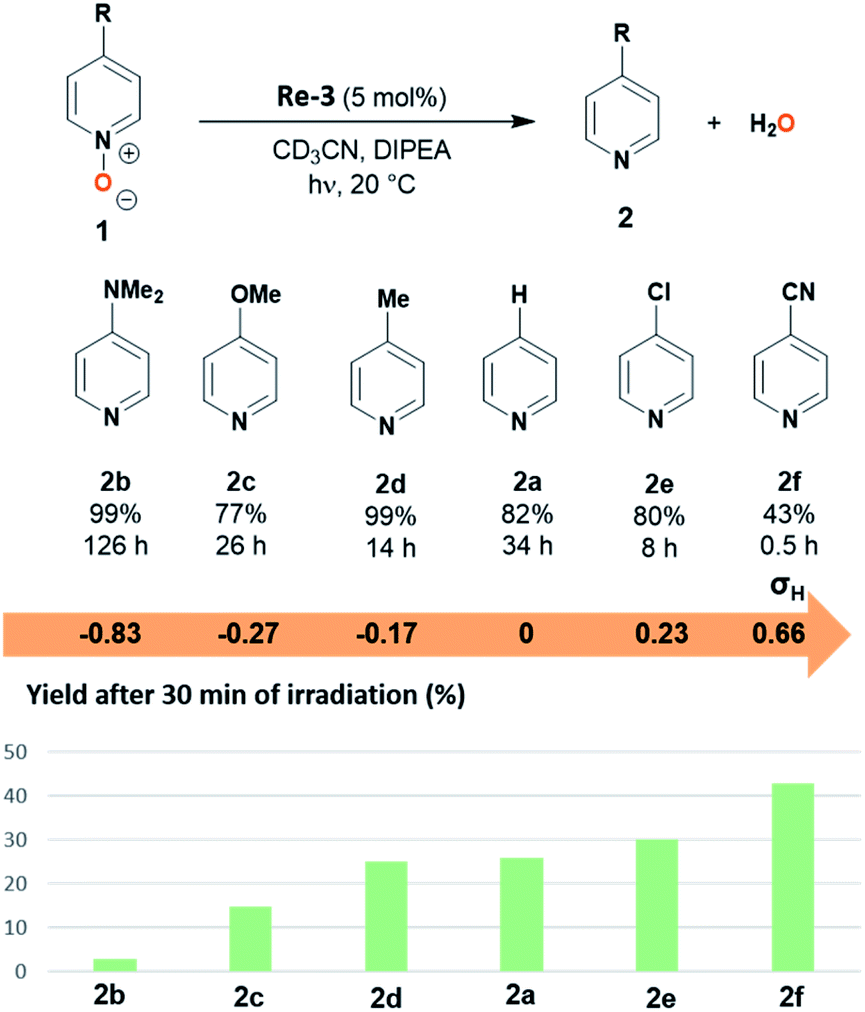 | ||
| Fig. 4 Photocatalytic reduction of para-substituted pyridine N-oxides. (top) Scope of substrates (yield, time of irradiation until maximum yield). Corresponding Hammett constants of the substituents. Reaction conditions: pyridine N-oxide (100 µmol), Re-3 (5 µmol, 5 mol%), DIPEA (570 µmol, 5.7 equiv.), CD3CN (0.5 mL), 20 °C. Yields were determined by 1H NMR analysis of the crude reaction mixture. (bottom) Yields of the photocatalytic deoxygenation of para-substituted pyridine N-oxides after 30 minutes of irradiation. | ||
This is the first photocatalytic system able to perform such a deoxygenation reaction without additional energy sources. In contrast, strong reductants based on Hantzch esters or hydrazine were used by the groups of Konev and Wangelin19c and Lee.19d
In order to demonstrate the selectivity of this reaction and its synthetic utility, we explored the deoxygenation of a more complex substrate, a 2,6-substituted pyridine N-oxide. Pyridine N-oxide indeed features an enhanced reactivity compared to the reduced pyridine, enabling the functionalization of the 2,6-positions by C–H activation and, as an example, Hiyama et al. recently reported the synthesis of 2,6-substituted pyridine 3, based on the oxidation of o-picoline to form the 2-methylpyridine N-oxide 4.30 A Ni-catalyzed coupling reaction leads to 2,6 substituted pyridine N-oxide 5, whose deoxygenation presents multiple challenges that are chemoselectivity and steric hindrance. In their publication, the Hiyama group used PCl3 as a reducing agent to perform such a deoxygenation. We were very pleased to observe that using our system, Re-3 catalyzed the deoxygenation with 82% yield after 8 h of irradiation (Scheme 1). This demonstrates the validity of such an approach for mild deprotection strategies, which may be extended to other substrates.
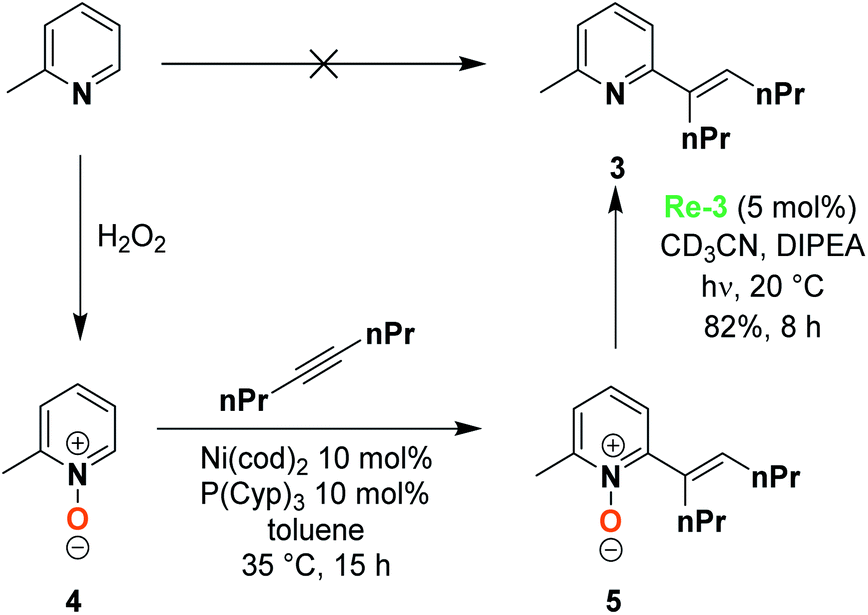 | ||
| Scheme 1 2-Methylpyridine functionalization via the N-oxide strategy. | ||
Conclusions
In summary, a new photochemical method has been developed to deoxygenate N–O bonds with radically different reduction potentials, over a 2.8 V window. [Re(4,4′-tBu-bpy)(CO)3Cl] (Re-3) is indeed capable of reducing both N2O under ambient conditions, and pyridine N-oxides 1 and 4 in good to excellent yields. Those results open new perspectives concerning the photocatalytic deoxygenation of nitrogen oxide-containing compounds. Further mechanistic studies on catalyst Re-3 are underway in our laboratory. We believe that these will provide new clues to understanding N–O bonds deoxygenation chemistry.Data availability
Crystallographic data for [compound number] has been deposited at the CCDC under 2056048 and 2056049 and can be obtained from https://www.ccdc.cam.ac.uk/. The datasets supporting this article have been uploaded as part of the supplementary material.Author contributions
MK and AO performed the investigations. PT performed the XRD analyses. EN, LAD and TC supervised the project. All authors contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the CEA, CNRS, ERC (Consolidator Grant no. 818260), and Institut de France for funding. M. K. was supported by a Master fellowship from the LABEX Charmmmat followed by a PhD fellowship from ADEME. We thank Thierry Bernard (CEA) for his assistance in conception and realization of the photochemistry reactor.Notes and references
- A. Mosier, J. K. Syers and J. R. Freney, Agriculture and the nitrogen cycle: Assessing the impacts of fertilizer use on food production and the environment, Island Press, 2013 Search PubMed.
- (a) N. Lehnert, H. T. Dong, J. B. Harland, A. P. Hunt and C. J. White, Nat. Rev. Chem., 2018, 2, 278–289 CrossRef CAS; (b) H. Tian, R. Xu, J. G. Canadell, R. L. Thompson, W. Winiwarter, P. Suntharalingam, E. A. Davidson, P. Ciais, R. B. Jackson, G. Janssens-Maenhout, M. J. Prather, P. Regnier, N. Pan, S. Pan, G. P. Peters, H. Shi, F. N. Tubiello, S. Zaehle, F. Zhou, A. Arneth, G. Battaglia, S. Berthet, L. Bopp, A. F. Bouwman, E. T. Buitenhuis, J. Chang, M. P. Chipperfield, S. R. S. Dangal, E. Dlugokencky, J. W. Elkins, B. D. Eyre, B. Fu, B. Hall, A. Ito, F. Joos, P. B. Krummel, A. Landolfi, G. G. Laruelle, R. Lauerwald, W. Li, S. Lienert, T. Maavara, M. MacLeod, D. B. Millet, S. Olin, P. K. Patra, R. G. Prinn, P. A. Raymond, D. J. Ruiz, G. R. van der Werf, N. Vuichard, J. Wang, R. F. Weiss, K. C. Wells, C. Wilson, J. Yang and Y. Yao, Nature, 2020, 586, 248–256 CrossRef CAS PubMed.
- (a) J. Rockstrom, W. Steffen, K. Noone, A. Persson, F. S. Chapin 3rd, E. F. Lambin, T. M. Lenton, M. Scheffer, C. Folke, H. J. Schellnhuber, B. Nykvist, C. A. de Wit, T. Hughes, S. van der Leeuw, H. Rodhe, S. Sorlin, P. K. Snyder, R. Costanza, U. Svedin, M. Falkenmark, L. Karlberg, R. W. Corell, V. J. Fabry, J. Hansen, B. Walker, D. Liverman, K. Richardson, P. Crutzen and J. A. Foley, Nature, 2009, 461, 472–475 CrossRef PubMed; (b) W. de Vries, J. Kros, C. Kroeze and S. P. Seitzinger, Curr. Opin. Environ. Sustain., 2013, 5, 392–402 CrossRef; (c) W. Steffen, K. Richardson, J. Rockstrom, S. E. Cornell, I. Fetzer, E. M. Bennett, R. Biggs, S. R. Carpenter, W. de Vries, C. A. de Wit, C. Folke, D. Gerten, J. Heinke, G. M. Mace, L. M. Persson, V. Ramanathan, B. Reyers and S. Sorlin, Science, 2015, 347, 1259855 CrossRef PubMed.
- (a) Electrochemical dinitrogen activation: To find a sustainable way to produce ammonia, S. Chen, S. Perathoner, C. Ampelli and G. Centi, in Horizons in sustainable industrial chemistry and catalysis, ed. S. Albonetti, S. Perathoner and E. A. Quadrelli, Elsevier, 2019, vol. 178, pp. 31–46 Search PubMed; (b) O. Elishav, B. Mosevitzky Lis, E. M. Miller, D. J. Arent, A. Valera-Medina, A. Grinberg Dana, G. E. Shter and G. S. Grader, Chem. Rev., 2020, 120, 5352–5436 CrossRef CAS PubMed.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, eaar6611 CrossRef PubMed.
- (a) Y. Yamazaki, H. Takeda and O. Ishitani, J. Photochem. Photobiol., C, 2015, 25, 106–137 CrossRef CAS; (b) Y. Kuramochi, O. Ishitani and H. Ishida, Coord. Chem. Rev., 2018, 373, 333–356 CrossRef CAS; (c) C. Chauvier and T. Cantat, ACS Catal., 2017, 7, 2107–2115 CrossRef CAS.
- (a) C. Costentin, M. Robert and J. M. Saveant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC; (b) H. Takeda, C. Cometto, O. Ishitani and M. Robert, ACS Catal., 2016, 7, 70–88 CrossRef.
- D. R. Lide, Crc handbook of chemistry and physics, CRC press, 2004 Search PubMed.
- (a) V. N. Parmon, G. I. Panov, A. Uriarte and A. S. Noskov, Catal. Today, 2005, 100, 115–131 CrossRef CAS; (b) K. Severin, Chem. Soc. Rev., 2015, 44, 6375–6386 RSC.
- R. Zeng, M. Feller, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2017, 139, 5720–5723 CrossRef CAS PubMed.
- L. Anthore-Dalion, E. Nicolas and T. Cantat, ACS Catal., 2019, 9, 11563–11567 CrossRef CAS.
- T. Ming, R. de Richter, S. Shen and S. Caillol, Environ. Sci. Pollut. Res., 2016, 23, 6119–6138 CrossRef CAS PubMed.
- R. Deeba, F. Molton, S. Chardon-Noblat and C. Costentin, ACS Catal., 2021, 11, 6099–6103 CrossRef CAS.
- T. Kubota and H. Miyazaki, Bull. Chem. Soc. Jpn., 1966, 39, 2057–2062 CrossRef CAS.
- Y. L. Wang and L. M. Zhang, Synthesis, 2015, 47, 289–305 CrossRef CAS.
- E. Howard and W. F. Olszewski, J. Am. Chem. Soc., 1959, 81, 1483–1484 CrossRef CAS.
- S. Chandrasekhar, C. R. Reddy, R. J. Rao and J. M. Rao, Synlett, 2002, 349–351 CrossRef CAS.
- H. P. Kokatla, P. F. Thomson, S. Bae, V. R. Doddi and M. K. Lakshman, J. Org. Chem., 2011, 76, 7842–7848 CrossRef CAS PubMed.
- (a) P. Xu and H. C. Xu, Synlett, 2019, 30, 1219–1221 CrossRef CAS; (b) Y. Fukazawa, A. E. Rubtsov and A. V. Malkov, Eur. J. Org. Chem., 2020, 3317–3319 CrossRef CAS; (c) M. O. Konev, L. Cardinale and A. Jacobi von Wangelin, Org. Lett., 2020, 22, 1316–1320 CrossRef CAS PubMed; (d) K. D. Kim and J. H. Lee, Org. Lett., 2018, 20, 7712–7716 CrossRef CAS PubMed.
- J. Hawecker, J. M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536–538 RSC.
- (a) J. Hu, J. Wang, T. H. Nguyen and N. Zheng, Beilstein J. Org. Chem., 2013, 9, 1977–2001 CrossRef PubMed; (b) Y. Pellegrin and F. Odobel, C. R. Chim., 2017, 20, 283–295 CrossRef CAS.
- K. Kalyanasundaram, J. Chem. Soc., Faraday Trans., 1986, 82, 2401–2415 RSC.
- V. W.-W. Yam, V. C.-Y. Lau and K.-K. Cheung, Organometallics, 2002, 14, 2749–2753 CrossRef.
- (a) J. M. Smieja and C. P. Kubiak, Inorg. Chem., 2010, 49, 9283–9289 CrossRef CAS PubMed; (b) J. M. Smieja, E. E. Benson, B. Kumar, K. A. Grice, C. S. Seu, A. J. Miller, J. M. Mayer and C. P. Kubiak, PNAS, 2012, 109, 15646–15650 CrossRef CAS PubMed; (c) E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC; (d) E. E. Benson and C. P. Kubiak, Chem. Commun., 2012, 48, 7374–7376 RSC.
- As suggested by a referee, an alternative explanation would involve a change in the redox potentials of the catalysts leading to a change in the efficiency of the electron transfer.
- As suggested by a referee, addition of chloride anions also improves the photocatalytic activity, in agreement with the findings of Lehn et al. in the photocatalytic reduction of CO2. See ref. 27 and ESI† for more details.
- J. Hawecker, J. M. Lehn and R. Ziessel, Helv. Chim. Acta, 1986, 69, 1990–2012 CrossRef CAS.
- L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS PubMed.
- L. P. Hammett, J. Am. Chem. Soc., 2002, 59, 96–103 CrossRef.
- K. S. Kanyiva, Y. Nakao and T. Hiyama, Angew. Chem., Int. Ed. Engl., 2007, 46, 8872–8874 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Data relating to the characterization data of materials and products, general methods, optimization studies, experimental procedures, gas chromatography, NMR spectra, and CIF X-ray structures. CCDC 2056048 and 2056049. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01974k |
| This journal is © The Royal Society of Chemistry 2021 |