
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiomimetic enterobactin analogue mediates iron-uptake and cargo transport into E. coli and P. aeruginosa†
Robert
Zscherp‡
a,
Janetta
Coetzee‡
bc,
Johannes
Vornweg
 a,
Jörg
Grunenberg
a,
Jennifer
Herrmann
bc,
Rolf
Müller
bc and
Philipp
Klahn
*a
a,
Jörg
Grunenberg
a,
Jennifer
Herrmann
bc,
Rolf
Müller
bc and
Philipp
Klahn
*a
aInstitute of Organic Chemistry, Technische Universität Braunschweig, Hagenring 30, D-38106 Braunschweig, Germany. E-mail: p.klahn@tu-bs.de
bDepartment for Microbial Natural Products, Helmholtz Institute for Pharmaceutical Research Saarland (HIPS), Helmholtz Center for Infection Research and Department of Pharmacy at Universität des Saarlandes, Campus Building E 8.1, D-66123 Saarbrücken, Germany
cGerman Center for Infection Research (DZIF), Site Hannover-Braunschweig, Germany
First published on 17th June 2021
Abstract
The design, synthesis and biological evaluation of the artificial enterobactin analogue EntKL and several fluorophore-conjugates thereof are described. EntKL provides an attachment point for cargos such as fluorophores or antimicrobial payloads. Corresponding conjugates are recognized by outer membrane siderophore receptors of Gram-negative pathogens and retain the natural hydrolyzability of the tris-lactone backbone. Initial density-functional theory (DFT) calculations of the free energies of solvation (ΔG(sol)) and relaxed Fe–O force constants of the corresponding [Fe-EntKL]3− complexes indicated a similar iron binding constant compared to natural enterobactin (Ent). The synthesis of EntKL was achieved via an iterative assembly based on a 3-hydroxylysine building block over 14 steps with an overall yield of 3%. A series of growth recovery assays under iron-limiting conditions with Escherichia coli and Pseudomonas aeruginosa mutant strains that are defective in natural siderophore synthesis revealed a potent concentration-dependent growth promoting effect of EntKL similar to natural Ent. Additionally, four cargo-conjugates differing in molecular size were able to restore growth of E. coli indicating an uptake into the cytosol. P. aeruginosa displayed a stronger uptake promiscuity as six different cargo-conjugates were found to restore growth under iron-limiting conditions. Imaging studies utilizing BODIPYFL-conjugates, demonstrated the ability of EntKL to overcome the Gram-negative outer membrane permeability barrier and thus deliver molecular cargos via the bacterial iron transport machinery of E. coli and P. aeruginosa.
Introduction
The development of bacterial resistance towards antimicrobial drugs is an intrinsic part of bacterial evolution and this, in turn, necessitates the continuous development of novel drugs able to kill these life-threatening multi-drug resistant human pathogens.1–3 Facing the current spread of bacterial resistance against clinically used antibiotics, the development of novel antimicrobial drugs and innovative concepts is of vast importance to counteract this serious threat for public health.1,4,5Four of the six ESKAPE pathogens,6,7 represent bacterial species considered as significant threat for public health due to a very high occurrence of multi-drug resistance, are Gram-negative bacteria. In addition, the pathogens recently prioritized by the WHO with a critical, high or medium need for the development of novel antimicrobial drugs are primarily Gram-negative bacteria, including β-lactam-resistant Escherichia coli and Pseudomonas aeruginosa or fluoroquinolone-resistant Salmonella and Shigella species.8 However, the development of novel antimicrobial drugs against Gram-negative bacteria is challenging, not only due to the presence of drug exporters as also found in Gram-positive bacteria, but because many antibacterial compounds fail to overcome the Gram-negative outer membrane. As a consequence, many potential drugs displaying antibacterial activity against Gram-positive bacteria remain inactive against Gram-negative bacteria, although their biological targets are generally present, as they fail to translocate over the cell envelope barrier.9
An innovative approach to enable translocation of antimicrobial drugs or reporter molecules over the Gram-negative cell envelope barrier and to develop novel antimicrobial drugs against these pathogens is the conjugation and hybridization with siderophores.4,10–12 Siderophores are small molecule iron chelators,13,14 produced and secreted by bacteria, fungi and plants to ensure their supply with iron, an essential growth factor for all living organisms.15 Beyond iron uptake, siderophores have multiple other biological functions in bacteria,16–18e.g. they are important virulence factors contributing to pathogenesis.17,19 After coordination of Fe(III), the resulting ferri-siderophores are recognized by specific siderophore receptors, transmembrane proteins embedded in the outer membrane, and are actively transported into the cytosol of bacteria.4,20–23 Siderophore drug conjugates can enter bacterial cells via the same pathway through siderophore receptor mediated uptake.1,4,24–32
The general concept of this Trojan Horse approach is derived from natural antetypes. The sideromycins,33 such as albomycins34–36 or the microcins E492 (ref. 37–40) and H47 (ref. 41) are utilized by different bacteria to defend their ecological niche against competing bacteria. These natural siderophore drug conjugates reveal a remarkable increase in antibacterial activity compared to the parental free antibiotics. Inspired by this natural concept researchers have developed artificial siderophore drug conjugates based on synthetically modified siderophore analogues,42–55 leading to active drug accumulation and tremendously increased antibacterial activity compared to the parental drugs.42,43,48 Furthermore, siderophore drug conjugates can expand the activity of Gram-positive-only drugs towards Gram-negative pathogens.50,51,56,57 Additionally, this strategy allows for the design of narrow-spectrum antibiotics,10,49,57 selectively targeting specific virulent pathogens, thereby reducing the selection pressure on the bacterial resistome. Importantly, the siderophore-β-lactam hybrid cefiderocol developed by Shionogi Pharmaceuticals was recently approved by the FDA for the treatment of pneumonia caused by Gram-negative bacteria.58
In this context, the tris-catechol siderophore enterobactin (Ent, Scheme 1A)14 displaying an extraordinarily high Fe(III) binding constant (Kd = 10−49 M)59,60 as well as its glycosylated derivatives, the salmochelins61 evading lipocalin-2 inactivation,62,63 are of high interest.
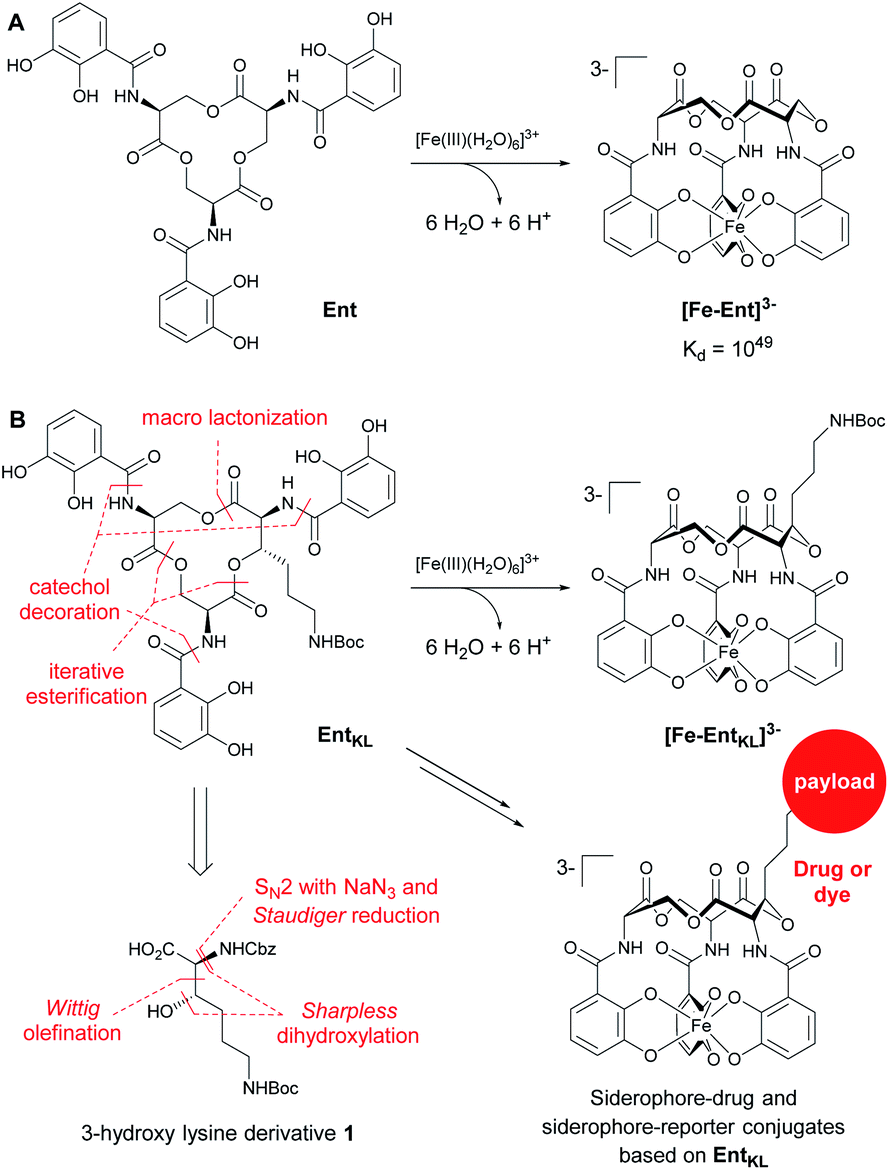 | ||
| Scheme 1 (A) Structures of enterobactin (Ent) and ferri-enterobactin ([Fe-Ent]3−); (B) retrosynthetic analysis and design concept of EntKL. | ||
Ent and salmochelins are biosynthesized by Gram-negative bacteria of the family Enterobacteriaceae, such as E. coli, Klebsiella pneumonia, Salmonella typhimurium, Yersinia enterocolitica and Shigella flexneri, to ensure their iron supply.64 However, Ent has also been found to be produced by Gram-positive Streptomyces species65 and it serves as a xenosiderophore for the opportunistic human pathogen P. aeruginosa, where internalization is mediated by the outer membrane siderophore receptor PfeA.66–69Ent and salmochelins also play a crucial role during infection18,70 and they have been demonstrated to sequester iron from human transferrin.17,19 Previously reported artificial Ent analogues used for the development of siderophore drug conjugates or siderophore-based imaging tools71,72 were based on modifications at one of the catechol units42,43,46,49,52,73–75 or the natural tris-serine lactone backbone was substituted by artificial moieties to generate an attachment point for payloads in the backbone of the siderophore.48,76–78 Although a co-crystal structure of [Fe-Ent]3− with its E. coli siderophore receptors of FepA79,80 and IroN80–82 is not available, recognition of [Fe-Ent]3− at other proteins such as FeuA,83–85 VctP,86 PfeA69 or lipocalin-2 (ref. 83) suggests that the attachment of larger payloads at the catechol units might interfere with [Fe-Ent]3− receptor recognition.
Therefore, we envisaged the synthesis of the novel, biomimetic enterobactin analogue EntKL (Scheme 1B). Our synthesis is based on the incorporation of the 3-hydroxy lysine derivative 1 into Ent backbone, aiming to generate an attachment point for antimicrobial payloads in the backbone out of the recognition site of the siderophore and simultaneously retain the natural hydrolyzability of the tris-lactone backbone.52,87
Result and discussion
Retrosynthetically, the final siderophore could be derived from a modified asymmetric tris-lactone backbone by decoration with catechol units via amide coupling (Scheme 1B). As the tin-template mediated trans-esterification approach utilized earlier, by Shanzer,88,89 later by Gutierrez90 and Raymond91 for the synthesis of the enterobactin tris-serine lactone backbone, leads exclusively to the formation of the thermodynamic, symmetric product, it is not applicable for the synthesis of the envisaged asymmetric tris-lactone backbone. Therefore, we decided to follow an approach of iterative assembly of a linear trimer containing the 3-hydroxy lysine derivative 1 with subsequent macro lactonization as used in the early synthesis of Ent by Corey,92 Rastetter93 and Rogers94 in order to generate the desired asymmetric backbone of EntKL.In order to estimate if the envisaged substituent at the asymmetric tris-lactone backbone would negatively impact the solution thermodynamics and/or the kinetic stability of the Fe(III) complex [Fe-EntKL]3−, we computed the free energies of solvation ΔG(sol) as well as the relevant (relaxed) Fe–O force constants for [Fe-Ent]3− and two stereoisomers of [Fe-EntKL]3− bearing an axial or equatorial substituent (Table 1) following the protocol of Baramov et al.95 (see ESI†).
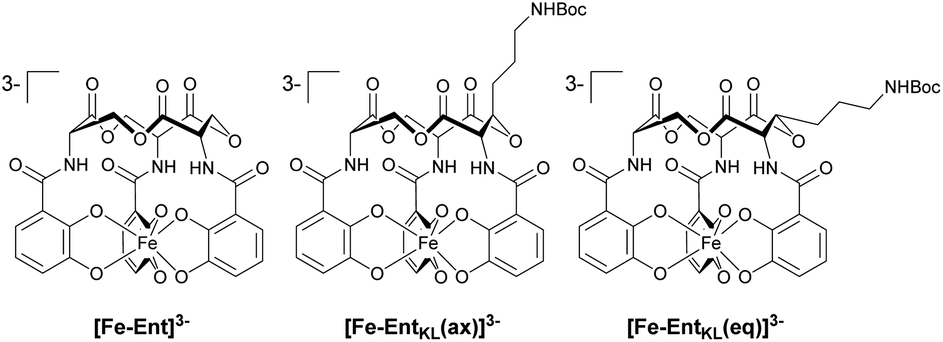
In a first step, we tried to reproduce the experimentally well-known free energy of complexation (ΔG(sol): −66.8 kcal mol−1 (ref. 59 and 60)) for [Fe-Ent]3−.
Our calculated value of −64.5 kcal mol−1 for ΔG(sol) [Fe-Ent]3− is very close to the literature ref. 59 and 60 positively validating our applied in silico procedure. Interestingly, the artificial ferri-enterobactin analogues seem to be even better iron binders than the unsubstituted [Fe-Ent]3− by several kcal mol−1. Furthermore, the value for [Fe-EntKL(eq)]3− was calculated to be energetically more favored as compared to [Fe-EntKL(ax)]3− by a value of 3.5 kcal mol−1.
Nevertheless, the absolute values need to be considered with caution and we hypothesize that both artificial enterobactin analogues, EntKL(eq) and EntKL(ax), lead to complexes which are at least of similar thermodynamic stability compared to [Fe-Ent]3− and that there is no significant energetical advantage of one analogue over the other. While siderophore Fe(III) complexes are thermodynamically very stable, they are kinetically labile. This is in contrast to, for example, their chromic counterparts, which are both thermodynamically and kinetically stable. In order to find out if this kinetic lability holds true also for our artificial ferri-enterobactin analogues, we calculated the relaxed Fe–O force constants, a method which was recently successfully applied in several other weakly bound systems. According to our computational results, all three complexes seem to be kinetically labile with relaxed Fe–O force constants well below 1 N cm−1 (see Table 1), indicating very weak bonds with less or no covalency.96–100 (Note, that the averaged Cr–O value for [CrEnt]3− is 1.25 N cm; unpublished results J. G.) Furthermore, our calculated Fe–O values are very similar. In summary, all three complexes seem to be kinetically labile, while being thermodynamically stable, as found earlier for [Fe-Ent]3− by Raymond and co-workers.101,102
Thus, expecting no negative impact of an axial backbone substituent on the Fe(III) complex stability, we developed a robust, scalable synthetic route to the required 3-hydroxy lysine derivative 1 starting from commercially available 4-aminobutanol (2) as outlined in Scheme 2. A sequence of Boc protection, Swern oxidation of the resulting N-Boc-4-aminobutanol (3) to aminal 4 and final Wittig olefination in presence of methyl (triphenyl-phosphoranylidene) acetate (5) at 100 °C led to formation of the E-configured α,β-unsaturated methyl ester 6 in excellent diastereoselectivity of E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z = 100:1 and 89% yield over 3 steps with only a single chromatographic purification step being required. An asymmetric sharpless dihydroxylation using AD-mix-α finally gave diol 7 in 83% yield. The formation of the corresponding cyclic sulfite in the presence of thionyl chloride and triethyl amine at 0 °C and subsequent α-inversion in the presence of sodium azide afforded α-azido methyl ester 8 in 64% yield. At this step the absolute configuration at the C3 position introduced by the sharpless dihydroxylation was determined to be S-configured with an enantiomeric excess of 94%ee using Mosher's method.103
:Z = 100:1 and 89% yield over 3 steps with only a single chromatographic purification step being required. An asymmetric sharpless dihydroxylation using AD-mix-α finally gave diol 7 in 83% yield. The formation of the corresponding cyclic sulfite in the presence of thionyl chloride and triethyl amine at 0 °C and subsequent α-inversion in the presence of sodium azide afforded α-azido methyl ester 8 in 64% yield. At this step the absolute configuration at the C3 position introduced by the sharpless dihydroxylation was determined to be S-configured with an enantiomeric excess of 94%ee using Mosher's method.103
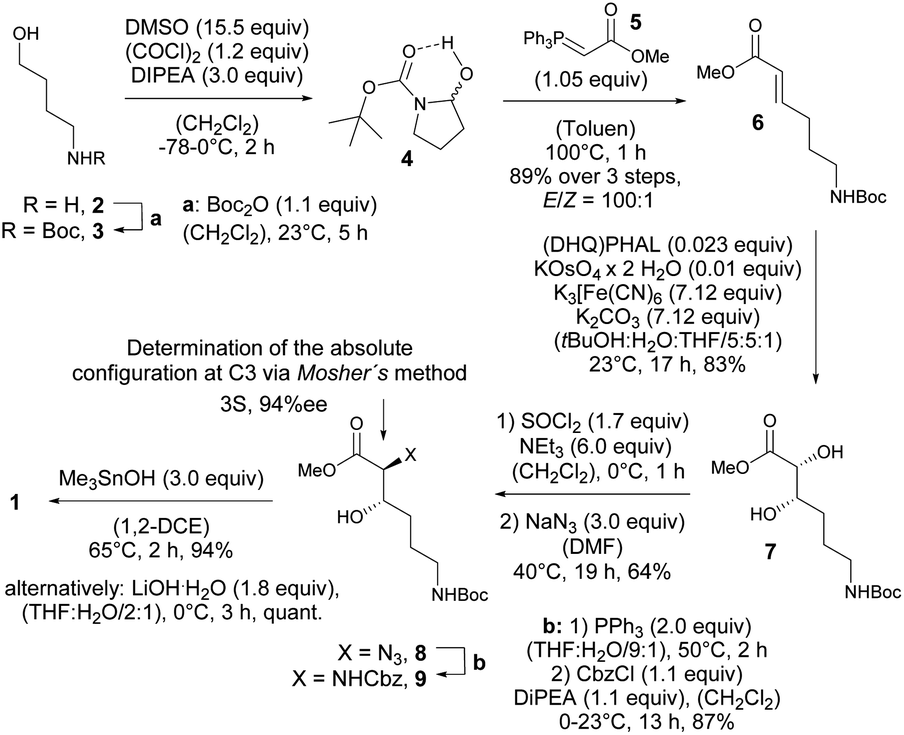 | ||
| Scheme 2 Synthesis of 3-hydroxy lysine derivative 1. | ||
Finally, a Staudinger reduction and subsequent Cbz protection generated the methyl ester 9 in 87% yield, which was saponified in the presence of trimethyltin hydroxide in 1,2-dichloro ethane at 65 °C using Nicolaou's protocol104 yielding the desired 3-hydroxy lysine derivative 1 in 94%.
With 1 in hands, we achieved the assembly of the linear trimer 17 as outlined in Scheme 3. First, the 2-methylanthraquinyl ester 11 was formed with 2-bromomethylanthraquinone 10 (MaqBr) in the presence of DBU in 55% yield.93 A two times consecutive sequence of Steglich esterification with Cbz-Ser(TBS)-OH 12 and subsequent desilylation in the presence of a mixture of aqueous, concentrated HF solution and TBAF led to the Maq-protected linear trimer 16 obtained in 64% yield over 4 steps. Ester 16 was then photolytically cleaved at 360 nm in the presence of N-methyl morpholine (NMM) in a mixture of iso-propanol and chloroform giving the unprotected linear trimer 17 as precursor for the backbone cyclization in 65% yield.93
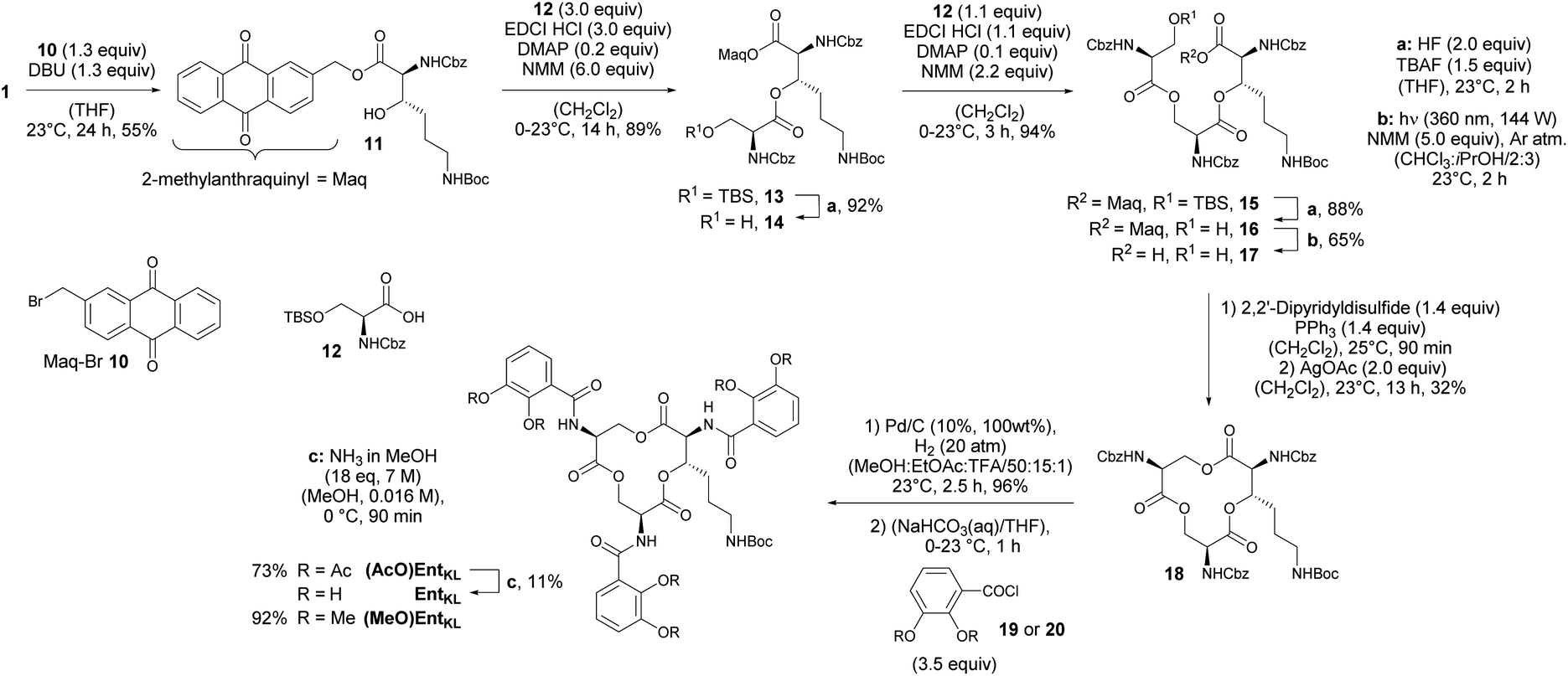 | ||
| Scheme 3 Assembly of a linear trimer, cyclization and final catechol decoration. | ||
As expected for the formation of a 12-membered ring with several axial substituents, the cyclization of 17 to the tris-lactone 18 was challenging.
The activation of the carboxylic acid via several reagents predominantly led to elimination under formation of the corresponding dihydro amino acid (Table 2).
| Entry | Conditions | Result |
|---|---|---|
| a Elimination. b Bad reproducibility. | ||
| 1 | MNBA (5.0 eq.), DMAP (8.0 eq.), (CH2Cl2), 0–23 °C, 20 h | 13%a |
| 2 | PPh3 (1.5 eq.), DIAD (1.3 eq.), (THF), 0–23 °C, 24 h | 14%a |
| 3 | (1) Corey's disulfane92 (1.4 eq.), PPh3 (1.4 eq.), (CH2Cl2), 23 °C, 10 min; (2) AgBF4 (5.0 eq.), (PhH), 25 °C, 16 h | 5–50%a,b |
| 4 | (1) 2,2′-Dipyridyldisulfide (1.4 eq.), PPh3 (1.4 eq.) in (CH2Cl2), 30 °C, 90 min; (2) AgOAc (2.0 eq.), (PhH), 30 °C, 3 h | 32%a |
After extensive screening of different methods for the macro lactonization, a first success with a low yield of 13% was achieved using Shiina's reagent105 (Entry 1, Table 2). Any other screened coupling reagent such as different carbodiimides, PyBOP, T3P (propylphosphonic anhydride) or Yamaguchi's reagent106 could not furnish the macro lactonization, favouring elimination. The activation of the alcohol through intramolecular Mitsunobu reaction gave similarly a low yield of 14% (Entry 2, Table 2). Finally, the highest yield of 50% for tris-lacton 18 was obtained using Gerlach's modification107 of the Corey–Nicolaou macro lactonization,108 first formation of the intermediate thioester using Corey's disulfane and subsequent silver-catalyzed cyclization at 23 °C in benzene (Entry 3, Table 2).
However, as the yield of this reaction was hardly reproducible, we finally applied 2,2′-dipyridyldisulfide to form the intermediate thioester and furnished macro lactonization in the presence of silver acetate giving 32% yield of the desired tris-lactone 18 reliably (Entry 4, Table 2).
The final quantitative, hydrogenolytic Cbz deprotection required 20 atm of hydrogen gas in a mixture of methanol, ethyl acetate and TFA. Subsequent acylation under Schotten–Baumann conditions with 2,3-diacetoxybenzoic acid chloride 19 or 2,3-dimethoxybenzoic acid chloride 20, respectively, led to the formation of (AcO)EntKL and (MeO)EntKL. EntKL could be obtained in low yield from (AcO)EntKL by mild saponification in diluted methanolic ammonia.
However, as it has been reported that acetylated siderophore-prodrugs can be activated in situ and are favorable to prevent the inactivation of catecholates by enzymatic methylation,48,76,109,110 we employed (AcO)EntKL as siderophore-prodrug and (MeO)EntKL as starting point for the synthesis of corresponding negative control probes, unable to bind iron or mediate uptake in our approach. In order to determine if the synthesized enterobactin derivatives are internalized by E. coli and P. aeruginosa during iron limitation, growth recovery assays were done with mutant strains that lack the ability to biosynthesize siderophores, and therefore require external siderophores to be able to grow under iron-limiting conditions. Importantly, iron-limiting conditions are typically found in vivo at the site of infections being part of the host immune response to prevent bacterial growth.58,111,112E. coli K-12 ΔentA grew to OD600 ≈ 0.35 in 50% MHB II medium (37 °C, t = 24 h), and this value decreased to <0.05 when 200 μM 2,2′-bipyridine (DP) was added to the media in order to simulate iron-limiting conditions. Low-micromolar concentrations of Ent restored growth, and the E. coli cultures reached OD600 ≈ 0.1, 0.15 and 0.3 in the presence of 1.0 μM, 10 μM and 15 μM Ent, respectively (Fig. 1A).
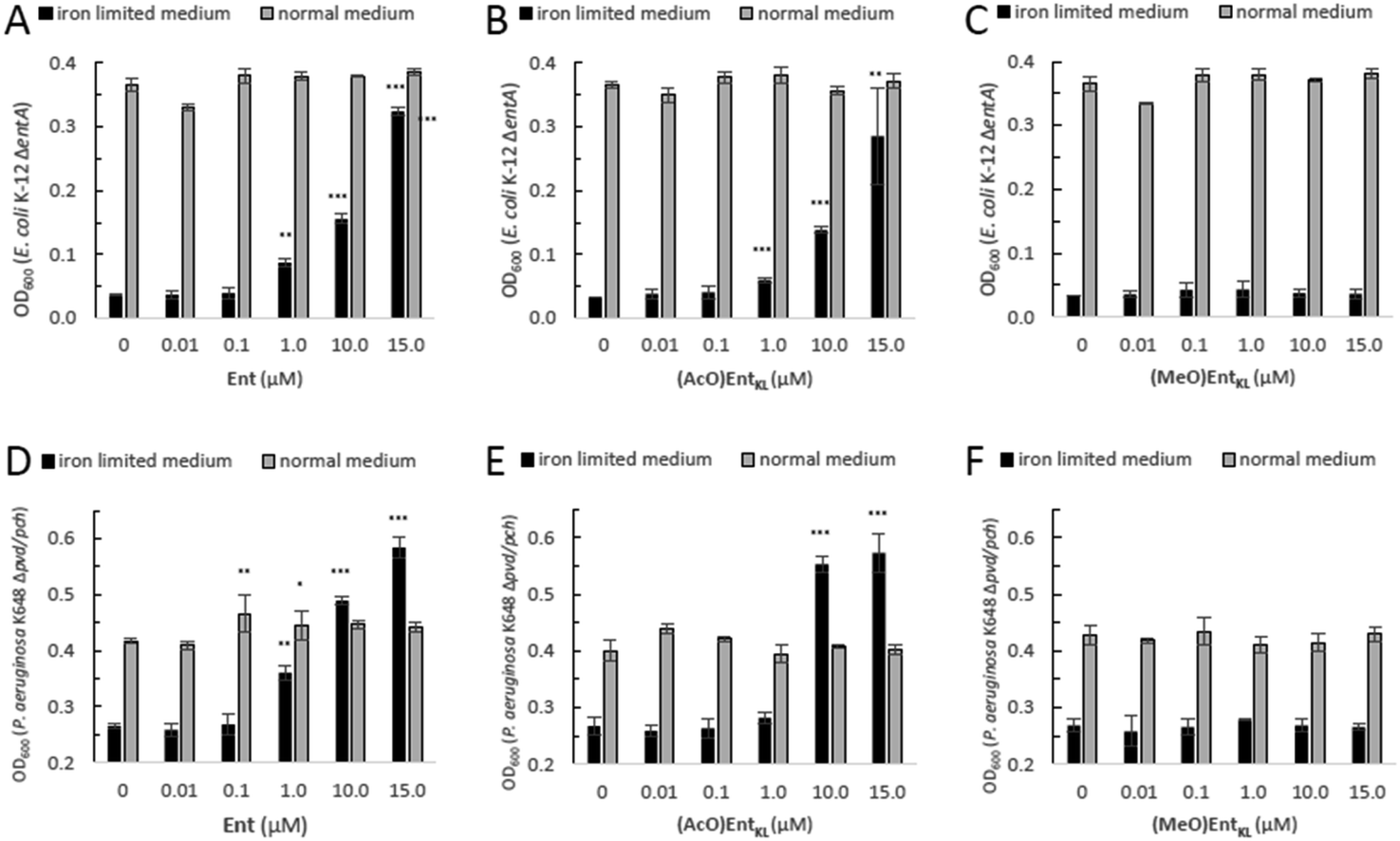 | ||
| Fig. 1 E. coli K-12 ΔentA and P. aeruginosa K648 Δpvd/pch growth recovery assays employing Ent in comparison to (AcO)EntKL and (MeO)EntKL (50% MHB II, ±200 or 600 μM DP (2,2′-bipyridine), t = 24 h, 37 °C). Gray bars: OD600 of bacteria cultured in the absence of DP. Black bars: OD600 of bacteria cultured in the presence of 200 μM (E. coli) or 600 μM (P. aeruginosa) DP. (A) Ent promotes growth recovery of E. coli. (B) (AcO)EntKL affords growth recovery of E. coli. (C) (MeO)EntKL shows no growth recovery of E. coli. (D) Ent promotes growth recovery of P. aeruginosa. (E) (AcO)EntKL affords growth recovery of P. aeruginosa. (F) (MeO)EntKL shows no growth recovery of P. aeruginosa. Each bar indicates the average of three independent replicates (two wells per replicate) and the error bars are the standard deviation of the mean. As P. aeruginosa shows a growth of <0.25 OD in the absence of an external siderophore, Y-axis is shown starting at an OD600 of 0.2. Statistical significance is indicated as a p-value < 0.05and was determined by an unpaired t-test using GraphPad Prism 9 (version 9.1.1). ***(P-value: <0.001), **(P-value: <0.005) and *(P-value: <0.05). | ||
Intriguingly, the addition of (AcO)EntKL led to restore the growth of E. coli K-12 ΔentA in the presence of DP in a concentration dependent-manner and with a similar efficiency as the natural siderophore Ent (Fig. 1A and B). A significant increase in growth was observed starting at 1 μM concentration, while at 10 μM and 15 μM the cultures grew to OD600 ≈ 0.15 and 0.3, respectively, indicating the ability of (AcO)EntKL to be internalized into the cytosol of E. coli. Similarly, P. aeruginosa K648 Δpvd/pch grew to OD600 ≈ 0.4 (37 °C, t = 24 h) and this value decreased to <0.25 in the presence of 600 μM DP. Supplementation of the iron-reduced growth medium with 1.0 μM of Ent resulted in the restoration of P. aeruginosa growth to OD600 ≈ 0.35 (Fig. 1D).
At higher concentrations of Ent of 10 μM and 15 μM the growth promoting response was even exceeding the growth in absence of DP, with P. aeruginosa K648 Δpvd/pch growing to OD600 ≈ 0.5 and 0.6, respectively. Again, (AcO)EntKL was also able to restore the growth of P. aeruginosa K648 Δpvd/pch under iron-limiting conditions in a concentration-dependent manner (Fig. 1D and E). While the growth promoting effect of (AcO)EntKL at a concentration of 1.0 μM was slightly smaller compared to Ent, the growth promoting was clearly surpassing the effect of Ent at 10 μM. In contrast to the uptake of Ent in E. coli, Ent never reaches the cytosol of P. aeruginosa as iron release takes place in the periplasm through cleavage of the siderophore backbone by the periplasmic esterase PfeE.113 This indicates that the compounds are accepted as substrates by PfeE. As expected, the permethylated probe (MeO)EntKL, was not able to restore growth of neither E. coli K-12 ΔentA nor P. aeruginosa K648 Δpvd/pch (Fig. 1C and F).
Next, we synthesized a series of cargo-conjugates of (AcO)EntKL and (MeO)EntKL attaching fluorophores and dyes that differ in their size in order to investigate whether (AcO)EntKL is able to deliver different cargo molecules to E. coli and P. aeruginosa. Therefore, we cleaved the Boc group at the amino handle in the presence of TFA in dichloromethane and reacted the resulting free amine in the presence of N-hydroxy succinimide (NHS) esters of the corresponding fluorophores and dyes (SulfoCy5-NHS, BODIPYFL-NHS and MG-NHS) obtaining the conjugates (AcO)EntKL-BODIPYFL, (AcO)EntKL-MG and (AcO)EntKL-SulfoCy5 as well as their respective negative control probes (MeO)EntKL-BODIPYFL, (MeO)EntKL-MG and (MeO)EntKL-SulfoCy5 in moderate yields as outlined in Scheme 4. Similarly, initial Boc cleavage in the presence of TFA and subsequent installation of a PEG4-N3 chain via NHS ester coupling gave access to the corresponding (AcO)EntKL-PEG4-N3 and (MeO)EntKL-PEG4-N3. Final copper(I)-mediated azide–alkyne click reaction with alkyne functionalized BODIPYs (BODIPY-alkyne and BODIPYFL-alkyne) furnished the conjugates (AcO)EntKL-PEG4-BODIPY and (AcO)EntKL-PEG4-BODIPYFL as well as their respective negative control probes (MeO)EntKL-PEG4-BODIPY and (MeO)EntKL-PEG4-BODIPYFL in moderate to good yields (Scheme 4). Finally, we synthesized (AcO)EntKL-PEG3-MG and (MeO)EntKL-PEG3-MGvia a sequence of initial Boc cleavage, installation of a PEG3-NHBoc chain via NHS ester coupling, final Boc cleavage and reaction of the free amine with MG-NHS in moderate yields.
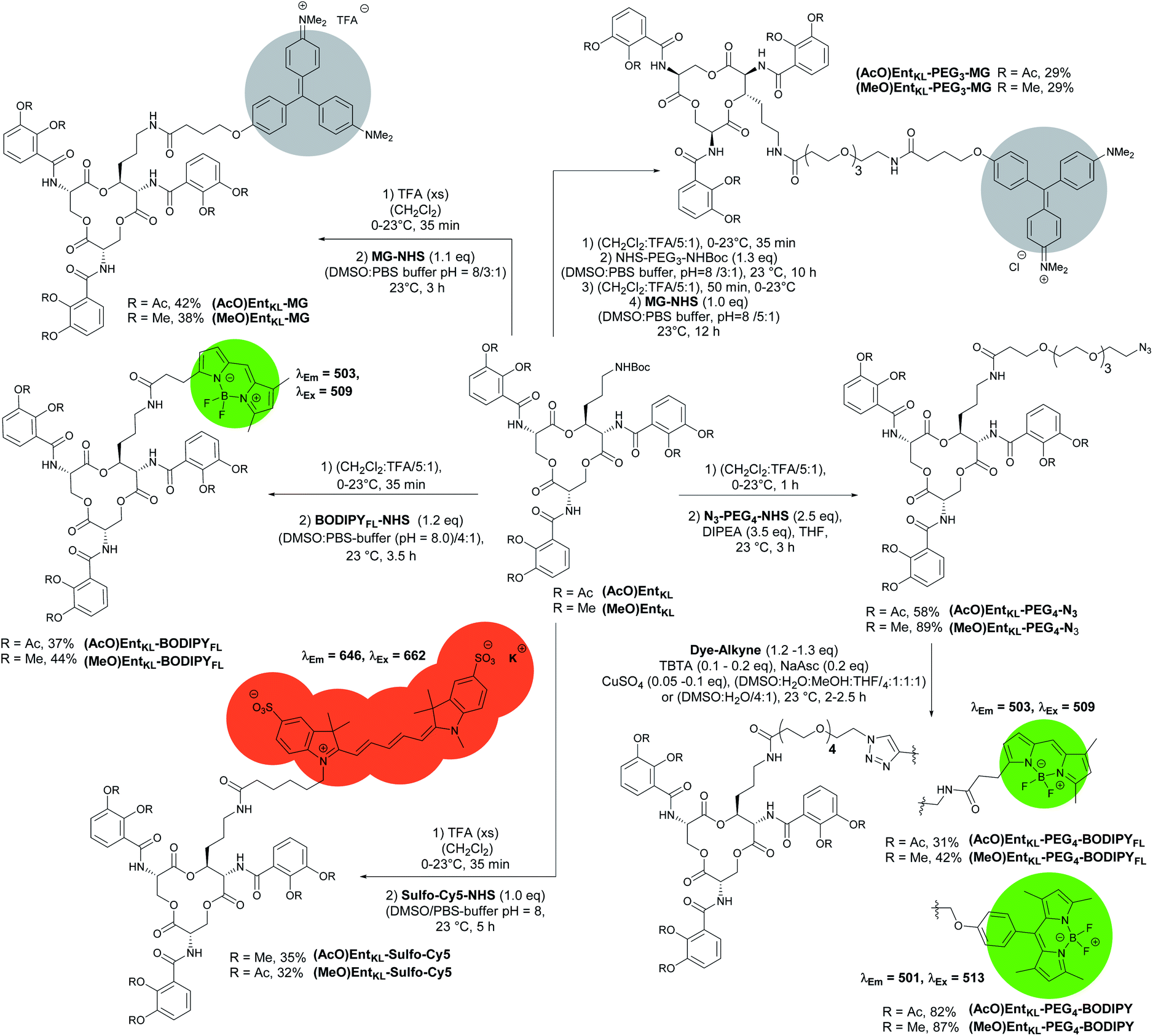 | ||
| Scheme 4 Synthesis of different EntKL-cargo conjugates bearing different fluorophores and chromophores as payload. | ||
When incubating E. coli K-12 ΔentA under iron-limiting conditions with (AcO)EntKL-PEG4-BODIPY, (AcO)EntKL-PEG4-BODIPYFL, (AcO)EntKL-BODIPYFL and (AcO)EntKL-SulfoCy5 led to a concentration-dependent growth recovery (Fig. 2A–D) clearly indicating the uptake of these cargo-conjugates to the cytosol of E. coli. The two conjugates (AcO)EntKL-MG and (AcO)EntKL-PEG3-MG bearing a malachite green derivative as cargo molecule did not lead to a growth recovery in E. coli (Fig. 2E and F). Furthermore, high concentrations of 10 μM and 15 μM seemed to reduce overall growth in E. coli K-12 ΔentA (Fig. 1E and F) as well as in the E. coli wild-type strain (see ESI: Fig. S7 and S8†). This might be due to the fact that these compounds are not taken up by the bacteria and therefore, they further reduce the available iron in the surrounding medium. Although, the malachite green derivative attached within (AcO)EntKL-MG and (AcO)EntKL-PEG3-MG has a large diameter of approximately 13.7 Å (extracted from an energy minimized structure by Chem3D® Ultra 15.1.0.144), size seems not to be the exclusive parameter for uptake. In comparison, (AcO)EntKL-SulfoCy5, bearing the rigid SulfoCy5 fluorophore with a length diameter of approximately 19.9 Å (extracted from an energy minimized structure by Chem3D® Ultra 15.1.0.144) of its indocyanine-backbone, led to a growth recovery under same conditions. Looking at the crystal structure of the enterobactin specific siderophore receptor FepA,79 the transmembrane pore opening upon recognition of Ent seems to have an elliptical inner diameter between 20-30 Å, large enough to harbor any of the reported conjugated discussed here. Therefore, additional parameters beyond size seem to determine the uptake of a bound cargo at the outer membrane receptor.
 | ||
| Fig. 2 E. coli K-12 ΔentA (A–F) and P. aeruginosa K648 Δpvd/pch (G–L) growth recovery assays employing (AcO)EntKL-PEG4-BODIPY, (AcO)EntKL-BODIPYFL, (AcO)EntKL-PEG4-BODIPYFL, (AcO)EntKL-SulfoCy5, (AcO)EntKL-MG and (AcO)EntKL-PEG3-MG (50% MHB II, ± 200 or 600 μM DP (2,2′-bipyridine), t = 24 h, 30 °C). Gray bars: OD600 of bacteria cultured in the absence of DP. Black bars: OD600 of bacteria cultured in the presence of 200 μM (E. coli) or 600 μM (P. aeruginosa) DP. (A) (AcO)EntKL-PEG4-BODIPY promotes growth recovery of E. coli. (B) (AcO)EntKL-SulfoCy5 promotes growth recovery of E. coli. (C) (AcO)EntKL-BODIPYFL promotes growth recovery of E. coli. (D) (AcO)EntKL-PEG4-BODIPYFL promotes growth recovery of E. coli. (E) (AcO)EntKL-MG is not able to restore growth of E. coli. (F) (AcO)EntKL-PEG3-MG is not able to restore growth of E. coli. As P. aeruginosa shows a growth of <0.25 OD in the absence of an external siderophore, Y-axis is shown starting at an OD600 of 0.2. (G) (AcO)EntKL-PEG4-BODIPY promotes growth recovery of P. aeruginosa. (H) (AcO)EntKL-SulfoCy5 promotes growth recovery of P. aeruginosa. (I) (AcO)EntKL-BODIPYFL promotes growth recovery of P. aeruginosa. (J) (AcO)EntKL-PEG4-BODIPYFL promotes growth recovery of P. aeruginosa. (K) (AcO)EntKL-MG promotes growth recovery of P. aeruginosa. (L) (AcO)EntKL-PEG3-MG promotes growth recovery of P. aeruginosa. Each bar indicates the average of three independent replicates (two wells per replicate) and the error bars are the standard deviation of the mean. Fig. S3–S8 (see ESI)† contain the assay results for the corresponding negative control probes (MeO)EntKL-PEG4-BODIPY, (MeO)EntKL-BODIPYFL, (MeO)EntKL-PEG4-BODIPYFL, (MeO)EntKL-SulfoCy5, (MeO)EntKL-MG and (MeO)EntKL-PEG3-MG, none of which were able to restore growth in neither E. coli nor or P. aeruginosa. Statistical significance is indicated as a p-value < 0.05 and was determined by an unpaired t-test using GraphPad Prism 9 (version 9.1.1). ***(P-value: <0.001), **(P-value: <0.005) and *(P-value: <0.05). | ||
However, size exclusion might still play a role at the ABC-type transporter mediating uptake of hydrolyzed fragments across the inner membrane.
When incubating the conjugates with P. aeruginosa K648 Δpvd/pch under iron-limiting media conditions all cargo conjugates led to clear concentration-dependent growth recovery, including (AcO)EntKL-MG and (AcO)EntKL-PEG3-MG, indicating the uptake of these cargo-conjugates to the periplasm of P. aeruginosa (Fig. 2G–L). These results are in line with the findings of Nolan and co-workers42 reporting that P. aeruginosa exhibits greater promiscuity for the uptake compared to E. coli. Looking at the crystal structure of PfeA,69 the outer membrane siderophore receptor of P. aeruginosa, a similar elliptical inner diameter between 25–35 Å can be assumed. Consistently, none of the corresponding permethylated negative control probes led to a growth recovery neither in E. coli nor P. aeruginosa (see ESI, Fig. S3–S8†). The overall growth recovery response to the added compounds was significantly higher for P. aeruginosa K648 Δpvd/pch compared to E. coli K-12 ΔentA, under non-iron-limiting conditions (Fig. 3B). The comparison of the growth recovery over all tested compounds in E. coli revealed a similar effciency of all conjugates compared to the natural siderophore Ent (Fig. 3A).
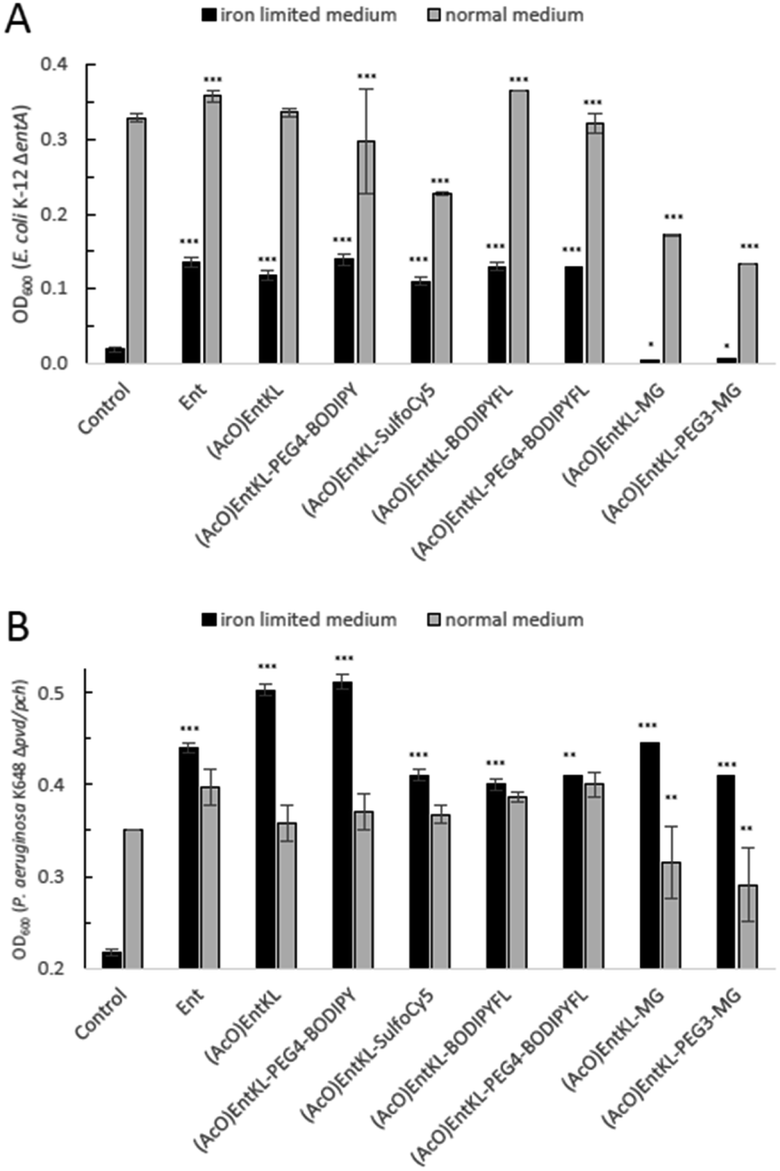 | ||
| Fig. 3 Comparison of E. coli K-12 ΔentA (A) and P. aeruginosa K648 Δpvd/pch (B) growth recovery assays employing 10 μM concentration of (AcO)EntKL-PEG4-BODIPY, (AcO)EntKL-BODIPYFL, (AcO)EntKL-PEG4-BODIPYFL, (AcO)EntKL-SulfoCy5, (AcO)EntKL-MG and (AcO)EntKL-PEG3-MG under iron-limiting and non-limiting conditions (50% MHB II, ±200 or 600 μM DP (2,2′-bipyridine), t = 24 h, 37 °C). Gray bars: OD600 of bacteria cultured in the absence of DP. Black bars: OD600 of bacteria cultured in the presence of 200 μM (E. coli) or 600 μM (P. aeruginosa) DP. Each bar indicates the average of three independent replicates (two wells per replicate) and the error bars are the standard deviation of the mean. As P. aeruginosa shows a growth of <0.25 OD in the absence of an external siderophore, Y-axis is shown starting at an OD600 of 0.2. Statistical significance is indicated as a p-value < 0.05and was determined by an unpaired t-test using GraphPad Prism 9 (version 9.1.1). ***(P-value: <0.001), **(P-value: <0.005) and *(P-value: <0.05). | ||
A similar picture was observed for the growth recovery in P. aeruginosa. However, for some of the compounds such as (AcO)EntKL and (AcO)EntKL-PEG4-BODIPY an even improved performance compared to Ent was observed (Fig. 3B). Furthermore, we were able to demonstrate that growth of P. aeruginosa mutants could be restored with (AcO)EntKL in the presence of comparably high concentrations (0.01 μM, 0.1 μM, 1.0 μM and 10 μM) of the human iron scavenger protein apo-transferrin (Kd = 10−22 M)114 (see ESI, Fig. S9†).
Further, an additional growth recovery assay was conducted with (AcO)EntKL in the presence of high concentrations (1.0 μM, 10.0 μM, 50 μM and 100 μM) bovine serum albumin (see ESI, Fig. S9†). It is worth to mention, that growth recovery was achieved with (AcO)EntKL. Albumin is responsible for the transport of lipophilic compounds and it is therefore able to bind Ent115 These results indicate the potential ability of our designed enterobactin derivative (AcO)EntKL to compete with human iron-binding and lipophilic transport serum proteins.
Next, we incubated (AcO)EntKL-BODIPYFL, (MeO)EntKL-BODIPYFL, (AcO)EntKL-PEG4-BODIPYFL and (MeO)EntKL-PEG4-BODIPYFL at different concentrations with E. coli K-12 ΔentA, P. aeruginosa K648 Δpvd/pch and their corresponding wild-type strains under iron-limiting conditions in order to proof whether uptake of the probes leads to fluorescence labelling of the bacteria, providing additional evidence for the uptake into the bacteria.115
To our delight, all assessed bacteria were fluorescently labelled when treated with either (AcO)EntKL-PEG4-BODIPYFL or (AcO)EntKL-BODIPYFL at concentrations of 10 μM (Fig. 4A, B, D and E, see ESI, Fig. S11, S13, S16 and S18†), further confirming the uptake into the wild-type and mutant strains of E. coli and P. aeruginosa. Weaker fluorescence was observed at 1.0 μM concentration of the fluorophore conjugates on all tested strains (see ESI, Fig. S11, S13, S16, S18 and Table S3†). In general, the fluorescence uptake into both mutant strains was higher compared to the wild types, and E. coli showed a significantly higher uptake than P. aeruginosa (see ESI, Table S3†).
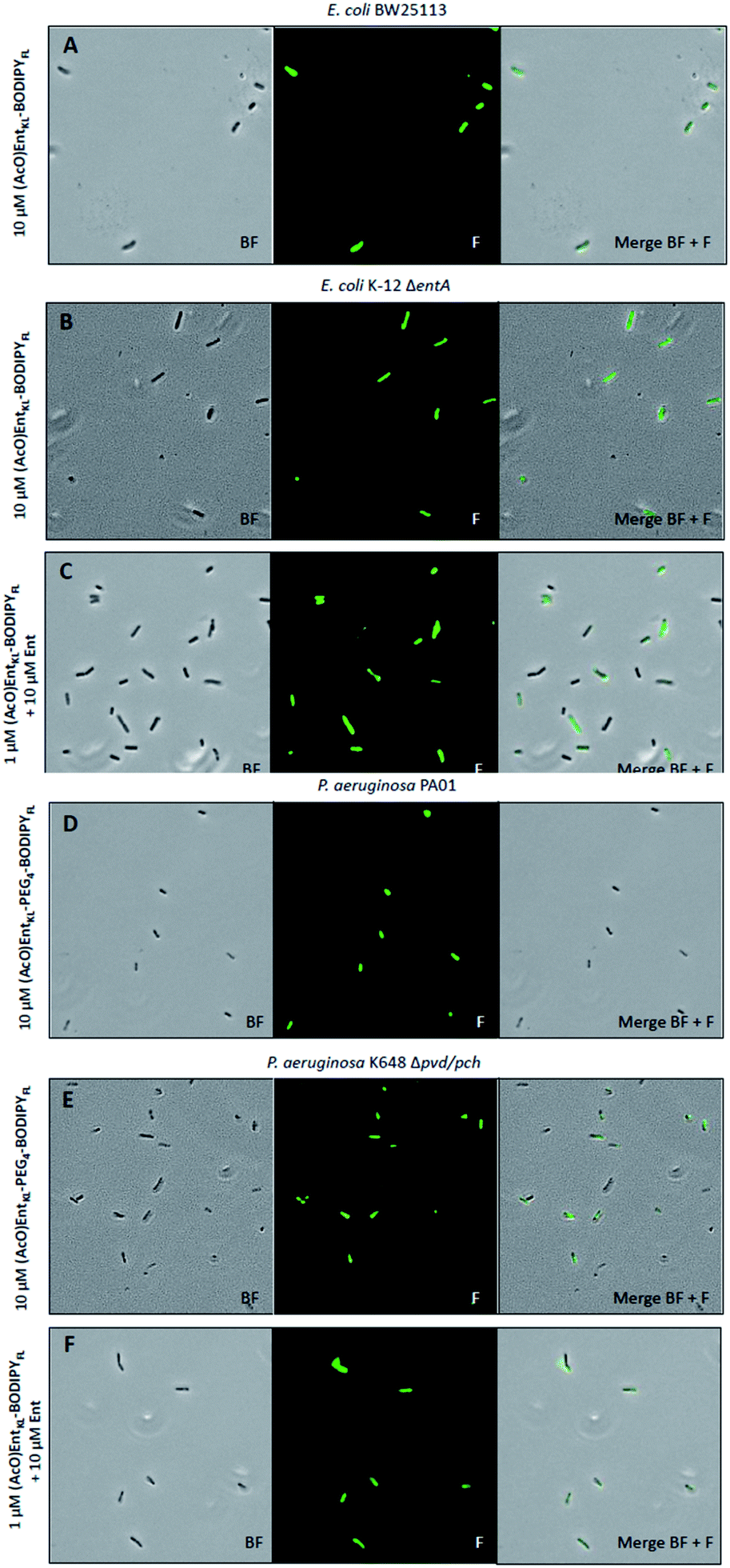 | ||
| Fig. 4 Fluorescence microscopy of E. coli BW25113, E. coli K-12 ΔentA, P. aeruginosa PA01 and P. aeruginosa K468 Δpch/pvd cultivated under iron-limiting conditions (50% MHB II, +200 or 600 μM DP (2,2′-bipyridine), t = 24 h, 37 °C) and treated with 10 μM of (AcO)EntKL-BODIPYFL (A and B), 10 μM of (AcO)EntKL-PEG4-BODIPYFL (D and E) or a mixture (AcO)EntKL-BODIPYFL (1 μM):Ent (10 μM) (C and F). All images were captured using a 40×/1.30 objective (overall magnification of 400×). BF: bright field, F: fluorescence GFP filter set (excitation: 480 nm, 20 nm bandwidth; emission: 527 nm, 15 nm bandwidth). For corrected total cell fluorescence (CTCF) of each experiment see Table S3 in the ESI.† | ||
Furthermore, we observed slight differences in the labeling performance of (AcO)EntKL-BODIPYFL and (AcO)EntKL-PEG4-BODIPYFL for the different bacteria. While (AcO)EntKL-BODIPYFL led to more prominent labelling of E. coli BW25113 and E. coli K-12 ΔentA, (AcO)EntKL-PEG4-BODIPYFL preferred of P. aeruginosa PA01 and P. aeruginosa K468 Δpvd/pch (see ESI, Table S3†). Consistently, no labelling was observed when cells were treated with either (MeO)EntKL-BODIPYFL, (MeO)EntKL-PEG4-BODIPYFL or non-conjugated BODIPYFL-alkyne alone (see ESI, Fig. S12, S14, S15, S17, S19 and S20†). Fluorescence labeling of E. coli K-12 ΔentA and P. aeruginosa K468 Δpvd/pch was also observed when treating the bacteria with mixtures of (AcO)EntKL-BODIPYFL:Ent/1:10, 1:1, 10:1 or 10:10 (Fig. S21 and S22†).
Interestingly, the fluorescence labelling for E. coli and P. aeruginosa mutant strains seems to be increased in the presence of Ent, as lower concentrations of 1.0 μM of the fluorophore conjugates led to strong fluorescence signals of similar intensity (Fig. 4C and F, see also ESI, Table S3†) compared to the 10-fold higher concentration in absence of Ent (Fig. 4B and E, see also ESI, Table S3†). We assume that the overall fitness of the bacteria is increased in the presence of additional siderophore, leading to a more efficient uptake of the fluorophore conjugates. These results underline the potential of EntKL-based reporter conjugates to serve as molecular tools for infection detection.
Finally, (AcO)EntKL, (MeO)EntKL and all derived cargo-conjugates were investigated for their antibacterial activity against the E. coli and P. aeruginosa strains and the compounds' cytotoxic activity against human HepG2 cells was assessed (see ESI, Table S4†). All tested compounds lack antibacterial and cytotoxic activity, which is important looking at (AcO)EntKL in the context of being potentially used as safe carrier molecule for the future development of antimicrobial siderophore drug conjugates that help to prevent fast resistance development. In addition, all compounds lack any cytotoxic activity against human HepG2 cells, enabling their possible application in mammalian cells. Notably, although the close derivative of Miller's artificial enterobactin analogue47(AcO)EntM displayed similar growth recovery efficacy in siderophore biosynthesis deficient mutants of E. coli and P. aeruginosa compared to (AcO)EntKL (see ESI, Fig. S10†), we found significant cytotoxic activity against human HepG2 cells with an IC50 value of 12.17 μM in our assay (see ESI, Table S4†).
Conclusions
In summary, we designed and synthesized the novel enterobactin derivative (AcO)EntKL, which retains the natural hydrolyzability of the tris-lactone scaffold, while providing an amino handle for easy attachment of cargos in the backbone. In silico studies at the DFT level of theory predicted a high thermodynamic stability combined with kinetic lability for the iron(III) complexes similarly as demonstrated earlier for the natural ferri-siderophore [Fe-Ent]3−.Growth recovery experiments with siderophore biosynthesis deficient mutants of E. coli and P. aeruginosa under iron-limiting conditions revealed an uptake of (AcO)EntKL and other EntKL-based derivatives into the tested bacteria, while, in contrast, permethylated probes based on (MeO)EntKL unable to bind iron, did not lead to any growth recovery.
Furthermore, imaging experiments under iron-limiting conditions utilizing E. coli and P. aeruginosa deficient mutants and wild-type strains showed labelling of the bacteria, further underlining the uptake of cargo conjugates.
As fluorophore uptake into both, E. coli K-12 ΔentA and P. aeruginosa K648 Δpvd/pch, was increased in the presence of supplemented Ent, we assume that overexpression of Ent would not contribute to any resistance against EntKL-based drug conjugates.
Since growth recovery was also observed in the presence of high concentrations of human apo-transferrin or albumin and no antimicrobial activity or cytotoxicity was observed, (AcO)EntKL holds potential to serve as good starting point for the assembly of antimicrobial siderophore drug conjugates to tackle infections caused by Gram-negative bacterial pathogens in humans.
Further investigations towards the determination of the size exclusion limit of siderophore receptors and the synthesis of first drug conjugates are ongoing.
Data availability
All data are available in the ESI.Author contributions
The research was conceived by P. K. The manuscript was written by P. K. with contributions of R. Z., J. C., J. G. and R. M. The ESI† was written by R. Z., P. K., J. C., J. H., J. G. and R. M. All compounds were synthesized by R. Z. Computations were planned by J. G. and conducted by J. V. Biological evaluation of the compounds was planned by J. H., R. M., J. C. and P. K. and conducted by J. C. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been carried out within the framework of the SMART BIOTECS alliance between the Technische Universität Braunschweig and the Leibniz Universität Hannover. This initiative is supported by the Ministry of Science and Culture (MWK) of Lower Saxony, Germany. Financially support by the DFG (Grant KL3012/2-1) and Fonds der Chemischen Industrie is gratefully acknowledged. The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies. The authors thank Prof. Dr Mark Brönstrup from the Helmholtz Centre of Infection Research for kindly providing bacterial strains used in this study. The authors thank the mass spectrometry unit of the Institute of Organic Chemistry at the TU Braunschweig, in particular Dr Ulrich Papke, the NMR spectroscopy unit of the Institute of Organic Chemistry, in particular Dr Kerstin Ibrom, for analytical support, Alexandra Amann for support in the biological evaluation of the compounds as well as Claire C. Jimidar, Charity S. G. Ganskow, Dr Elmira Ghabraie, Prof. Dr Stefan Schulz and Dr Anna Luisa Klahn for helpful discussion and proofreading of the manuscript.Notes and references
- P. Klahn and M. Brönstrup, Curr. Top. Microbiol. Immunol., 2016, 389, 365–417 Search PubMed.
- V. M. D'Costa, C. E. King, L. Kalan, M. Morar, W. W. L. Sung, C. Schwarz, D. Froese, G. Zazula, F. Calmels, R. Debruyne, G. B. Golding, H. N. Poinar and G. D. Wright, Nature, 2011, 477, 457–461 CrossRef PubMed.
- K. M. G. O'Connell, J. T. Hodgkinson, H. F. Sore, M. Welch, G. P. C. Salmond and D. R. Spring, Angew. Chem., Int. Ed., 2013, 52, 10706–10733 CrossRef PubMed.
- P. Klahn and M. Brönstrup, Nat. Prod. Rep., 2017, 34, 832–885 RSC.
- U. Theuretzbacher, K. Outterson, A. Engel and A. Karlen, Nat. Rev. Microbiol., 2020, 18, 275–285 CrossRef PubMed.
- H. W. Boucher, G. H. Talbot, D. K. Benjamin, J. Bradley, R. J. Guidos, R. N. Jones, B. E. Murray, R. A. Bonomo and D. Gilbert, Clin. Infect. Dis., 2013, 56, 1685–1694 CrossRef PubMed.
- H. W. Boucher, G. H. Talbot, J. S. Bradley, J. E. Edwards, D. Gilbert, L. B. Rice, M. Scheld, B. Spellberg and J. Bartlett, Clin. Infect. Dis., 2009, 48, 1–12 CrossRef PubMed.
- WHO, Prioritization of pathogens to guide discovery, research and development of new antibiotics for drug-resistant bacterial infections, including tuberculosis, World Health Organization, Geneva, WHO/EMP/IAU/2017.12, 2017 Search PubMed.
- G. Zhou, Q. Shi, X. Huang and X. Xie, Int. J. Mol. Sci., 2015, 16, 21711–21733 CrossRef CAS PubMed.
- Y.-M. Lin, M. Ghosh, P. A. Miller, U. Möllmann and M. J. Miller, BioMetals, 2019, 32, 425–451 CrossRef CAS PubMed.
- A. V. Cheng and W. M. Wuest, ACS Infect. Dis., 2019, 5, 816–828 CrossRef PubMed.
- M. J. Miller and R. Liu, Acc. Chem. Res., 2021, 54, 1646–1661 CrossRef CAS PubMed.
- R. C. Hider and X. Kong, Nat. Prod. Rep., 2010, 27, 637–657 RSC.
- K. N. Raymond, E. A. Dertz and S. S. Kim, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3584–3588 CrossRef CAS PubMed.
- M. Sánchez, L. Sabio, N. Gálvez, M. Capdevila and J. M. Dominguez-Vera, IUBMB Life, 2017, 69, 382–388 CrossRef PubMed.
- T. C. Johnstone and E. M. Nolan, Dalton Trans., 2015, 44, 6320–6339 RSC.
- V. I. Holden and M. A. Bachman, Metallomics, 2015, 7, 986–995 CrossRef CAS PubMed.
- P. Saha, B. S. Yeoh, R. A. Olvera, X. Xiao, V. Singh, D. Awasthi, B. C. Subramanian, Q. Chen, M. Dikshit, Y. Wang, C. A. Parent and M. Vijay-Kumar, J. Immunol., 2017, 198, 4293–4303 CrossRef PubMed.
- J. Behnsen and M. Rafatellu, MBio, 2016, 7, e01906-16 CrossRef PubMed.
- V. Braun and M. Braun, FEBS Lett., 2002, 529, 78–85 CrossRef CAS PubMed.
- P. E. Klebba, Front. Biosci., 2003, 8, 1422–1436 CrossRef PubMed.
- L. Ma, W. Kaserer, R. Annamalai, D. C. Scott, B. Jin, X. Jiang, Q. Xiao, H. Maymani, L. M. Massis, L. C. S. Ferreira, S. M. C. Newton and P. E. Klebba, J. Biol. Chem., 2007, 282, 397–406 CrossRef CAS PubMed.
- C. R. Smallwood, L. Jordan, V. Trinh, D. W. Schuerch, A. Gala, M. Hanson, Y. Shipelskiy, A. Majumdar, S. M. C. Newton and P. E. Klebba, J. Gen. Physiol., 2014, 144, 71–80 CrossRef CAS PubMed.
- A. S. Skwarecki, S. Milewski, M. Schielmann and M. J. Milewska, Nanomedicine, 2016, 12, 2215–2240 CrossRef CAS PubMed.
- C. Ji, R. E. Juárez-Hernández and M. J. Miller, Future Med. Chem., 2012, 4, 297–313 CrossRef CAS PubMed.
- M. G. P. Page, Ann. N. Y. Acad. Sci., 2013, 1277, 115–126 CrossRef CAS PubMed.
- K. Li, W. H. Chen and S. D. Bruner, BioMetals, 2016, 29, 377–388 CrossRef CAS PubMed.
- K. H. Negash, J. K. S. Norris and J. T. Hodgkinson, Molecules, 2019, 24, 3314 CrossRef CAS PubMed.
- I. J. Schalk, Clin. Microbiol. Infect., 2018, 24, 801–802 CrossRef CAS PubMed.
- V. Braun, A. Pramanik, T. Gwinner, M. Köberle and E. Bohn, BioMetals, 2009, 22, 3–13 CrossRef CAS PubMed.
- H. Budzikiewicz, Curr. Top. Med. Chem., 2001, 1, 73–82 CrossRef CAS PubMed.
- A. Górska, A. Sloderbach and M. P. Marszałł, Trends Pharmacol. Sci., 2014, 35, 442–449 CrossRef PubMed.
- T. A. Wencewicz and M. J. Miller, in Antibacterials. Topics in Medicinal Chemistry, ed. J. Fisher, S. Mobashery and M. J. Miller, Springer, Cham, 2017, vol. 26 Search PubMed.
- V. Braun, K. Gunthner, K. Hantke and L. Zimmermann, J. Bacteriol., 1983, 156, 308–315 CrossRef CAS PubMed.
- A. Hartmann, H.-P. Fiedler and V. Braun, Eur. J. Biochem., 1979, 99, 517–524 CrossRef CAS PubMed.
- A. Pramanik and V. Braun, J. Bacteriol., 2006, 188, 3878–3886 CrossRef CAS PubMed.
- E. M. Nolan and C. T. Walsh, Biochemistry, 2008, 47, 9289–9299 CrossRef CAS PubMed.
- S. Duquesne, D. Destoumieux-Garzón, J. Peduzzi and S. Rebuffat, Nat. Prod. Rep., 2007, 24, 708–734 RSC.
- D. Destoumieux-Garzón, J. Peduzzi, X. Thomas, C. Djediat and S. Rebuffat, BioMetals, 2006, 19, 181–191 CrossRef PubMed.
- X. Thomas, D. Destoumieux-Garzón, J. Peduzzi, C. Afonso, A. Blond, N. Birlirakis, C. Goulard, L. Dubost, R. Thai, J. C. Tabet and S. Rebuffat, J. Biol. Chem., 2004, 279, 28233–28242 CrossRef CAS PubMed.
- J. D. Palmer, B. M. Mortzfeld, E. Piattelli, M. W. Silby, B. A. McCormick and V. Bucci, ACS Infect. Dis., 2020, 6, 672–679 CrossRef CAS PubMed.
- T. Zheng, J. L. Bullock and E. M. Nolan, J. Am. Chem. Soc., 2012, 134, 18388–18400 CrossRef CAS PubMed.
- T. Zheng and E. M. Nolan, J. Am. Chem. Soc., 2014, 136, 9677–9691 CrossRef CAS PubMed.
- T. A. Wencewicz, U. Möllmann, T. E. Long and M. J. Miller, BioMetals, 2009, 22, 633–648 CrossRef CAS PubMed.
- M. J. Miller, A. J. Walz, H. Zhu, C. Wu, G. Moraski, U. Möllmann, E. M. Tristani, A. L. Crumbliss, M. T. Ferdig, L. Checkley, R. L. Edwards and H. I. Boshoff, J. Am. Chem. Soc., 2011, 133, 2076–2079 CrossRef CAS PubMed.
- W. Neumann and E. M. Nolan, J. Biol. Inorg Chem., 2018, 23, 1025–1036 CrossRef CAS PubMed.
- H. Kong, W. Cheng, H. Wei, Y. Yuan, Z. Yang and X. Zhang, Eur. J. Med. Chem., 2019, 182, 111615 CrossRef CAS PubMed.
- C. Ji, P. A. Miller and M. J. Miller, J. Am. Chem. Soc., 2012, 134, 9898–9901 CrossRef CAS PubMed.
- P. Chairatana, T. Zheng and E. M. Nolan, Chem. Sci., 2015, 6, 4458–4471 RSC.
- M. Ghosh, Y.-M. Lin, P. A. Miller, U. Möllmann, W. Boggess and M. J. Miller, ACS Infect. Dis., 2018, 4, 1529–1535 CrossRef CAS PubMed.
- M. Ghosh, P. A. Miller, U. Möllmann, W. D. Claypool, V. A. Schroeder, W. R. Wolter, M. Suckow, H. Yu, S. Li, W. Huang, J. Zajicek and M. J. Miller, J. Med. Chem., 2017, 60, 4577–4583 CrossRef CAS PubMed.
- W. Neumann, M. Sassone-Corsi, M. Raffatellu and E. M. Nolan, J. Am. Chem. Soc., 2018, 140, 5193–5201 CrossRef CAS PubMed.
- W. Neumann and E. M. Nolan, J. Biol. Inorg Chem., 2018, 23, 1025–1036 CrossRef CAS PubMed.
- T. A. Wencewicz, T. E. Long, U. Möllmann and M. J. Miller, Bioconjugate Chem., 2013, 24, 473–486 CrossRef CAS PubMed.
- S. Wittmann, M. Schnabelrauch, I. Scherlitz-Hofmann, U. Möllmann, D. Ankel-Fuchs and L. Heinisch, Bioorg. Med. Chem., 2002, 10, 1659–1670 CrossRef CAS PubMed.
- M. Ghosh, P. A. Miller and M. J. Miller, J. Antibiot., 2019, 73, 152–157 CrossRef PubMed.
- R. Liu, P. A. Miller, S. B. Vakulenko, N. K. Stewart, W. C. Boggess and M. J. Miller, J. Med. Chem., 2018, 61, 3845–3854 CrossRef CAS PubMed.
- M. G. P. Page, Clin. Infect. Dis., 2019, 69, S529–S537 CrossRef CAS PubMed.
- R. C. Scarrow, D. J. Ecker, C. Ng, S. Liu and K. N. Raymond, Inorg. Chem., 1991, 30, 900–906 CrossRef CAS.
- L. D. Loomis and K. N. Raymond, Inorg. Chem., 1991, 30, 906–911 CrossRef CAS.
- S. I. Müller, M. Valdebenito and K. Hantke, BioMetals, 2009, 22, 691–695 CrossRef PubMed.
- M. A. Fischbach, H. Lin, D. R. Liu and C. T. Walsh, Nat. Chem. Biol., 2006, 2, 132–138 CrossRef CAS PubMed.
- C. Guo, L. K. Steinberg, M. Cheng, J. H. Song, J. P. Henderson and M. L. Gross, ACS Chem. Biol., 2020, 15, 1154–1160 CrossRef CAS PubMed.
- S. M. Payne and I. B. Neilands, Crit. Rev. Microbiol., 1988, 16, 81–111 CrossRef CAS PubMed.
- H. P. Fiedler, P. Krastel, J. Müller, K. Gebhardt and A. Zeeck, FEMS Microbiol. Lett., 2001, 196, 147–151 CrossRef CAS PubMed.
- B. Ghysels, U. Ochsner, U. Möllman, L. Heinisch, M. Vasil, P. Cornelis and S. Matthijs, FEMS Microbiol. Lett., 2005, 246, 167–174 CrossRef CAS PubMed.
- K. Poole, L. Young and S. Neshat, J. Bacteriol., 1990, 172, 6991–6996 CrossRef CAS PubMed.
- C. R. Dean, S. Neshat and K. Poole, J. Bacteriol., 1996, 178, 5361–5369 CrossRef CAS PubMed.
- L. Moynié, S. Milenkovic, G. L. A. Mislin, V. Gasser, G. Malloci, E. Baco, R. P. Mccaughan, M. G. P. Page, I. J. Schalk, M. Ceccarelli and J. H. Naismith, Nat. Commun., 2019, 10, 3673 CrossRef PubMed.
- W. Zhu, M. G. Winter, L. Spiga, D. P. Beiting, L. V Hooper, S. E. Winter, W. Zhu, M. G. Winter, L. Spiga, E. R. Hughes, R. Chanin, A. Mulgaonkar, L. V Hooper and S. E. Winter, Cell Host Microbe, 2020, 27, 1–13 CrossRef PubMed.
- M. Petrik, C. Zhai, H. Haas and C. Decristoforo, Clin. Transl. Imaging, 2017, 5, 15–27 CrossRef PubMed.
- R. Nosrati, S. Dehghani, B. Karimi, M. Yousefi, S. M. Taghdisi, K. Abnous, M. Alibolandi and M. Ramezani, Biosens. Bioelectron., 2018, 117, 1–14 CrossRef CAS PubMed.
- A. A. Lee, Y. S. Chen, E. Ekalestari, S. Ho, N. Hsu, T. Kuo and T. A. Wang, Angew. Chem., Int. Ed. Engl., 2016, 55, 12338–12342 CrossRef CAS PubMed.
- M. B. Nodwell and R. Britton, ACS Infect. Dis., 2020, 7, 153–161 CrossRef PubMed.
- A. Sargun, T. C. Johnstone, H. Zhi, M. Raffatellu and E. Nolan, Chem. Sci., 2021, 12, 4041–4056 RSC.
- K. Ferreira, H. Y. Hu, V. Fetz, H. Prochnow, B. Rais, P. P. Müller and M. Brönstrup, Angew. Chem., Int. Ed., 2017, 56, 8272–8276 CrossRef CAS PubMed.
- M. Schnabelrauch, S. Wittmann, K. Rahn, U. Möllmann, R. Reissbrodt and L. Heinisch, BioMetals, 2000, 13, 333–348 CrossRef CAS PubMed.
- C. Y. Zamora, A. G. E. Madec, W. Neumann, E. M. Nolan and B. Imperiali, Bioorg. Med. Chem., 2018, 26, 5314–5321 CrossRef CAS PubMed.
- S. K. Buchanan, B. S. Smith, L. Venkatramani, D. Xia, L. Esser, M. Palnitkar, R. Chakraborty, D. Van Der Helm and J. Deisenhofer, Nat. Struct. Biol., 1999, 6, 56–63 CrossRef CAS PubMed.
- W. Rabsch, W. Voigt, R. Reissbrodt, R. M. Tsolis and A. J. Bäumler, J. Bacteriol., 1999, 181, 3610–3612 CrossRef CAS PubMed.
- K. Hantke, G. Nicholson, W. Rabsch and G. Winkelmann, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3677–3682 CrossRef CAS PubMed.
- T. A. Russo, C. D. McFadden, U. B. Carlino-MacDonald, J. M. Beanan, T. J. Barnard and J. R. Johnson, Infect. Immun., 2002, 70, 7156–7160 CrossRef CAS PubMed.
- T. C. Johnstone and E. M. Nolan, J. Am. Chem. Soc., 2017, 139, 15245–15250 CrossRef CAS PubMed.
- F. Peuckert, M. Miethke, A. G. Albrecht, L. O. Essen and M. A. Marahiel, Angew. Chem., Int. Ed., 2009, 48, 7924–7927 CrossRef CAS PubMed.
- F. Peuckert, A. L. Ramos-Vega, M. Miethke, C. J. Schwörer, A. G. Albrecht, M. Oberthür and M. A. Marahiel, Chem. Biol., 2011, 18, 907–919 CrossRef CAS PubMed.
- X. Liu, Q. Du, Z. Wang, S. Liu, N. Li, Y. Chen, C. Zhu, D. Zhu, T. Wei, Y. Huang, S. Xu and L. Gu, FEBS Lett., 2012, 586, 1240–1244 CrossRef CAS PubMed.
- R. J. Abergel, A. M. Zawadzka, T. M. Hoette and K. N. Raymond, J. Am. Chem. Soc., 2009, 131, 12682–12692 CrossRef CAS PubMed.
- A. Shanzer, J. Libman, S. Lifson and C. E. Felder, J. Am. Chem. Soc., 1986, 108, 7609–7619 CrossRef CAS PubMed.
- A. Shanzer and J. Libman, J. Chem. Soc., Chem. Commun., 1983, 846–847 RSC.
- R. J. A. Ramirez, L. Karamanukyan, S. Ortiz and C. G. Gutierrez, Tetrahedron Lett., 1997, 38, 749–752 CrossRef CAS.
- M. Meyer, J. R. Telford, S. M. Cohen, D. J. White, J. Xu, K. N. Raymond and R. V March, J. Am. Chem. Soc., 1997, 119, 10093–10103 CrossRef CAS.
- E. J. Corey and S. Bhattacharyya, Tetrahedron Lett., 1977, 3919–3922 CrossRef CAS.
- W. H. Rastetter, T. J. Erickson and M. C. Venuti, J. Org. Chem., 1981, 45, 3579–3590 CrossRef.
- H. J. Rogers, J. Chem. Soc., Perkin Trans. 1, 1995, 3073–3075 RSC.
- T. Baramov, B. Schmid, H. Ryu, J. Jeong, K. Keijzer, L. von Eckardstein, M. H. Baik and R. D. Süssmuth, Chem.–Eur. J., 2019, 25, 6955–6962 CrossRef CAS PubMed.
- K. Brandhorst and J. Grunenberg, J. Chem. Phys., 2010, 132, 184101 CrossRef.
- M. D. Walter, J. Grunenberg and P. S. White, Chem. Sci., 2011, 2, 2120–2130 RSC.
- J. Grunenberg and N. Goldberg, J. Am. Chem. Soc., 2000, 122, 6045–6047 CrossRef CAS.
- J. Grunenberg, Chem. Sci., 2015, 6, 4086–4088 RSC.
- J. Grunenberg, Angew. Chem., 2017, 129, 7394–7397 CrossRef.
- S. S. Isied, G. Kuo and K. N. Raymond, J. Am. Chem. Soc., 1976, 98, 1763–1767 CrossRef CAS PubMed.
- A. Avdeef, S. R. Sofen, T. L. Bregante and K. N. Raymond, J. Am. Chem. Soc., 1978, 100, 5362–5370 CrossRef CAS.
- J. A. Dale, D. L. Dull and H. S. Mosher, J. Org. Chem., 1969, 34, 2543–2549 CrossRef CAS.
- K. C. Nicolaou, A. A. Estrada, M. Zak, S. H. Lee and B. S. Safina, Angew. Chem., Int. Ed., 2005, 44, 1378–1382 CrossRef CAS PubMed.
- I. Shiina, R. Ibuka and M. Kubota, Chem. Lett., 2002, 286–287 CrossRef CAS.
- J. Inanaga, K. Hirata, H. Saeki, T. Katsuki and M. Yamaguchi, Bull. Chem. Soc. Jpn., 1979, 52, 1989–1993 CrossRef CAS.
- H. Gerlach and A. Thalmann, Helv. Chim. Acta, 1974, 57, 2661–2663 CrossRef CAS.
- E. J. Corey and K. C. Nicolaou, J. Am. Chem. Soc., 1974, 96, 5614–5616 CrossRef CAS.
- U. Möllmann, L. Heinisch, A. Bauernfeind, T. Köhler and D. Ankel-Fuchs, BioMetals, 2009, 22, 615–624 CrossRef PubMed.
- N. Ohi, B. Aoki, T. Kuroki, M. Matsumoto, K. Kojima and T. Nehashi, J. Antibiot., 1987, 40, 22–28 CrossRef CAS PubMed.
- N. Sukhbaatar and T. Weichhart, Pharmaceuticals, 2018, 11, 137 CrossRef CAS PubMed.
- T. Ganz, Int. J. Hematol., 2018, 107, 7–15 CrossRef CAS PubMed.
- Q. Perraud, P. Cantero, B. Roche and V. Gasser, Mol. Cell. Proteomics, 2020, 19, 589–607 CrossRef CAS PubMed.
- P. Aisen and I. Listowsky, Annu. Rev. Biochem., 1980, 49, 357–393 CrossRef CAS PubMed.
- J. B. Neilands and K. Konopka, Biochemistry, 1984, 23, 2122–2127 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02084f |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |