
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactions of diborenes with terminal alkynes: mechanisms of ligand-controlled anti-selective hydroalkynylation, cycloaddition and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e002.gif) C triple bond scission†
C triple bond scission†
Lukas Englert‡
ab,
Uwe Schmidt‡ab,
Michael Dömling‡ab,
Max Passargusab,
Tom E. Stennettab,
Alexander Hermannab,
Merle Arrowsmith ab,
Marcel Härterichab,
Jonas Müssigab,
Alexandra Phillippsab,
Dominic Prieschlab,
Anna Rempelab,
Felix Rohmab,
Krzysztof Radackiab,
Fabian Schorrab,
Torsten Thiessab,
J. Oscar C. Jiménez-Halla*c and
Holger Braunschweig*ab
ab,
Marcel Härterichab,
Jonas Müssigab,
Alexandra Phillippsab,
Dominic Prieschlab,
Anna Rempelab,
Felix Rohmab,
Krzysztof Radackiab,
Fabian Schorrab,
Torsten Thiessab,
J. Oscar C. Jiménez-Halla*c and
Holger Braunschweig*ab
aInstitute for Inorganic Chemistry, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: h.braunschweig@uni-wuerzburg.de
bInstitute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
cDepartamento de Química, Universidad de Guanajuato, Noria Alta S/N, 36050 Guanajuato, Mexico. E-mail: jjimenez@ugto.mx
First published on 21st June 2021
Abstract
The reactions of terminal acetylenes with doubly Lewis base-stabilised diborenes resulted in different outcomes depending on the nature of the ligands at boron and the conformation of the diborene (cyclic versus acyclic). N-heterocyclic carbene (NHC)-stabilised diborenes tended to undergo anti-selective hydroalkynylation at room temperature, whereas [2 + 2] cycloaddition was observed at higher temperatures, invariably followed by a C–N bond activation at one NHC ligand, leading to the ring-expansion of the initially formed BCBC ring and formation of novel boron-containing heterocycles. For phosphine-stabilised diborenes only [2 + 2] cycloaddition was observed, followed by a rearrangement of the resulting 1,2-dihydro-1,2-diborete to the corresponding 1,3-isomer, which amounts to complete scission of both the B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) B double and C
B double and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C triple bonds of the reactants. The elusive 1,2-isomer was finally trapped by using a cyclic phosphine-stabilised diborene, which prevented rearrangement to the 1,3-isomer. Extensive density functional theory (DFT) calculations provide a rationale for the selectivity observed.
C triple bonds of the reactants. The elusive 1,2-isomer was finally trapped by using a cyclic phosphine-stabilised diborene, which prevented rearrangement to the 1,3-isomer. Extensive density functional theory (DFT) calculations provide a rationale for the selectivity observed.
Introduction
Transition-metal-catalysed reactions of terminal alkynes with olefins provide an atom-efficient way of forming new C–C bonds. Depending on the choice of catalyst and the nature of the terminal alkyne and alkene substrates, several types of reactivity can be targeted, including the hydroalkynylation of the CC double bond,1–8 [2 + 2] cycloaddition reactions to yield cyclobutenes,9–12 or ene-yne metathesis to form conjugated dienes.13–20 While the reactions of terminal alkynes with alkenes require a catalyst to overcome high activation barriers, the analogous reactions with heavier group 14 alkene species proceed spontaneously owing to the much higher energy of their HOMO relative to that of alkenes. Therefore, disilenes, digermenes and distannenes undergo spontaneous [2 + 2] cycloadditions with terminal alkynes to form the corresponding 1,2-disila-,21–26 1,2-digerma-27–30 and 1,2-distannacyclobut-3-enes.31,32 Only in the case of the acenaphthene-bridged distannene reported by Wesemann and co-workers was the cycloaddition with trimethylsilylacetylene found to be reversible.31 Experimental mechanistic studies on these cycloaddition reactions suggested a stepwise mechanism via a biradical or zwitterionic intermediate rather than a concerted process, which would be symmetry-forbidden.23,25–27,33
Several spontaneous reactions with alkynes have been reported with group 13 alkene analogues. Similarly to the heavier group 14 congeners, the addition of alkynes to Tokitoh's dialumene-benzene adduct led to the formation of 1,2-dialuminacyclobut-3-enes (Scheme 1a),34 while Power's diaryldigallene underwent twofold cycloaddition with two equivalents of phenylacetylene to yield a 1,4-digallacyclohexa-2,5-diene (Scheme 1b).35 More recently, Inoue and co-workers reported that a doubly N-heterocyclic carbene (NHC)-stabilised 1,2-disilyldialumene undergoes both [2 + 2] cycloaddition and syn-selective hydroalkynylation with phenylacetylene (Scheme 1c).36 We have shown that under photolytic conditions the doubly phosphine-stabilised diborene I undergoes a cycloaddition/rearrangement reaction with 2-butyne to yield the phosphine-stabilised homoaromatic 1,3-dihydro-1,3-diborete II, presumably resulting from rearrangement of a 1,2-dihydro-1,2-diborete intermediate (Scheme 1d).37 In a similar manner the addition of propyne to our 2,3-diborabutatriene III also yielded an aromatic 1,3-diborete, IV, stabilised by two alkyl radical groups derived from cyclic (alkyl)(amino)carbene ligands (Scheme 1e), while the addition of acetylene led to the formation of the first neutral 1,4-diborabenzene, V (Scheme 1f).37
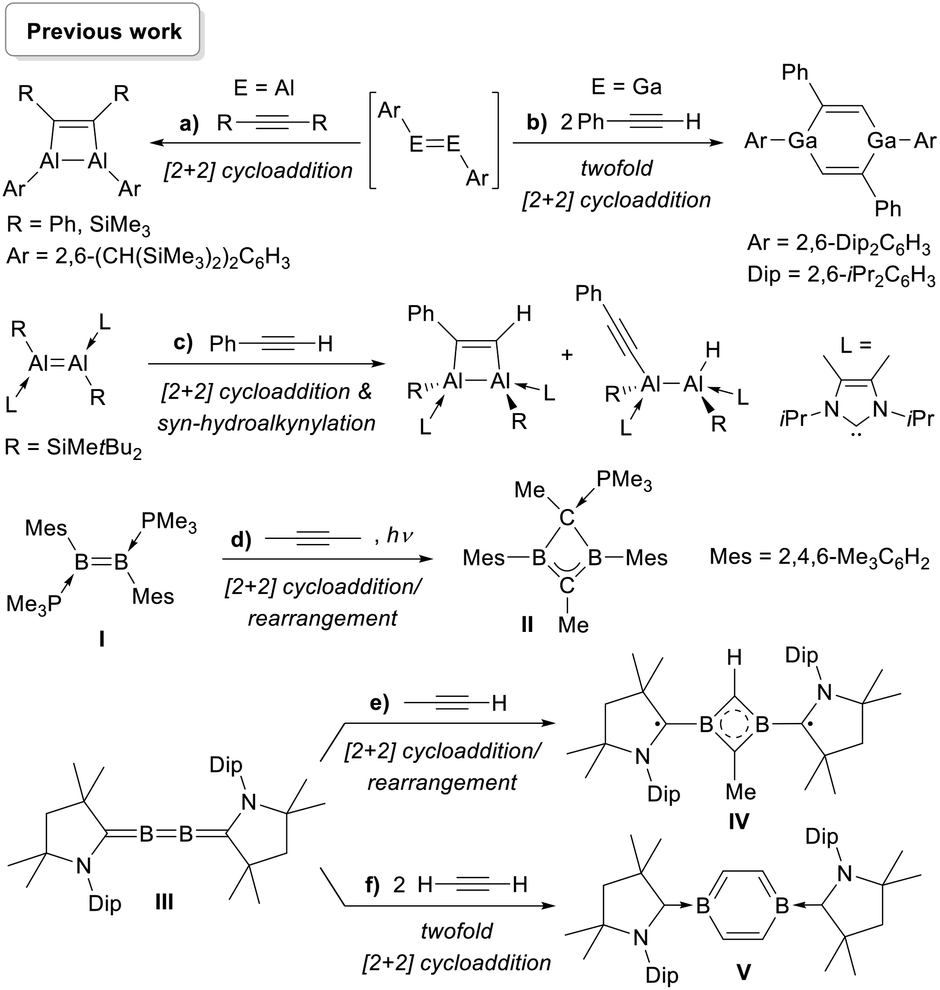 | ||
| Scheme 1 Spontaneous reactions of group 13 alkene analogues with alkynes. | ||
In this work we show that, depending on the nature of the ligand environment at boron and the reaction conditions, the addition of terminal alkynes to diborenes may result in unprecedented hydroalkynylation of the BB double bond or [2 + 2] cycloaddition (Scheme 2), with or without rearrangement of the resulting 1,2-dihydro-1,2-diborete. Computational mechanistic analyses provide a rationale for the experimentally observed selectivity.
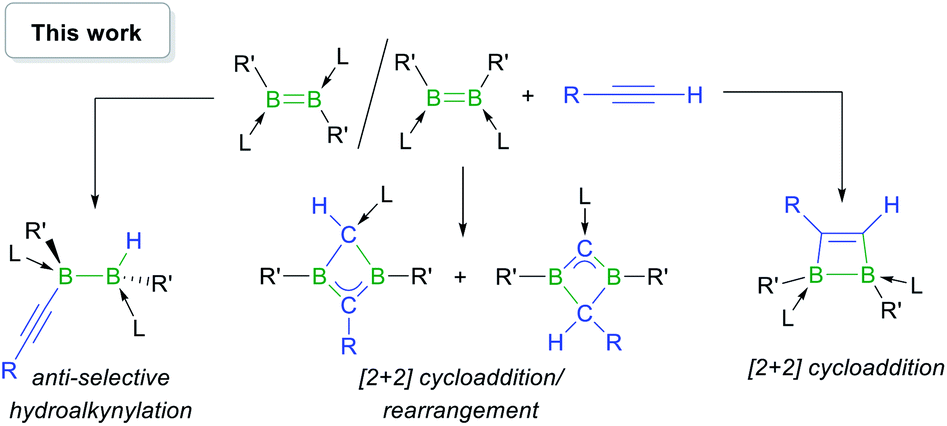 | ||
| Scheme 2 Reactivity of base-stabilised diborenes with terminal acetylenes. L = phosphine or NHC; R,R′ = anionic substituents. | ||
Results and discussion
Reactivity of terminal alkynes with NHC-stabilised diborenes
The reaction of the doubly NHC-stabilised divinyldiborene 1 (ref. 38) with acetylene in benzene at room temperature yielded a colourless solution with a single, very broad, 11B NMR resonance around −19 ppm (Scheme 3). While multiple attempts to isolate this compound failed because of its oily consistency, the reaction of 1 with ferrocenylacetylene (FcCCH) resulted in the formation of a compound, 2-Fc, with a similar 11B NMR resonance at −18.6 ppm, in ca. 98% selectivity as determined by 11B NMR-spectroscopic analysis of the reaction mixture (Scheme 3).§2-Fc was identified by X-ray crystallographic analysis (Fig. 1) as the hydroalkynylation product of the BB double bond, resulting in a doubly NHC-stabilised 1-alkynyl-2-hydrodiborane.
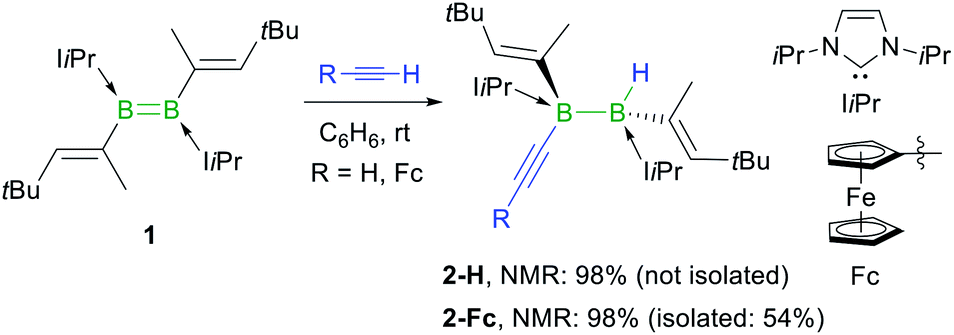 | ||
| Scheme 3 Anti-selective hydroalkynylation of NHC-stabilised diborene 1. Relative stereochemistry shown only. NMR yields determined by 11B NMR spectroscopy. | ||
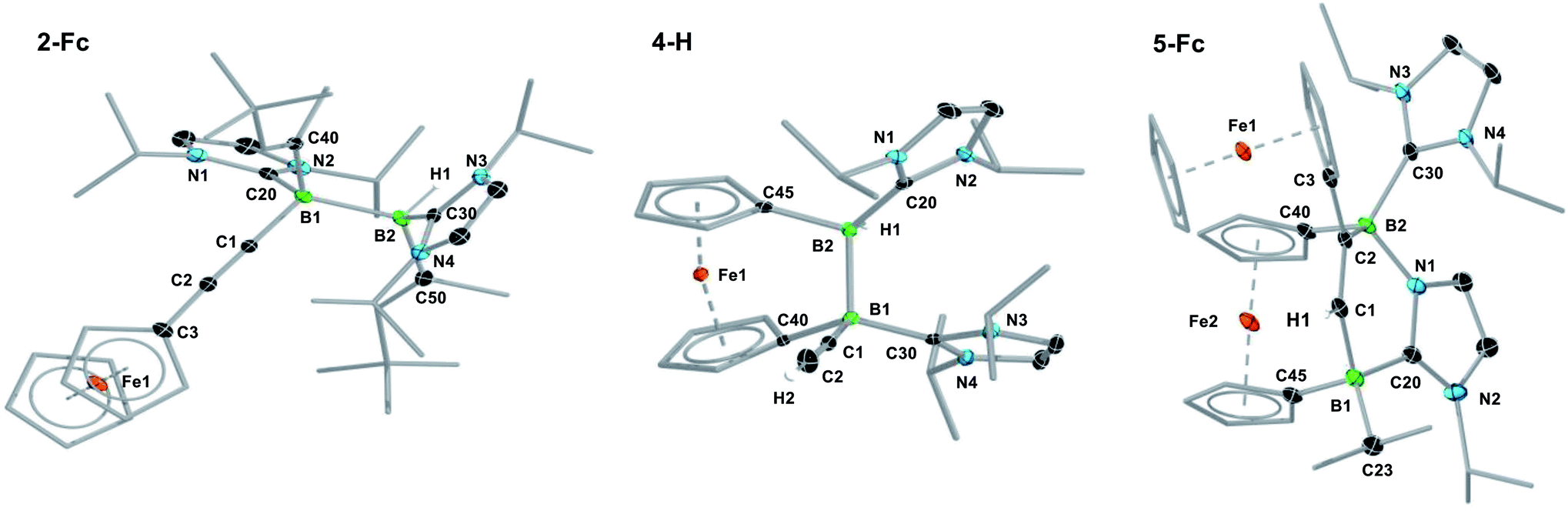 | ||
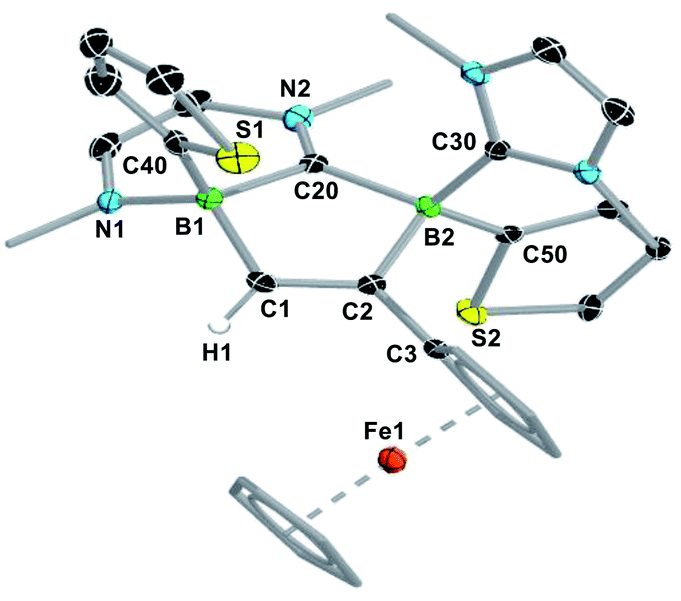
| Fig. 1 Crystallographically-derived molecular structures of (from left to right) 2-Fc, 4-H and 5-Fc. Atomic displacement ellipsoids set at 50% probability. Ellipsoids of ligand periphery and hydrogen atoms omitted for clarity, except for alkene-, alkyne- and boron-bound hydrogen atoms. Selected bond lengths (Å) and angles (°) for 2-Fc: B1–B2 1.826(3), B1–C1 1.594(3), B2–H1 1.144(19), B1–C20 1.663(3), B2–C30 1.621(3), C1–C2 1.206(2), torsion angles (C1–B1–B2–H1) 166(1), (C20–B1–B2–C30) 158.63(15), (C40–B1–B2–C50) 166.52(15); for 4-H: B1–B2 1.835(2), B1–C1 1.592(2), B2–H1 1.147(17), B1–C20 1.612(2), B2–C30 1.629(2), C1–C2 1.201(2), torsion (C1–B1–B2–H1) 179.9(9), (C20–B1–B2–C30) −55.00(17), (C40–B1–B2–C50) 45.41(14), Cp tilt angle (bridging ferrocenediyl) 16.3; for 5-Fc: B1–C1 1.620(4), C1–C2 1.350(3), C2–B2 1.618(3), B2–N1 1.583(3), N1–C20 1.356(3), C20–B1 1.642(4), C20–N2 1.361(3), B2–C30 1.655(3), Cp tilt angle (bridging ferrocenediyl) 2.5. | ||
The fact that only one resonance is observed in the 11B NMR spectrum despite the unsymmetrical substitution pattern of the diborane suggests that the resonances of the two boron nuclei overlap, i.e. that the hydride and the acetylide ligand have a very similar electronic influence on the boron nuclei. The 1H NMR spectrum shows the formation of a single diastereomer. The presence of the boron-bound hydride was confirmed by a BH singlet in the 1H{11B} NMR spectrum at 2.45 ppm. The hydroacetylenation of B–B multiple bonds offers a new atom-efficient route to alkynylboranes, which are traditionally synthesised by salt metathesis of halo- or alkoxyboranes with lithium or Grignard acetylides,39–41 or by the catalytic dehydrocoupling of terminal alkynes with hydroboranes.42,43 The solid-state structure of 2-Fc shows that the addition of the C–H bond occurred with anti-selectivity, similarly to the hydrogenation,44 transfer hydrogenation45 and hydroboration of diborenes.46,47 This is rather unexpected since the 1,2-addition of the C–H bond to the BB double bond should occur in a syn fashion, similar to the hydroalkynylation of Inoue's dialumene with phenylacetylene (Scheme 1c).36 The mechanistic analyses presented later in this paper (vide infra) shed some light on this unexpected anti-selectivity.
In a similar manner, the room-temperature reaction of acetylene, phenylacetylene and ferrocenylacetylene with the dibora[2]ferrocenophane 3 (ref. 48) resulted in hydroalkynylation of the BB double bond, yielding the ferrocenediyl-bridged and doubly NHC-stabilised 1-alkynyl-2-hydrodiboranes 4-R (R = H, Ph), with ≥85% selectivity according to 11B NMR spectra of the reaction mixtures (Scheme 4a).§ Like 2-Fc, compounds 4-R show only one 11B NMR resonance at ca. −18 ppm despite their unsymmetrical nature. The BH resonance was detected in the 1H{11B} NMR spectra as a 1H singlet at ca. 3.2 ppm, while the terminal acetylenic proton of 4-H appeared as a 1H singlet at 2.70 ppm. Yellow single crystals of 4-H suitable for X-ray diffraction analysis confirmed the 1,2-anti-addition of the C–H bond, as already observed in 2-Fc (Fig. 1). The B–B bond length of 4-H (1.835(2) Å) is comparable to that of 2-Fc (1.826(3) Å) despite the additional strain imposed by the bridging ferrocenediyl ligand.
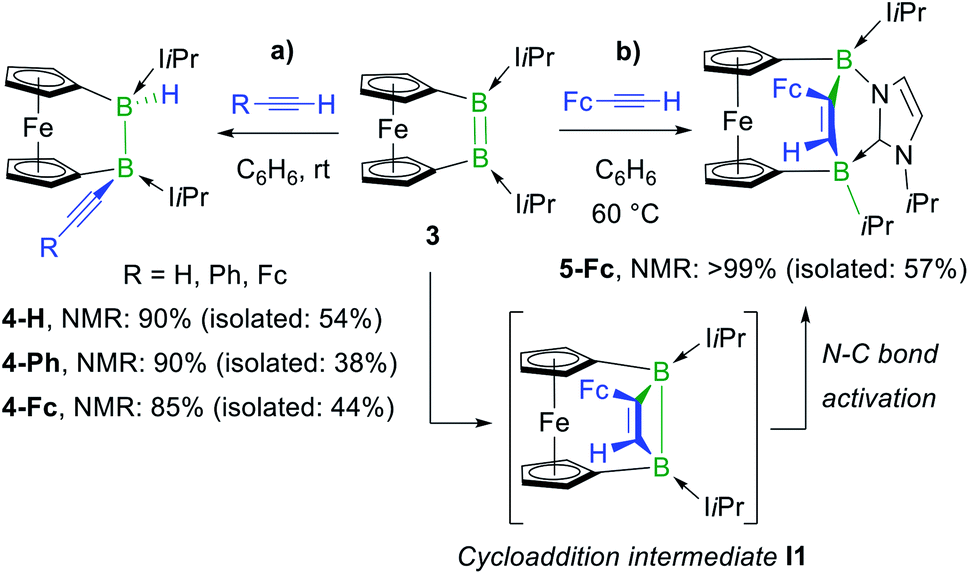 | ||
| Scheme 4 Divergent reactivity of dibora[2]ferrocenophane 3 with terminal acetylenes and postulated cycloaddition intermediate I1 in square brackets. Relative stereochemistry shown only. | ||
In contrast, the reaction of 3 with ferrocenylacetylene at 60 °C afforded the 1,2-diboraalkene 5-Fc as the sole product (Scheme 4b),§ which formally results from the [2 + 2] cycloaddition of the acetylene to the diborene, yielding intermediate I1, followed by the insertion of the B–B bond into an exocyclic C–N bond at one NHC ligand. A similar exocyclic C–N bond activation at an NHC ligand has been observed in the reaction of a doubly NHC-stabilised diiododisilene with a thiolate.49 The 11B NMR spectrum of 5-Fc shows two broad resonances at −7.7 and −14.7 ppm, the former corresponding to the iPr-bearing B1 and the latter to the N-bound B2. The proton of the bridging alkene moiety appears in the 1H NMR spectrum as a deshielded 1H singlet at 8.02 ppm. The solid-state structure of 5-Fc (Fig. 1) shows the formation of a six-membered 2,5-dihydro-1,2,5-azadiborinine ring, which adopts a boat conformation with the boron atoms at the apices, fused at C20–N1 with the imidazol-2-ylidene ring of the former NHC ligand. The C1–C2 bond length of 1.350(4) Å denotes a typical CC double bond, while the N2–C20, C20–N1 and N1–B2 bond lengths of 1.361(3), 1.356(4) and 1.583(4) Å, respectively, are all within the range of partial double bonds, indicating some degree of π delocalisation over the N2–C20–N1–B2 moiety. The fact that the cycloaddition intermediate I1 cannot be isolated and undergoes rapid ring-expansion by activation of the N–CiPr bond is likely due to the double ring strain in I1, imposed by both the four-membered 1,2-dihydro-1,2-diborete ring and the ferrocenediyl bridge. The temperature-dependent selectivity of the reaction of 3 with ferrocenylacetylene indicates that 4-Fc is the kinetic product while 5-Fc is the thermodynamic product. Attempts to obtain the analogous compounds 5-H and 5-Ph by heating the reaction mixtures of 3 with acetylene and phenylacetylene at 60 °C exclusively yielded 4-H and 4-Ph, respectively. These appear, therefore, to be both the thermodynamic and kinetic products. However, work-up of one room-temperature reaction of 3 with phenylacetylene yielded, besides 4-Ph, a few crystals of 5-Ph, the solid-state structure of which is analogous to that of 5-Fc (see Fig. S53†), thus indicating that the activation energy of the cycloaddition reaction must actually be relatively close to that of the hydroalkynylation reaction in this case.
The reactions of the dithienyl- and difuryldiborenes 6Tn and 6Fu,47 respectively, with phenylacetylene and tert-butylacetylene proved extremely unselective regardless of the reaction conditions applied, yielding mixtures of up to six different products with 11B NMR shifts spanning the −25 to +3 ppm range. In contrast, the analogous reactions with ferrocenylacetylene at room temperature in benzene proved relatively selective, yielding the orange-coloured [1,3]diborolo[3,2-b][1,4,2]diazaborinine derivatives 7Tn-Fc and 7Fu-Fc as the major products with ca. 65% and 80% selectivity, respectively, as determined by 11B NMR spectroscopy (Scheme 5), alongside several minor products including the hydroalkynylation products (δ11B ≈ −20 ppm).§7Tn-Fc and 7Fu-Fc can be viewed as resulting from the cycloaddition of FcCCH to the diborene, yielding the intermediate 1,2-dihydro-1,2-diborete I2 (Scheme 5), followed by ring expansion of one NHC ligand through the addition of the B–B bond of I2 to the carbene carbon atom and insertion of one boron atom into the Ccarbene–N bond. This type of NHC ring expansion has previously been observed for a number of NHC-stabilised diboranes,50–52 and is probably favoured in this case by the release of ring strain upon expansion of the B2C2 to the B2C3 ring. The 11B NMR resonances of 7Tn-Fc appear at −4.6 and −11.5 ppm, those of 7Fu-Fc at −6.0 and −12.7 ppm. The 1H NMR BCH proton of the 1,2-diboraalkene moiety of 7Tn-Fc appears as a broad resonance at 7.23 ppm. The 1H NMR spectrum of crude 7Fu-Fc shows two distinct BCH resonances at 7.89 and 7.78 ppm in a 82![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :18 ratio. Since the cycloaddition step must take place in a syn fashion, only the (S,R)/(R,S) diastereomer of 7Fu-Fc should be formed. The presence of two distinct BCH resonances therefore suggests the formation of the two regioisomers A and B of 7Fu-Fc (Scheme 5).
:18 ratio. Since the cycloaddition step must take place in a syn fashion, only the (S,R)/(R,S) diastereomer of 7Fu-Fc should be formed. The presence of two distinct BCH resonances therefore suggests the formation of the two regioisomers A and B of 7Fu-Fc (Scheme 5).
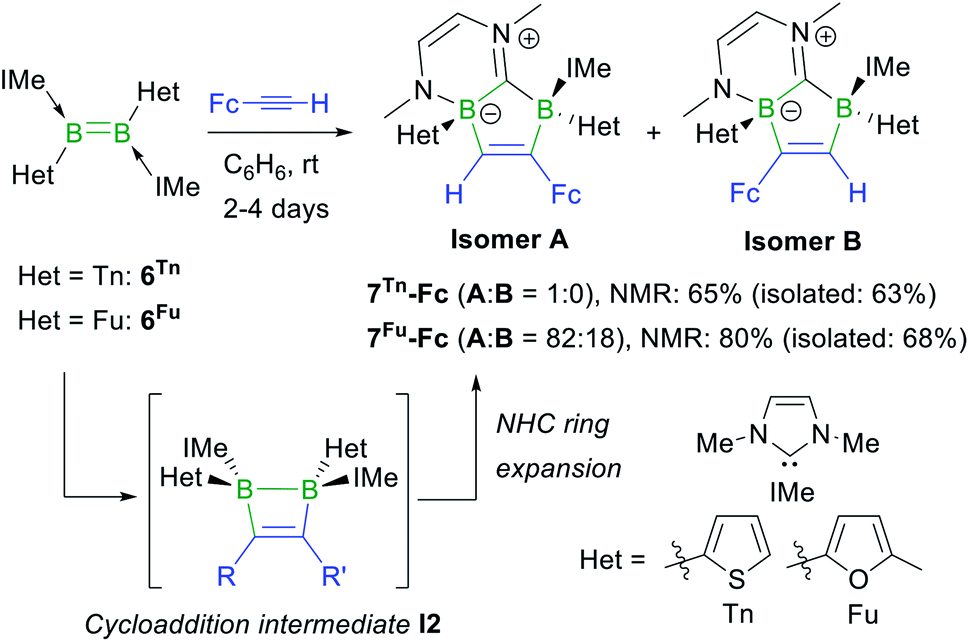 | ||
| Scheme 5 Reaction of ferrocenylacetylene with 6Tn and 6Fu, and postulated intermediate I2 in square brackets. Relative stereochemistry shown only. | ||
An X-ray crystallographic analysis of the regioisomer A of 7Tn-Fc (Fig. 3) confirms the ring expansion of the original 1,2-dihydro-1,2-diborete and of one NHC ligand, leading to the fused C5B2N2 heterocycle. As expected from the suggested reaction pathway viaI2, the structure is that of the (S,R)/(R,S) diastereomer. The C1–C2 bond length of 1.341(3) Å is that of a typical CC double bond. Furthermore, the short C20–N2 bond length of 1.305(3) Å is indicative of a CN double bond, making this compound a zwitterion.
Reactivity of terminal alkynes with phosphine-stabilised diborenes
Having shown that NHC-stabilised diborenes undergo both hydroalkynylation and [2 + 2] cycloaddition with terminal acetylenes, depending on the substitution pattern of the diborene and/or kinetic versus thermodynamic reaction control, our attention turned to phosphine-stabilised diborenes. Previous work by our group had shown that diborene I reacts with 2-butyne under photolytic conditions to generate the homoaromatic 1,3-dihydro-1,3-diborete II (Scheme 1d).37 In a similar manner, diborene I reacted with 1-hexyne, trimethylsilyl- (=TMS), p-tolyl- (=Tol) and ferrocenylacetylene at 80 °C over a period of several days to yield the 1,3-dihydro-1,3-diboretes 8-Bu, 8-TMS, 8-Tol and 8-Fc, respectively, with 85–98% selectivity as determined by 11B NMR spectroscopy (Scheme 6).§ This complete scission of BB multiple and CC triple bonds is also observed in the reaction of a NHC-stabilised diboryne with alkynes, which leads to the formation of highly reactive antiaromatic 1,3-diboretes.53 As described previously, the formation of 8-R is likely to proceed via a 1,2-dihydro-1,2-diborete I3, which spontaneously rearranges to the thermodynamically more stable 1,3 isomer,54 with loss of one PMe3. The existence of I3 is supported by the fortuitous isolation of a few crystals of 9-Tol (Scheme 6 and Fig. 2), which results from the intramolecular C–H activation of a mesityl-ortho-methyl group at the B–B bond of I3. In all cases the major or sole product obtained was the analogue of II, tautomer C, in which the remaining PMe3 ligand is bound to the positively charged, former terminal carbon of the acetylene. The 11B NMR shifts of 8-R are dependent upon the nature of the substituent R, increasing from 25.6 to 38.8 ppm, in the order of R = Me < nBu < p-tolyl < Fc < SiMe3, i.e. with the steric demands of R. Concomitantly, the 31P NMR shifts decrease from 23.2 to 15.6 ppm following the same order. For 8-Bu and 8-Tol a second minor product was observed in solution (<10%), in which the 11B NMR resonance is shifted significantly downfield to 54.7 and 51.4 ppm, respectively, compared to C, while the 31P NMR resonance is shifted more than 20 ppm upfield to −3.6 and −2.7 ppm, respectively. These compounds were identified by NMR spectroscopy and X-ray crystallographly as tautomer D, in which the proton at the phosphine-bound carbon atom has migrated to the R-bound carbon atom of the BCBC ring.
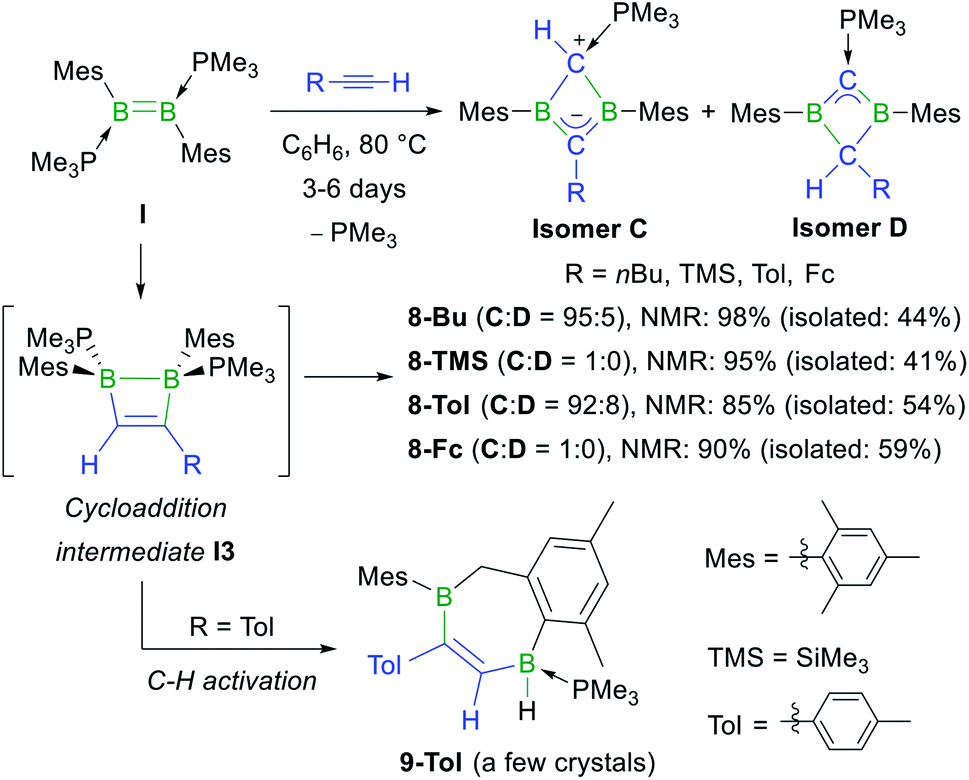 | ||
| Scheme 6 Reaction of phosphine-stabilised diborene I with terminal acetylenes, postulated intermediate I3 in square brackets, and C–H activation product 9-Tol deriving from I3. Relative stereochemistry shown only. | ||
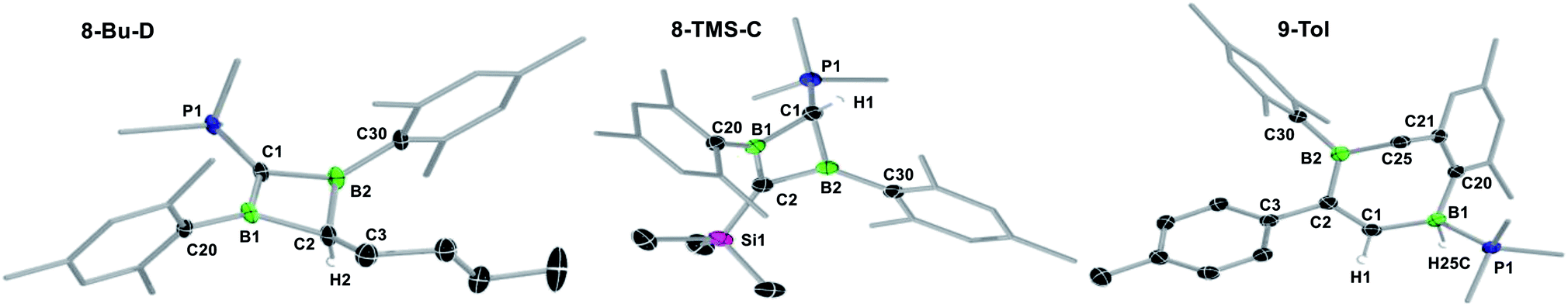 | ||
| Fig. 2 Crystallographically-derived molecular structures of (from left to right) 8-Bu-D, 8-TMS-C and 9-Tol. Atomic displacement ellipsoids set at 50% probability. Ellipsoids of ligand periphery and hydrogen atoms omitted for clarity, except for C1/C2- and boron-bound hydrogen atoms. Selected bond lengths (Å) and angles (°) for 8-Bu-D: B1–C1 1.501(3), C1–B2 1.509(3), B2–C2 1.615(3), C2–B1 1.613(3), B1⋯B2 1.992(3), C1–P1 1.698(2), B1–C1–B2 82.88(16), C1–B2–C2 96.40(17), B1–C2–B2 76.20(15), C2–B1–C1 96.78(16), Σ(∠B1) 359.78(18), Σ(∠B2) 359.78(18), Σ(∠C1) 355.10(16), torsion angles (C1–B1–C20–C21) 95.8(3), (C1–B2–C30–C31) −91.6(3); for 8-TMS-C: B1–C1 1.6580(19), C1–B2 1.619(2), B2–C2 1.492(2), C2–B1 1.485(2), B1⋯B2 1.869(2), C1–P1 1.7455(14), B1–C1–B2 69.52(9), C1–B2–C2 99.21(11), B1–C2–B2 77.76(11), C2–B1–C1 100.67(11), Σ(∠B1) 359.36(15), Σ(∠B2) 358.22(12), Σ(∠C2) 351.35(11), torsion angles (C1–B1–C20–C21) −133.95(14), (C1–B2–C30–C31) −108.61(16); for 9-Tol: B1–C1 1.603(2), C1–C2 1.356(2), C2–B2 1.558(2), B1–H1 1.159(17), B1–P1 1.9510(16). | ||
 | ||
| Fig. 3 Crystallographically-derived molecular structure of 7Tn-Fc. Atomic displacement ellipsoids set at 50% probability. Ellipsoids of ligand periphery and hydrogen atoms omitted for clarity, except for the diboraalkene-bound proton. Selected bond lengths (Å): B1–C1 1.603(3), C1–C2 1.341(3), C2–B2 1.664(3), B2–C20 1.644(3), C20–B1 1.631(3), B1–N1 1.559(3), C20–N2 1.305(3). | ||
X-ray crystallographic analysis of 8-R-C (R = TMS, Tol, Fc) and 8-Bu-D (Fig. 2 and S54 and S55 in the ESI†) show a similar butterfly conformation to other structurally related 1,3-diboretes,37,55–58 with a puckering angle between the two CB2 planes of around 30°. The sterics of the substituent R influence the distortion of the BCBC heterocycle and the degree of rotation of the mesityl ligands, both aimed at minimising the steric repulsion between the mesityl and R groups. Thus the B⋯B distance decreases from 1.992(3) Å in the least sterically encumbered derivative 8-Bu-D to 1.869(2) Å in the most sterically encumbered derivative 8-TMS-C. Concomitantly, the absolute values of the torsion angles (C1–B1–C20–C21) and (C1–B2–C30–C31) increase from 95.8(3) and 91.6(3)°, respectively, in 8-Bu-D to 133.95(14) and 108.61(16)°, respectively, in 8-TMS-C. The B–C bonds to the sp3-hybridised carbon atom (1.613(3)–1.658(2) Å) are within the upper range of B–C single bonds, whereas the B–C bonds to the sp3-hybridised carbon atom (1.483(2)–1.509(3) Å) are within the range of partial double bonds, typical of a 2π-homoaromatic structure. For 8-Fc-C, the π electron density seems to be delocalised over the entire CpCB2 moiety as shown by the near-coplanar arrangement of the Cp ring and CB2 unit, and the short C–CCp partial double bond (1.455(3) Å). It is noteworthy that the P1–C1 bond 8-Bu-D (1.698(2) Å) is actually within the range of PC double bonds (Ph3PC(CH3)2: 1.696(6) Å).59
In the hope of isolating the elusive 1,2-dihydro-1,2-diborete cycloaddition product we finally examined the reactivity of the cyclic diborene 10 (ref. 60) with terminal acetylenes. Indeed, the bridging benzylphosphine ligand of 10 should prevent rearrangement of the 1,2-diborete to the 1,3-isomer and the absence of NHC ligands or acidic protons should prevent further ligand activation reactions. As expected, diborene 10 underwent a [2 + 2] cycloaddition with trimethylsilylacetylene, resulting in the formation of the polycyclic 1,2-dihydro-1,2-diborete 11-TMS with ca. 98% selectivity (Scheme 7).§ The 11B NMR spectrum of 11-TMS shows two broad resonances very close to each other at −16.0 and −16.5 ppm, while the 31P NMR spectrum displays two broad doublets at 8.7 and −12.9 ppm for the PhCH2PCy2 and PMe3 phosphorus nuclei, respectively (3JP–P = 107 Hz). The proton of the bridging alkene moiety appears as a 1H NMR doublet at 8.16 ppm (3JH–P = 6.0 Hz). The analogous reaction with ferrocenylacetylene at room temperature proved far less selective, 11B and 31P NMR spectra evidencing the formation of 11-Fc (δ11B = −17.5, −18.0 ppm) with ca. 45% selectivity, alongside several unidentified species. 11-TMS is only the third structurally characterised 1,2-dihydro-1,2-diborete. Like Kaufman's 1,2-diamino-1,2-dihydro-1,2-benzodiborete (B–B 1.727(2) Å), obtained from the reductive cyclisation of the o-(BClNiPr2)2C6H4 precursor,61 the B2C2 ring of 11-TMS is slightly twisted (torsion angles (C1,B1,B2,C2) 9.9(2); (B1,C1,C2,B2) 13.3(3)°) due to the strain of the fused ring system. In contrast, Siebert's 1,2-diamino-1,2-dihydro-1,2-diborete (B–B 1.75 Å) is fully planar.62 Furthermore, the B–B bond in 11-TMS is significantly elongated (1.832(5) Å) due to the additional coordination of the two phosphine ligands (Fig. 4).
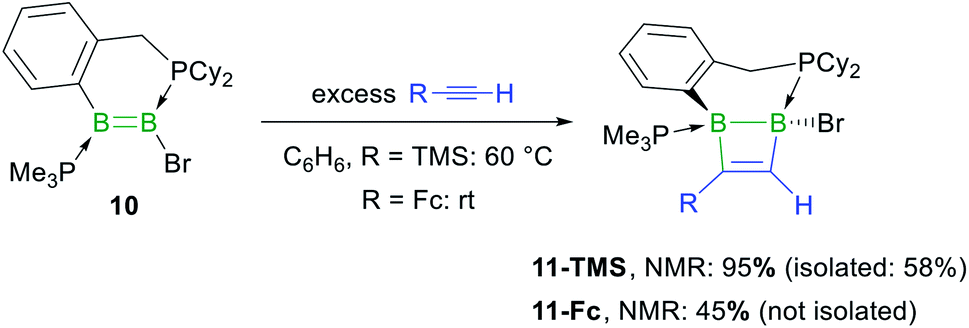 | ||
| Scheme 7 Cycloaddition of terminal acetylenes to the phosphine-stabilised cyclic diborene 10. Relative stereochemistry shown only. | ||
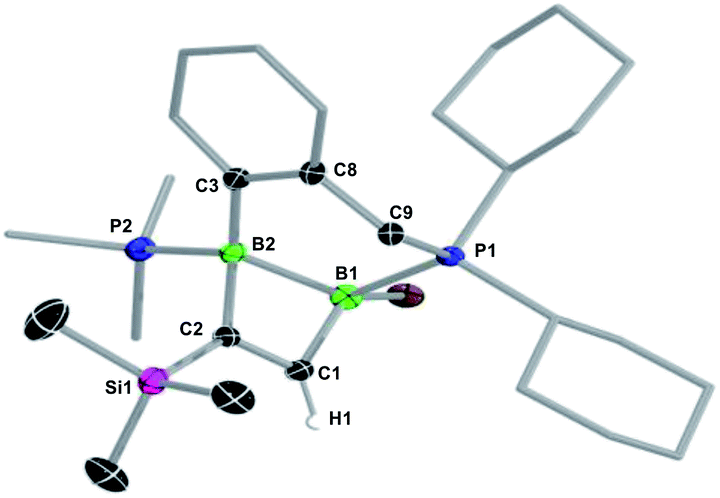 | ||
| Fig. 4 Crystallographically-derived molecular structure of 11-TMS. Atomic displacement ellipsoids set at 50% probability. Ellipsoids of ligand periphery and hydrogen atoms omitted for clarity. Selected bond lengths (Å) and angles (°): B1–B2 1.832(5), B2–C2 1.629(5), C2–C1 1.355(5), C1–B1 1.566(5), B1–P1 1.930(4), B2–P2 1.922(4), C1–B1–B2 83.4(2), B1–B2–C2 78.5(2), B2–C2–C1 98.5(3), C2–C1–B1 97.2(3), torsion angles (C1,B1,B2,C2) 9.9(2), (B1,C1,C2,B2) 13.3(3). | ||
Computational mechanistic analyses
In order to gain more insight into the selectivity of these reactions we performed a computational mechanistic study, derived from theoretical calculations at the (SMD:benzene)ω-B97XD/def2-tzvpp//ω-B97XD/def2-svp level (see ESI† for further computational details).¶ The model diborenes chosen for this study were the doubly IMe-stabilised 1,2-bis(1-methylvinyl)diborene R1, representative of NHC-stabilised diborenes, and the doubly PMe3-stabilised 1,2-diphenyldiborene R2, representative of phosphine-stabilised diborenes, while propyne was chosen as the model alkyne.The overall calculated reaction profiles for the NHC-stabilised diborene R1 (Fig. 5, red pathways) show that the hydroalkynylation product anti-P1C–H is the kinetic product (ΔG‡ = 18.9 kcal mol−1), the reaction being highly exothermic (ΔG1 = −34.0 kcal mol−1). The [2 + 2] cycloaddition proceeds via an unsymmetrical zwitterionic late transition state, TS1B2C2, in which the B–CMe bond is already pre-formed, similarly to what has been postulated for some cycloadditions of alkynes to disilenes.25,33 The [2 + 2] cycloaddition product P1B2C2, for which the energy barrier is only 1.6 kcal mol−1 higher than for the hydroalkynylation process, is the thermodynamic product, being 5.9 kcal mol−1 more stable than anti-P1C–H. This is in good agreement with the experimental observations that hydroalkynylation is favoured at room temperature for diborenes 1 and 3, whereas cycloaddition is favoured at elevated temperatures for 3. The fact that hydroalkynylation is not observed with diborenes 6Tn/Fu suggests that the barriers of the two processes may be inverted in this case.
 | ||
| Fig. 5 Energy profiles for the [2 + 2] cycloaddition and 1,2-addition of the C–H bond of propyne to the model doubly IMe-stabilised 1,2-bis(1-methylvinyl)diborene R1 (red pathways) and the doubly PMe3-stabilised 1,2-diphenyldiborene R2 (blue pathways) in benzene at room temperature, calculated at the (SMD:benzene)ω-B97XD/def2-tzvpp//ω-B97XD/def2-svp level of theory. Energies in parentheses in kcal mol−1. | ||
Given that the hydroalkynylation of R1 shown in Fig. 5 produces the syn-addition product (syn-P1C–H) rather than the experimentally observed anti-addition product (anti-P1C–H), the syn-to-anti conversion mechanism was also investigated (see Fig. S59 in the ESI†). The only energetically viable route at 25 °C proceeds through a NHC decoordination–recoordination mechanism with a mild overall barrier (ΔG‡R = 12.0 kcal mol−1), via a hydride-bridged diborane intermediate, Intbridge (Scheme 8). The resulting product, anti-P1C–H, is more stable than syn-P1C–H by 3.9 kcal mol−1.|| The absence of syn-to-anti isomerisation in Inoue's dialumene hydroalkynylation product (Scheme 1c)36 despite the much weaker Al–CNHC bonds, which should in theory favour the dissociation of one NHC ligand required for the isomerisation (bond dissociation energies at 298 K: D°B–C = 107 kcal mol−1, D°Al–C = 64 kcal mol−1) ,61,63 may be due to the fact that the resulting singly NHC-stabilised 1-alkynyl-2-hydrodialane is less likely to stabilise itself through a three-centre-two-electron hydride bridge, as is common for hydrodiboranes.64
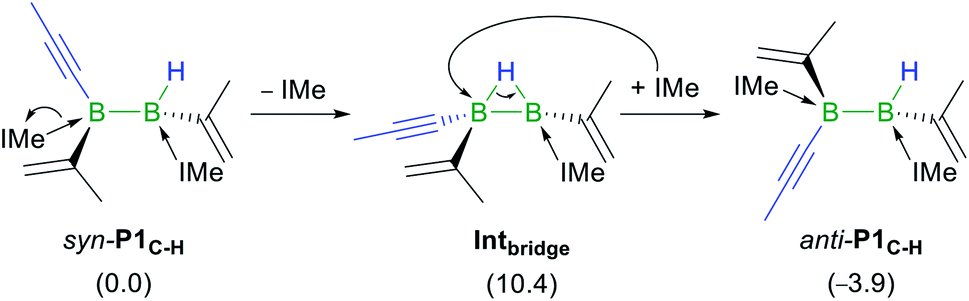 | ||
| Scheme 8 Mechanism of the syn-to-anti isomerisation of P1C–H. Energies in parentheses in kcal mol−1. | ||
The overall calculated reaction profiles for the phosphine-stabilised diborene R2 (Fig. 5, blue pathways) show that the cycloaddition product, P2B2C2, which is precursor to the final 1,3-dihydro-1,3-diborete P2BCBC, is both the kinetic (ΔG‡ = 16.6 kcal mol−1) and the thermodynamic product (ΔG2 = −40.5 kcal mol−1), being 9.1 kcal mol−1 more stable than the hydroalkynylation product anti-P2C–H. This is again in agreement with the experimental observations that [2 + 2] cycloaddition is favoured in all cases for diborenes I and 10. The [2 + 2] cycloaddition again proceeds via an unsymmetrical zwitterionic late transition state, TS2B2C2, in which the B–CMe bond is already pre-formed.**
The rearrangement of P2B2C2 to P2BCBC was also investigated computationally (Fig. 6). Initially, PMe3 dissociates from the boron atom at the β position to the methyl substituent of the 1,2-dihydro-1,2-diborete (ΔG‡ = 10.9 kcal mol−1) (TS3) and ΔG3 = 8.4 kcal mol−1 (Int3)).†† To compensate for the loss of electron density, the B2C2 ring distorts from a planar ring to a nonplanar rhombic cluster, in which the sp2-hybridised boron atom B1 can interact with the CC double bond (B1⋯C1 1.84 Å). A subsequent 1,2-shift of B2 in a tetrahedron-like transition state TS4 (ΔG‡ = 16.2 kcal mol−1) reduces the bond order and generates a new nonplanar rhombic cluster, Int4 (ΔG4 = 0.1 kcal mol−1, i.e.Int3 and Int4 are energetically equivalent isomers, but not in equilibrium with each other), with a long C⋯C interaction (1.77 Å). Int4 then converts into the tautomer C of the 1,3-dihydro-1,3-diborete P2BCBC by a PMe3 decoordination-recoordination mechanism from B2 to C1 via the phosphine-free intermediate Int5 (ΔG5 = 9.5 kcal mol−1, ΔG‡R = 10.6 kcal mol−1).‡‡ The total energy barrier from P2B2C2 to TS4 (ΔG‡ = 24.6 kcal mol−1) constitutes the rate-limiting step of the overall reaction from R2 and propyne to P2B2C2.65 This explains the elevated temperature (80 °C) required over a period of several days for reaction completion. In the final step, C tautomerises to the slightly more stable isomer D (ΔGC–D = −2.6 kcal mol−1). Preliminary calculations suggest that this step is likely to be an intermolecular proton exchange between the positively-charged C1 carbon of one molecule and the negatively-charged C2 carbon of a second molecule (see Fig. S60 in the ESI for details†). The fact that C is the major or sole tautomer observed in the experimentally-realised systems 8-R (R = Bu, Tol, SiMe3, Fc) is consistent with the increased steric bulk of the mesityl (experimental) boron substituent relative to the phenyl substituents present in the computational model, the former being far more likely to hinder an intermolecular approach. The intermolecular proton transfer would therefore only be possible in the least sterically hindered analogues, 8-Bu and 8-Tol, for which a small proportion of D is indeed observed.
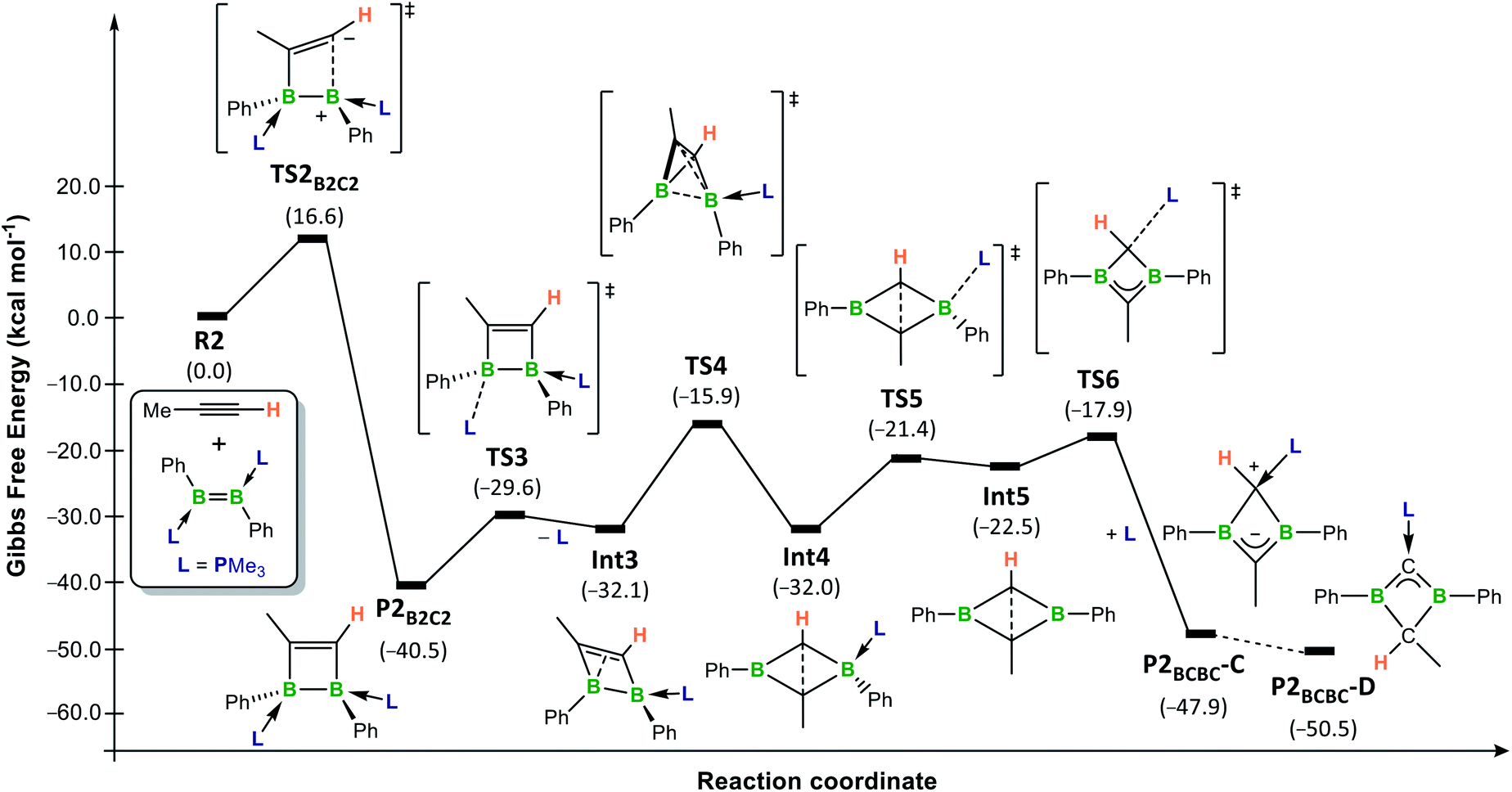 | ||
| Fig. 6 Energy profile for the conversion of model diborene R2 and propyne into the tautomers C and D of P2BCBC in benzene at room temperature, calculated at the (SMD:benzene)ω-B97XD/def2-tzvpp//ω-B97XD/def2-svp level of theory. Energies in parentheses in kcal mol−1. | ||
Conclusions
The reactions of terminal alkynes with doubly base-stabilised diborenes of the form R(L)BB(L)R (L = Lewis base, R = anionic substituent) result in different outcomes, largely depending on the relative predominance of kinetic or thermodynamic control, the nature of the Lewis base L and the conformation (acyclic or cyclic) of the diborene. With NHC-stabilised diborenes the kinetic product tends to be the hydroalkynylation product, which is obtained with exclusive anti selectivity. Computational analyses show that this anti selectivity results from the rearrangement of the expected syn addition product through decoordination–recoordination of one NHC ligand and the formation of a singly NHC-stabilized hydride-bridged diborane intermediate. The thermodynamic product is the 1,2-dihydro-1,2-diborete, resulting from a [2 + 2] cycloaddition reaction between the BB double and CC triple bonds. In all cases, however, the strained B2C2 ring undergoes spontaneous ring expansion by activating either an endo- or exocyclic C–N bond at one NHC ligand, thereby providing access to novel fused B, N-heterocycles.
With phosphine-stabilised diborenes experimental and computational data show that the 1,2-dihydro-1,2-diborete cycloaddition product is both the kinetic and thermodynamic product. For acyclic diborenes, this 1,2-dihydro-1,2-diborete invariably rearranges to a thermodynamically more stable, homoaromatic 1,3-dihydro-1,3-diborete, effectively resulting in the scission of both the original BB double and CC triple bonds. Calculations show that this rearrangement proceeds via a four-step mechanism, with loss of one phosphine ligand and migration of the other phosphine from boron to the adjacent carbon atom. Depending on the substitution of the terminal alkyne precursor, two tautomers of this phosphine-stabilised 1,3-dihydro-1,3-diborete are obtained, differing by the attachment of the former acetylenic proton to either of the carbon atoms of the BCBC ring. Computational results suggest a bimolecular mechanism for this tautomerisation, which would indeed account for the observed selectivity. The elusive 1,2-dihydro-1,2-diborete cycloaddition product was finally stabilised by employing a cyclic diborene, the bridging benzylphosphine ligand of which prevents the 1,2-to-1,3-rearrangement.
Author contributions
H. B. supervised the study. L. E., U. S., M. D., M. P., T. E. S., A. H., A. P. and F. R. carried out the synthetic work. M. A., M. H., J. M., D. P., A. R., K. R., F. S. and T. T. carried out the X-ray crystallographic analyses. J. O. C. J.-H. carried out the computational studies. M. A. prepared the manuscript. L. E., U. S., M. D., T. E. S., A. H., M. A. and J. O. C. J.-H. prepared the ESI. All authors read and commented on the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We gratefully acknowledge the European Research Council for funding under the European Union Horizon 2020 Research and Innovation Program (grant agreement no. 669054).Notes and references
- D. F. Fernández, J. L. Mascareñas and F. López, Chem. Soc. Rev., 2020, 49, 7378 RSC.
- R. Khan, J. Chen and B. Fan, Adv. Synth. Catal., 2020, 362, 1564 CrossRef CAS.
- Z.-X. Wang and B.-J. Li, J. Am. Chem. Soc., 2019, 141, 9312 CrossRef PubMed.
- Z.-X. Wang, X.-Y. Bai and B.-J. Li, Synlett, 2017, 28, 509 CrossRef CAS.
- Y. Shen, B. Huang, J. Zheng, C. Lin, Y. Liu and S. Cui, Org. Lett., 2017, 19, 1744 CrossRef CAS PubMed.
- S. Hosseyni, C. A. Smith and X. Shi, Org. Lett., 2016, 18, 6336 CrossRef CAS PubMed.
- M. Shirakura and M. Suginome, J. Am. Chem. Soc., 2009, 131, 5060 CrossRef CAS PubMed.
- M. Shirakura and M. Suginome, Org. Lett., 2009, 11, 523–526 CrossRef CAS PubMed.
- M. E. Muratore, A. Homs, C. Obradors and A. M. Echavarren, Chem.–Asian J., 2014, 9, 3066 CrossRef CAS PubMed.
- V. Lopez-Carrillo and A. M. Echavarren, J. Am. Chem. Soc., 2010, 132, 9292 CrossRef CAS PubMed.
- W. Tam, J. Goodreid and N. Cockburn, Curr. Org. Synth., 2009, 6, 219 CrossRef CAS.
- T. Mitsudo, H. Naruse, T. Kondo, Y. Ozaki and Y. Watanabe, Angew. Chem., Int. Ed. Engl., 1994, 33, 580–581 CrossRef.
- V. Saini, B. J. Stokes and M. S. Sigman, Angew. Chem., Int. Ed., 2013, 52, 11206 CrossRef CAS PubMed.
- C. Fischmeister and C. Bruneau, Beilstein J. Org. Chem., 2011, 7, 156 CrossRef CAS PubMed.
- S. K. Chattopadhyay, S. Karmakar, T. Biswas, K. C. Majumdar, H. Rahaman and B. Roy, Tetrahedron, 2007, 63, 3919 CrossRef CAS.
- H. Villar, M. Frings and C. Bolm, Chem. Soc. Rev., 2007, 36, 55 RSC.
- S. T. Diver and A. J. Giessert, Chem. Rev., 2004, 104, 1317 CrossRef CAS PubMed.
- C. Storm Poulsen and R. Madsen, Synthesis, 2003, 1, 1 Search PubMed.
- A. Kinoshita, N. Sakakibara and M. Mori, J. Am. Chem. Soc., 1997, 119, 12388 CrossRef CAS.
- B. M. Trost and M. K. Trost, J. Am. Chem. Soc., 1991, 113, 1852 CrossRef.
- M. Majumdar, I. Bejan, V. Huch, A. J. P. White, G. R. Whittell, A. Schaefer, I. Manners and D. Scheschkewitz, Chem.–Eur. J., 2014, 20, 9225 CrossRef CAS PubMed.
- A. Meltzer, M. Majumdar, A. J. P. White, V. Huch and D. Scheschkewitz, Organometallics, 2013, 32, 6844 CrossRef CAS.
- S. E. Gottschling, M. C. Jennings and K. M. Baines, Can. J. Chem., 2005, 83, 1568 CrossRef CAS.
- M. Ichinohe, T. Matsuno and A. Sekiguchi, Chem. Commun., 2001, 2, 183 RSC.
- D. J. De Young and R. West, Chem. Lett., 1986, 6, 883 CrossRef.
- Y. Nakadaira, R. Sato and H. Sakurai, Chem. Lett., 1985, 643 CrossRef CAS.
- K. L. Hurni and K. M. Baines, Chem. Commun., 2011, 47, 8382 RSC.
- Y. Ishida, A. Sekiguchi and Y. Kabe, J. Am. Chem. Soc., 2003, 125, 11468 CrossRef CAS PubMed.
- N. Fukaya, M. Ichinohe and A. Sekiguchi, Angew. Chem., Int. Ed., 2000, 39, 3881 CrossRef CAS.
- S. A. Batcheller and S. Masamune, Tetrahedron Lett., 1988, 29, 3383 CrossRef CAS.
- J. Schneider, J. Henning, J. Edrich, H. Schubert and L. Wesemann, Inorg. Chem., 2015, 54, 6020 CrossRef CAS PubMed.
- V. Y. Lee, T. Fukawa, M. Nakamoto, A. Sekiguchi, B. L. Tumanskii, M. Karni and Y. Apeloig, J. Am. Chem. Soc., 2006, 128, 11643 CrossRef CAS PubMed.
- K. K. Milnes, L. C. Pavelka and K. M. Baines, Chem. Soc. Rev., 2016, 45, 1019 RSC.
- T. Agou, T. Nagata and T. Tokitoh, Angew. Chem., Int. Ed., 2013, 52, 10818 CrossRef CAS PubMed.
- Z. Zhu, X. Wang, M. M. Olmstead and P. P. Power, Angew. Chem., Int. Ed., 2009, 48, 2027 CrossRef CAS PubMed.
- P. Bag, A. Porzelt, P. J. Altmann and S. Inoue, J. Am. Chem. Soc., 2017, 139, 14384 CrossRef CAS PubMed.
- M. Arrowsmith, J. Böhnke, H. Braunschweig, M. A. Celik, C. Claes, W. C. Ewing, I. Krummenacher, K. Lubitz and C. Schneider, Angew. Chem., Int. Ed., 2016, 55, 11271 CrossRef CAS PubMed.
- A. Hermann, F. Fantuzzi, M. Arrowsmith, T. Zorn, I. Krummenacher, B. Ritschel, K. Radacki, B. Engels and H. Braunschweig, Angew. Chem., Int. Ed., 2020, 59, 15717 CrossRef CAS PubMed.
- J. Jiao and Y. Nishihara, J. Organomet. Chem., 2012, 721–722, 3 CrossRef CAS.
- D. E. Kaufmann and N. Ocal, Sci. Synth., 2004, 6, 635 CAS.
- H. C. Brown and M. Srebnik, Organometallics, 1987, 6, 629 CrossRef CAS.
- E. A. Romero, R. Jazzar and G. Bertrand, Chem. Sci., 2017, 8, 165 RSC.
- C. I Lee, J. Zhou and O. V. Ozerov, J. Am. Chem. Soc., 2013, 135, 3560 CrossRef PubMed.
- J. H. Müssig, M. Thaler, R. D. Dewhurst, V. Paprocki, J. Seufert, J. D. Mattock, A. Vargas and H. Braunschweig, Angew. Chem., Int. Ed., 2019, 58, 4405 CrossRef PubMed.
- M. Dömling, M. Arrowsmith, U. Schmidt, L. Werner, A. C. Castro, J. O. C. Jiménez-Halla, R. Bertermann, J. H. Müssig, D. Prieschl and H. Braunschweig, Angew. Chem., Int. Ed., 2019, 58, 9782 CrossRef PubMed.
- T. Brückner, T. E. Stennett, M. Heß and H. Braunschweig, J. Am. Chem. Soc., 2019, 141, 14898 CrossRef PubMed.
- H. Braunschweig, R. D. Dewhurst, C. Hörl, A. K. Phukan, F. Pinzner and S. Ullrich, Angew. Chem., Int. Ed., 2014, 53, 3241 CrossRef CAS PubMed.
- H. Braunschweig, I. Krummenacher, C. Lichtenberg, J. D. Mattock, M. Schäfer, U. Schmidt, C. Schneider, T. Steffenhagen, S. Ullrich and A. Vargas, Angew. Chem., Int. Ed., 2017, 56, 889 CrossRef CAS PubMed.
- Y. Wang, H. P. Hickox, Y. Xie, P. Wei, H. F. Schaefer III and G. H. Robinson, J. Am. Chem. Soc., 2017, 139, 16109 CrossRef CAS PubMed.
- T. Thiess, S. K. Mellerup and H. Braunschweig, Chem.–Eur. J., 2019, 25, 13572 CrossRef CAS PubMed.
- M. Eck, S. Würtemberger-Pietsch, A. Eichhorn, J. H. J. Berthel, R. Bertermann, U. S. D. Paul, H. Schneider, A. Friedrich, C. Kleeberg, U. Radius and T. B. Marder, Dalton Trans., 2017, 46, 3661 RSC.
- S. Pietsch, U. Paul, I. A. Cade, M. J. Ingleson, U. Radius and T. B. Marder, Chem.–Eur. J., 2015, 21, 9018 CrossRef CAS PubMed.
- T. Brückner, M. Arrowsmith, M. Heß, K. Hammond, M. Müller and H. Braunschweig, Chem. Commun., 2019, 55, 6700 RSC.
- M. L. McKee, Inorg. Chem., 2000, 39, 4206 CrossRef CAS PubMed.
- T. Brückner, M. Arrowsmith, M. Heß, K. Hammond, M. Müller and H. Braunschweig, Chem. Commun., 2019, 55, 6700 RSC.
- M. Pilz, J. Allwohn, W. Massa and A. Berndt, Angew. Chem., Int. Ed., 1990, 29, 399 CrossRef.
- H. Meyer, G. Baum, W. Massa, S. Berger and A. Berndt, Angew. Chem., Int. Ed., 1987, 26, 546 CrossRef.
- H. Meyer, G. Baum, W. Massa and A. Berndt, Angew. Chem., Int. Ed., 1987, 26, 798 CrossRef.
- H. Schmidbaur, A. Schier, B. Milewski-Mahrla and U. Schubert, Chem. Ber., 1982, 115, 722 CrossRef CAS.
- T. E. Stennett, J. D. Mattock, I. Vollert, A. Vargas and H. Braunschweig, Angew. Chem., Int. Ed., 2018, 57, 4098 CrossRef CAS PubMed.
- D. E. Kaufmann, R. Boese and A. Scheer, Chem. Ber., 1994, 127, 2349 CrossRef CAS.
- M. Hildenbrand, H. Pritzkow and W. Siebert, Angew. Chem., Int. Ed. Engl., 1985, 24, 759 CrossRef.
- Y. R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, FL, 2007 Search PubMed.
- H. Beall and D. F. Gaines, Boron Hydrides, in Encyclopedia of Physical Science and Technology, 3rd edn, Academic Press, 2003, p. 301 Search PubMed.
- S. Kozuch and J. M. L. Martin, ChemPhysChem, 2011, 12, 1413 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental details, characterisation data for all reported compounds and details of the DFT calculations. CCDC 2076364–2076374. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02081a |
| ‡ These authors contributed in equal parts to the paper. |
| § The relatively low isolated yields of many of the reaction products presented herein, despite the good to excellent selectivities observed by 11B NMR spectroscopy (see 11B NMR spectra of reaction mixtures prior to work-up, Fig. S39–S52 in the ESI†), is due to a combination of (a) the small reaction scales (typically 20–50 mg of diborene), (b) the very high solubility of the products in pentane at −30 °C (11B NMR spectra of the mother liquor after recrystallization always showed large amounts of product remaining in solution), and (c) the difficulty of separating the products from remaining excess alkyne. |
| ¶ Four other basis sets of similar quality and size were tested, and the same trend was followed. There is a minor discrepancy with Pople's 6-311+G(d,p) basis set. |
| || Although anti-P1C–H may be obtained directly from the hydroalkynylation of the cis-conformer of R1, this route was discarded due to the high rotation barrier of the BB double bond. |
| ** The origin of the ligand-controlled selectivity for cycloaddition versus hydroalkynylation seems to be steric rather than electronic control. See Fig. S61 in ESI† and associated text. |
| †† Dissociation of the other phosphine is unlikely since the analogous intermediate is 4 kcal mol−1 higher in energy than Int3. Moreover, a comparison of the B–P distances in P2B2C2 suggests that the first phosphine (B–P 1.94 Å) is more labile than the second one (1.92 Å). |
| ‡‡ PMe3 (NBO charge: +1.24) coordinates to the more negatively charged carbon (CH: −0.72 versus CMe: −0.53) of the diborete ring. |
| This journal is © The Royal Society of Chemistry 2021 |