
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aldehyde-catalyzed epoxidation of unactivated alkenes with aqueous hydrogen peroxide†
Ierasia
Triandafillidi
ab,
Maroula G.
Kokotou
a,
Dominik
Lotter
b,
Christof
Sparr
 *b and
Christoforos G.
Kokotos
*a
*b and
Christoforos G.
Kokotos
*a
aLaboratory of Organic Chemistry, Department of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis 15771, Athens, Greece. E-mail: ckokotos@chem.uoa.gr
bDepartment of Chemistry, University of Basel, St. Johanns-Ring 19, Basel 4056, Switzerland. E-mail: christof.sparr@unibas.ch
First published on 21st June 2021
Abstract
The organocatalytic epoxidation of unactivated alkenes using aqueous hydrogen peroxide provides various indispensable products and intermediates in a sustainable manner. While formyl functionalities typically undergo irreversible oxidations when activating an oxidant, an atropisomeric two-axis aldehyde capable of catalytic turnover was identified for high-yielding epoxidations of cyclic and acyclic alkenes. The relative configuration of the stereogenic axes of the catalyst and the resulting proximity of the aldehyde and backbone residues resulted in high catalytic efficiencies. Mechanistic studies support a non-radical alkene oxidation by an aldehyde-derived dioxirane intermediate generated from hydrogen peroxide through the Payne and Criegee intermediates.
Introduction
Epoxides are versatile intermediates for the synthesis of countless irreplaceable compounds.1 Since the introduction of various versatile metal-catalyzed epoxidations pioneered by Sharpless, Jacobsen, Katsuki and others,2 alkene epoxidation has become one of the most studied reactions in organic synthesis, both in industry and academia.2 After the successful developments employing metal catalysts, much effort has been devoted to the development of protocols where a more sustainable metal-free catalyst is employed. The organocatalytic epoxidation has flourished1a,1b,2 in particular with ketone,2 acid,3 nitrile4 or iminium salt2 catalysts (Scheme 1, A). Pioneered by Adam,5 Curci,6 Yang,7 Denmark,8 Shi,9 Miller,10 Page11 and others, a variety of oxidants, such as Oxone, tert-butyl hydroperoxide (TBHP) or less commonly hydrogen peroxide (H2O2) were utilized to in situ generate the key dioxirane, peracid, imidoperoxoic acid, or oxaziridine intermediates.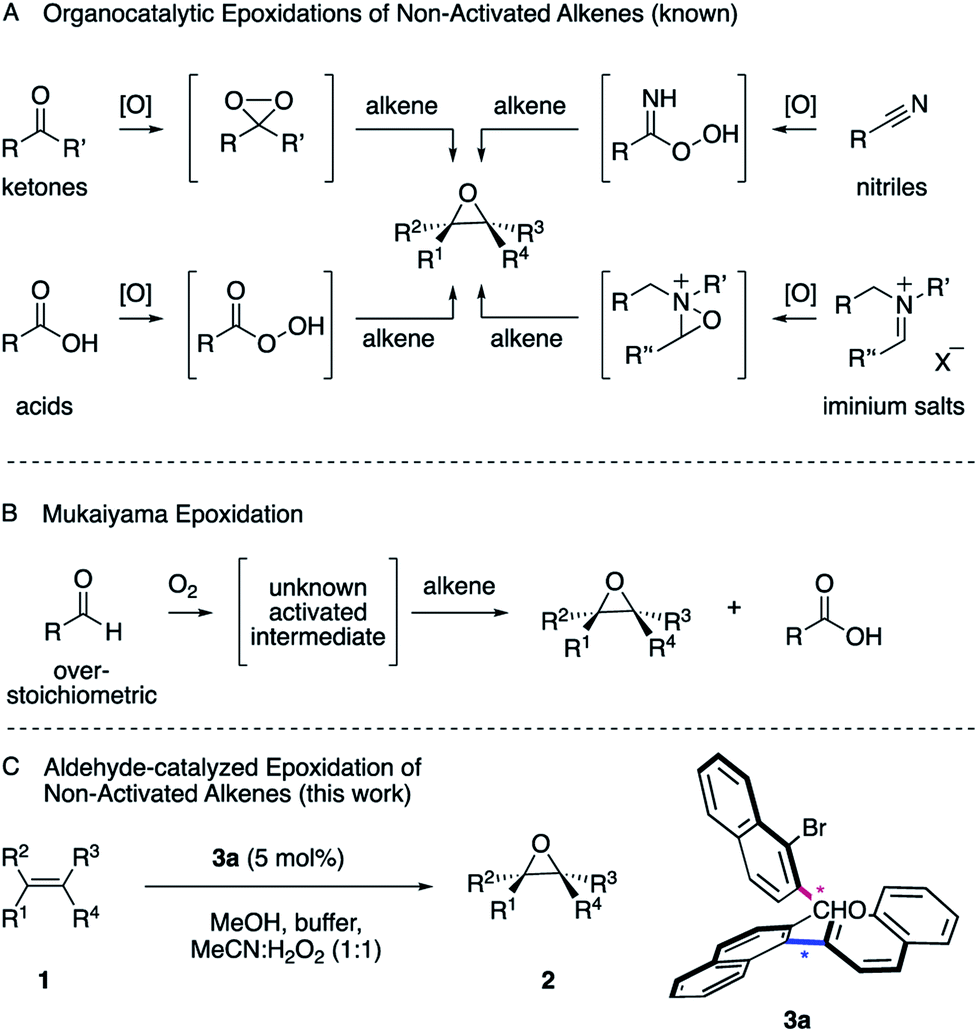 | ||
| Scheme 1 Established epoxidations and conceptualization. | ||
Over the last few years, the epoxidation of alkenes with environmentally friendly aqueous H2O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 as the oxidant is drawing increasing attention, since it is inexpensive, relatively safe and affords water as byproduct. However, hydrogen peroxide by itself is a poor oxidant for organic oxidations2,13 and thus has to be activated by a catalyst to form a reactive intermediate in order to enable the epoxidation of unactivated alkenes. As one of the most common functionalities in organic chemistry, a wide range of aldehydes are readily available and have been employed as catalysts.14 However, one of the most interesting characteristics is its auto-oxidation, where the aldehyde reacts with molecular oxygen, leading to a variety of activated intermediates.15 For instance, in the versatile Mukaiyama epoxidation, overstoichiometric amounts of aldehydes have been employed as the mediators of the reaction (Scheme 1, B).16–18 As a result, a particularly mild and selective process allowed the epoxidation of an exceptionally broad range of alkenes at room temperature employing molecular oxygen as the terminal oxidant. The process can thereby be performed with16 or without transition metal catalysts17 or in the presence of N-hydroxyphthalimide as the catalyst.18 The mechanisms of these epoxidations were studied in great detail. Nonetheless, the nature of the active oxygen species remains a subject of debate.16 Considering the broad utility of the Mukaiyama epoxidation and the ideal attributes of aqueous H2O2 as oxidant for sustainable synthesis, we anticipated the development of an aldehyde-catalyzed epoxidation of unactivated alkenes by accommodating the formyl functionality within a spatially well-defined site of an atropisomeric multiaxis system. We describe herein, that the atropisomeric two-axis aldehyde catalyst 3a that was identified as an efficient catalyst for the activation of aqueous hydrogen peroxide, allows the synthesis of a broad range of epoxides 2 from a variety of unactivated alkenes 1 with notable turnover (Scheme 1, C).
12 as the oxidant is drawing increasing attention, since it is inexpensive, relatively safe and affords water as byproduct. However, hydrogen peroxide by itself is a poor oxidant for organic oxidations2,13 and thus has to be activated by a catalyst to form a reactive intermediate in order to enable the epoxidation of unactivated alkenes. As one of the most common functionalities in organic chemistry, a wide range of aldehydes are readily available and have been employed as catalysts.14 However, one of the most interesting characteristics is its auto-oxidation, where the aldehyde reacts with molecular oxygen, leading to a variety of activated intermediates.15 For instance, in the versatile Mukaiyama epoxidation, overstoichiometric amounts of aldehydes have been employed as the mediators of the reaction (Scheme 1, B).16–18 As a result, a particularly mild and selective process allowed the epoxidation of an exceptionally broad range of alkenes at room temperature employing molecular oxygen as the terminal oxidant. The process can thereby be performed with16 or without transition metal catalysts17 or in the presence of N-hydroxyphthalimide as the catalyst.18 The mechanisms of these epoxidations were studied in great detail. Nonetheless, the nature of the active oxygen species remains a subject of debate.16 Considering the broad utility of the Mukaiyama epoxidation and the ideal attributes of aqueous H2O2 as oxidant for sustainable synthesis, we anticipated the development of an aldehyde-catalyzed epoxidation of unactivated alkenes by accommodating the formyl functionality within a spatially well-defined site of an atropisomeric multiaxis system. We describe herein, that the atropisomeric two-axis aldehyde catalyst 3a that was identified as an efficient catalyst for the activation of aqueous hydrogen peroxide, allows the synthesis of a broad range of epoxides 2 from a variety of unactivated alkenes 1 with notable turnover (Scheme 1, C).
Results and discussion
We initiated our epoxidation studies using several atropisomeric two-, three- and four-axis systems 3a–3e, which were recently prepared.19 To our delight, 5 mol% of catalyst 3a provided excellent activity for the epoxidation of α-methyl-styrene (1a) using H2O2 as the oxidant (Scheme 2), which is in stark contrast to literature precedents where overstoichiometric amounts of aldehyde were required.16–18 We next compared this remarkable finding for the epoxidation of unactivated alkenes with the other atropisomeric aldehydes comprising two to four stereogenic axes, as well as commercially available aldehydes under otherwise identical conditions (2 eq. H2O2:MeCN in t-BuOH/aq. 0.6 M K2CO3; 4 × 10−5 M EDTA tetrasodium salt, pH = 11). When aldehyde 3a was employed as the catalyst, the desired epoxide 2a was formed in 74% yield. Interestingly, the diastereomeric aldehyde 3b provided epoxide 2a in significantly lower yield (34%), underscoring the prerequisite spatial positioning of aryl and bromide moieties to augment catalyst turnover. Furthermore, the diastereomeric catalysts 3c and 3d with three stereogenic axes as well as the atropisomeric four-axis system 3e also gave considerably lower yields. While the relative configuration of 3a notably impacts activity conclusively by governing the conformation of catalytic intermediates, the enantioselectivity remained low for all cases (≤10% ee). The catalytic activity trends were also confirmed with the commercially available aldehydes without rotationally restricted axes (3f–i). Interestingly, an enhanced yield was also observed for ortho-bromonaphthaldehyde (3i) as compared to parent 2-naphthaldehyde (3h), supporting the notion that a bromide in proximity to the active site is beneficial for the reaction outcome. Nonetheless, the atropisomeric two-axis aldehyde 3a outperformed all other catalysts, highlighting the impact of bromine and aryl groups in proximity to the active site to improve the catalytic activity and turnover.
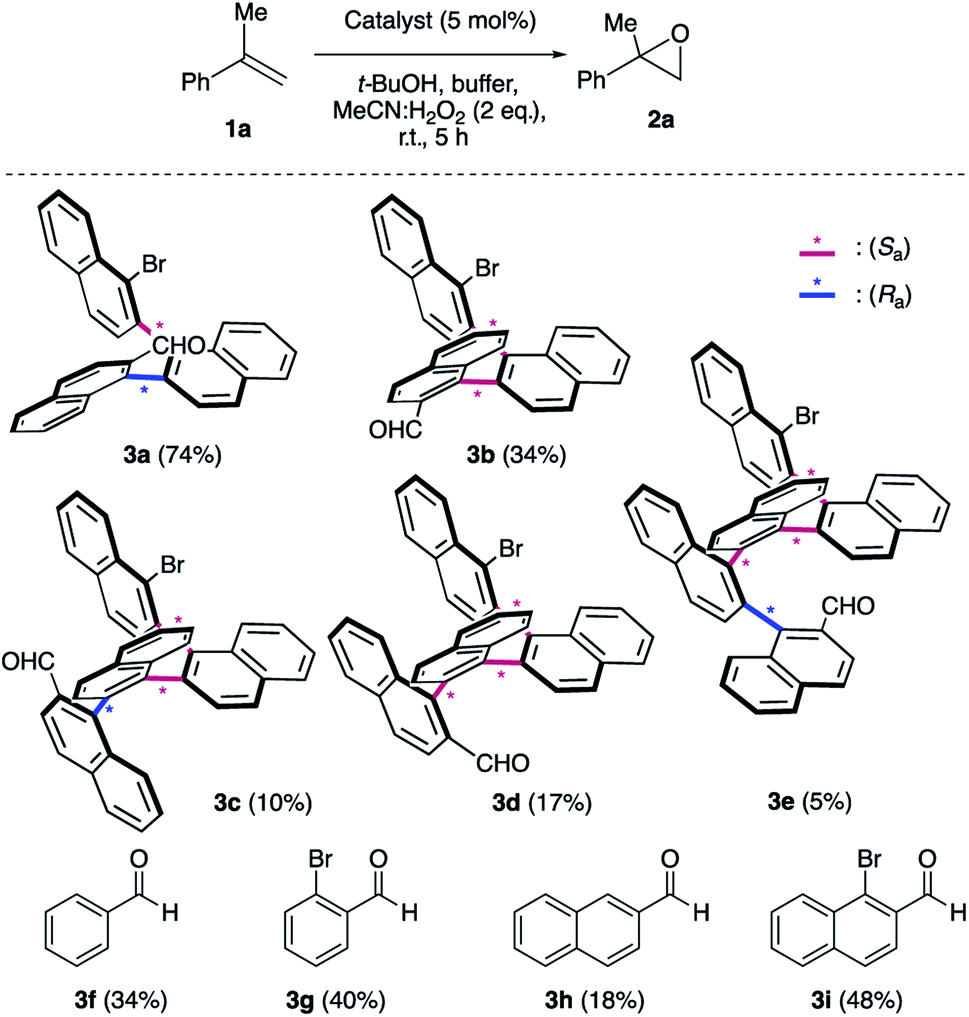 | ||
| Scheme 2 Survey of aldehyde catalysts. aK2CO3/EDTA buffer (pH = 11). The yields were determined by 1H NMR using an internal standard. | ||
Having identified the unique characteristics of catalyst 3a, we focused on optimizing the reaction conditions (Table 1). With 5 mol% of 3a in the presence of 6 eq. H2O2 and aqueous buffer in t-BuOH, the desired α-methyl styrene oxide (2a) was formed in 98% yield (entry 2). Furthermore, control experiments without catalyst, oxidant, buffer or MeCN confirmed that all components are necessary for the epoxidation (Table 1, entry 3–6). In agreement with our previous findings,20a,20b a lower pH provided the desired product in compromised yield, validating that pH = 11 is ideal for the generation of Payne's intermediate20b,21 (peroxycarboximidic acid V from MeCN and H2O2, Table 1, entry 7 and mechanism in Scheme 5). The importance of the generation of Payne's intermediate for the success of our protocol is also highlighted by the fact that in the absence of MeCN, no epoxidation is taking place (Table 1, entry 6). However, Payne's intermediate alone cannot promote the reaction to completion as can be extracted by the results of Table 1, entry 3. Using different solvents (entries 8–12), methanol was determined as the most effective medium for the reaction, forming the desired α-methyl styrene oxide in quantitative yield (entry 12). On the other hand, a decrease of the catalyst loading to 1 mol% led to a reduced yield of 83% (entry 13). Furthermore, after performing the epoxidation (Table 1, entry 12), the catalyst 3a was recovered intact (94% recovery by 1H-NMR, 82% recovery after column chromatography). To our knowledge, this is the first example, where an aldehyde is employed in substoichiometric amounts as the catalyst for an epoxidation reaction (5 mol% versus usually 10–50 mol% of an activated ketone in literature7–10 or 5–10 equivalents of pivaldehyde16–18).
|
|
||||
|---|---|---|---|---|
| Entry | 3a [mol%] | Solvent | Deviation | Yield (%) |
| a K2CO3/EDTA buffer (pH = 11). Yield determined by 1H NMR using an internal standard. | ||||
| 1 | 5.0 | t-BuOH | — | 74% |
| 2 | 5.0 | t-BuOH | 6 eq. H2O2 | 98% |
| 3 | 0 | t-BuOH | — | 9% |
| 4 | 5.0 | t-BuOH | No H2O2 | 0% |
| 5 | 5.0 | t-BuOH | No buffer | 0% |
| 6 | 5.0 | t-BuOH | No MeCN | 0% |
| 7 | 5.0 | t-BuOH | pH = 10 | 41% |
| 8 | 5.0 | EtOAc | — | 67% |
| 9 | 5.0 | MeCN | — | 82% |
| 10 | 5.0 | CH2Cl2 | — | 15% |
| 11 | 5.0 | CHCl3 | — | 12% |
| 12 | 5.0 | MeOH | — | quant. |
| 13 | 1.0 | MeOH | — | 83% |

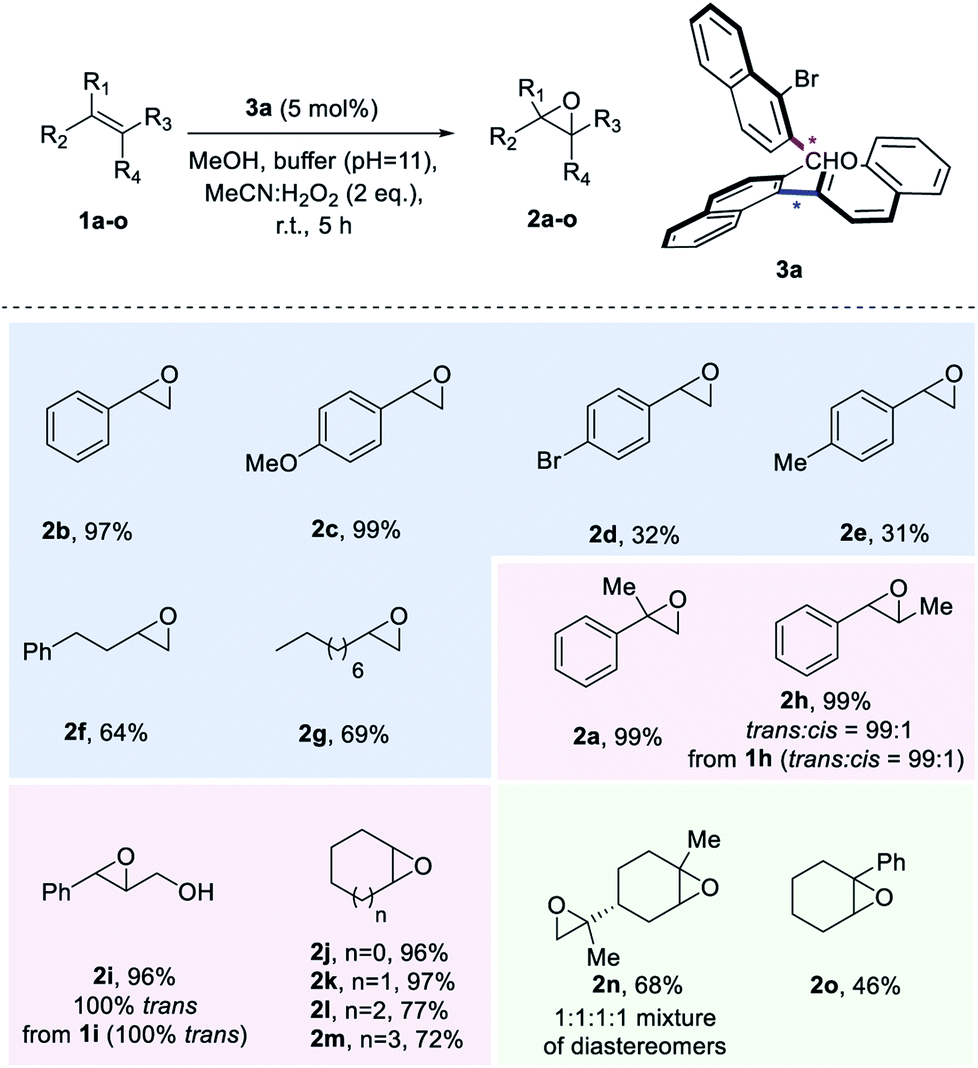 | ||
| Scheme 3 Substrate Scope. aK2CO3/EDTA buffer (pH = 11). Yields of products after isolation by column chromatography. | ||
 | ||
| Scheme 4 Mechanistic and radical clock experiments. | ||
 | ||
| Scheme 5 Proposed mechanism. | ||
With the optimized reaction conditions defined, we set out to explore the substrate scope of the aldehyde-catalysed epoxidation of unactivated alkenes (Scheme 3). A variety of monosubstituted styrenes were first tested, providing excellent isolated yields for the unfunctionalized and methoxy-substituted styrenes (2b and 2c), but low yields for 2d and 2e (in the case of 2e, 28% of the corresponding diol was also isolated). In contrast to known procedures which lead to low amounts of epoxide when terminal aliphatic alkenes are employed, synthetically meaningful yields were obtained for corresponding epoxides 2f and 2g. Gratifyingly, when studying disubstituted alkenes, geminal and vicinal disubstituted olefins afforded an excellent outcome [2a, 2h (99:1 trans:cis from 99:1 trans:cis1h) and 2i (only trans from only trans2i)] and also cyclic disubstituted alkenes provided epoxides in up to 97% isolated yield (2j–2m). To explore the limitations of the method, also trisubstituted unactivated alkene substrates were investigated, leading to a 68% yield for a double epoxidation to product 2n and an inferior performance for aryl-substituted 2o. Unfortunately, tetrasubstituted alkenes or linear cis alkenes are not viable substrates for this method, as in the former case, the desired epoxide is not formed, while in the latter case, low yields (around 16%) are observed.
Due to the novelty of aldehyde catalysis for epoxidations with hydrogen peroxide, mechanistic studies to probe the reaction pathway of this epoxidation were carried out (Scheme 4). In agreement with analogous studies on the auto-oxidation of aldehydes,15a,15b,15d,16i,18b control experiments for the epoxidation of 1a to 2a support a mechanism involving an auto-oxidation of aldehyde 3a. When the reaction was performed under an argon atmosphere (Scheme 4), only 11% product formation was observed, emphasizing the involvement of oxygen in the reaction in agreement with the epoxidation occurring through the Payne intermediate (see Table 1, entry 3). In contrast, almost quantitative yields were obtained by performing the reaction under open air or a balloon of oxygen, verifying the notion that oxygen is a constituent of the mechanism. According to literature,15a,15b,15d,16i,18b oxygen is required for the generation of the Criegee intermediate IV.22 Indeed, adding 2 mol% of mCPBA in the mechanistic experiment that was performed in the absence of oxygen, the epoxidation reaction can be restored to a good extent (42% yield, see below). Furthermore, if TEMPO or BHT were added to the reaction as a radical trap, no epoxidation product was formed, verifying the generation of radical intermediates in the autooxidation of the catalyst. When the reaction was performed in the dark, the yield of the desired epoxide 2a remained constant, proving that the reaction mechanism does not follow a photochemical pathway. In order to exclude the possibility that the epoxidation is promoted by an in situ generated peracid, we substituted catalyst 3a by its acid or alternatively by PhCOOH and observed a very low yield, which again can be assigned to the reactivity of the Payne intermediate. This is in agreement with literature,10 where an acid in combination with MeCN/H2O2 cannot promote the epoxidation reaction. These results in combination with the almost quantitative recovery of 3a from the reaction mixture, verify that a peracid is not responsible for the epoxidation, since in that case, 3a could not have been recovered intact and should have been oxidized to the corresponding acid,23 which is not capable of performing the epoxidation. In order to test the nature of the active intermediate that enables the epoxidation of the unactivated alkenes, we performed the epoxidation with the radical clock 1p. Epoxide 2p was observed in 19% yield, along with 19% of diol 2p′ and 62% of unreacted 1p, without any ring-opened product detected. This finding supports that the active oxidant is of non-radical nature, and that the radical steps during autooxidation of the aldehyde precede the epoxidation step.
A proposed mechanism consistent with the findings consequently involves the initial autooxidation of a fraction of the aldehyde 3a under aerobic conditions, forming radical intermediate I (Scheme 5).15a,15b,15d,16i,18b The peroxy-radical intermediate II and peracid III is subsequently formed with oxygen.15 The addition of peracid III to aldehyde 3a provides the Criegee intermediate IV.15b,22 At pH = 11, the Payne intermediate V is generated,4,20b,21 which is in equilibrium with VI from a reaction with peracid III. The Criegee and Payne intermediates (IV + V) result in a postulated species VII, which breaks up to form the key dioxirane intermediate VIII, the acetamide byproduct and regenerates peracid III. Another plausible pathway is the transformation of intermediate IV and V, to form III and VII′, which can also give rise to dioxirane VIII and acetamide. The dioxirane intermediate VIII as the active oxidant then reacts with unactivated alkene 1a to form epoxide 2a, regenerating aldehyde 3a to close the catalytic cycle. The crucial role of the buffer for the generation of intermediate V is highlighted in the control experiments, as well as the inability of V alone to push the reaction to completion. Oxygen from air is required for the autooxidation of the aldehyde to form intermediate IV. A small amount of peracid is necessary to promote the catalytic cycle via the generation of IV and not to perform the epoxidation, as highlighted by the recovery of 3a after reaction completion and the reinstatement of the epoxidation yield when catalytic mCPBA was added to the control experiment under argon. A finding consistent with the formation of dioxirane VIII is that 3a under Shi-like conditions with Oxone as the oxidant, simulating the formation of the dioxirane-intermediate, provided the desired epoxide 2a in good yield (65%).24
Considering this mechanistic hypothesis, we examined the possible pathways of the reaction by High Resolution Mass Spectrometry (HRMS, see ESI† for details) and thereby detected several of the intermediates of the proposed pathway. Adding TEMPO as the radical scavenger to the reaction mixture, the adducts of I and II (trapped with TEMPO) were identified, supporting that the autoxidation step is occurring. The Criegee intermediate IV and both intermediates V and VI were also detected, corroborating the formation of the Payne intermediate, when using the appropriate aqueous buffer (pH = 11). Moreover, also the central alkene oxidant dioxirane VIII was detected by HRMS. In order to rule out artifacts, the measurements were also repeated using CD3CN or 2-bromobenzaldehyde (3g) to detect similar intermediates, such as the corresponding Criegee intermediate and the dioxirane by HRMS, thus further underpinning the proposed mechanism.24 Intermediate VII was observed in the case of 3g with CD3CN, while VII′ was detected with 3a in the presence of TEMPO. In accord with these findings, both short-lived intermediates provide exploratory support of an aldehyde-catalyzed epoxidation by means of dioxirane formation.
Conclusions
In conclusion, we have developed a sustainable and efficient method for the epoxidation of unactivated alkenes utilizing an atropisomeric multiaxis aldehyde as the catalyst in combination with hydrogen peroxide as the oxidant. This study reveals the feasibility of aldehyde-catalyzed epoxidations and the mechanistic studies shed first light on this unique reactivity. To our knowledge this is the first example of an epoxidation reaction that employs an aldehyde in catalytic amounts. Ongoing studies aim to rationalize the individual requirements for catalyst design to further increase catalytic efficiency and to accomplish stereoselectivity.Data availability
Experimental data are available from the authors upon request.Author contributions
I. T., C. S. and C. G. K. conceived the study, designed the experiments, and analyzed the data. I. T. performed the experiments and M. G. K. the HRMS studies. D. L. provided catalysts 3b–3e. The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. G. K., I. T. and M. G. K. gratefully acknowledge the Hellenic Foundation for Research and Innovation (HFRI) for financial support through a grant, which is financed by 1st Call for H. F. R. I. Research Projects to Support Faculty Members & Researchers and the procurement of high-cost research equipment grant (grant number 655). I. T. and M. G. K. would like to thank State Scholarships Foundation (IKY) for financial support through a post-doctoral research scholarship funded by the “Supporting Post-Doctoral Researchers-Call B” (MIS 5033021), action of the Operational Programme “Human Resources Development, Education and Lifelong Learning” and co-funded by the European Social Fund (ESF) and Greek National Resources. The COST Action C–H Activation in Organic Synthesis CA15106 is acknowledged for supporting the Short-Term Scientific Mission of I. T. to the University of Basel and Dr D. Kaldre for helpful discussions. The Swiss National Science Foundation is gratefully acknowledged for financial support (C. S. and D. L.).Notes and references
- For selected reviews, see: (a) O. A. Wong and Y. Shi, Chem. Rev., 2008, 108, 3958 CrossRef CAS; (b) Y. Zhu, Q. Wang, R. G. Cornwall and Y. Shi, Chem. Rev., 2014, 114, 8199 CrossRef CAS; For books, see: (c) A. K. Yudin, Aziridines and Epoxides in Organic Synthesis, Wiley-VCH, Weinheim, 2006 CrossRef; (d) G. Centi, S. Perathoner and S. Abate, Modern Heterogeneous Oxidation Catalysis: Design, Reactions and Characterization, Wiley-VCH, Weinheim, 2009 Search PubMed.
- For a recent review, see: I. Triandafillidi, D. I. Tzaras and C. G. Kokotos, ChemCatChem, 2018, 10, 2521 CrossRef CAS.
- For the initial introduction of m-CBPA: N. Prileschajew, Ber. Dtsch. Chem. Ges., 1909, 42, 4811 CrossRef CAS.
- G. B. Payne, P. H. Deming and P. H. Williams, J. Org. Chem., 1961, 26, 659 CrossRef CAS.
- For selected examples, see: (a) W. Adam, R. Curci and J. O. Edwards, Acc. Chem. Res., 1989, 22, 205 CrossRef CAS; (b) W. Adam, P. B. Rao, H.-G. Degen, A. Levai, T. Patonay and C. R. Saha-Moller, J. Org. Chem., 2002, 67, 259 CrossRef CAS PubMed.
- For selected examples, see: R. Mello, M. Fiorentino, O. Sciacovelli and R. Curci, J. Org. Chem., 1988, 53, 3890 CrossRef CAS.
- For selected examples, see: (a) D. Yang, Y.-C. Yip, M.-W. Tang, M.-K. Wong, J.-H. Zheng and K.-K. Cheng, J. Am. Chem. Soc., 1996, 118, 491 CrossRef CAS; (b) D. Yang, M.-K. Wong, Y.-C. Yip, X.-C. Wang, M.-W. Tang, J.-H. Zheng and K.-K. Cheng, J. Am. Chem. Soc., 1998, 120, 5943 CrossRef CAS.
- For selected examples, see: (a) S. E. Denmark, Z. Wu, C. M. Crudden and H. Matsuhashi, J. Org. Chem., 1997, 62, 8288 CrossRef CAS PubMed; (b) S. E. Denmark and H. Matsuhashi, J. Org. Chem., 2002, 67, 3479 CrossRef CAS.
- For selected examples, see: (a) Y. Tu, Z.-X. Wang and Y. Shi, J. Am. Chem. Soc., 1996, 118, 9806 CrossRef CAS; (b) Z.-X. Wang, Y. Tu, M. Frohn, J.-R. Zhang and Y. Shi, J. Am. Chem. Soc., 1997, 119, 11224 CrossRef CAS; (c) C. P. Burke and Y. Shi, J. Org. Chem., 2007, 72, 4093 CrossRef CAS PubMed; (d) N. Nieto, I. J. Munslow, H. Fernadez-Perez and A. Vidal-Ferran, Synlett, 2008, 18, 2856 Search PubMed.
- For selected examples, see: (a) G. Peris, C. E. Jakobsche and S. J. Miller, J. Am. Chem. Soc., 2007, 129, 8710 CrossRef CAS PubMed; (b) C. E. Jakobsche, G. Peris and S. J. Miller, Angew. Chem., Int. Ed., 2008, 47, 6707 CrossRef CAS PubMed; (c) P. A. Lichtor and S. J. Miller, Nat. Chem., 2012, 4, 990 CrossRef CAS.
- For selected examples, see: (a) P. C. B. Page, G. A. Rassias, D. Bethell and M. B. Schilling, J. Org. Chem., 1998, 63, 2774 CrossRef CAS PubMed; (b) P. C. B. Page, M. M. Farah, B. R. Buckley and A. J. Blacker, J. Org. Chem., 2007, 72, 4424 CrossRef CAS PubMed.
- The environmental impact of H2O2 is a matter of debate: (a) Hydrogen Peroxide, Toxicological Overview, Public Health England, 2009, https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/337708/Hydrogen_Peroxide_Toxicological_Overview_phe_v1.pdf Search PubMed; (b) T. Mahaseth and A. Kuzminov, Mutat. Res., 2017, 773, 274 CrossRef CAS; (c) S. Fukuzumi, Y. -M. Lee and W. Nam, Chin. J. Catal., 2021, 42, 1241 CrossRef CAS; (d) Y. Xue, Y. Wang, Z. Pan and K. Sayama, Angew. Chem., Int. Ed., 2021, 60, 10469 CrossRef CAS PubMed.
- (a) G. Goor, J. Glenneberg and S. Jacobi, Hydrogen Peroxide, Wiley-VCH, Weinheim, 2012 Search PubMed. For reviews see: (b) G. Grigoropoulou, J. H. Clark and J. A. Elings, Green Chem., 2003, 5, 1 RSC; (c) G. De Faveri, G. Ilyashenko and M. Watkinson, Chem. Soc. Rev., 2011, 40, 1722 RSC.
- For a recent review, see: Q. Wang, Q. Gu and S. You, Angew. Chem., Int. Ed., 2019, 58, 6818 CrossRef CAS.
- For selected examples, see: (a) J. R. McNesby and C. A. Heller Jr, Chem. Rev., 1954, 54, 325 CrossRef CAS; (b) L. Vanoye, A. Favre-Réguillon, A. Aloui, R. Philippe and C. de Bellefon, RSC Adv., 2013, 3, 18931 RSC; (c) K. Miyamoto, J. Yamashita, S. Narita, Y. Sakai, K. Hirano, T. Saito, C. Wang, M. Ochiai and M. Uchiyama, Chem. Commun., 2017, 53, 9781 RSC; (d) A. Maity, S.-M. Hyun and D. C. Powers, Nat. Chem., 2018, 10, 200 CrossRef CAS PubMed.
- (a) T. Yamada, T. Takai, O. Rhode and T. Mukaiyama, Chem. Lett., 1991, 20, 1 CrossRef; (b) T. Yamada, T. Takai, O. Rhode and T. Mukaiyama, Bull. Chem. Soc. Jpn., 1991, 64, 2109 CrossRef CAS; (c) W. W. Nam, H. J. Kim, S. H. Kim, R. Y. N. Ho and J. S. Valentine, Inorg. Chem., 1996, 35, 1045 CrossRef CAS PubMed; (d) B. B. Wentzel, P. A. Gosling, M. C. Feiters and R. J. M. Nolte, J. Chem. Soc., Dalton Trans., 1998, 2241 RSC; (e) B. B. Wentzel, P. L. Alsters, M. C. Feiters and R. J. M. Nolte, J. Org. Chem., 2004, 69, 3453 CrossRef CAS; (f) A. C. Serra and A. Gonsalves, Tetrahedron Lett., 2011, 52, 3489 CrossRef CAS; (g) Y. Li, X. T. Zhou, S. Y. Chen, R. C. Luo, J. Jiang, Z. X. Liang and H. B. Ji, RSC Adv., 2015, 5, 30014 RSC; (h) J. Wang, M. Yang, W. Dong, Z. Jin, J. Tang, S. Fan, Y. Lu and G. Wang, Catal. Sci. Technol., 2016, 6, 161 RSC; (i) W. Zhou, J. Zhou, Y. Chen, A. Cui, F. Sun, M. He, Z. Xu and Q. Chen, Appl. Catal., A, 2017, 542, 191 CrossRef CAS.
- (a) K. Kaneda, S. Haruna, T. Imanaka, M. Hamamoto, Y. Nishiyama and Y. Ishii, Tetrahedron Lett., 1992, 33, 6827 CrossRef CAS; (b) K. R. Lassila, F. J. Waller, S. E. Werkheiser and A. L. Wressell, Tetrahedron Lett., 1994, 35, 8077 CrossRef CAS; (c) C. Lehtinen and G. Brunow, Org. Process Res. Dev., 1999, 3, 101 CrossRef CAS; (d) F. Loeker and W. Leitner, Chem.–Eur. J., 2000, 6, 2011 CrossRef CAS.
- (a) F. Minisci, C. Gambarotti, M. Pierini, O. Porta, C. Punta, F. Recupero, M. Lucarini and V. Mugnaini, Tetrahedron Lett., 2006, 47, 1421 CrossRef CAS; (b) R. Spaccini, L. Liguori, C. Punta and H. R. Bjorsvik, ChemSusChem, 2012, 5, 261 CrossRef CAS PubMed.
- D. Lotter, A. Castrogiovanni, M. Neuburger and C. Sparr, ACS Cent. Sci., 2018, 4, 656 CrossRef CAS PubMed.
- (a) D. Limnios and C. G. Kokotos, J. Org. Chem., 2014, 79, 4270 CrossRef CAS PubMed; (b) E. Voutyritsa, A. Theodorou, M. G. Kokotou and C. G. Kokotos, Green Chem., 2017, 19, 1291 RSC; (c) D. Limnios and C. G. Kokotos, ACS Catal., 2013, 3, 2239 CrossRef CAS; For examples on the use of trifluoromethyl ketones for oxidation reaction from other groups, see: (d) Q.-Q. Cheng, Z. Zhou, H. Jiang, J. H. Siitonen, D. H. Ess, X. Zhang and L. Kürti, Nat. Catal., 2020, 3, 386 CrossRef CAS; (e) M. Cai, K. Xu, Y. Li, Z. Nie, L. Zhang and S. Luo, J. Am. Chem. Soc., 2021, 143, 1078 CrossRef CAS.
- (a) G. B. Payne and P. H. Williams, J. Org. Chem., 1961, 26, 651 CrossRef CAS; (b) L. A. Arias, S. Adkins, C. J. Nagel and R. D. Bach, J. Org. Chem., 1983, 48, 888 CrossRef CAS; (c) S. Ye, J. J. Hu and D. Yang, Angew. Chem., Int. Ed., 2018, 57, 10173 CrossRef CAS.
- (a) J. Criegee, Justus Liebigs Ann. Chem., 1948, 560, 127 CrossRef. This Criegee intermediate is not to be confused with the Criegee zwitterion formed in alkene ozonolysis. (b) Z. Hassan, M. Stahlberger, N. Rosenbaum and S. Bräse, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202014974.
- (a) N. Shapiro and A. Vigalok, Angew. Chem., Int. Ed., 2008, 47, 2849 CrossRef CAS PubMed; (b) Z. E. Hamami, L. Vanoye, P. Fongarland, C. de Bellefon and A. Favre-Reguillon, J. Flow Chem., 2016, 6, 206 CrossRef CAS.
- For detailed mechanistic studies and results, see ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data, optimisation studies, mechanistic and HRMS studies. See DOI: 10.1039/d1sc02360h |
| This journal is © The Royal Society of Chemistry 2021 |