
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAssembly of multicyclic isoquinoline scaffolds from pyridines: formal total synthesis of fredericamycin A†
Fang-Xin
Wang
 a,
Jia-Lei
Yan
a,
Zhixin
Liu
a,
Tingshun
Zhu
a,
Yingguo
Liu
a,
Shi-Chao
Ren
a,
Wen-Xin
Lv
a,
Zhichao
Jin
b and
Yonggui Robin
Chi
*ab
a,
Jia-Lei
Yan
a,
Zhixin
Liu
a,
Tingshun
Zhu
a,
Yingguo
Liu
a,
Shi-Chao
Ren
a,
Wen-Xin
Lv
a,
Zhichao
Jin
b and
Yonggui Robin
Chi
*ab
aDivision of Chemistry & Biological Chemistry, School of Physical & Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore
bState Key Laboratory Breeding Base of Green Pesticide and Agricultural Bioengineering, Key Laboratory of Green Pesticide and Agricultural Bioengineering, Ministry of Education, Guizhou University, Huaxi District, Guiyang 550025, China. E-mail: robinchi@ntu.edu.sg
First published on 28th June 2021
Abstract
The construction of an isoquinoline skeleton typically starts with benzene derivatives as substrates with the assistance of acids or transition metals. Disclosed here is a concise approach to prepare isoquinoline analogues by starting with pyridines to react with β-ethoxy α,β-unsaturated carbonyl compounds under basic conditions. Multiple substitution patterns and a relatively large number of functional groups (including those sensitive to acidic conditions) can be tolerated in our method. In particular, our protocol allows for efficient access to tricyclic isoquinolines found in hundreds of natural products with interesting bioactivities. The efficiency and operational simplicity of introducing structural complexity into the isoquinoline frameworks can likely enable the collective synthesis of a large set of natural products. Here we show that fredericamycin A could be obtained via a short route by using our isoquinoline synthesis as a key step.
Isoquinolines and their derivatives are common structural motifs in numerous natural products. Among them, the analogues of isoquinolines fused with rings from the benzene side such as 8-hydroxyisoquinolin-1[2H]-one (Fig. 1a) have been found in hundreds of natural products with interesting bioactivities.1 For example, fredericamycin A and the related family members, isolated from Streptomyces griseus, show both antimicrobial and anti-tumor activities.2 Ericamycin is a natural product isolated in the culture of Streptomyces varius n. sp. with anti-staphylococcal activities.3 Due to the widespread presence of isoquinolines in both natural and synthetic molecules, numerous approaches have been developed to assemble this class of scaffolds.4 The dominated strategies reported to date focus on forming the new pyridine ring of isoquinolines (Fig. 1b, left part). Classic methods include Bischler–Napieralski isoquinoline synthesis,4a,b Pictet–Gams isoquinoline synthesis,4a and Pomeranz–Fritsch reaction.4a These reactions, proven to be useful since as early as 1893,5 have their own merits and limitations. For instance, high reaction temperature (e.g. reflux in toluene) and strong acids are typically required and thus functional group tolerance can become challenging. On the other side, the introduction of structural complexities and substitution patterns is constrained as the substrates have to be pre-settled to favor the formation of pyridine moieties. Here we report a new approach to prepare isoquinoline scaffolds by constructing a new benzene ring (Fig. 1b, right part).6 Our method starts with pyridine derivatives as the substrates to react with readily available β-ethoxy α,β-unsaturated carbonyl compounds. The reaction cascade involves five main plausible mechanistic processes (Michael addition, Dieckmann condensation, elimination, aromatization and in situ methylation) to furnish isoquinoline-based products with medium to good yields. The tricyclic isoquinoline-containing products might serve as formal common starting points for rapid total synthesis of a large number of natural products, such as those exemplified in Fig. 1a. In the present study, we demonstrate that starting from the tricyclic isoquinoline adduct 6a prepared using our method, fredericamycin A can be synthesized in 8 steps (Fig. 1c). Our strategy for isoquinoline assembly offers complementary and in certain cases better solutions not readily provided by the classic methods. We expect our method to find impressive applications in concise modular synthesis of complex natural products and molecular libraries, especially those bearing isoquinoline units fused with additional cyclic structures.
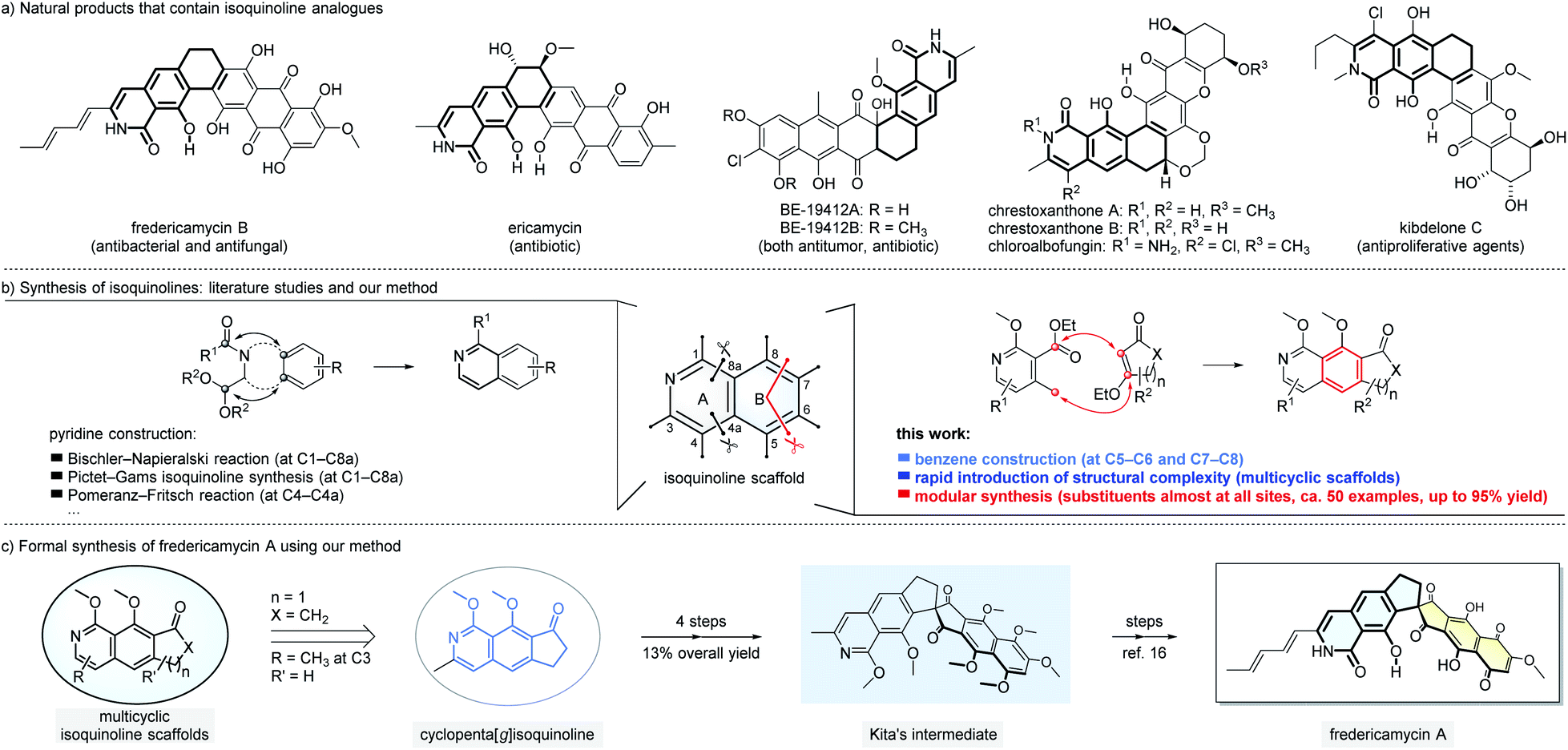 | ||
| Fig. 1 Isoquinoline analogues and their synthesis. | ||
Our design and initial studies are illustrated in Scheme 1.7 We first used pyridine 1a to react with α-substituted cycloenones (2a–2d), in the hope of obtaining isoquinoline 3a as the target product (Scheme 1a). The use of 2a and 2b was inspired by studies from Tamura, in which α-Br in 1,4-naphthoquinone was used as a leaving group to form an aromatic ring.8 Unfortunately, no product was formed and most of the starting materials were recovered. When SPh (2c) or SOPh (2d) was incorporated at the α site of the cycloenone, side products 4a and 4b were isolated respectively in moderate yields. The Michael products 4a and 4b could not be further transformed into our desired cyclic product 3a under various conditions. We then studied the use of β-substituted cycloenones (2e–2g) to react with 1a (Scheme 1b). No reactions were observed when 2e or 2f was used. To our delight, when the halogen of 2e/2f was replaced with a methoxy unit (OCH3, substrate 2g), an encouraging amount of annulation product 3a was detected (10% yield). A side product 5a was also obtained (5% yield) in this initial study and it couldn't be further transformed into the annulation product 3a under various alkaline conditions. It is noteworthy that, while β-alkoxy cycloenones (specifically, only β-alkoxy cyclohexenones) have been used in Staunton–Weinreb annulation9 to prepare fused aromatic compounds, no examples for those containing a heterocyclic aromatic ring were reported.10 Even for the construction of an aromatic ring without any heteroatom, low yields (mostly ranging from 0 to 30%) often occurred for this type of annulation starting with β-alkoxy cycloenones,9 which severely hampered its usage in Staunton–Weinreb annulation for the total synthesis of natural products. Our initial results showcased the possibility of direct assembly of isoquinoline scaffolds from β-methoxy cyclopentenone for the first time, though also in a low yield of 10%.
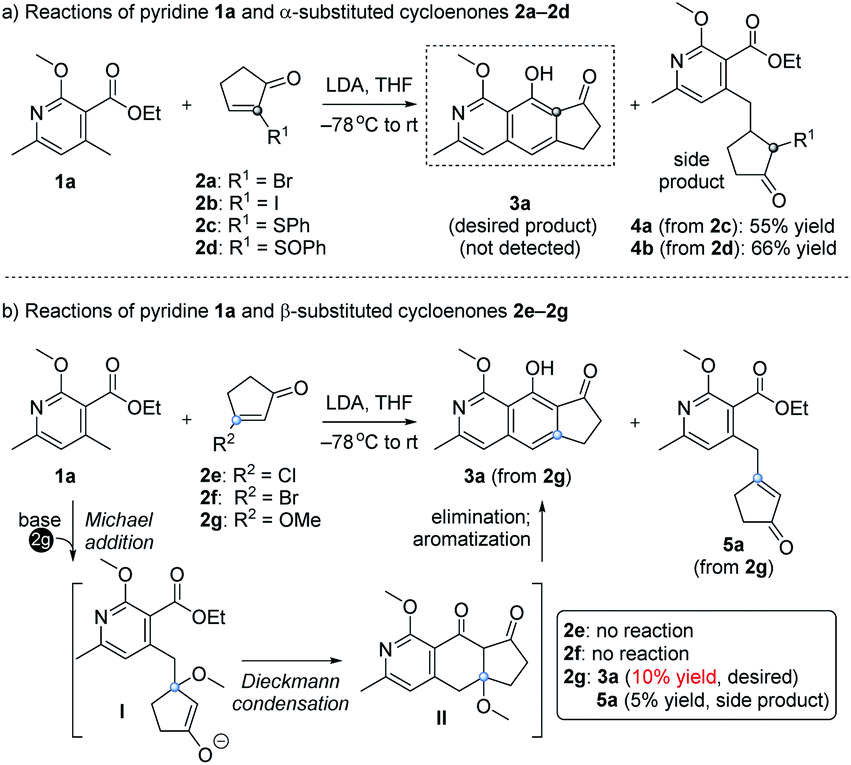 | ||
| Scheme 1 Proposed routes and initial studies for isoquinoline synthesis. | ||
With the initial results in hand, we performed additional condition optimization (Table 1). The best conditions (Table 1, entry 1) involve the deprotonation of 1a with LDA at −78 °C, warming the reaction mixture to room temperature in 10 minutes after the completion of slow addition of 2h, and most importantly, in situ methylation of the hydroxyl group of phenol 3a, which accounts for the relatively high yield of adducts 6a (72% yield). Without methylation, the phenol adduct 3a was isolated only in 14% yield (entry 2).11 The β-methoxy cyclopentenone 2g could also react to give 6a in a lower yield of 65% (entry 3). Other bases [such as triethylenediamine (DABCO), diazabicyclo[5.4.0]undec-7-ene (DBU), 4-dimethylaminopyridine (DMAP), lithium bis(trimethylsilyl)amide (LiHMDS) and potassium bis(trimethylsilyl)amide (KHMDS)] gave poorer results with yields ranging from 0 to 42% (entry 4). When THF was changed to other solvents, lower yields (<41%) were obtained (entry 5). Revising the ratio of 1a to 2h from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5 to 1.5:1 delivered 6a in 39% to 54% yields (entries 6–8). Lower reaction temperature (e.g. −78 °C) could not improve the outcome of this cascade transformation, but gave 23% yield of 6a together with 16% yield of recovered starting material 2h (entry 9). Long exposure to low temperature in step 1 could also lead to a considerable amount of the undesired elimination product 5a (ca. 29% yield), which was decomposed under the following methylation conditions (step 2). No product was observed in the absence of the methoxy group in 1a as it could stabilize the transition state via the formation of a metallate complex (entry 10).
:1.5 to 1.5:1 delivered 6a in 39% to 54% yields (entries 6–8). Lower reaction temperature (e.g. −78 °C) could not improve the outcome of this cascade transformation, but gave 23% yield of 6a together with 16% yield of recovered starting material 2h (entry 9). Long exposure to low temperature in step 1 could also lead to a considerable amount of the undesired elimination product 5a (ca. 29% yield), which was decomposed under the following methylation conditions (step 2). No product was observed in the absence of the methoxy group in 1a as it could stabilize the transition state via the formation of a metallate complex (entry 10).
|
|
||
|---|---|---|
| Entry | Variation from standard conditions | Yieldb (%) |
| a Standard conditions: 1a (0.2 mmol) and LDA (0.2 mmol) reacted in THF at −78 °C for 1 h; 2h (0.1 mmol) was added dropwise to the mixture before warming up to rt in 10 min. The reaction was quenched by the addition of saturated aqueous solution of NH4Cl after completion monitored by TLC. After the removal of solvents, the crude residue was treated directly with TBAB (0.2 eq.), NaOH (2.0 eq.) in water (1 mL), and Me2SO4 (4.0 eq.) in CH2Cl2 (1 mL). b Isolated yield. c Recovered starting material 2h: 16% yield. | ||
| 1 | None | 72 |
| 2 | Without methylation | 14 |
| 3 | OCH3 instead of OEt in 2h | 65 |
| 4 | DABCO, DBU, DMAP, LiHMDS and KHMDS instead of LDA | 0–42 |
| 5 | Other solvents in step 1 | <41 |
| 6 |
1a:2h = 1:1 |
39 |
| 7 |
1a:2h = 1.5:1 |
54 |
| 8 |
1a:2h = 1:1.5 |
42 |
| 9c | −78 °C for step 1 | 23 |
| 10 | H instead of OCH3 in 1a | 0 |
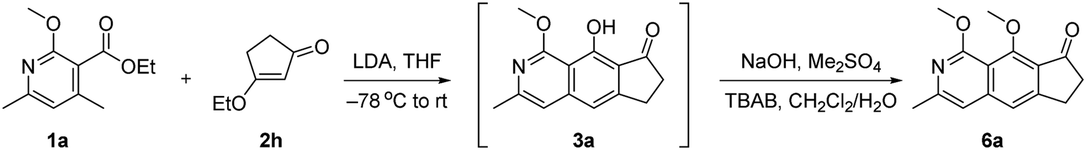
With the optimal reaction conditions in hand, we next examined the scope of the pyridine derivatives 1. As we can see from Scheme 2, substrates with the aliphatic substituents at C3 could afford the corresponding tricyclic isoquinoline products (6a and 6b) in acceptable yields. Besides, the incorporation of an aromatic ring at this site (6c–6j) also works well for this transformation, wherein electron-rich aromatic rings (6c–6g) could give higher yields than the corresponding electron-deficient ones (6h–6j). It should be noted that the relatively lower yield of 44% for 6h was partially due to the slow reaction rate as the recovered starting material was always detected in this transformation. When it comes to C4 substitution, the isoquinoline products with broad structural diversities such as alkyl (6k), alkenyl (6l–6n),12 alkynyl (6o), benzyl derivatives with different substituents on the phenyl ring (6p–6t), heteroaromatic ring (6u) and thioether (6v) could be obtained in 57–93% yields. Moreover, substrates bearing acid-hydrolyzable functionalities (6w) and with a relatively bulky secondary substituent (6x) also worked well under the optimized reaction conditions. Next, we examined the possibility of introducing a side chain at C5. To our delight, the substrate with an ethyl group instead of the methyl group on the aromatic ring reacted smoothly to deliver the corresponding isoquinoline 6y in 89% yield. Further study revealed that the exposure of the bicyclic substrate 5,6,7,8-tetrahydroisoquinoline derivative to the optimized reaction conditions could furnish the polycyclic product 6z in 92% yield. Finally, we relocated the nitrogen atom in the pyridine ring. The experimental results indicated that the substrate with nitrogen atom located at C3 can't react to form the corresponding isoquinoline 6aa, possibly due to the mismatched dipole orientation. When the nitrogen atom was sited at the ortho-position of the methyl group in the aromatic ring, quinoline 6ab could not be detected either under the optimized reaction conditions. The control experiments showcased the decisive influence of the location of nitrogen atom in the aromatic ring on the reactivity of this cascade transformation.
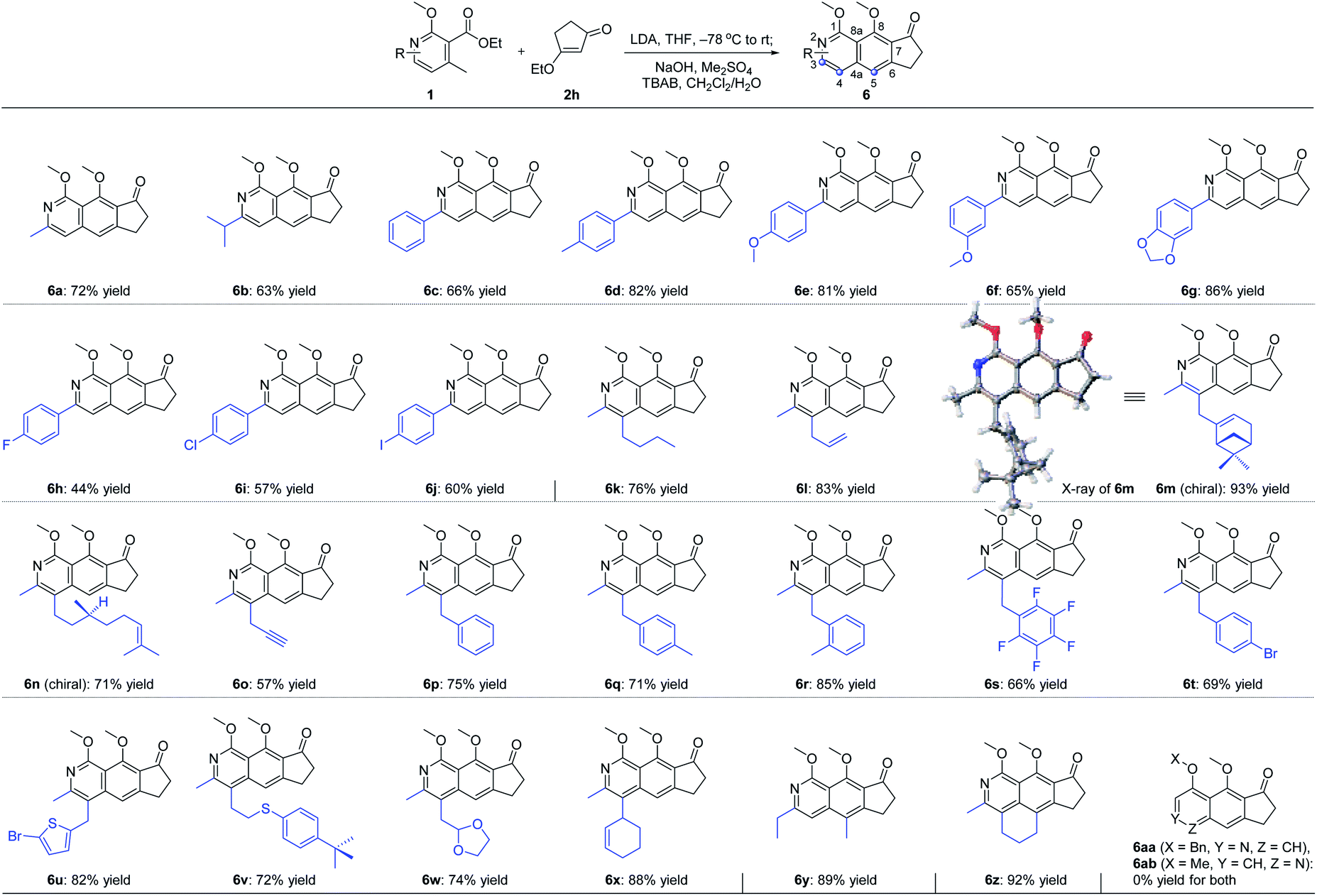 | ||
| Scheme 2 Scope of pyridine derivatives. | ||
For the five-membered cycloenone derivatives 2 (Scheme 3), substrates with different substituents at the α′ position work well for this transformation (6ac–6ak),12 of which the incorporation of a quaternary carbon center (6aj) and a heteroatom (6ak) at this site was included. The introduction of an allyl group at the β′ position in cyclopentenone proved to be viable for this transformation, delivering 6al in 64% yield. More encouragingly, when the sterically hindered substrate with a quaternary carbon center located at the γ site was exposed to the optimized reaction conditions, the isoquinoline 6am was obtained in 65% yield. This is challenging, considering the fact that the reacting site is just adjacent to a sterically bulky all-carbon quaternary stereocenter. Bicyclic 3-ethoxy-1H-inden-1-one is also suitable for this cascade transformation, giving the tetracyclic 10H-indeno[1,2-g]isoquinolin-10-one derivative 6an in 89% yield. When it comes to six-membered cycloenone derivatives (6ao–6au), substrates with substituents at α′ and β′ positions all worked smoothly to provide the corresponding isoquinoline products in moderate to high yields. Notably, Kita reported a 5-step reaction sequence to get the tricyclic benzo[g]isoquinoline-derived product 6as starting from the 1a analogue in an overall yield of 22%.6b Using our developed method, 6as could be easily obtained in 53% yield from 1a. Unexpectedly, a side product 6av was isolated in moderate yield when it comes to the γ-substituted substrate. Further study revealed that cyclohept-2-en-1-one with a medium-sized ring (6aw), lactone (6ax), and lactam (6ay) all worked well for this annulation cascade, which significantly expanded the substrate scope of this powerful cascade transformation.
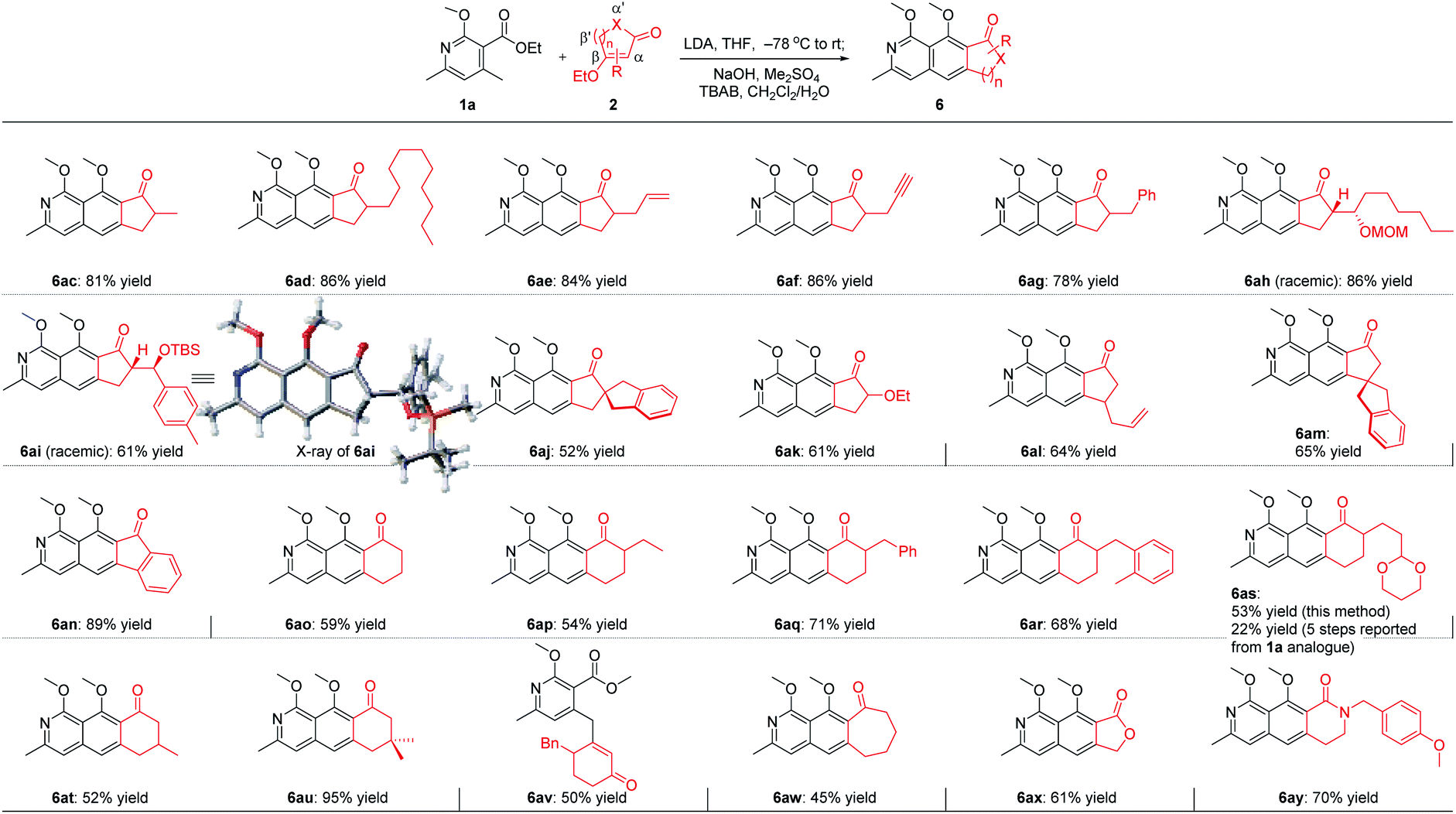 | ||
| Scheme 3 Scope of cycloenone derivatives and more. | ||
Finally, fredericamycin A was selected further as the target molecule to verify the flexibility of our method in the total synthesis of natural products, especially those containing 8-hydroxyisoquinolin-1[2H]-one units.13 Since its first isolation in 1981, fredericamycin A attracted much attention from the synthetic community due to its interesting chemical structure and significant anti-tumor activity.2,14,15 The synthetic route was inspired by the expeditious work from Bach.16a As shown in Scheme 4, we started our synthetic attempts with our developed multifold reaction sequence of pyridine 1a and β-ethoxy enone 2h, delivering the corresponding methyl ether 6a on a gram scale. To the best of our knowledge, this is the first example of isoquinoline synthesis directly starting from a pyridine derivative in a single step. The aromatic ketone 6a was subjected to a Mukaiyama aldol/pinacol rearrangement cascade with cyclobutene 7 to give spiro diketone 8 in 42% yield.7,16 After oxidation with DDQ, the pivotal synthon 9 was obtained in 62% yield.7 It should be noted that the addition of p-TsOH is necessary for this transformation as a sluggish reaction rate was detected in the absence of an acid. Meanwhile, a four-step access of phthalidyl chloride 10 was developed starting from a commercially available benzoic acid derivative.7,17 For the crucial Hauser–Kraus annulation18 between fragments 9 and 10, we found that the coupling product 11 was not stable and thus protected directly as the corresponding methyl ether. After extensive screening of reaction conditions,7 LiOtBu turned out to be the only efficient base for this annulation. Mechanistically, the intermolecular Michael addition of segments 9 and 10 was followed by successive transformations involving Dieckmann condensation of enolate V, extrusion of chloride anions from the diketone VI, and last aromatization of the advanced intermediate VII to afford the hexacyclic diphenol 11 with the full skeleton embedded in fredericamycin A. As far as we know, this is the first example of 3-halophthalide as the Hauser donor instead of the classic sulfonyl- or cyano-containing substrates in Hauser–Kraus annulation, as 3-halophthalide was previously reported not suitable for this annulation.18aIn situ methylation of the newly formed phenol hydroxyls delivered Kita's intermediate 12 in 51% yield in 2 steps. A further 4-step sequence ensured the accomplishment of fredericamycin A.19 The overall synthetic route clearly showcased the power of ingenious introduction of multifold reaction cascades to realize the best performance from the point of step economy.
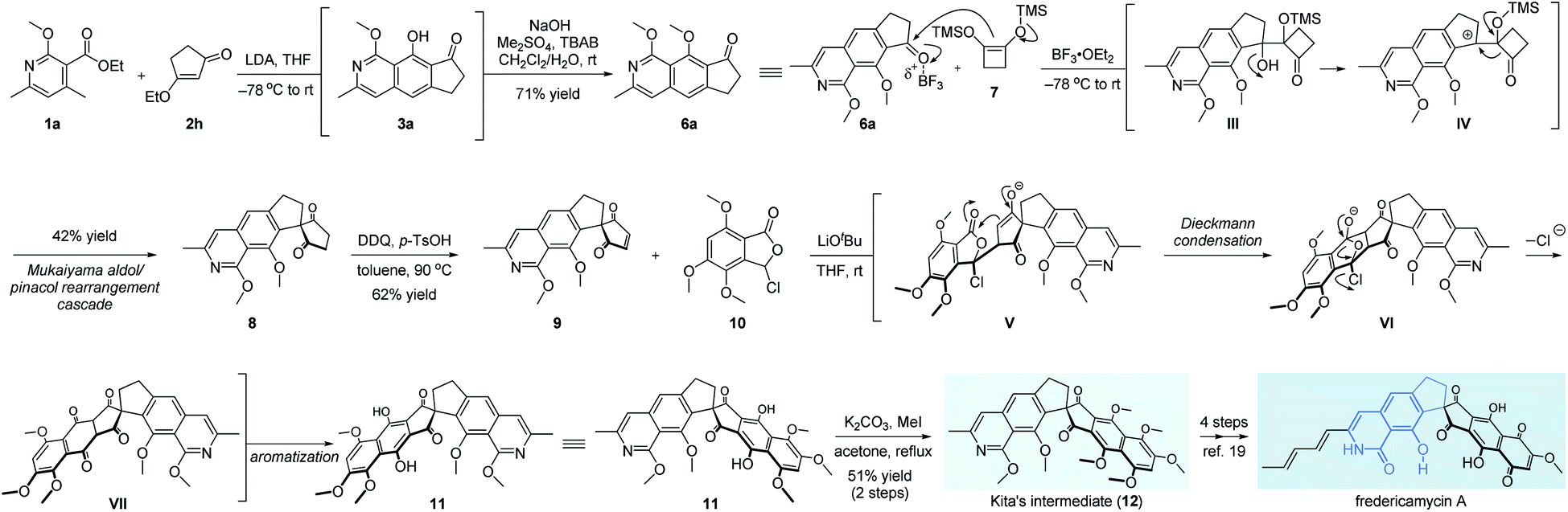 | ||
| Scheme 4 Formal synthesis of fredericamycin A. | ||
Conclusions
In summary, we have developed a concise approach for the rapid assembly of multicyclic isoquinoline scaffolds from pyridines and β-ethoxy α,β-unsaturated carbonyl compounds. In addition to operational simplicity, an intriguing feature of our strategy lies in the facile introduction of substituents and substitution patterns. Specifically, except for a very few cases (such as 6av), substituents can be installed at all possible sites of our tricyclic isoquinoline molecules. This flexibility of rich substituent incorporation can likely enable framework-based construction of a large class of natural products and their analogues. In the present study, we demonstrate that fredericamycin A can be prepared in a short route, involving tricyclic isoquinoline construction, Mukaiyama aldol/pinacol rearrangement cascade, and Hauser–Kraus-type annulation as key steps. We expect our studies to find use in rapid and scalable preparation of complex natural products and their analogues, providing libraries of sophisticated molecules for bioactivity investigations.Author contributions
F.-X. W. conducted most of the experiments; T. Z. and Y. L. initiated the study and chose the natural product target; J.-L. Y., Z. L., T. Z., Y. L., S.-C. R. and W.-X. L. performed some of the studies; Z. J. contributed to designs and discussions; Y. R. C. conceptualized and directed the project. All authors contributed to discussions and manuscript preparation.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We thank Dr Yongxin Li (NTU) for assistance with X-ray structural analysis. We gratefully acknowledge the support from the Instrumental Analysis Centre of Shenzhen University (China). We acknowledge financial support from the Singapore National Research Foundation under its NRF Investigatorship (NRF-NRFI2016-06) and Competitive Research Program (NRF-CRP22-2019-0002); the Ministry of Education, Singapore, under its MOE AcRF Tier 1 Award (RG7/20, RG5/19, and RG1/18), MOE AcRF Tier 2 (MOE2019-T2-2-117), and MOE AcRF Tier 3 Award (MOE2018-T3-1-003); the Agency for Science, Technology and Research (A*STAR) under its A*STAR AME IRG Award (A1783c0008 and A1783c0010); GSK-EDB Trust Fund; Nanyang Research Award Grant, Nanyang Technological University; the National Natural Science Foundation of China (21772029, 21801051, 21807019, 21961006, 22071036, 22061007, 82360589, and 81360589), Frontiers Science Center for Asymmetric Synthesis and Medicinal Molecules, Department of Education, Guizhou Province [Qianjiaohe KY number (2020)004]; the 10 Talent Plan (Shicengci) of Guizhou Province ([2016]5649); the Science and Technology Department of Guizhou Province ([2018]2802 and [2019]1020); the Program of Introducing Talents of Discipline to Universities of China (111 Program, D20023) at Guizhou University.Notes and references
- For selected reviews on the biological activities of isoquinoline alkaloids, see:
(a) M. Iranshahy, R. J. Quinn and M. Iranshahi, RSC Adv., 2014, 4, 15900–15913 RSC
; (b) Z.-X. Qing, J.-L. Huang, X.-Y. Yang, J.-H. Liu, H.-L. Cao, F. Xiang, P. Cheng and J.-G. Zeng, Curr. Med. Chem., 2018, 25, 5088–5114 CrossRef CAS PubMed
- For the biological activities of fredericamycin A, see: D. J. Warnick-Pickle, K. M. Byrne, R. C. Pandey and R. J. White, J. Antibiot., 1981, 34, 1402–1407 CrossRef CAS PubMed
- For the biological activities of ericamycin, see: M. Shimura, S. Kondo, T. Nishio, E. Akita, K. Satoh and T. Hara, J. Antibiot., 1966, 19, 51–55 CAS
- For the selected reviews on isoquinoline synthesis, see:
(a)
Name Reactions in Heterocyclic Chemistry, ed. J. J. Li and E. J. Corey, John Wiley & Sons, Hoboken, NJ, 2005, pp. 376–385 Search PubMed
- For the first discovery of Bischler–Napieralski isoquinoline synthesis, see:
(a) A. Bischler and B. Napieralski, Ber. Dtsch. Chem. Ges., 1893, 26, 1903–1908 CrossRef
- For limited examples of isoquinoline synthesis from pyridine via multiple steps, see:
(a) D. L. J. Clive and J. Sedgeworth, J. Heterocycl. Chem., 1987, 24, 509–511 CrossRef CAS
- For details, see the ESI.†.
- Y. Tamura, A. Wada, M. Sasho and Y. Kita, Chem. Pharm. Bull., 1983, 31, 2691–2697 CrossRef CAS
- For a review on Staunton–Weinreb annulation, see: C. D. Donner, Tetrahedron, 2013, 69, 3747–3773 CrossRef CAS
- For one example of annulation involving pyridine and cyclohexenone without substitution at the β site to form pyridine-containing products, see: M. G. Charest, C. D. Lerner, J. D. Brubaker, D. R. Siegel and A. G. Myers, Science, 2005, 308, 395–398 CrossRef CAS PubMed
- This is interesting as the phenol adduct 3a itself is stable enough when exposed to air, though a sharp decrease in the yield was observed after purification through column chromatography. If this is a general phenomenon for some (if not all) phenol adducts, low yields previously reported in the reactions involving β-alkoxy cycloenones might be easily explained as all the phenol products in ref. 9 were isolated without in situ protection.
- The absolute configuration of 6m and the relative configuration of 6ai were determined by X-ray crystallographic analysis. CCDC 2061702 (6m) and CCDC 2061701 (6ai)†..
- For the selected examples on the total synthesis of 8-hydroxyisoquinolin-1[2H]-one-containing natural products in the past 10 years, see:
(a) D. L. Sloman, J. W. Bacon and J. A. Porco, J. Am. Chem. Soc., 2011, 133, 9952–9955 CrossRef CAS PubMed
- For the isolation and characterization of fredericamycin A, see:
(a) R. C. Pandey, M. W. Toussaint, R. M. Stroshane, C. C. Kalita, A. A. Aszalos, A. L. Garretson, T. T. Wei, K. M. Byrne, R. F. Geoghegan and R. J. White, J. Antibiot., 1981, 34, 1389–1401 CrossRef CAS PubMed
- For synthetic reviews on fredericamycin A, see:
(a) D. L. J. Clive, Y. Tao, A. Khodabocus, Y.-J. Wu, A. G. Angoh, S. M. Bennett, C. N. Boddy, L. Bordeleau, M. Cantin, D. R. Cheshire, D. Kellner, G. Kleiner, D. S. Middleton, C. J. Nichols, S. R. Richardson, L. Set and P. G. Vernon, Stud. Nat. Prod. Chem., 1995, 16, 27–74 CAS
- For similar reactions on the Mukaiyama aldol/pinacol rearrangement cascade, see:
(a) J. A. Wendt, P. J. Gauvreau and R. D. Bach, J. Am. Chem. Soc., 1994, 116, 9921–9926 CrossRef CAS
- J. C. Evans, R. C. Klix and R. D. Bach, J. Org. Chem., 1988, 53, 5519–5527 CrossRef CAS
- For a review on Hauser annulation, see:
(a) D. Mal and P. Pahari, Chem. Rev., 2007, 107, 1892–1918 CrossRef CAS PubMed
- In Kita's two reports on the synthesis of fredericamycin A, the overall synthetic routes contain 27 and 25 steps, respectively. For details, see:
(a) Y. Kita, K. Higuchi, Y. Yoshida, K. Iio, S. Kitagaki, S. Akai and H. Fujioka, Angew. Chem., Int. Ed., 1999, 38, 683–686 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2061702 and 2061701. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02442f |
| This journal is © The Royal Society of Chemistry 2021 |