
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCellular delivery of dinucleotides by conjugation with small molecules: targeting translation initiation for anticancer applications†
Natalia
Kleczewska‡
 a,
Pawel J.
Sikorski‡
a,
Zofia
Warminska
ab,
Lukasz
Markiewicz
a,
Renata
Kasprzyk
abc,
Natalia
Baran
ad,
Karina
Kwapiszewska
e,
Aneta
Karpinska
e,
Jaroslaw
Michalski
e,
Robert
Holyst
e,
Joanna
Kowalska
*c and
Jacek
Jemielity
*a
a,
Pawel J.
Sikorski‡
a,
Zofia
Warminska
ab,
Lukasz
Markiewicz
a,
Renata
Kasprzyk
abc,
Natalia
Baran
ad,
Karina
Kwapiszewska
e,
Aneta
Karpinska
e,
Jaroslaw
Michalski
e,
Robert
Holyst
e,
Joanna
Kowalska
*c and
Jacek
Jemielity
*a
aCentre of New Technologies, University of Warsaw Banacha 2c, 02-097 Warsaw, Poland. E-mail: j.jemielity@cent.uw.edu.pl
bCollege of Inter-Faculty Individual Studies in Mathematics and Natural Sciences, University of Warsaw, Banacha 2c, 02-097 Warsaw, Poland
cDivision of Biophysics Institute of Experimental Physics, Faculty of Physics University of Warsaw, Pasteura 5, 02-093 Warsaw, Poland. E-mail: jkowalska@fuw.edu.pl
dFaculty of Biology University of Warsaw, I. Miecznikowa 1, 02-096 Warsaw, Poland
eInstitute of Physical Chemistry Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland
First published on 29th June 2021
Abstract
Targeting cap-dependent translation initiation is one of the experimental approaches that could lead to the development of novel anti-cancer therapies. Synthetic dinucleoside 5′,5′-triphosphates cap analogs are potent antagonists of eukaryotic translation initiation factor 4E (eIF4E) in vitro and could counteract elevated levels of eIF4E in cancer cells; however, transformation of these compounds into therapeutic agents remains challenging – they do not easily penetrate into cells and are susceptible to enzymatic cleavage. Here, we tested the potential of several small molecule ligands – folic acid, biotin, glucose, and cholesterol – to deliver both hydrolyzable and cleavage-resistant cap analogs into cells. A broad structure–activity relationship (SAR) study using model fluorescent probes and cap–ligand conjugates showed that cholesterol greatly facilitates uptake of cap analogs without disturbing the interactions with eIF4E. The most potent cholesterol conjugate identified showed apoptosis-mediated cytotoxicity towards cancer cells.
Introduction
Nucleoside and dinucleoside oligophosphates, and their synthetic analogs, are a class of biologically active compounds which often act as modulators of metabolic pathways and protein activity in cells and, therefore, have great therapeutic potential.1 However, in vivo applications of these compounds are largely limited due to poor cellular permeability arising from high polarity and a negative net charge at physiological pH.2 To date, the cell-permeability issue for nucleotides has been fully addressed only for nucleoside monophosphates,3,4 which can be disguised as pronucleotides that contain charge-masking phosphate modifications that undergo removal after delivery into cells.5–7 However, such an approach is not suitable for (di)nucleoside di- and triphosphates because fully charge-masked pyrophosphate bonds are chemically unstable and undergo rapid hydrolysis under aqueous conditions. Therefore, alternative approaches have been developed for the delivery of nucleoside oligophosphates into cells, including application of nanogels,8 nanoparticles,9 and naturally-inspired or artificial molecular transporters.10,11 Meier et al. have recently developed cell-permeable nucleoside diphosphate and triphosphate prodrugs (DiPPro and TriPPro, respectively) that carry highly lipophilic masking groups exclusively at the terminal phosphate, which has yielded a significant breakthrough in this field.12–14 However, the issue of delivering dinucleoside oligophosphates and other structurally related compounds into cells remains largely unaddressed.15 Here, we explore whether ligand-mediated cellular delivery of dinucleoside oligophosphates can resolve this problem.As model dinucleotides with highly promising biological activity in vitro but limited applications in vivo due to permeability issues, we used 7-methylguanosine 5′ cap analogs. 7-Methylguanosine triphosphate is attached to the 5′ end of all eukaryotic messenger RNAs (mRNAs), forming the so called 5′ cap.16,17 This cap protects mRNA from premature degradation and participates in initiation of translation by interacting with eukaryotic translation initiation factor 4E (eIF4E). eIF4E is one of the components of translation initiation complex eIF4F, which guides ribosome recruitment to mRNA during protein biosynthesis.18 eIF4E has been found to act as a regulator of cap-dependent translation that strongly affects translation of selected intracellular mRNAs. These transcripts (“weak” mRNAs) usually have long structured 5′-untranslated regions (UTRs) and, interestingly, many encode proteins responsible for angiogenesis (VEGF-A), cell proliferation (c-myc), cell survival (Bcl-2), and other aspects of oncogenesis.19 Increased eIF4E expression is associated with poor prognosis in cancer patients20 and induces cancer transformation of primary epithelial cells and fibroblasts.21,22 In contrast, downregulation of eIF4E levels inhibits melanoma proliferation and invasion.23 Therefore, various therapeutic strategies for targeting eIF4E have been proposed to combat carcinogenesis.24,25 These include antisense oligonucleotides and siRNAs to modulate eIF4E mRNA levels,26,27 aptamers which target eIF4E,28,29 and small molecules to disrupt the stability of the eIF4F complex either by preventing eIF4E binding to the mRNA 5′ cap or by hindering association of eIF4E with the eIF4G scaffold protein.30,31
 | ||
| Fig. 1 (A) mRNA 5′ cap structure, (B) ligand selection process methodology using fluorescent probes, (C) mode of action for translation inhibitors (cap-ligands) and their cellular delivery and interaction with eIF4E. The folic acid – receptor-mediated endocytosis pathway and passive diffusion for cholesterol are displayed. Created with https://www.BioRender.com. | ||
Synthetic cap analogs have been studied as potential eIF4E antagonists for nearly three decades,32 but their activity was limited by poor cell permeability. Of these, 7-alkylaryl 5′-monophosphates were identified as potent eIF4E antagonists. To enable cellular uptake, the compounds have been converted to several prodrugs including tryptamine phosphoramidates of N7-benzyl GMP (4Ei-1) and 7-Cl-phenylethylene GMP that release active drugs inside cells.33–35 However, this strategy is applicable only to monophosphorylated cap analogs, which in vitro have much lower activity than their di-, and triphosphate counterparts.36
A particularly promising group of cap-derived eIF4E antagonists are oligophosphorylated mono- and dinucleotide cap analogs that have high affinity for eIF4E and are resistant to degradation by the cap-specific pyrophosphatase DcpS.37,38 Numerous DcpS-resistant dinucleotide cap analogs have been designed by replacing one or more oxygen atoms in the triphosphate bridge with another atom or group of atoms (e.g. compounds carrying non-bridging γ-O-to-S, γ-O-to-BH3, β-O-to-BH3,39,40 bridging β-γ-O-to-CH2 or β-γ-O-to-NH,41–43 or 5′-O-to-S [5′-PSL]44) and have shown superior potency and stability in rabbit reticulocyte lysates. However, the potential use of these compounds has never been demonstrated in vivo.
Here, we sought to develop a ligand-based approach for the delivery of dinucleotide cap analogs into cells that would also be applicable also to other biologically relevant dinucleoside oligophosphates. As potential transporters, we evaluated several small molecule ligands previously identified as transporting vehicles for various types of (macro)biomolecules (Fig. 1). The tested ligands included folic acid, which uses a receptor-mediated endocytosis pathway;45 biotin, which is dominantly taken up by high-affinity biotin transporters;46 glucose, which enters cells through facilitated diffusion;47 and cholesterol, which facilitates passive diffusion of small molecules into cells.48 To select the most active ligands and ideal cell culture models, we first studied simple fluorescent probes using flow cytometry, confocal microcopy, and fluorescence correlation spectroscopy (FCS). Based on those studies we synthesized several cap analogs decorated with select ligands and a fluorescent dye to verify that the ligands are capable of transporting negatively charged dinucleoside oligophosphates into cells. After confirming the proof of concept, we synthesized a series of cap analogs differing in susceptibility to DcpS and conjugated those to the most potent ligands to examine their biological activity both in vitro and towards breast cancer cells. As a result, we identified several compounds with good cellular permeability, high activity, and stability in vitro, and ability to induce apoptosis in cancer cells.
Results and discussion
Synthesis of fluorescent probes and their permeability assessment
We first verified the ability of select ligands to transport polar molecules into cells and choose optimal cell culture models for further experiments. Probes 1–4 were thus designed consisting of the non-permeable fluorescent dye fluorescein (FAM) conjugated to folic acid, biotin, glucose, or cholesterol via a polyether linker (Fig. 2A).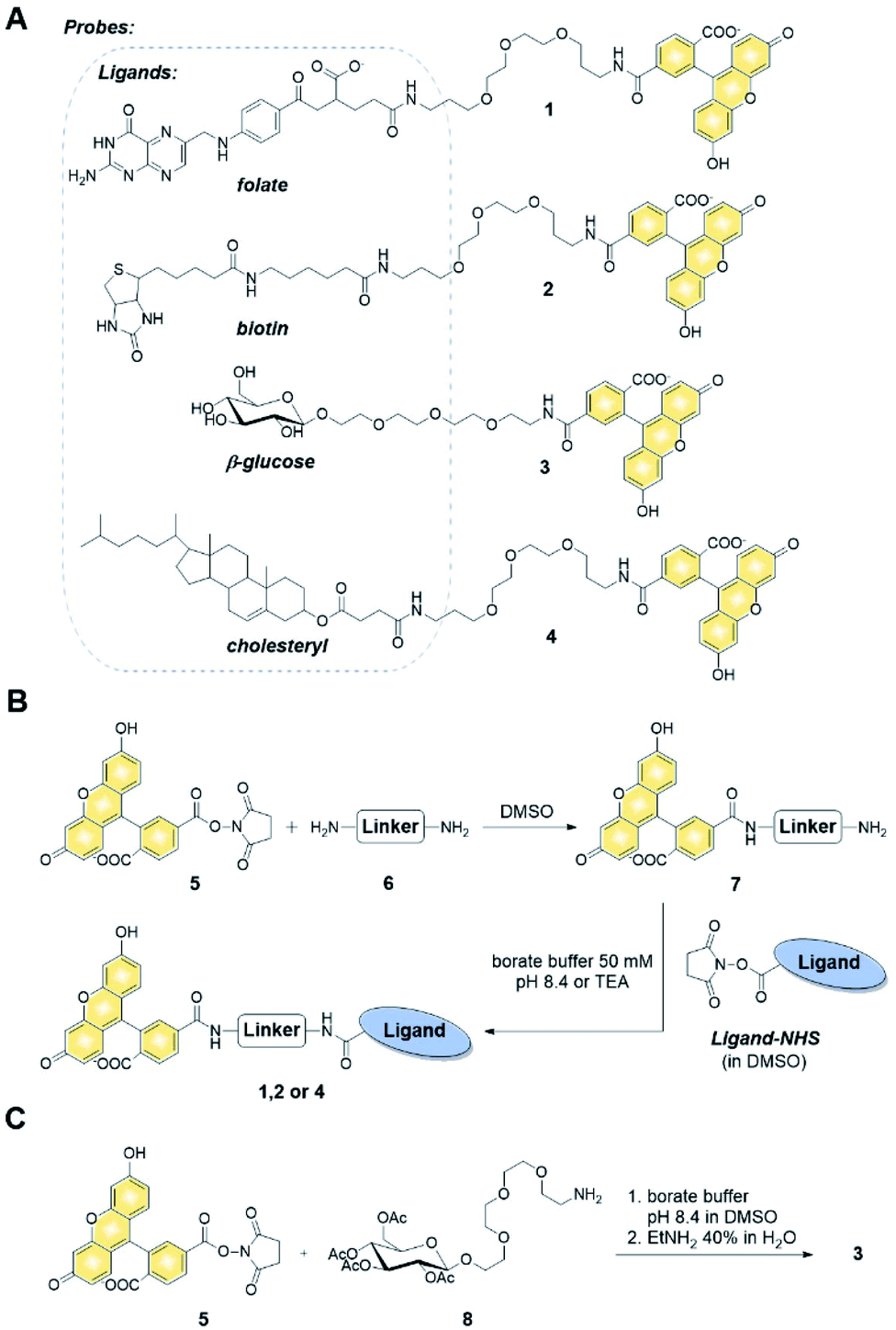 | ||
| Fig. 2 Chemical synthesis of probes 1–4 (A) structures of probes 1-4, (B) synthesis of compounds 1, 2 and 4via NHS chemistry: linker: 4,7,10-trioxa-1,13-diaminotridecane (6), ligand-NHS: folate-NHS, biotin-NHS, cholesteryl hemisuccinate-NHS, (C) synthesis of compound 3. | ||
The probes were synthesized by taking advantage of N-hydroxysuccinimide chemistry (NHS) and previously reported procedures (Fig. 2B and C; ESI†).49–51 The cellular permeability of the probes was evaluated in three human cancer cell lines: MDA-MB-231, which overexpress folate receptor (FR) α for endosomal uptake of folic acid;52 SK-RB-3, which expresses the SMVT1 receptor for biotin46 and the GLUT-1 transporter for glucose;53 and A549, which expresses none of these receptors.54 Potential uptake of probes 1–4 was initially assessed by flow cytometry. Cells were incubated with the probes for 2 h followed by flow cytometry analysis of the cells to quantify FAM emission. Folic acid-conjugated probe 1 efficiently increased FAM emission of MDA-MB-231 cells, but not A549 cells (Fig. 3A), suggesting that delivery via receptor-mediated endocytosis might occur indeed in the former case. Neither the biotin-conjugated probe 2 nor β-glucose-conjugated probe 3 affected the FAM emission in any of the studied cell lines (Fig. 3B and C). In contrast, the cholesterol-conjugated probe 4 increased FAM emission for all studied cells lines (Fig. 3D), suggesting either internalization via a diffusion-based mechanism or unspecific binding to cell membrane.
 | ||
| Fig. 3 Initial selection of cell-permeable ligands. (A–D) Specific cell lines were incubated with FAM-labeled probes and their cellular uptake were assessed by flow cytometry. Histograms represent overlaid flow cytometry data as a percentage of unstained and FAM-ligand (50 nM) – stained cells. (A) Probe 1 uptake in MDA-MB-231 cells compared to A549 cells. Probe 2–4 uptake in SK-BR-3 and MDA-MB-231 cells compared to A549 cells, respectively (B–D). Normalized FCS autocorrelation curves obtained in MDA-MB-231 cells incubated with (E) FAM-PEG, probe 1 (F) or 4 (G). Confocal imaging/FLIM of a cell incubated with 50 nM probe 1 (H) or 4 (I). FLIM image colored according to the scheme: blue – fluorescence lifetime < 2.4 ns (autofluorescence), red – fluorescence lifetime > 2.6 ns (probe). | ||
To further qualitatively and quantitatively evaluate the uptake of probes 1 and 4 into MDA-MB-231 cells we used FCS method, which verifies probe localization and estimates the size of fluorescent entities. FCS enables the measurement of the diffusion coefficients for various fluorescently-labeled probes in the cell.55 By knowing the hydrodynamic radius of the studied probe and employing a cytoplasm viscosity model for various length scales, one can predict the theoretical coefficient for the probe freely diffusing in the cytoplasm and compare it to experimental values.56–58 If the value measured in the cells is lower than the predicted value, it indicates binding of the probe to the cellular components. To that end, the hydrodynamic radii of probes 1 and 4 (Tables S1 and S2†) and cytoplasmic viscosity of MDA-MB-231 cells were first established by FCS (Table S3 and Fig. S1†).
The cellular uptake of these probes, along with linker-functionalized fluorescein (FAM-PEG), were then studied by FCS. MDA-MB-231 cells were incubated for 1 h with 1, 4, or FAM-PEG and subjected to FCS measurements based on which the autocorrelation curves were calculated (Fig. 3E–G). For compounds 1 and 4, FCS autocorrelation curves were fitted with a two-component free diffusion model (Table 1, Table S1†) while for the FAM-PEG probe, no autocorrelation of fluorescence signal was detected. For both probes, the diffusion coefficients for the first, faster component (D1) were in line with values expected for free diffusion of the probes in cytosol (187 μm2 s−1 and 205 μm2 s−1, respectively, Table 1), whereas the slower components (D2) showed high variability from 1 μm2 s−1 to 14 μm2 s−1. Such D2 values are characteristic for large proteins and small membrane structures.
| Probe | FAM-PEG-FA (1) | FAM-PEG-CHOL (4) | m7GpppA-FAM-FA (15) | m7GpppA-FAM-CHOL (17) |
|---|---|---|---|---|
| a D cyto – diffusion coefficient in the cytoplasm. | ||||
| D cyto [μm2 s−1], predicted for free probe in cytosol | 187 | 205 | 96.6 | 58.5 |
| D 1 [μm2 s−1], faster component in FCS | 184 ± 56 | 203 ± 29 | 87.0 ± 43.6 | 58.2 ± 12.4 |
| D 2 [μm2 s−1], slower component in FCS | 1–9 high variability | 3–14 high variability | 0.1–9.8 high variability | 0.1–5 high variability |
No components with D < 0.1 μm2 s−1, which is typically interpreted as active transport of endosomes, was observed for any of the probes. To verify localization of probes 1 and 4, fluorescent lifetime imaging (FLIM) was performed (Fig. 3H and I). FLIM, in contrast to typical confocal microscopy, enables differentiation of emission coming from the probe (characterized by long fluorescence lifetimes, t > 2.6 ns) from autofluorescence of the cell (t < 2.4 ns). These observations confirmed the presence of compounds 1 and 4 in MDA-MB-231 cells and revealed mostly cytoplasmatic localization for both. Overall, these data indicate that both folic acid and cholesterol are good candidates for studying cap analog delivery into cancer cells. The initial evaluation in different cultured cells suggested that cholesterol-conjugated probe 4 is taken up by a receptor-independent mechanism, whereas uptake of probe 1 requires the presence of folic acid receptors on the cell surface. However, the FCS measurements did not provide any further details differentiating these two potential mechanisms.
Synthesis of fluorescent cap–ligand conjugates (m7GpppA-FAM-ligand) and their cellular permeability
We next tested whether folic acid and cholesterol may facilitate delivery of dinucleotide cap analogs into cells. To that end, we synthesized a series of doubly functionalized cap analogs that were labeled with fluorescein and conjugated to either folic acid, biotin, or cholesterol (15, 16, and 17, respectively; compound 16 being used as a negative control). The label and ligand were attached at the position N6 of adenine by means of NHS and CuAAC chemistry, respectively, and a multivalent polyamine linker (Scheme 1 and ESI†). | ||
Scheme 1 Chemical synthesis of fluorescently labeled cap conjugates. Conditions and reagents: (a) H2O, NaOH to adjust to pH = 8, 72 h, 65 °C, (b) ZnCl2, DMF, 24 h, rt, (c) 6-carboxyfluorescein-N-hydroxysuccinimide (FAM-NHS), borate buffer (50 mM), pH = 8.4, DMSO, (d) for 15 (with folate azide) and 16 (with biotin azide): CuSO4x5H2O, NaAsc, DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O, (e) for 17 (with cholesteryl-TEG-azide): Cu(II)-TBTA, NaAsc, triethylammonium acetate pH = 7, DMSO. :H2O, (e) for 17 (with cholesteryl-TEG-azide): Cu(II)-TBTA, NaAsc, triethylammonium acetate pH = 7, DMSO. | ||
Flow cytometry analysis showed that the cap analog conjugated with folic acid (15) potentially penetrated into MDA-MB-231 cells (FR+), but not A549 cells (FR−) (Fig. 4A and B). This is consistent with the results obtained for probe 1, and reveals that the presence of negative charge in the small molecule does not prevent transport by folic acid receptors.
 | ||
| Fig. 4 Cellular uptake of FAM labeled cap analog probes by (A) MDA-MB-231 and (B) A549 cells assessed by flow cytometry. Histograms represent overlaid flow cytometry data of unstained and m7GpppA-FAM-ligand (50 nM)-stained cells. (C) Probe 15 uptake by MDA-MB-231 cells (50 nM) in the presence of increasing concentrations of folic acid. Histograms represent overlaid flow cytometry data as a percentage of unstained and m7GpppA-FAM-FA-stained cells. (D) Uptake of FAM labeled cap analog conjugates by A549 and MDA-MB-231 cells assessed by confocal microscopy. (E and F) Normalized FCS autocorrelation curves for MDA-MB-231 cells incubated with probes 15 and 17 and their diffusion coefficients in cytoplasm derived from FCS data. Confocal imaging/FLIM of a cell incubated with 50 nM probe 15 (H) and 17 (G). FLIM image colored according to the scheme: blue – fluorescence lifetime < 2.4 ns (autofluorescence), red – fluorescence lifetime > 2.6 ns (probe). | ||
Compound 17, bearing the cholesterol moiety, penetrated into both the MDA-MB-231 and A549 cell lines (Fig. 4A and B), which also agrees with the results obtained for probe 4. As expected, based on the results for probe 2, neither cell line took up biotin-conjugated compound 16 (Fig. 4A and B).
To additionally verify that cellular uptake of compound 15 is dependent on FR expression on the cell membrane, we performed a flow-cytometry based competition assay (Fig. 4C). The uptake of compound 15 was inhibited in a dose-dependent manner by free folic acid in the cell culture medium, indicating an FR-specific mechanism. The uptake of compounds 15, 16, and 17 was independently studied by confocal microscopy (Fig. 4D) and verified by checking the orthogonal axis views (Fig. S2†). The results were generally consistent with the flow cytometry data, with compound 15 permeating only into MDA-MB-231 (FR+) cells, compound 17 permeating into both cell lines, and compound 16 being non-permeable.
FCS measurements performed in MDA-MB-231 cells (Fig. 4E and F) revealed primarily cytoplasmic localization for both compounds 15 and 17. The diffusion coefficients (D1) of the major, faster components were in line with values expected for free diffusion of the probes in the cytosol (Table 1, Table S1†), suggesting that the most abundant fluorescent species in the cell is a compound successfully delivered to the cytoplasm. The slower components (D2) showed high variability from 0.1 μm2 s−1 to 9.8 μm2 s−1, which is characteristic for large proteins and small membrane structures. Components with D < 0.1 μm2 s−1, typically assigned to endosomes, have not been found in the FCS signals for any of the studied probes.
Cap analog conjugates with folic acid and cholesterol inhibit cap-dependent translation in vitro
After identifying the ligands capable of transporting the cap analogs into cells, we focused on optimizing the structure of the cap residue to maximize its translation inhibitory properties. In particular, we focused on tailoring the ligand attachment site for optimal interaction with eIF4E and decreasing susceptibility to cleavage by DcpS – the pyrophosphatase that rapidly degrades native cap residues in the cell. Therefore, we synthesized a series of cap analogs carrying amino-alkyl linkers at either the N6-position of adenosine or the ribose moiety of guanosine and optionally introduced a γ-phosphorothioate (γ-PS) modification that prevents the cleavage by DcpS40 (compounds 21, 22, 28, and 29, Scheme 2). Phosphorothioate cap analogs (22, 29) exist as two P-diastereomers (D1/D2) that were difficult to separate by RP HPLC. Since the initial results showed that the affinities of the diastereomers for eIF4E were very similar (Fig. S3†), we decided to use diastereomeric mixtures of 22 and 29 in further studies. | ||
| Scheme 2 Chemical syntheses and applied conditions and reagents: (a) ZnCl2, DMF, (b) NHS-activated ligand (a = FA – folate, b = Chol – cholesteryl hemisuccinate), borate buffer pH = 8.4, DMSO. Phosphorothioates (18, 22, 24, 26, 29, 31) were obtained as a mixture of diastereoisomers (D1/D2). | ||
Folic acid or cholesterol were then attached to these cap analogs using NHS-chemistry47,48 to obtain eight different cap–ligand conjugates (23a–b, 24a–b, 30a–b, and 31a–b). The conjugates were purified by RP HPLC and their m/z values were confirmed by high-resolution mass spectrometry (HR-MS, ESI†).
We next assessed the affinity of these conjugates to eIF4E. To this end, a recently developed fluorescence intensity binding assay based on a pyrene-labeled probe (PyFLINT-B) was applied.59 The PyFLINT-B assay is based on measuring changes of pyrene fluorescence in a pyrene-labeled probe upon its competitive displacement from eIF4E by the studied compounds and allows for fast and reliable measurement of apparent dissociation constants, based on which equilibrium dissociation constants (KD) can be calculated. The KD values of all studied ligands, along with the reference compounds m7GpppA and m7GpppG, are shown in Table 2, while the binding curves are shown in the ESI (Fig. S2†). The affinity of m7GpppG for eIF4E as determined by PyFLINT-B was 2-fold higher than that of m7GpppA, which is in agreement with previous findings by other methods.60 The introduction of an aminohexyl linker at the N6 position of adenosine (compound 21) resulted in about 2-fold decrease in affinity for eIF4E (KD 0.88 ± 0.15 μM for m7GpppA-HDA (21) vs. KD 0.43 ± 0.08 μM for m7GpppA). A similar thermodynamic effect was observed upon introduction of the linker at the ribose moiety of guanosine (compound 28) (KD 0.485 ± 0.092 μM for m7GpppG-L6N (28) vs. KD 0.216 ± 0.035 μM for m7GpppG).
| No. | Compound | K D [μM] | IC50 [μM] (translation inhibition)b | IC50 [μM] (cytotoxicity)c |
|---|---|---|---|---|
| a Dissociation constants (KD) for eIF4E–cap analog complexes determined using the PyFLINT-B method at 20 °C. Data shown are means ± SD of three independent experiments; for compounds 23b, 30b, and 31b, only one repetition was performed. b From cap-dependent translation inhibition in rabbit reticulocyte lysate (RRL). c From resazurin cell viability assay in MDA-MB-231 cells. d Value in brackets is for K562 cells. e The values were not determined due to limited available material. | ||||
| 21 | m7GpppA | 0.429 ± 0.079 | 78. ± 26 | ≫30 |
| m7GpppA-HDA | 0.884 ± 0.148 | 23.9 ± 5.5 | nd. | |
| 22 | m7GpSppA-HDA-D1/D2 | 0.433 ± 0.102 | 10.8 ± 1.7 | nd. |
| 23a | m7GpppA-HDA-FA | 0.39 ± 0.09 | 28.1 ± 4.2 | ≫ 30 |
| 23b | m7GpppA-HDA-CHOL | 0.051 | 12.6 ± 2.1 | 8.28 ± 1.87 |
| 24a | m7GpSppA-HDA-FA D1/D2 | 0.30 ± 0.06 | 29.9 ± 3.3 | ≫30 |
| 24b | m7GpSppA-HDA-CHOL D1/D2 | nd.e | nd.e | 5.20 ± 0.78 |
| m7GpppG | 0.216 ± 0.035 | 39.6 ± 13.7 | ≫30 | |
| 28 | m7GpppG-L6N-2′-O/3′-O | 0.485 ± 0.092 | 10.9 ± 1.4 | nd. |
| 29 | m7GpSppG-L6N-2′-O/3′-O D1/D2 | 0.242 ± 0.045 | 6.0 ± 0.8 | nd. |
| 30a | m7GpppG-L6N -2′-O/3′-O-FA | 0.12 ± 0.02 | 16.1 ± 3.5 | ≫30 |
| 30b | m7GpppG-L6N -2′-O/3′-O-CHOL | 0.043 | 26.1 ± 5.0 | 10.90 ± 2.81 |
| 31a | m7GpSppG-L6N -2′-O/3′-O-FA D1/D2 | 0.10 ± 0.02 | 4.2 ± 0.6 | ≫30 |
| 31b | m7GpSppG- L6N 2′-O/3′-O-CHOL D1/D2 | 0.026 | 20.5 ± 5.7 | 6.52 ± 1.73 μM [6.04 ± 1.58]d |
| Folic acid | >10 | ≫500 | ≫30 | |
| Cholesterol | 0.94 | 99. ± 20. | ≫30 | |
The presence of the γ-PS moiety increased affinity to eIF4E by about 2-fold (KD 0.88 ± 0.15 μM for m7GpppA-HDA (21) vs. KD 0.43 ± 0.10 μM for m7GpSppA-HDA D1/D2 (22) and KD 0.485 ± 0.092 μM for m7GpppG-L6N (28) vs. KD 0.242 ± 0.045 μM for m7GpSppG-L6N-2′-O/3′-O D1/D2 (29)). Interestingly, the introduction of appropriate ligands (FA or Chol; compounds 23a to 31b) alleviated the destabilizing effect introduced by the aminoalkyl linker, suggesting it was at least partly caused by electrostatic repulsion of the positively charged amino group with positively charged surfaces in eIF4E. KD values for compounds bearing the folate modification were from 1.5-fold to 4-fold higher than corresponding unconjugated caps (e.g. KD 0.433 ± 0.102 μM for m7GpSppA-HDA-D1/D2 (22) vs. KD 0.30 ± 0.06 μM for m7GpSppA-HDA-FA D1/D2 (24a)). Unexpectedly, the incorporation of the cholesterol moiety resulted in even greater stabilization of the cap-eIF4E complex ranging from 9-fold to 17-fold improvement (e.g. KD 0.242 ± 0.045 μM for m7GpSppG-L6N-2′-O/3′-O D1/D2 (29) vs. KD 0.026 μM for m7GpSppG-L6N-2′-O/3′-O-CHOL D1/D2 (31b)). This might be due to additional hydrophobic interactions with eIF4E provided by the cholesteryl moiety.
In order to evaluate the ability of cap analogs to target eIF4E in a more complex biological system, we studied their potency to inhibit cap-dependent translation in rabbit reticulocyte lysate (RRL) programmed with capped luciferase-encoding mRNA.61 We have previously shown that the ability of cap analogs to inhibit translation in RRL correlates qualitatively with their affinity for eIF4E, but may also be affected by DcpS-like activity, which is also present in the lysate.62 Therefore, the compounds were tested in an experimental set up in which both factors could be taken into account. Namely, the compounds were incubated in RRL for 1 h followed by the addition of mRNA and luciferase activity measurement after another hour. It was expected that 1 h pre-incubation in RRL will relatively decrease the inhibitory potency of cap analogs susceptible to DcpS, compared to analogs that are resistant to DcpS.62
The IC50 values reasonably correlated with KD values from PyFLINT-B assay (Table 2), albeit with some important exceptions. For instance, the introduction of hexamine linkers resulted in stronger inhibition compared to unmodified reference compounds [IC50 23.9 ± 5.5 μM for m7GpppA-HDA (21) vs. IC50 78.1 ± 25.6 μM for m7GpppA and IC50 10.9 ± 1.4 μM for m7GpppG-L6N (28) vs. IC50 39.6 ± 13.7 μM for m7GpppG; Table 2, Fig. S4],† despite their lower affinity for eIF4E. This observation confirms that factors other than affinity for eIF4E, and DcpS-susceptibility in particular, influence the inhibitory properties under the conditions of our experiment. DcpS exhibits a tight cap-binding mode, consequently affinity and susceptibility of cap analogs to DcpS is usually decreased upon functionalization of ribose or nucleobase moieties.40,63,64
Therefore, it is plausible that the incorporation of a linker at the N6-position of adenine or the 2′-O/3′-O-position of ribose in the cap decreased its susceptibility to DcpS, thereby stabilizing it in the lysate. The presence of γ-PS conferred high inhibitory potency for all tested compounds. Compounds 22 and 29 bearing this modification had lower IC50 values than their unmodified counterparts (IC50 23.9 ± 5.5 μM for m7GpppA-HDA (21) vs. IC50 10.8 ± 1.7 μM for m7GpSppA-HDA D1/D2 (22) and IC50 10.9 ± 1.4 μM for m7GpppG-L6N (28) vs. IC50 6.0 ± 0.8 μM for m7GpSppG-L6N-2′-O/3′-O D1/D2 (29)). This observation is in agreement with our previous studies showing that a non-bridging γ-O-to-S modification in the triphosphate bridge decreases the susceptibility of cap analogs to DcpS (in addition to stabilizing the cap-eIF4E complex).40 Interestingly, despite a significant increase in affinity for eIF4E, the presence of ligands did not enhance inhibitory properties of cap analogs in RRL compared to corresponding compounds with aminohexyl linkers. Compounds 23a, 23b, 30a, and 31a had IC50 values almost unchanged compared to values for the cap analogs with aminoalkyl linkers, whereas compounds 24a, 30b, and 31b were up to 3-fold weaker inhibitors. Overall, all cap–ligand conjugates studied exhibited sufficient potency and stability in RLL to be considered as suitable candidates for evaluation in cell culture experiments.
Cholesterol cap analogs influence cell viability and their potency depends on a 5′,5′-triphosphate bridge modification
Knowing the in vitro properties of the synthesized conjugates, we next analyzed their cytotoxicity towards a breast cancer cell line (MDA-MB-231). We first verified whether the non-fluorescent cap analogs entered the cells. To that end, an immunofluorescent assay using a cap-specific antibody was employed.10 Visualization of immunostained cells clearly showed that cap analog conjugates were taken up by MDA-MB-231 (FR+) cells regardless of the attached ligand (Fig. 5A). | ||
| Fig. 5 Cellular response to cap analog conjugates. (A) Verification of cellular delivery of cap conjugates. MDA-MB-231 cells after 2 h treatment with 0.5 μM compound were fixed and stained with anti-cap antibody. (B) Viability of MDA-MB-231 cells treated with different concentrations of selected compounds. After 72 h incubation, cells viability was assessed with the resazurin assay. Data points were normalized to mock treated cells for each time point and represent mean value ± SD from at least two independent biological replications with two technical replicates per treatment. (C) IC50 values of cholesterol cap analog conjugates calculated based on the cell viability assay presented in (B), bars represent mean value ± SD. Statistical significance: * p < 0.05, ** p < 0.01 (Welch's t test). (D) Viability of K562 cells treated with different concentrations of selected compounds. After 72 h incubation, cells viability was assessed with the resazurin assay. Data points were normalized to mock treated cells for each time point and represent mean value ± SD two independent biological replications with two technical replicates per treatment. (E) Apoptosis assessed by Annexin V/propidium iodide staining. K562 cells were treated with 5 μM and 20 μM 31b for 20 h and examined by flow cytometry. Viable cells are in the (I) lower left quadrant, early apoptotic are in the (II) lower right quadrant, late apoptotic cells are in the (III) upper right quadrant and non-viable necrotic cells are in the (IV) upper left quadrant. (F) Apoptotic cell fraction calculated based on the Annexin V/propidium iodide staining presented in (E). Bars represent mean value ± SD from at least three independent biological replications. Statistical significance: * p < 0.05, *** p < 0.001 (Welch's t test). | ||
Next, cytotoxicity of the compounds (23a, 23b, 24a, 24b, 30a, 30b, 31a, 31b, m7GpppA, m7GpppG, free folic acid, and free cholesterol moiety) towards MDA-MB-231 cells was determined by resazurin assay. Only the cholesterol conjugates significantly influenced cell viability (Fig. 5B and S5†). The compounds carrying the γ-PS moiety in the 5′,5′-triphosphate bridge (24b and 31b) had cytotoxic effects about 2-fold stronger than unmodified counterparts (IC50 8.3 ± 1.9 μM for 23bvs. IC50 5.2 ± 0.8 μM for 24b and IC50 10.9 ± 2.8 μM for 30bvs. IC50 6.0 ± 1.6 μM for 31b), clearly showing that stabilizing the cap against DcpS is beneficial for the biological activity of the cap analogs in living cells (Fig. 5C). Next, we checked whether cholesterol cap analogs exhibit cytotoxic activity towards other cancer cell lines. To that end, a leukemia cancer cell line (K562) was treated with cap analog 31b. Resazurin assay confirmed the cytotoxicity of this compound towards the K562 cell line (Fig. 5D; IC50 6.5 ± 1.7 μM) at a level comparable to MDA-MB-231 cells (IC50 6.0 ± 1.6 μM; Table 2).
Cholesterol cap analogs induce apoptosis in a leukemia cell line
Most anticancer drugs currently used in clinical oncology exploit apoptotic mechanisms to trigger cancer cell death.65 Therefore, to verify whether cholesterol-conjugated cap analogs induce apoptosis in cancer cells, K562 cells were incubated with the most potent compound (31b), followed by a more detailed cell viability analysis. The ability of 31b to induce apoptosis was determined by flow cytometry using Annexin V and PI (propidium iodide) staining. This analysis detects changes in plasma membrane integrity during cell apoptosis that result in the appearance of phosphatidylserine on the outer leaflet of the membrane.Annexin V has a strong, calcium-dependent affinity for phosphatidylserine and, therefore, can be used as a marker of apoptosis. PI is a DNA binder and can be used to detect genomic DNA released from necrotic cells. Double staining with PI and Annexin V enables differentiation and quantification of living, apoptotic, and necrotic cells. Fig. 5E shows representative dot plots from Annexin V/PI staining, whereas Fig. 5F shows the percentages of apoptotic cells. These data reveal that compound 31b indeed induces apoptosis in a dose-dependent manner (30.5 ± 6.5% and 60.8 ± 3.2% apoptosis in the presence 5 μM and 20 μM cap analog, respectively).
Conclusions
We synthesized a series of fluorescent probes and doubly functionalized cap analogs, which allowed us to examine the potential of four pre-selected ligands (folic acid, biotin, glucose, and cholesterol) to transport polar, negatively charged molecules into cells. Only folic acid and cholesterol facilitated cellular uptake of fluorescent probes and cap analogs in the studied models. The lack of observable cellular uptake for glucose and biotin conjugates may result from insufficient transport efficiency for these ligands in the studied cell lines or disturbance of the transport by cargo attachment.We next optimized the structure of the cap moiety for conjugation with folic acid and cholesterol to maximize its translation inhibitory properties. To that end, we synthesized a set of cap-derived eIF4E antagonists with folic acid and cholesterol attached in two different positions of cap analogs (at the nucleobase and the ribose moiety) and optionally carrying a γ-PS modification that prevents cap degradation by DcpS. We tested the cytotoxicity of these compounds towards cancer cells. Both cholesterol and folate conjugates were efficiently taken up by cancer cells, but cytotoxic effects were only observed for compounds conjugated with cholesterol. The cytotoxicity of these conjugates was notably enhanced by introducing the γ-PS modification in the 5′,5′-triphosphate bridge. The possible explanations of this phenomenon include either strong specific binding to folic acid receptors and resulting entrapment of the cap analogs in residual endosomal structures or competitive binding of the conjugates to other folate recognizing proteins, which prevents binding by eF4E. If this is the case, the issue may be resolved in the future by application of cleavable conjugates that release free cap structures following cell entry. Moreover, the designed cap analogs can serve as a platform to test the potential of other ligands to deliver impermeable compounds to mammalian cells. Importantly, dual labeling of cap analogs enables not only intracellular visualization and tracking but also opens up new possibilities to test combinations of cell penetrating ligands, which could lead to the development of superior delivery strategies such as a dual labeling approach that could bypass the “endosome escape” issue. Further studies, including in vivo evaluation on animal models, are required to fully assess the anticancer activity and selectivity of cap–cholesterol conjugates.
Data availability
All data needed to evaluate the conclusions are present in the paper and in the associated supplementary material.Author contributions
JK, JJ, NK, and PJS designed the study. NK and ZW performed the syntheses. PJS, NB, and LM performed biological assays. RK performed the binding assay. KK, AK, JM, RH performed FCS and FLIM studies. NK, PS, JK, and JJ prepared the first draft of the manuscript. All authors reviewed, edited, and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Centre Poland (UMO-2016/20/S/ST5/00364 to NK and 2018/31/B/ST5/03821 to JK), the Foundation for Polish Science (TEAM/2016-2/13 to JJ) and the National Centre for Research and Development Poland (LIDER/10/0033/L-9/17/NCBR/2018 to KK). Figures were created with https://www.BioRender.com.Notes and references
- M. K. Yates and K. L. Seley-Radtke, Antiviral Res., 2019, 162, 5–21 CrossRef CAS PubMed.
- J. Lipfert, S. Doniach, R. Das and D. Herschlag, Annu. Rev. Biochem., 2014, 83, 813–841 CrossRef CAS PubMed.
- C. Meier, M. Lorey, E. De Clercq and J. Balzarini, J. Med. Chem., 1998, 41, 1417–1427 CrossRef CAS PubMed.
- I. Lefebvre, C. Perigaud, A. Pompon, A.-M. Aubertin, J.-L. Girardet, A. Kirn, G. Gosselin and J.-L. Imbach, J. Med. Chem., 1995, 38, 3941–3950 CrossRef CAS PubMed.
- U. Pradere, E. C. Garnier-Amblard, S. J. Coats, F. Amblard and R. F. Schinazi, Chem. Rev., 2014, 114, 9154–9218 CrossRef CAS PubMed.
- F. Pertusat, M. Serpi and C. McGuigan, Antivir. Chem. Chemother., 2012, 22, 181–203 CrossRef PubMed.
- A. Kraszewski, M. Sobkowski and J. Stawinski, Front. Chem., 2020, 8, 595738 CrossRef CAS PubMed.
- S. Vinogradov, A. Zeman, E. Batrakova and A. Kabanov, J. Controlled Release, 2005, 107, 143–157 CrossRef CAS PubMed.
- H. Hillaireau, T. Le Doan, M. Appel and P. Couvreur, J. Controlled Release, 2006, 116, 346–352 CrossRef CAS PubMed.
- M. Zochowska, A.-C. Piguet, J. Jemielity, J. Kowalska, E. Szolajska, J.-F. Dufour and J. Chroboczek, Nanomedicine, 2015, 11, 67–76 CrossRef CAS PubMed.
- Z. Zawada, A. Tatar, P. Mocilac, M. Buděšínský and T. Kraus, Angew. Chem., Int. Ed., 2018, 57, 9891–9895 CrossRef CAS PubMed.
- C. Meier, Antiviral Chem. Chemother., 2017, 25, 69–82 CrossRef PubMed.
- T. Gollnest, T. D. de Oliveira, D. Schols, J. Balzarini and C. Meier, Nat. Commun., 2015, 6, 8716 CrossRef CAS PubMed.
- S. Warnecke and C. Meier, J. Org. Chem., 2009, 74, 3024–3030 CrossRef CAS PubMed.
- S. F. Dowdy, Nat. Biotechnol., 2017, 35, 222–229 CrossRef CAS PubMed.
- J. D. Lewis and E. Izaurflde, Eur. J. Biochem., 1997, 247, 461–469 CrossRef CAS PubMed.
- M. Warminski, P. J. Sikorski, J. Kowalska and J. Jemielity, Top. Curr. Chem., 2017, 375, 16 CrossRef PubMed.
- A. Batool, S. Aashaq and K. I. Andrabi, J. Cell. Biochem., 2019, 120, 14201–14212 CrossRef CAS PubMed.
- S. Uttam, C. Wong, T. J. Price and A. Khoutorsky, Front. Genet., 2018, 9, 470 CrossRef CAS PubMed.
- B. Culjkovic and K. L. Borden, J. Oncol., 2009, 2009, 1–12 CrossRef PubMed.
- A. Lazaris-Karatzas, K. S. Montine and N. Sonenberg, Nature, 1990, 345, 544–547 CrossRef CAS PubMed.
- S. Avdulov, S. Li, V. Michalek, D. Burrichter, M. Peterson, D. M. Perlman, J. C. Manivel, N. Sonenberg, D. Yee, P. B. Bitterman and V. A. Polunovsky, Cancer Cell, 2004, 5, 553–563 CrossRef CAS.
- C. E. Joyce, A. G. Yanez, A. Mori, A. Yoda, J. S. Carroll and C. D. Novina, Cancer Res., 2017, 77, 613–622 CrossRef CAS PubMed.
- J. Chu and J. Pelletier, Cold Spring Harbor Perspect. Biol., 2017, 10, a032995 CrossRef PubMed.
- A. Fan and P. P. Sharp, J. Med. Chem., 2021, 64, 2436–2465 CrossRef CAS PubMed.
- J. R. Graff, B. W. Konicek, T. M. Vincent, R. L. Lynch, D. Monteith, S. N. Weir, P. Schwier, A. Capen, R. L. Goode, M. S. Dowless, Y. Chen, H. Zhang, S. Sissons, K. Cox, A. M. McNulty, S. H. Parsons, T. Wang, L. Sams, S. Geeganage, L. E. Douglass, B. L. Neubauer, N. M. Dean, K. Blanchard, J. Shou, L. F. Stancato, J. H. Carter and E. G. Marcusson, J. Clin. Invest., 2007, 117, 2638–2648 CrossRef CAS PubMed.
- F. Zhou, M. Yan, G. Guo, F. Wang, H. Qiu, F. Zheng, Y. Zhang, Q. Liu, X. Zhu and L. Xia, Med. Oncol., 2011, 28, 1302–1307 CrossRef CAS PubMed.
- W. M. Guo, K. W. Kong, C. J. Brown, S. T. Quah, H. L. Yeo, S. Hoon and Y. Seow, Mol. Ther.--Nucleic Acids, 2014, 3, e217 CrossRef CAS PubMed.
- S. Miyakawa, RNA, 2006, 12, 1825–1834 CrossRef CAS PubMed.
- N. J. Moerke, H. Aktas, H. Chen, S. Cantel, M. Y. Reibarkh, A. Fahmy, J. D. Gross, A. Degterev, J. Yuan, M. Chorev, J. A. Halperin and G. Wagner, Cell, 2007, 128, 257–267 CrossRef CAS PubMed.
- R. Cencic, D. R. Hall, F. Robert, Y. Du, J. Min, L. Li, M. Qui, I. Lewis, S. Kurtkaya, R. Dingledine, H. Fu, D. Kozakov, S. Vajda and J. Pelletier, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1046–1051 CrossRef CAS PubMed.
- E. Darzynkiewicz, I. Ekiel, S. M. Tahara, L. S. Seliger and A. J. Shatkin, Biochemistry, 1985, 24, 1701–1707 CrossRef CAS.
- Z. Ahmad, B. A. Jacobson, M. W. McDonald, N. Vattendahl Vidal, G. Vattendahl Vidal, S. Chen, M. Dillenburg, A. M. Okon, M. R. Patel, C. R. Wagner and R. A. Kratzke, Cancer Chemother. Pharmacol., 2020, 85, 425–432 CrossRef CAS PubMed.
- C. R. Wagner, V. V. Iyer and E. J. McIntee, Med. Res. Rev., 2000, 20, 417–451 CrossRef CAS.
- B. Ghosh, A. O. Benyumov, P. Ghosh, Y. Jia, S. Avdulov, P. S. Dahlberg, M. Peterson, K. Smith, V. A. Polunovsky, P. B. Bitterman and C. R. Wagner, ACS Chem. Biol., 2009, 4, 367–377 CrossRef CAS PubMed.
- J. Zuberek, J. Jemielity, A. Jablonowska, J. Stepinski, M. Dadlez, R. Stolarski and E. Darzynkiewicz, Biochemistry, 2004, 43, 5370–5379 CrossRef CAS PubMed.
- M. Ziemniak, M. Strenkowska, J. Kowalska and J. Jemielity, Future Med. Chem., 2013, 5, 1141–1172 CrossRef CAS PubMed.
- M. Kalek, J. Jemielity, Z. M. Darzynkiewicz, E. Bojarska, J. Stepinski, R. Stolarski, R. E. Davis and E. Darzynkiewicz, Bioorg. Med. Chem., 2006, 14, 3223–3230 CrossRef CAS PubMed.
- J. Kowalska, A. Wypijewska del Nogal, Z. M. Darzynkiewicz, J. Buck, C. Nicola, A. N. Kuhn, M. Lukaszewicz, J. Zuberek, M. Strenkowska, M. Ziemniak, M. Maciejczyk, E. Bojarska, R. E. Rhoads, E. Darzynkiewicz, U. Sahin and J. Jemielity, Nucleic Acids Res., 2014, 42, 10245–10264 CrossRef CAS PubMed.
- J. Kowalska, M. Lewdorowicz, J. Zuberek, E. Grudzien-Nogalska, E. Bojarska, J. Stepinski, R. E. Rhoads, E. Darzynkiewicz, R. E. Davis and J. Jemielity, RNA, 2008, 14, 1119–1131 CrossRef CAS PubMed.
- A. M. Rydzik, M. Kulis, M. Lukaszewicz, J. Kowalska, J. Zuberek, Z. M. Darzynkiewicz, E. Darzynkiewicz and J. Jemielity, Bioorg. Med. Chem., 2012, 20, 1699–1710 CrossRef CAS PubMed.
- J. Jemielity, J. Kowalska, A. M. Rydzik and E. Darzynkiewicz, New J. Chem., 2010, 34, 829 RSC.
- A. M. Rydzik, M. Warminski, P. J. Sikorski, M. R. Baranowski, S. Walczak, J. Kowalska, J. Zuberek, M. Lukaszewicz, E. Nowak, T. D. W. Claridge, E. Darzynkiewicz, M. Nowotny and J. Jemielity, Nucleic Acids Res., 2017, 45, 8661–8675 CrossRef CAS PubMed.
- B. A. Wojtczak, P. J. Sikorski, K. Fac-Dabrowska, A. Nowicka, M. Warminski, D. Kubacka, E. Nowak, M. Nowotny, J. Kowalska and J. Jemielity, J. Am. Chem. Soc., 2018, 140, 5987–5999 CrossRef CAS PubMed.
- Y. Lu and P. S. Low, Adv. Drug Delivery Rev., 2012, 64, 342–352 CrossRef.
- W. X. Ren, J. Han, S. Uhm, Y. J. Jang, C. Kang, J.-H. Kim and J. S. Kim, Chem. Commun., 2015, 51, 10403–10418 RSC.
- E. C. Calvaresi and P. J. Hergenrother, Chem. Sci., 2013, 4, 2319 RSC.
- D. Irby, C. Du and F. Li, Mol. Pharm., 2017, 14, 1325–1338 CrossRef CAS PubMed.
- M. Warminski, P. J. Sikorski, Z. Warminska, M. Lukaszewicz, A. Kropiwnicka, J. Zuberek, E. Darzynkiewicz, J. Kowalska and J. Jemielity, Bioconjugate Chem., 2017, 28, 1978–1992 CrossRef CAS PubMed.
- A. F. Trindade, R. F. M. Frade, E. M. S. Maçôas, C. Graça, C. A. B. Rodrigues, J. M. G. Martinho and C. A. M. Afonso, Org. Biomol. Chem., 2014, 12, 3181–3190 RSC.
- S. C. Kinsky, J. E. Loader and A. L. Benson, J. Immunol. Methods, 1983, 65, 295–306 CrossRef CAS.
- J. D. Seitz, J. G. Vineberg, E. Herlihy, B. Park, E. Melief and I. Ojima, Bioorg. Med. Chem., 2015, 23, 2187–2194 CrossRef CAS PubMed.
- R. L. Aft, F. W. Zhang and D. Gius, Br. J. Cancer, 2002, 87, 805–812 CrossRef CAS PubMed.
- D. Ren, F. Kratz and S.-W. Wang, Biochem. Eng. J., 2014, 89, 33–41 CrossRef CAS PubMed.
- R. Macháň and T. Wohland, FEBS Lett., 2014, 588, 3571–3584 CrossRef PubMed.
- T. Kalwarczyk, N. Ziȩbacz, A. Bielejewska, E. Zaboklicka, K. Koynov, J. Szymański, A. Wilk, A. Patkowski, J. Gapiński, H.-J. Butt and R. Hołyst, Nano Lett., 2011, 11, 2157–2163 CrossRef CAS PubMed.
- T. Kalwarczyk, K. Kwapiszewska, K. Szczepanski, K. Sozanski, J. Szymanski, B. Michalska, P. Patalas-Krawczyk, J. Duszynski and R. Holyst, J. Phys. Chem. B, 2017, 121, 9831–9837 CrossRef CAS PubMed.
- K. Kwapiszewska, T. Kalwarczyk, B. Michalska, K. Szczepański, J. Szymański, P. Patalas-Krawczyk, T. Andryszewski, M. Iwan, J. Duszyński and R. Hołyst, Sci. Rep., 2019, 9, 5906 CrossRef PubMed.
- R. Kasprzyk, B. J. Starek, S. Ciechanowicz, D. Kubacka, J. Kowalska and J. Jemielity, Chem. -Eur. J., 2019, 25, 6728–6740 CrossRef CAS PubMed.
- A. Niedzwiecka, J. Marcotrigiano, J. Stepinski, M. Jankowska-Anyszka, A. Wyslouch-Cieszynska, M. Dadlez, A.-C. Gingras, P. Mak, E. Darzynkiewicz, N. Sonenberg, S. K. Burley and R. Stolarski, J. Mol. Biol., 2002, 319, 615–635 CrossRef CAS.
- J. Jemielity, T. Fowler, J. Zuberek, J. Stepinski, M. Lewdorowicz, A. Niedzwiecka, R. Stolarski, E. Darzynkiewicz and R. E. Rhoads, RNA, 2003, 9, 1108–1122 CrossRef CAS PubMed.
- J. Kowalska, M. Lukaszewicz, J. Zuberek, M. Ziemniak, E. Darzynkiewicz and J. Jemielity, Bioorg. Med. Chem. Lett., 2009, 19, 1921–1925 CrossRef CAS PubMed.
- M. Gu, C. Fabrega, S.-W. Liu, H. Liu, M. Kiledjian and C. D. Lima, Mol. Cell, 2004, 14, 67–80 CrossRef CAS PubMed.
- A. Wypijewska del Nogal, M. D. Surleac, J. Kowalska, M. Lukaszewicz, J. Jemielity, M. Bisaillon, E. Darzynkiewicz, A. L. Milac and E. Bojarska, FEBS J., 2013, 280, 6508–6527 CrossRef CAS PubMed.
- G. Pistritto, D. Trisciuoglio, C. Ceci, A. Garufi and G. D'Orazi, Aging, 2016, 8, 603–619 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02143e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |