
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct electrochemical hydrodefluorination of trifluoromethylketones enabled by non-protic conditions †
John R.
Box
 ,
Alexander P.
Atkins
and
Alastair J. J.
Lennox
*
,
Alexander P.
Atkins
and
Alastair J. J.
Lennox
*
School of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK. E-mail: a.lennox@bristol.ac.uk
First published on 6th July 2021
Abstract
CF2H groups are unique due to the combination of their lipophilic and hydrogen bonding properties. The strength of H-bonding is determined by the group to which it is appended. Several functional groups have been explored in this context including O, S, SO and SO2 to tune the intermolecular interaction. Difluoromethyl ketones are under-studied in this context, without a broadly accessible method for their preparation. Herein, we describe the development of an electrochemical hydrodefluorination of readily accessible trifluoromethylketones. The single-step reaction at deeply reductive potentials is uniquely amenable to challenging electron-rich substrates and reductively sensitive functionality. Key to this success is the use of non-protic conditions enabled by an ammonium salt that serves as a reductively stable, masked proton source. Analysis of their H-bonding has revealed difluoromethyl ketones to be potentially highly useful dual H-bond donor/acceptor moieties.
The difluoromethyl group (CF2H) has attracted significant recent attention in medicinal chemistry,1,2 which complements the well-documented importance and growing use of fluorine in small molecule pharmaceuticals.3–6 The CF2H group is an H-bond donor7,8 that is also lipophilic,9,10 a unique combination that positions it as an increasingly valuable tool within drug-discovery.11 CF2H has been used as a bioisostere of OH and SH in serine and cystine moieties, respectively, as well as NH2 groups, where greater lipophilicity and rigidity provide advantages to pharmacokinetics and potency.12–14
The hydrogen-bond acidity of CF2H groups is exceptionally dependent on the atom or group to which it is appended (Fig. 1A).1,2 The H-bond acidity of alkyl-CF2H groups is half that of O–CF2H and even a quarter of SO2–CF2H groups.1 This mode of control allows the H-bonding strength and, therefore its function, to be finely tuned. While much research has focused on the synthesis, behaviour and use of XCF2H groups, where X = O, S, SO, SO2, Ar, it is surprising that the corresponding carbonyl containing moiety (X = CO) has remained relatively elusive in these contexts. Not only would difluoromethyl ketones (DFMK) be expected to provide a relatively strong H-bond, but the carbonyl unit provides a complementary, yet proximal mode of intermolecular interaction (Fig. 1B). Indeed, the dual action of neighbouring H-bond donor and acceptor functionalities provides the fundamental basis for many biological systems, including in the secondary structure assembly mechanisms for proteins and DNA/RNA nucleobase pairing, as well as in enzyme/substrate complexes. Indeed, the DFMK functionality has demonstrated important utility in biological applications, including anti-malarial and -coronaviral properties.15 Finally, the carbonyl provides a useful synthetic handle for further derivatization.
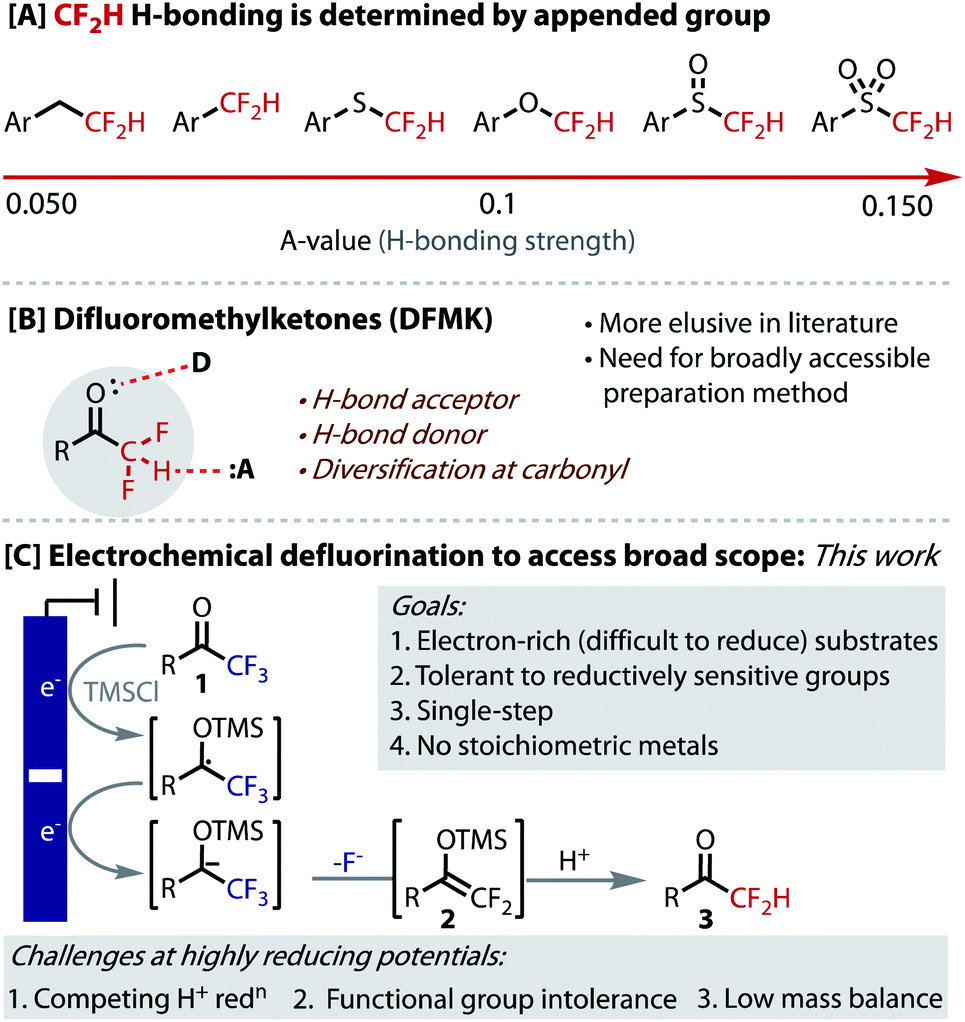 | ||
| Fig. 1 H-Bonding in DFMKs and their synthesis via hydrodefluorination. | ||
While some progress has been made on the synthesis of DFMKs,16 there still remains a need for a general and more broadly accessible route to their preparation. Current strategies for DFMK preparation require multi-step processes, expensive reagents, installation of activating groups, or are inherently low yielding.15a,16–25 The hydrodefluorination of trifluoromethyl ketones (1) potentially represents the most accessible strategy, as the starting materials are most readily prepared through a high-yielding trifluoroacetylation of C–H or C–X bonds.26–29 In 2001, Prakash demonstrated the viability of this approach using 2 equivalents of magnesium metal as stoichiometric reductant to drive the defluorination, with a second hydrolysis step (HCl (3–5 M) or fluoride, overnight stirring) to reveal the product.30 The scope in this 2-step process (6 substrates) reflects the limitations of using a reductant, such as Mg, that has a fixed reduction potential, as well as incompatibilities arising from Mg/halide exchange with aryl halides. Similar limitations with the use of electron-rich substrates were revealed in related contributions from Uneyama.31
In order to access more electron-rich and reductively challenging substrates, such as those containing medicinally relevant heterocycles, we postulated that electrochemical reduction could be employed (Fig. 1C). Electrosynthesis is becoming an increasingly valuable enabling technology and has seen a recent resurgence due to the precise control, unique selectivity, and the potential scalability and sustainability benefits that it offers.32–36 This strategy would avoid the undesirable use of stoichiometric metals and the ‘deep-reduction’ potentials required are readily accessed by simply selecting the applied potential. Pioneering early work from Uneyama on the cathodic formation of silylenol ether intermediate 2, suggested this approach could be viable.37,38 The fundamental challenge in designing a practical, single-step process under highly reducing potentials (<−2.0 V vs. Fc/Fc+), is to avoid the reduction of the proton source, which would otherwise compete to generate H2 gas and leave the starting material untouched. Uneyama does not demonstrate hydrodefluorination, presumably due to this problem. Additional challenges posed by ‘deep-reduction’ include a lack of tolerance for reduction-sensitive functionality (alkene, C–X bonds etc.), low mass balance due to substrate decomposition and the undesirable use of sacrificial metal anodes.39 Solving these problems should provide generally applicable, safe and scalable conditions for the hydrodefluorination of readily accessible trifluoromethyl ketones (1).
Given the electron-rich nature of indoles, their ubiquity in bioactive compounds, and their ease of functionalisation, we chose indole 1a as the model substrate for optimisation. The highly reductive potentials required will render it a challenging substrate, which should lead to more general conditions suitable for other important substrate classes. Indeed, when we applied the Mg conditions of Prakash to this substrate, no silyl enol ether intermediate (2a) was observed, nor product 3a, and the starting material remained completely untouched (Table 1, entry 1). Moving to an electrochemical set-up, the use of a sacrificial Mg anode in an undivided cell again returned no defluorinated product (entry 2). The applied potential was sufficiently negative to reduce the evolving Mg2+ ions, and so the substrate was again left untouched.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Conditions different from above | Reductant | Proton source | 1a /% | (2a) 3aa/% |
| a 19F NMR yields. b TMSCl only added to cathodic chamber. c TMSCl added to both cathodic and anodic chambers. | |||||
| 1 | Mg 0 , THF, no electricity (Prakash conditions for3) | Mg0 | — | 100 | (0) n/a |
| 2b | Undivided cell, TBAPF6 | Sacrificial Mg anode | — | 100 | (0) n/a |
| 3b | Pb:C (cath:an), 0 oC, 30 mA (Uneyama conditions for2) | TBABr (4 eq.) | — | 33 | (32) 0 |
| 4b | — | TBABr (2 eq.) | (a) Acetic acid; (b) oxalic acid. | 51; 100 | 0; 0 |
| 5b | — | TBABr (2 eq.) | Dimethylurea | 82 | 0 |
| 6b | — | TBABr (2 eq.) | TEAPF6 (4 eq.) | 49 | 45 |
| 7 | TMSCl (0 eq.) | TBABr (2 eq.) | TEAPF6 (4 eq.) | 83 | 0 |
| 8b | TMSCl (6 eq.) | TBABr (2 eq.) | TEAPF6 (4 eq.) | 49 | 49 |
| 9c | TMSCl (3 + 3 eq.) | TBABr (2 eq.) | TEAPF 6 (4 eq.) | 0 | 97 |
| 10c | Entry 9, but Pt:Gr (cath:An) | TBABr (2 eq.) | TEAPF6 (4 eq.) | 0 | 94 |
| 11c | Entry 9, but Ni:Pt (cath:An) | TBABr (2 eq.) | TEAPF6 (4 eq.) | 0 | 83 |
| 12c | Entry 9, but Stainless Steel:Pt (cath:An) | TBABr (2 eq.) | TEAPF6 (4 eq.) | 0 | 85 |
| 13c | Entry 9, but Gr:Pt (cath:An) | TBABr (2 eq.) | TEAPF6 (4 eq.) | 0 | 18 |
The electrochemical conditions of Uneyama for preparing silylenol ethers (2) were applied to our indole 1a (entry 3). Unsurprisingly, no hydrodefluorinated product was observed, however intermediate 2a was formed in a 32% yield. In an effort to improve this yield we explored several solvents, reductants, additives and electrode materials, all of which were conducted in a divided cell at constant current and ambient temperature.40 In addition, as we were keen to develop a single-step protocol, by avoiding the second hydrolysis step that can readily form homo-coupled aldol side products,38 we surveyed a range of added proton sources for in situ delivery of 3a. The addition of carboxylic acids, such as acetic or oxalic acid (entry 4), gave no desired product, as the competing reduction of protons to H2 gas dominated. Dimethylurea was recently used as a proton source in an electrochemical ‘deep-reduction’,41 but it returned no trace of intermediate 2a or product 3a (entry 5). We hypothesized that increasing the conductivity of the system, with additional tetraalkylammonium salts (from 2 to 4 eq.), the formation of intermediate 2a may be facilitated by avoiding large cell potentials. While this change did facilitate a lower cell potential, we discovered these salts behaved as reductively stable yet competent masked proton donors: 4 eq. NEt4PF6 gave 45% yield of product 3a, with no sign of intermediate 2a (entry 6). The detection of triethylamine in solution suggests donation through a Hoffmann elimination.42 With the exception of NMe4+, other tetraalkylammonium salts were also competent proton donors (NEt4+ > NBu4+ > NPr4+).
A critical improvement to the yield was observed when the use of the radical anion trapping agent, TMSCl, was optimised. With no TMSCl, 3a was not observed (entry 7), and a loading of 6 equivalents saw little improvement over 3 equivalents (entry 8 vs. 6). Experiments hitherto described were conducted with TMSCl added only to the cathodic chamber (entries 2–8). Only when the 6 equivalents was split between both chambers was a drastic improvement observed (entry 9), giving an optimised yield of 97%. Notably, the increase in conversion still occurred with only 2 F, implying that a lower steady-state concentration may be important in the cathode chamber. To test this hypothesis, TMSCl was slowly added to the catholyte by syringe-pump addition over the course of the reaction, which gave a similar yield of 94%.40 Although intermediate 2a is transient and was never observed, the importance of TMSCl to trap and stabilise reduced 1a was revealed by DFT (B3LYP/6-311+g(d)) calculations,40 which suggested a thermodynamically highly challenging reaction in its absence.
The oxidation of bromide to tribromide occurs on the anode, which is an ideal counter-electrode process: not only is bromide an inexpensive and metal-free sacrificial reductant, but as the produced Br3− is anionic, it does not rapidly migrate to the cathodic chamber, preventing unwanted side reactions.43 The generated Br3− can even be used in follow-up bromination reactions.44 An increase in the applied cell potential during the reaction signifies the consumption of Br−, and the oxidation of Br3− to Br2 (Fig. 2).45 Despite needing 3 equivalents of Br− to form 2 equivalents of Br3− after 2 F, the loading of Br− could be reduced to 2 equivalents without affecting yield. No over-reduction of 3a to the monofluoromethyl ketone was observed, which is significant considering the small difference in reduction potentials.40 This emphasises the importance of a flat chronopotentiometry trace that is achieved with Br− oxidation. Other reductants were found to be sub-optimal, including diisopropylamine and oxalic acid.40
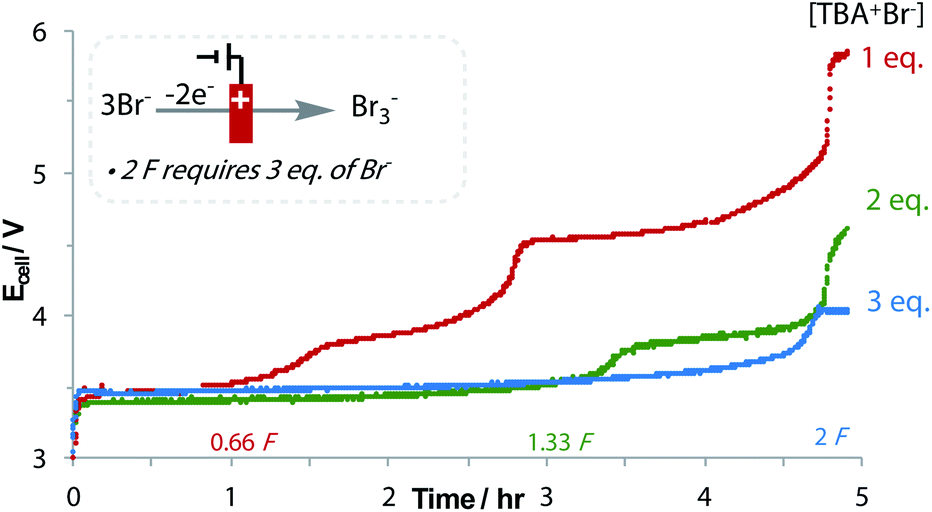 | ||
| Fig. 2 Reaction of 1a to 3a with 3 different Br− concentrations. | ||
A graphite anode performed equally well as platinum for the counter electrode reaction (entry 10). Only marginally reduced yields were observed with nickel and stainless-steel cathodes (entries 11 and 12), however, a drastic decrease in the yield was observed with a graphite cathode (entry 13), possibly due to substrate grafting.39
We proceeded to explore the substrate scope with our optimized conditions, Fig. 3. As expected, our electrochemical conditions were suitable for the hydrodefluorination of electron-poor acetophenone derivatives (1b, 1c). However, unlike with the use of Mg,30 substrates containing electron donating substituents are now well tolerated (1d–k). In addition, no hydrodebromination was observed for 1b, highlighting the selectivity and orthogonality granted by the use of our Mg-free, non-protic conditions. A selection of extended π-systems was tolerated, producing pyridyl 3l, biphenyl 3m, benzothiophene 3n, primary amine 3o, and pyrimidines 3p and 3q and in moderate to excellent yields. Chromoionophore dye 1r and stilbene 1s and were transformed in excellent yield, demonstrating tolerance to reductively sensitive alkenes, which would otherwise hydrogenate under protic electrochemical conditions.46 Anthracenyl 1t and naphthyl substrates 1u and 1v all transformed efficiently in good to excellent yields, the latter of which underwent direct double hydrodefluorination. 4.5% over-reduction was observed in the double defluorination product, 3v, which was the only instance where this side-product was observed in greater than 1% quantities.40 The good mass-balance and faradaic efficiency is notable considering the delocalization of charge around extended π-systems increases the likelihood of grafting.47
 | ||
| Fig. 3 Isolated yields of DFMKs tested under the reaction conditions at 0.5 mmol scale. NMR yields in parentheses. aReaction run at 10 mA; breaction run in IKA Divided ProSyn: quantitative yield based on RSM; c5 mmol scale, Ni foil:Gr (cath:an); disolated as the corresponding ketone following purification on silica.49 | ||
The model indole substrate 1a gave an excellent yield of DFMK at 0.5 mmol scale, which gave equally high yields when scaled up 10-fold (5 mmol), thereby demonstrating the robustness and practicality of the technique. We were also able to successfully prepare 3a in a commercially available divided cell set-up.40 Alternative groups on nitrogen, including Boc, perfluoropyridyl and benzyl (3w–y), as well as the free indole 3z, were well tolerated and gave moderate to good yields of 3. Tosyl and acetyl groups on nitrogen were less well tolerated.40 As with the acetophenones, indoles with electron donating (1aa) and withdrawing (1ab) groups proceeded to product. Methoxy demethylation of 3aa should lead to the corresponding phenol,48 which is difficult to prepare using other methodologies due to competing side-reactions. Halide substitution also successfully yielded DFMKs (3ac–ag). The inclusion of the aryl-iodide functionality is especially notable due to its facile reduction; when a silver cathode was used to convert 1ag, hydrodeiodination was observed, but which was absent under our non-protic conditions with a Pt cathode. Increased steric bulk around the reacting center in thiophenyl and phenyl-substituted substrates 1ah and 1ai had no negative influence and gave good yields of product.
Heterocyclic trifluoromethylketones were successfully hydrodefluorinated under the standard conditions, including indole 3aj, carbazole 3ak, pyrrole 3al, pyridine 3am, and pyrazoles 3an and 3ao, the latter of which leads to a compound with anti-malarial activity.15a Alkyl trifluoromethylketones are more difficult to reduce compared to aromatic trifluoromethylketones, and are therefore challenging substrates to hydrodefluorinate, and impossible to convert using other methods. Nevertheless, oleyl 1ap, cyclohexyl 1aq and ethyl 1ar substrates were all amenable to the conditions, although the smaller alkyl products were cumbersome to isolate due to their volatility. The non-protic optimized conditions ensured no loss of mass-balance at these enhanced reduction potentials (|Ecell| = ca. 3.4–3.7 V for alkyl substrates vs. ca. 2.3–2.7 V for acetophenones and indoles). Finally, we tested the conditions on trifluoroacetamide 1as, thioester 1at and imines 1au and 1av. For each of these, the corresponding product was returned in moderate to good yields. Despite some complications in their isolation, these results are notable considering their difference in structure and lack of precedent. Unsuccessful substrates included a nitro-substituted indole, which was insoluble in the reaction medium, and hydrated TFMKs.40
We tested a variety of substrates with the Mg-mediated conditions reported by Prakash to gauge the level of complementary between the methods.30 While acetophenone derivatives 1k and 1am were amenable to reduction with Mg, bromide substitution in 1b was unsurprisingly not tolerated with Grignard formation dominating. Indoles – 1a, 1ai, pyrazole – 1an, alkyl – 1aq, 1ar and anilide – 1as based trifluoromethylketones were untouched by Mg in all cases, with starting materials recovered only.
To explore the value of the DFMK moiety in synthesis, we derivatized it in a variety of ways, Fig. 4. Resubjecting the product 3a to our non-protic hydrodefluorination conditions led to monofluorinated product 4, providing an alternative to the use of electrophilic fluorine sources.50 Reduction of the ketone in 3ae to the methyl ether and alcohol successfully gave products, 5 and 6, respectively. The dithiane of 3a, which is a useful synthetic intermediate, was formed in excellent yield (7). A Corey–Chaykovsky methenylation gave epoxide 8 in good yield. A Horner–Wadsworth–Emmons reaction transformed the carbonyl to give alkene 9. Nucleophilic attack of the ketone was demonstrated with a trifluoromethylation reaction to give highly fluorinated alcohol 10. Orthogonal reactivity was also demonstrated with a Suzuki–Miyaura cross-coupling that gave biaryl 11. Interestingly, deuterium was not exchanged into 3a when stirred in a mixture of D2O and MeCN, providing evidence for a less favourable enolization.
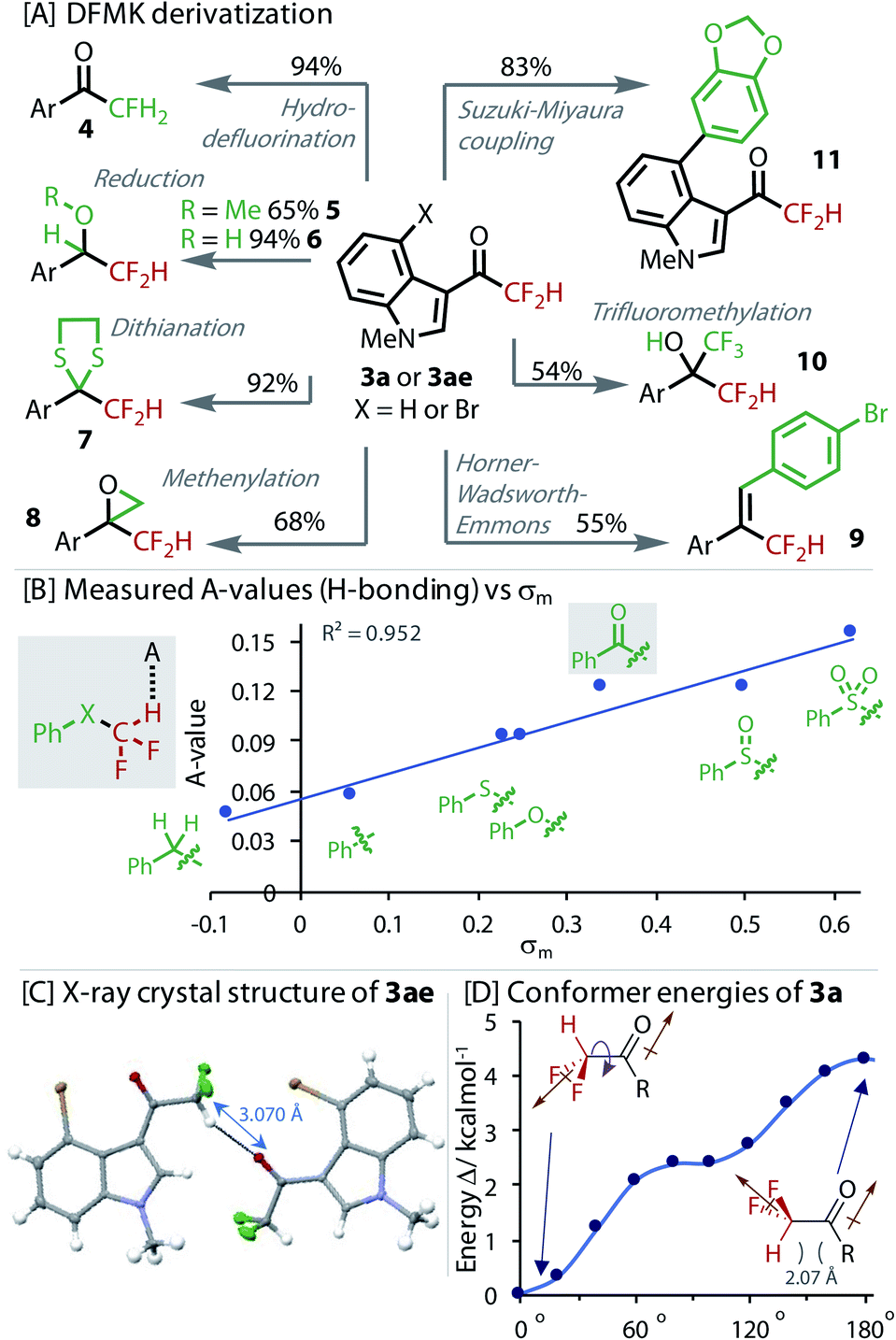 | ||
| Fig. 4 [A] Derivatization of DFMKs. X = H (3a) for 4, 7, and 8, X = Br (3ae) for others; [B] H-bond strength (A-value) correlated to σm Hammett parameter; [C] intermolecular H-bond revealed in X-ray crystal structure of 3ae; [D] DFT calculated (B3LYP/6-311+g(d)) relative energies of conformers with rotation around HC–CO bond. Brown arrows indicate direction of dipole. | ||
The H-bond strength (A-value) was measured for a series of phenyl substituted X–CF2H derivatives using the NMR method from Abraham, Fig. 4B.51–53 These experiments confirmed the sensitivity of the H-bonding ability to the identity of X. DFMK 3g and sulfoxide–CF2H were found to be comparable H-bond donors, which were only marginally less than the sulfone–CF2H. The H-bond strength correlated best with the σm parameter, reflecting the strong influence of inductive effects. Multiple regression analysis showed that any contribution of σp was statistically insignificant (P value = 0.33).
Analysis of the X-ray crystal structure of 3ae, showed an inter-molecular H-bond between the CF2H and a carbonyl from a neighbouring molecule (Fig. 4C). DFT was used to calculate the relative conformer energy with rotation about the (O)C–CF2H dihedral bond (Fig. 4D). The lowest energy conformer eclipsed the H with the carbonyl, implying the possibility of an energy lowering intra-molecular H-bond. However, analysis of the other derivatives in the set (C(O)CH3, C(O)CFH2 and C(O)CF3) revealed that the alignment of dipoles was the dominant effect (brown arrows, Fig. 4D).40 The absence of an unusually low or even negative A-value also provides evidence against an intramolecular H-bond.51 Interestingly, in the solid-state structure (Fig. 4C), the highest energy conformer (with dipoles aligned) is adopted, highlighting the stronger propensity of this moiety to engage in H-bonding interactions.
In conclusion, we have developed a mono-selective hydrodefluorination to access a broad scope of DFMKs, enabled by non-protic electrochemical conditions at deeply reducing potentials. These moieties have been studied and diversified and reveal themselves to be potentially useful dual H-bond donor/acceptor moieties. This is especially interesting considering the structurally related trifluoromethylketones are known reversible protease inhibitors;54,55 thus, the additional H-bonding moiety could enhance interaction within enzymatic active sites.15
Data availability
All underlying data are provided as ESI accompanying this paper.Author contributions
J. R. B. and A. P. A. performed the experimental work, J. R. B. conducted the computational calculations, J. R. B. and A. J. J. L. wrote the manuscript, A. J. J. L. conceived and directed the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Royal Society (University Research Fellowship, Enhancement award and Research Grant for Research Fellow to AJJL), EPSRC (EP/S018050/1) and Syngenta for funding. We would also like to acknowledge Dr Natalie Pridmore (University of Bristol) for the X-ray crystallography. We would like to dedicate this manuscript to Prof Shannon Stahl on the occasion of his 50th birthday.Notes and references
- Y. Zafrani, G. Sod-Moriah, D. Yeffet, A. Berliner, D. Amir, D. Marciano, S. Elias, S. Katalan, N. Ashkenazi, M. Madmon, E. Gershonov and S. Saphier, J. Med. Chem., 2019, 62, 5628–5637 CrossRef CAS PubMed.
- Y. Zafrani, D. Yeffet, G. Sod-Moriah, A. Berliner, D. Amir, D. Marciano, E. Gershonov and S. Saphier, J. Med. Chem., 2017, 60, 797–804 CrossRef CAS.
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed.
- M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS.
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- C. D. Sessler, M. Rahm, S. Becker, J. M. Goldberg, F. Wang and S. J. Lippard, J. Am. Chem. Soc., 2017, 139, 9325–9332 CrossRef CAS PubMed.
- J. A. Erickson and J. I. McLoughlin, J. Org. Chem., 1995, 60, 1626–1631 CrossRef CAS.
- Q. A. Huchet, B. Kuhn, B. Wagner, N. A. Kratochwil, H. Fischer, M. Kansy, D. Zimmerli, E. M. Carreira and K. Müller, J. Med. Chem., 2015, 58, 9041–9060 CrossRef CAS PubMed.
- B. Jeffries, Z. Wang, H. R. Felstead, J.-Y. Le Questel, J. S. Scott, E. Chiarparin, J. Graton and B. Linclau, J. Med. Chem., 2020, 63, 1002–1031 CrossRef CAS PubMed.
- Y. Zafrani, S. Saphier and E. Gershonov, Future Med. Chem., 2020, 12, 361–365 CrossRef CAS.
- B. Zheng, S. V. D'Andrea, L.-Q. Sun, A. X. Wang, Y. Chen, P. Hrnciar, J. Friborg, P. Falk, D. Hernandez, F. Yu, A. K. Sheaffer, J. O. Knipe, K. Mosure, R. Rajamani, A. C. Good, K. Kish, J. Tredup, H. E. Klei, M. Paruchuri, A. Ng, Q. Gao, R. A. Rampulla, A. Mathur, N. A. Meanwell, F. McPhee and P. M. Scola, ACS Med. Chem. Lett., 2018, 9, 143–148 CrossRef CAS.
- F. Narjes, K. F. Koehler, U. Koch, B. Gerlach, S. Colarusso, C. Steinkühler, M. Brunetti, S. Altamura, R. De Francesco and V. G. Matassa, Bioorg. Med. Chem. Lett., 2002, 12, 701–704 CrossRef CAS.
- D. E. Yerien, S. Barata-Vallejo and A. Postigo, Chem.–Eur. J., 2017, 23, 14676–14701 CrossRef CAS.
- (a) E. Camerino, D. M. Wong, F. Tong, F. Körber, A. D. Gross, R. Islam, E. Viayna, J. M. Mutunga, J. Li, M. M. Totrov, J. R. Bloomquist and P. R. Carlier, Bioorg. Med. Chem. Lett., 2015, 25, 4405–4411 CrossRef CAS PubMed; (b) A. Citarella, D. Gentile, A. Rescifina, A. Piperno, B. Mognetti, G. Gribaudo, M. T. Sciortino, W. Holzer, V. Pace and N. Micale, Int. J. Mol. Sci., 2021, 22, 1398 CrossRef CAS PubMed.
- G. Pattison, Eur. J. Org. Chem., 2018, 3520–3540 CrossRef CAS.
- I. Pravst, M. Zupan and S. Stavber, Synthesis, 2005, 3140–3146 CAS.
- D. J. Leng, C. M. Black and G. Pattison, Org. Biomol. Chem., 2016, 14, 1531–1535 RSC.
- M. F. Sowaileh, C. Han, R. A. Hazlitt, E. H. Kim, J. P. John and D. A. Colby, Tetrahedron Lett., 2017, 58, 396–400 CrossRef CAS.
- J. Phetcharawetch, N. M. Betterley, D. Soorukram, M. Pohmakotr, V. Reutrakul and C. Kuhakarn, Eur. J. Org. Chem., 2017, 6840–6850 CrossRef CAS.
- A. L. Trifonov, V. V. Levin, M. I. Struchkova and A. D. Dilman, Org. Lett., 2017, 19, 5304–5307 CrossRef CAS.
- M. Miele, A. Citarella, N. Micale, W. Holzer and V. Pace, Org. Lett., 2019, 21, 8261–8265 CrossRef CAS.
- T. H. Krane Thvedt, K. Kaasa, E. Sundby, C. Charnock and B. H. Hoff, Eur. J. Med. Chem., 2013, 68, 482–496 CrossRef CAS PubMed.
- T. H. Krane Thvedt, E. Fuglseth, E. Sundby and B. H. Hoff, Tetrahedron, 2009, 65, 9550–9556 CrossRef CAS.
- S. Yu, W. Plunkett, M. Kim, E. Wu and L. Pu, Org. Chem. Front., 2014, 1, 395–404 RSC.
- X. Creary, J. Org. Chem., 1987, 52, 5026–5030 CrossRef CAS.
- J.-P. Bégué and D. Bonnet-Delpon, Tetrahedron, 1991, 47, 3207–3258 CrossRef.
- The corresponding Friedel–Crafts C–H difluoroacetylation reaction that would lead directly to the product is unprecedented for arenes, and rare for indoles (for example, with gaseous difluoroacetonitrile, see E. K. Raja and D. A. Klumpp, Tetrahedron Lett., 2011, 52, 5170–5172, for two isolated examples with difluoroacetic anhydride, see F. Brüning, H. Nagae, D. Käch, K. Mashima and A. Togni, Chem.–Eur. J., 2019, 25, 10818 and H. Rueeger, R. Lueoend, O. Rogel, J. M. Rondeau, H. Möbitz, R. MacHauer, L. Jacobson, M. Staufenbiel, S. Desrayaud and U. Neumann, J. Med. Chem., 2012 55, 3364–3386, or with difluoroacetic acid at high temperature, see S.-J. Yao, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, J. Org. Chem., 2016, 81, 4226–4234). This strategy is more commonly used for the difluoroacetylation of O, N or S-centred nucleophiles, see for example, C. R. Jones, P. K. Baruah, A. L. Thompson, S. Scheiner and M. D. Smith, J. Am. Chem. Soc., 2012, 134, 12064–12071 CrossRef CAS PubMed.
- Difluoroacetylation reagents are more expensive than trifluoroacetylation reagents (e.g., difluoroacetic anhydride is over 170 times the cost of trifluoroacetic anhydride (April 2021, major UK supplier)), and if combined with a lithiated arene, undergo competing deprotonation of the CF2H group, see for example ref. 15a. Compared to DFMKs, trifluoromethylketones themselves are also more inexpensive and readily available.
- G. Surya Prakash, J. Hu and G. A. Olah, J. Fluorine Chem., 2001, 112, 355–360 CrossRef.
- H. Amii, T. Kobayashi, Y. Hatamoto and K. Uneyama, Chem. Commun., 1999, 1323–1324 RSC.
- D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386–12400 RSC.
- S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 CrossRef.
- A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC.
- K. Uneyama, K. Maeda, T. Kato and T. Katagiri, Tetrahedron Lett., 1998, 39, 3741–3744 CrossRef CAS.
- K. Uneyama, G. Mizutani, K. Maeda and T. Kato, J. Org. Chem., 1999, 64, 6717–6723 CrossRef CAS.
- D. M. Heard and A. J. J. Lennox, Angew. Chem., Int. Ed., 2020, 59, 18866–18884 CrossRef CAS PubMed.
- See ESI for details.†.
- B. K. Peters, K. X. Rodriguez, S. H. Reisberg, S. B. Beil, D. P. Hickey, Y. Kawamata, M. Collins, J. Starr, L. Chen, S. Udyavara, K. Klunder, T. J. Gorey, S. L. Anderson, M. Neurock, S. D. Minteer and P. S. Baran, Science, 2019, 363, 838–845 CrossRef CAS PubMed.
- H. Sun and S. G. DiMagno, J. Am. Chem. Soc., 2005, 127, 2050–2051 CrossRef CAS PubMed.
- This can be clearly observed by the characteristic orange colour of Br3−, which builds up over the course of the reaction, only mixing slowly into the cathodic chamber after the electrolysis is terminated.
- The generated [Br3−] collected in the anolyte was used to cleanly brominate phenyl trifluoroborate and acetophenone.
- F. Wang and S. S. Stahl, Angew. Chem., Int. Ed., 2019, 58, 6385–6390 CrossRef CAS PubMed.
- X. Liu, R. Liu, J. Qiu, X. Cheng and G. Li, Angew. Chem., Int. Ed., 2020, 59, 13962–13967 CrossRef CAS PubMed.
- S. N. Mailu, T. T. Waryo, P. M. Ndangili, F. R. Ngece, A. A. Baleg, P. G. Baker and E. I. Iwuoha, Sensors, 2010, 10, 9449–9465 CrossRef CAS PubMed.
- V. Bernard-Gauthier, A. Aliaga, A. Aliaga, M. Boudjemeline, R. Hopewell, A. Kostikov, P. Rosa-Neto, A. Thiel and R. Schirrmacher, ACS Chem. Neurosci., 2015, 6, 260–276 CrossRef CAS PubMed.
- G. Verniest, E. Van Hende, R. Surmont and N. De Kimpe, Org. Lett., 2006, 8, 4767–4770 CrossRef CAS PubMed.
- T. H. Krane Thvedt, E. Fuglseth, E. Sundby and B. H. Hoff, Tetrahedron, 2009, 65, 9550–9556 CrossRef CAS.
- M. H. Abraham, R. J. Abraham, W. E. Acree, A. E. Aliev, A. J. Leo and W. L. Whaley, J. Org. Chem., 2014, 79, 11075–11083 CrossRef CAS PubMed.
- M. H. Abraham, R. J. Abraham, J. Byrne and L. Griffiths, J. Org. Chem., 2006, 71, 3389–3394 CrossRef CAS PubMed.
- A = 0.0065 + 0.133Δδ, where Δδ = 1H NMR δ(DMSO) − δ(CDCl3).
- H. K. Nair and D. M. Quinn, Bioorg. Med. Chem. Lett., 1993, 3, 2619–2622 CrossRef CAS.
- C. P. Govardhan and R. H. Abeles, Arch. Biochem. Biophys., 1990, 280, 137–146 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2061359. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01574e |
| This journal is © The Royal Society of Chemistry 2021 |