
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect synthesis of pentasubstituted pyrroles and hexasubstituted pyrrolines from propargyl sulfonylamides and allenamides†
Changqing
Ye‡
a,
Yihang
Jiao‡
a,
Mong-Feng
Chiou
a,
Yajun
Li
 a and
Hongli
Bao
*ab
a and
Hongli
Bao
*ab
aKey Laboratory of Coal to Ethylene Glycol and Its Related Technology, State Key Laboratory of Structural Chemistry, Center for Excellence in Molecular Synthesis, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, 155 Yangqiao Road West, Fuzhou, Fujian 350002, People's Republic of China. E-mail: hlbao@fjirsm.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, People's Republic of China
First published on 8th June 2021
Abstract
Multisubstituted pyrroles are important fragments that appear in many bioactive small molecule scaffolds. Efficient synthesis of multisubstituted pyrroles with different substituents from easily accessible starting materials is challenging. Herein, we describe a metal-free method for the preparation of pentasubstituted pyrroles and hexasubstituted pyrrolines with different substituents and a free amino group by a base-promoted cascade addition–cyclization of propargylamides or allenamides with trimethylsilyl cyanide. This method would complement previous methods and support expansion of the toolbox for the synthesis of valuable, but previously inaccessible, highly substituted pyrroles and pyrrolines. Mechanistic studies to elucidate the reaction pathway have been conducted.
Pyrroles are molecules of great interest in a variety of compounds including pharmaceuticals, natural products and other materials. Pyrrole fragments for example are key motifs in bioactive natural molecules, forming the subunit of heme, chlorophyll and bile pigments, and are also found in many clinical drugs, including those in Fig. 1a.1 Although many classical methods of pyrrole synthesis, including the Paal–Knorr condensation,2 the Knorr reaction,3 the Hantzsch reaction,4 transition metal-catalyzed reactions,5 and multicomponent coupling reactions,6 have been developed over many years, the efficient synthesis of multisubstituted pyrroles is still challenging. In condensation syntheses of pyrroles, the major problems lie in the extended syntheses of complex precursors and limited substitution patterns that are allowed. Multicomponent reactions are superior when building pyrrole core structures with more substituents. Among these, the [2+2+1] cycloaddition reaction of alkynes and primary amines is attractive because of the readily available alkyne and amine substrates and the ability to construct fully substituted pyrroles.7 However, with the exception of some rare examples,8 most [2+2+1] cycloaddition reactions afford pyrroles with two or more identical substituents. The synthesis of multisubstituted pyrroles with all different substituents from simple starting materials therefore remains a major challenge.
 | ||
| Fig. 1 Previous reports and this work on propargylamides transformation. | ||
Easily accessible propargylamides are classical, privileged building blocks broadly utilized for the synthesis of a large variety of heterocyclic molecules such as pyrroles, pyridines, thiazoles, oxazoles and other relevant organic frameworks.9 For example, Looper10et al. reported the synthesis of 2-aminoimidazoles from propargyl cyanamides and Eycken11 reported a method starting from propargyl guanidines which undergo a 5-exo-dig heterocyclization as shown in Fig. 1b. Subsequently, Wan12et al. revealed the cyclization of N-alkenyl propargyl sulfonamides into pyrroles via sulfonyl migration. Inspired by these transformations and multi-substituted pyrrole synthesis, we report herein a direct synthesis of pentasubstituted pyrroles and hexasubstituted pyrrolines with all different substituents from propargyl sulfonylamides and allenamides.
Previously, Zhu,13 Ji14 and Qiu13b,15 reported efficient syntheses of 2-aminopyrroles from isocyanides. Ye16 and Huang17 independently developed gold-catalyzed syntheses of 2-amino-pentasubstituted pyrroles with ynamides. Despite the many advantages of these methods, they all afford protected amines, rather than free amines. The deprotection of these amines may cause problems in further transformations of the products. Our method delivers pyrroles with an unprotected free amino group and are often complementary to the previously well-developed classical methods.
Initially, the cyclization reaction of N-(1,3-diphenylprop-2-yn-1-yl)-N-ethylbenzenesulfonamide (1a) with trimethylsilyl cyanide (TMSCN) was carried out with Ni(PPh3)2Cl2 as a catalyst, a base (Cs2CO3) and DMF as a solvent. Different metal catalysts, such as Ni(PPh3)2Cl2, Pd(OAc)2, Cu(OAc)2, and Co(OAc)2 provided the desired product with similar yields (Table 1, entries 1–4), suggesting that this reaction probably does not benefit from a metal catalyst. The reaction without any metal catalyst was conducted and as suspected, the same yield of 2a was obtained (Table 1, entry 5). In order to further optimize the reaction conditions, different bases, such as KF, K3PO4, K2CO3, KOH, KOtBu, and Et3N were investigated but failed to deliver a better result (Table 1, entries 6–11). Screening of solvents indicated that DMF is optimal for this transformation (Table 1, entries 12–16).
|
|
||||
|---|---|---|---|---|
| Entry | Cat. | Base | Solvent | Yield |
| a Reaction conditions: 1a (0.1 mmol, 1 equiv.), TMSCN (0.3 mmol, 3 equiv.), cat. (0 or 10 mol%), base (0.3 mmol, 3 equiv.) and solvent (1 mL), at 80 °C for 10 h; isolated yield. | ||||
| 1 | Ni(PPh3)2Cl2 | Cs2CO3 | DMF | 67% |
| 2 | Pd(OAc)2 | Cs2CO3 | DMF | 65% |
| 3 | Cu(OAc)2 | Cs2CO3 | DMF | 65% |
| 4 | Co(OAc)2 | Cs2CO3 | DMF | 63% |
| 5 | Cs2CO3 | DMF | 65% | |
| 6 | KF | DMF | Trace | |
| 7 | K3PO4 | DMF | Trace | |
| 8 | K2CO3 | DMF | 48% | |
| 9 | KOH | DMF | 52% | |
| 10 | KOtBu | DMF | 46% | |
| 11 | Et3N | DMF | Trace | |
| 12 | Cs2CO3 | CH3CN | 18% | |
| 13 | Cs2CO3 | DME | 23% | |
| 14 | Cs2CO3 | Toluene | Trace | |
| 15 | Cs2CO3 | DCE | Trace | |
| 16 | Cs2CO3 | Dioxane | Trace | |
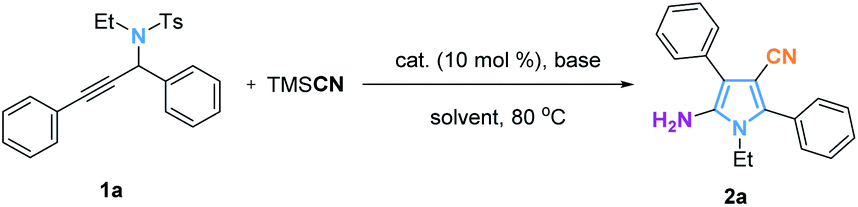
With the optimal reaction conditions in hand, we investigated the scope of this reaction. As shown in Fig. 2, the transformation tolerates a broad variety of substituted propargylamides (1). The R1 group could be an aryl group containing either electron-donating groups or electron-withdrawing groups, and the corresponding products (2b–2h) were obtained in yields of 62–80%. The substituent R1 could also be an alkyl group such as 1-hexyl in which case the reaction provided the corresponding pyrrole (2i) in 53% yield. Exploration of the R2 substituent was also conducted. Electron-rich and electron-deficient substituents in the aromatic ring of R2 gave the desired products (2j–2o) with yields of 70–81%. The product bearing a furyl group (2p) can be produced in 61% yield. However, when R2 group is an aliphatic group, the reaction failed to provide the desired product. Substituent groups R3, such as benzyl (2q) or 3,4-dimethoxyphenylethyl (2r) were also compatible in the reaction, providing the corresponding products in moderate yields. Significantly, this method has the potential to produce core structures (for example 2s) similar to that in Atorvastatin. Interestingly, when alkynyl substituted isoquinolines (1t–1v) were used as the substrates, the reactions smoothly afforded fused pyrrolo[2,1-α]isoquinoline derivatives (2t–2v), members of a class of compounds that are found widely in marine alkaloids and exhibit anticancer and antiviral activity.18
 | ||
| Fig. 2 Substrate scope of propargylamides. Reaction conditions: 1 (0.20 mmol, 1 equiv.), TMSCN (0.60 mmol, 3 equiv.), Cs2CO3 (0.60 mmol, 3 equiv.) and DMF (2 mL), at 80 °C for 10 h; isolated yield. | ||
Allenes are key intermediates in the synthesis of many complex molecules.19 As a subtype of allenes, allenamines are also useful as reaction intermediates.20 Although the transformation of allenamides to multisubstituted pyrroles has not been previously recorded, this reaction probably goes through the allenamide intermediates which can be derived from propargyl sulfonamides under basic conditions. To verify this hypothesis, the trisubstituted allenamide (3) was synthesized and subjected to the standard reaction conditions. A pyrrole (2a) was isolated in 82% yield from this reaction (Fig. 3). This result confirmed our assumption and raised a new question: is it possible to build hexasubstituted pyrrolines from tetrasubstituted allenamides? A range of tetrasubstituted allenamides21 was tested under the standard reaction conditions, and the hexasubstituted pyrrolines were obtained as is shown in Fig. 4. The R1 group could be an aryl substituent or an alkyl chain, and the corresponding products (5a–5e) were obtained with good yields. Various aryl groups with either electron-donating groups or electron-withdrawing groups in the aromatic ring of R2 provided the desired products (5f–5k) in 62–83% yields. In addition, the difluoromethyl group can also be replaced by a phenyl group, and the reaction provided the corresponding product 5l in 82% yield. It is worth noting that these pyrroline products are not easily accessible from other methods.
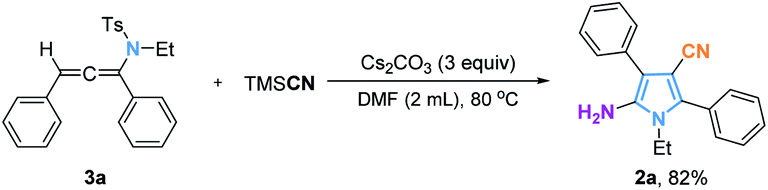 | ||
| Fig. 3 Synthesis of substituted pyrroles from allenes. | ||
 | ||
| Fig. 4 Substrate scope of tetrasubstituted allenamides. Reaction conditions: 4 (0.10 mmol, 1 equiv.), TMSCN (0.30 mmol, 3 equiv.), K2CO3 (0.30 mmol, 3 equiv.) and DMF (1 mL), at 80 °C for 10 h, isolated yield. | ||
Some synthetic applications of this method are shown in Fig. 5. The amide is a naturally occurring and ubiquitous functional group. When using benzoyl chloride to protect the free amino group of the fully-substituted pyrrole (2a), a bis-dibenzoyl amide (6) was obtained in the presence of a base, triethylamine while the monobenzoyl protected amide (7) was obtained in the presence of pyridine as the base (Fig. 5a). This method also provides a straightforward approach to pyrrole fused lactam structures (Fig. 5b). For examples, a five-membered lactam and a six-membered lactam were generated separately in a one pot reaction, directly from, (8 and 10), respectively. Taking advantage of this method, an analogue of the drug Atorvastatin was synthesized in 5 steps (Fig. 5c), demonstrating the synthetic value of the reaction.
 | ||
| Fig. 5 Synthetic applications. | ||
Mechanistic experiments were performed (Fig. 6) to explore the mechanism of the reaction. When 3 equivalents of TEMPO were added, the reaction was not inhibited and the desired product (2a) was formed in 62% yield (Fig. 6a). This result suggested that the reaction might not involve a radical process. To probe the reaction further, a kinetic study was conducted (Fig. 6b). According to this study, the propargylamide (1a) was completely converted to an allenamide (3a) in 10 min under the standard conditions. The multi-substituted pyrrole (2a) was then gradually produced from the intermediate allenamide and no other reaction intermediates were observed or identified. On the other hand, DFT calculations of substrates 3b and 4a were carried out at the B3LYP-D3(SMD)/Def2-TZVP//B3LYP-D3/Def2-SVP level of theory to identify the natural bond orbital (NBO) charges on the carbons of the allene moieties. NBO charges on the internal carbon in both 3b and 4a are 0.11 and 0.18, respectively (Fig. 6c) indicating that the nucleophilic addition of cyanide anion onto the internal carbon should be reasonable as opposed to its addition onto the terminal carbon. Pathways of the cyano addition to 3b were also calculated (Fig. 6d). The transition state of cyano addition on the internal carbon (TS1), is indeed much lower than addition on the terminal carbon (TS2). The intermediate of internal carbon addition int1, is more stable than int2, implying that the internal carbon addition pathway is not only kinetically but also thermodynamically favoured.
 | ||
| Fig. 6 Mechanistic studies and proposed mechanism. | ||
Based on the results of these mechanistic studies, a plausible reaction mechanism for the synthesis of pentasubstituted pyrroles and hexasubstituted pyrrolines is proposed and is shown in Fig. 6e. First, under basic conditions, the propargylamide isomerizes to an intermediate allenamide (A), which can be attacked nucleophilically by the cyanide anion to afford an intermediate imine (B) with release of the sulfonyl group. Then, the second cyanide anion attacks the imine to form an intermediate (C), which can undergo cyclization and protonation to afford the fully substituted pyrrole (2). Similarly, the hexasubstituted pyrroline product (5) can be obtained from double nucleophilic attack of the intermediate (A) by the cyanide ion.
Conclusions
In conclusion, we have developed a direct base-promoted synthesis of pentasubstituted pyrroles and hexasubstituted pyrrolines with all different substituents and a free amino group from propargylamides and allenamides with TMSCN. TMSCN serves as a C1 synthon for the construction of the pyrrole skeleton but also as a cyano group source. This method could complement the previously established classical methods.Data availability
The electronic supplementary information include experimental detail, computational data, NMR data, HRMS data and IR data.Author contributions
Y. J. and H. B. conceived the idea. C. Y. and Y. J. conducted the experiments and they contribute equally to this paper. M.-F. C. conducted the DFT calculation. C. Y., Y. J., and H. B. analyzed the data. C. Y., Y. L., and H. B. co-wrote the paper. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Professor Daqiang Yuan from our institute for X-ray crystallography analysis. This work was supported by the National Key R&D Program of China (Grant No. 2017YFA0700103), the NSFC (Grant No. 21922112, 21871258, and 22001251), the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB20000000).Notes and references
- (a) B. D. Roth, Prog. Med. Chem., 2002, 40, 1 Search PubMed; (b) V. Bhardwaj, D. Gumber, V. Abbot, S. Dhiman and P. Sharma, RSC Adv., 2015, 5, 15233 Search PubMed; (c) A. Kirschning, M. Ries, S. Domann, W. Martin, W. Albrecht, P. Arnold and S. Laufer, Bioorg. Med. Chem. Lett., 1997, 7, 903 Search PubMed; (d) J. LoVerme, A. Duranti, A. Tontini, G. Spadoni, M. Mor, S. Rivara, N. Stella, C. Xu, G. Tarzia and D. Piomelli, Bioorg. Med. Chem. Lett., 2009, 19, 639 Search PubMed; (e) S. Ushiyama, T. Yamada, Y. Murakami, S.-i. Kumakura, S.-i. Inoue, K. Suzuki, A. Nakao, A. Kawara and T. Kimura, Eur. J. Pharmacol., 2008, 578, 76 Search PubMed; (f) K. Arai, Y. Morikawa, N. Ubukata, H. Tsuruoka and T. Homma, J. Pharmacol. Exp. Ther., 2016, 358, 548 Search PubMed.
- C. Paal, Ber. Dtsch. Chem. Ges., 1885, 18, 367 Search PubMed.
- L. Knorr, Ber. Dtsch. Chem. Ges., 1884, 17, 1635 Search PubMed.
- A. Hantzsch, Ber. Dtsch. Chem. Ges., 1890, 23, 1474 Search PubMed.
- (a) A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084 Search PubMed; (b) Z. W. Gilbert, R. J. Hue and I. A. Tonks, Nat. Chem., 2016, 8, 63 Search PubMed; (c) I. Nakamura and Y. Yamamoto, Chem. Rev., 2004, 104, 2127 Search PubMed; (d) M. Gao, C. He, H. Chen, R. Bai, B. Cheng and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 6958 Search PubMed; (e) N. Yoshikai and Y. Wei, Asian J. Org. Chem., 2013, 2, 466 Search PubMed; (f) M. Zhang, X. Fang, H. Neumann and M. Beller, J. Am. Chem. Soc., 2013, 135, 11384 Search PubMed; (g) X. Li, M. Chen, X. Xie, N. Sun, S. Li and Y. Liu, Org. Lett., 2015, 17, 2984 Search PubMed; (h) D. Andreou, M. G. Kallitsakis, E. Loukopoulos, C. Gabriel, G. E. Kostakis and I. N. Lykakis, J. Org. Chem., 2018, 83, 2104 Search PubMed; (i) M.-B. Li, E. S. Grape and J.-E. Bäckvall, ACS Catal., 2019, 9, 5184 Search PubMed; (j) F.-L. Hong, Y.-B. Chen, S.-H. Ye, G.-Y. Zhu, X.-Q. Zhu, X. Lu, R.-S. Liu and L.-W. Ye, J. Am. Chem. Soc., 2020, 142, 7618 Search PubMed; (k) Y. Zhou, L. Zhou, L. T. Jesikiewicz, P. Liu and S. L. Buchwald, J. Am. Chem. Soc., 2020, 142, 9908 Search PubMed; (l) M. P. Doyle, K. Dong, A. Humeidi, W. Griffith, H. Arman and X. Xu, Angew. Chem., Int. Ed., 2021, 60, 13394 Search PubMed.
- (a) J. R. Chen, X. Q. Hu, L. Q. Lu and W. J. Xiao, Acc. Chem. Res., 2016, 49, 1911 Search PubMed; (b) V. Estevez, M. Villacampa and J. C. Menendez, Chem. Soc. Rev., 2014, 43, 4633 Search PubMed; (c) V. Estevez, M. Villacampa and J. C. Menendez, Chem. Soc. Rev., 2010, 39, 4402 Search PubMed; (d) F. Bellina and R. Rossi, Tetrahedron, 2006, 62, 7213 Search PubMed; (e) J. Shen, G. Cheng and X. Cui, Chem. Commun., 2013, 49, 10641 Search PubMed.
- (a) J. A. Malone, C. E. Toussel, F. R. Fronczek and R. Kartika, Org. Lett., 2019, 21, 3610 Search PubMed; (b) K. Kawakita, E. P. Beaumier, Y. Kakiuchi, H. Tsurugi, I. A. Tonks and K. Mashima, J. Am. Chem. Soc., 2019, 141, 4194 Search PubMed; (c) A. J. Pearce, X. Y. See and I. A. Tonks, Chem. Commun., 2018, 54, 6891 Search PubMed; (d) K. Matsui, M. Shibuya and Y. Yamamoto, Commun. Chem., 2018, 1, 21 Search PubMed; (e) Z. W. Davis-Gilbert, X. Wen, J. D. Goodpaster and I. A. Tonks, J. Am. Chem. Soc., 2018, 140, 7267 Search PubMed; (f) H.-C. Chiu, X. Y. See and I. A. Tonks, ACS Catal., 2018, 9, 216 Search PubMed; (g) W. Liu, H. Jiang and L. Huang, Org. Lett., 2010, 12, 312 Search PubMed; (h) Q. Li, A. Fan, Z. Lu, Y. Cui, W. Lin and Y. Jia, Org. Lett., 2010, 12, 4066 Search PubMed; (i) V. Cadierno, J. Gimeno and N. Nebra, Chem.–Eur. J., 2007, 13, 9973 Search PubMed; (j) D. Suzuki, Y. Nobe, Y. Watai, R. Tanaka, Y. Takayama, F. Sato and H. Urabe, J. Am. Chem. Soc., 2005, 127, 7474 Search PubMed; (k) R. Dhawan and B. A. Arndtsen, J. Am. Chem. Soc., 2004, 126, 468 Search PubMed; (l) N. Chatani and T. Hanafusa, J. Org. Chem., 1991, 56, 2166 Search PubMed; (m) X. Wan, D. Xing, Z. Fang, B. Li, F. Zhao, K. Zhang, L. Yang and Z. Shi, J. Am. Chem. Soc., 2006, 128, 12046 Search PubMed; (n) C. V. Galliford, J. S. Martenson, C. Stern and K. A. Scheidt, Chem. Commun., 2007, 631 Search PubMed.
- (a) Y. Cheng, C. K. Klein and I. A. Tonks, Chem. Sci., 2020, 11, 10236 Search PubMed; (b) M. Schlegel, P. Coburger and C. Schneider, Chem.–Eur. J., 2018, 24, 14207 Search PubMed; (c) D. Hong, Y. Zhu, Y. Li, X. Lin, P. Lu and Y. Wang, Org. Lett., 2011, 13, 4668 Search PubMed; (d) J. S. Yadav, B. V. Subba Reddy, M. Srinivas, C. Divyavani, S. Jeelani Basha and A. V. S. Sarma, J. Org. Chem., 2008, 73, 3252 Search PubMed.
- (a) V. A. Peshkov, O. P. Pereshivko, A. A. Nechaev, A. A. Peshkov and E. V. Van der Eycken, Chem. Soc. Rev., 2018, 47, 3861 Search PubMed; (b) K. Lauder, A. Toscani, N. Scalacci and D. Castagnolo, Chem. Rev., 2017, 117, 14091 Search PubMed; (c) S.-H. Kim-Lee, I. Alonso, P. Mauleón, R. G. Arrayás and J. C. Carretero, ACS Catal., 2018, 8, 8993 Search PubMed; (d) R. P. Singh, J. Das, M. Yousufuddin, D. Gout and C. J. Lovely, Org. Lett., 2017, 19, 4110 Search PubMed; (e) O. P. Pereshivko, V. A. Peshkov, J. Jacobs, L. V. Meervelt and E. V. Van der Eycken, Adv. Synth. Catal., 2013, 355, 781 Search PubMed; (f) Z. Jiang, P. Lu and Y. Wang, Org. Lett., 2012, 14, 6266 Search PubMed; (g) H. Song, Y. Liu and Q. Wang, Org. Lett., 2013, 15, 3274 Search PubMed; (h) G. Cheng, Y. Weng, X. Yang and X. Cui, Org. Lett., 2015, 17, 3790 Search PubMed.
- (a) R. L. Giles, J. D. Sullivan, A. M. Steiner and R. E. Looper, Angew. Chem., Int. Ed., 2009, 48, 3116 Search PubMed; (b) R. L. Giles, R. A. Nkansah and R. E. Looper, J. Org. Chem., 2010, 75, 261 Search PubMed.
- D. S. Ermolat'ev, J. B. Bariwal, H. P. Steenackers, S. C. De Keersmaecker and E. V. Van der Eycken, Angew. Chem., Int. Ed., 2010, 49, 9465 Search PubMed.
- (a) Y. Zhao, H. Wang, X. Li, D. Wang, X. Xin and B. Wan, Org. Biomol. Chem., 2016, 14, 526 Search PubMed; (b) X. Xin, H. Wang, X. Li, D. Wang and B. Wan, Org. Lett., 2015, 17, 3944 Search PubMed; (c) X. Xin, D. Wang, X. Li and B. Wan, Angew. Chem., Int. Ed., 2012, 51, 1693 Search PubMed.
- (a) G. Qiu, Q. Wang and J. Zhu, Org. Lett., 2017, 19, 270 Search PubMed; (b) P. Fontaine, G. Masson and J. Zhu, Org. Lett., 2009, 11, 1555 Search PubMed.
- X. Wang, X.-P. Xu, S.-Y. Wang, W. Zhou and S.-J. Ji, Org. Lett., 2013, 15, 4246 Search PubMed.
- D. Chen, Y. Shan, J. Li, J. You, X. Sun and G. Qiu, Org. Lett., 2019, 21, 4044 Search PubMed.
- A. H. Zhou, Q. He, C. Shu, Y. F. Yu, S. Liu, T. Zhao, W. Zhang, X. Lu and L. W. Ye, Chem. Sci., 2015, 6, 1265 Search PubMed.
- (a) L. Zhu, Y. Yu, Z. Mao and X. Huang, Org. Lett., 2015, 17, 30 Search PubMed; (b) Y. Wu, L. Zhu, Y. Yu, X. Luo and X. Huang, J. Org. Chem., 2015, 80, 11407 Search PubMed.
- R. J. Andersen, D. J. Faulkner, C. H. He, G. D. Van Duyne and J. Clardy, J. Am. Chem. Soc., 1985, 107, 5492 Search PubMed.
- (a) S. Ma, Acc. Chem. Res., 2009, 42, 1679 Search PubMed; (b) S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074 Search PubMed; (c) J. Ye and S. Ma, Acc. Chem. Res., 2014, 47, 989 Search PubMed.
- (a) L. L. Wei, H. Xiong and R. P. Hsung, Acc. Chem. Res., 2003, 36, 773 Search PubMed; (b) T. Lu, Z. Lu, Z. X. Ma, Y. Zhang and R. P. Hsung, Chem. Rev., 2013, 113, 4862 Search PubMed.
- (a) M. Taj Muhammad, Y. Jiao, C. Ye, M.-F. Chiou, M. Israr, X. Zhu, Y. Li, Z. Wen, A. Studer and H. Bao, Nat. Commun., 2020, 11, 416 Search PubMed; (b) Q. Zhang, M. T. Muhammad, M.-F. Chiou, Y. Jiao, H. Bao and Y. Li, Org. Lett., 2020, 22, 5261 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2024921 and 2024922. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02090k |
| ‡ These two authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |