
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemically switchable polymerization from surface-anchored molecular catalysts†
Miao
Qi‡
 a,
Haochuan
Zhang‡
a,
Qi
Dong‡
a,
Jingyi
Li
a,
Rebecca A.
Musgrave
b,
Yanyan
Zhao
a,
Nicholas
Dulock
a,
Dunwei
Wang
*a and
Jeffery A.
Byers
*a
a,
Haochuan
Zhang‡
a,
Qi
Dong‡
a,
Jingyi
Li
a,
Rebecca A.
Musgrave
b,
Yanyan
Zhao
a,
Nicholas
Dulock
a,
Dunwei
Wang
*a and
Jeffery A.
Byers
*a
aDepartment of Chemistry, Boston College, 2609 Beacon St., Chestnut Hill, MA 02467, USA. E-mail: dwang@bc.edu; jeffery.byers@bc.edu
bDepartment of Chemistry and Chemical Biology, Harvard University, 12 Oxford Street, Cambridge, Massachusetts 02138, USA
First published on 28th May 2021
Abstract
Redox-switchable polymerizations of lactide and epoxides were extended to the solid state by anchoring an iron-based polymerization catalyst to TiO2 nanoparticles. The reactivity of the molecular complexes and their redox-switching characteristics were maintained in the solid-state. These properties resulted in surface-initiated polymerization reactions that produced polymer brushes whose chemical composition is dictated by the oxidation state of the iron-based complex. Depositing the catalyst-functionalized TiO2 nanoparticles on fluorine-doped tin oxide resulted in an electrically addressable surface that could be used to demonstrate spatial control in redox-switchable polymerization reactions. By using a substrate that contained two electrically isolated domains wherein one domain was exposed to an oxidizing potential, patterns of surface-bound polyesters and polyethers were accessible through sequential application of lactide and cyclohexene oxide. The differentially functionalized surfaces demonstrated distinct physical properties that illustrated the promise for using the method to pattern surfaces with multiple, chemically distinct polymer brushes.
Introduction
Catalytic reactions that operate at the interface between homogeneous and heterogeneous catalysis has been a longstanding goal in chemical synthesis.1,2 The benefits of combining easily tunable and well-defined single site homogeneous catalysts with robust and more easily recycled heterogeneous catalysts has inspired many researchers to pursue strategies for heterogenizing well-defined transition metal-based homogeneous catalysts by anchoring them to solid supports.3–5 These hybrid catalysts often demonstrate superior catalytic properties compared to their homogeneous counterparts, including increased activity, increased catalyst lifetimes, and catalysts that can more readily be recycled.6 Transition metal-based complexes supported onto electrodes7–9 and semiconducting materials10,11 have shown great promise for evolving the capabilities of hybrid catalysts by combining the bulk properties of the solid support with the molecular properties of the transition metal-based complexes. In particular, rapid injection of electrons or holes into surface-anchored molecular catalysts has opened new opportunities for reactions relevant to energy storage and conversion.12,13Outside of the advances made in energy research, hybrid catalysts have not been extensively explored in other areas.14,15 In polymer chemistry, supporting molecular transition metal-based catalysts has enabled surface-initiated polymerization reactions, which have led to polymer brushes that have unique and tunable properties.16–18 By combining these innovations with photolithographic techniques, impressive spatial resolution down to the nanometer scale can be achieved.19–21 Despite these excellent achievements, the ability to construct surfaces patterned with multiple, different polymeric materials has been challenging. Surfaces decorated with polymer brushes with varying physical properties have many applications: they can be sensors,22,23 anti-fouling paints,24,25 and they can be used for several applications in the electronics industry.26,27 However, state of the art techniques to synthesize such materials involve multistep manipulations that require several high yielding surface modifications.28–30 These methods have also been limited to monomers that are polymerized through radical or olefin metathesis polymerization mechanisms. This limitation makes it more difficult to incorporate classes of polymers that do not have hydrocarbon backbones (e.g., polyesters, polyamides, polycarbonates, etc.). Alternatives to traditional methods may simplify fabrication methods for obtaining patterned surfaces and broaden monomer scope, both of which will further the utility of surface-initiated polymerization systems.
One such alternative is to utilize redox-switchable polymerization reaction systems31,32 for surface-initiated polymerization. Since the Gibson and Long groups developed the first instance of redox-switchable lactide polymerization,33 many groups have reported control over polymerization processes by altering catalyst oxidation state.33–39 Common strategies include installing redox active moieties (e.g. ferrocene) in ligand frameworks33 or using redox-active elements that also serve as the site for catalysis.34,37,38 Our group has been investigating a redox-switchable polymerization system based on bis(imino)pyridine iron bisalkoxide complexes that belongs to the latter class of catalysts. These complexes can toggle between catalyzing the ring-opening polymerization (ROP) of lactide and epoxides when either redox reagents or electrochemical stimuli are applied (Scheme 1).37,38,40 Such a unique selectivity provides opportunity for the synthesis of block copolymers38 and cross-linked polymer networks.41 Herein, we extend this concept for surface-initiated polymerization reactions so that it may be used for facile surface modifications. The method relies on supporting redox-switchable iron-based molecular polymerization catalysts to a semi-conducting titanium dioxide electrode surface containing electrically isolated zones that enable selective oxidation of the switchable catalyst (Scheme 1). We envision that such a system will enable in situ generation of different polymer patterns simply upon selective application of an electrical stimulus, thereby greatly reducing the steps required for forming sophisticated polymer patterns. Moreover, unlike most other surface-initiated polymerization systems, the anchored catalyst catalyzes ROP reactions,42,43 which expands the scope of monomers that typically engage in surface-initiated polymerization reactions. In addition to the advances made in surface-initiated polymerization reactions, the research disclosed here involves characterization of redox-active metal centers supported on metal oxide surfaces using a variety of physical and spectroscopic techniques. Such fundamental information is uncommon and will be invaluable for the further development of hybrid catalysts derived from supporting homogeneous catalysts on functional surfaces.
 | ||
| Scheme 1 Iron-based catalysts reported for the redox-switchable polymerization of lactide and epoxides38,40 are extended to the solid state by appending iron-based catalysts to titanium dioxide nanoparticles. Application of an electric stimulus to a conducting substrate containing electrically isolated zones coated with these particles enables patterning of surfaces with two different polymer brushes. | ||
Results and discussion
Synthesis and characterization of iron(II)-anchored nanoparticles
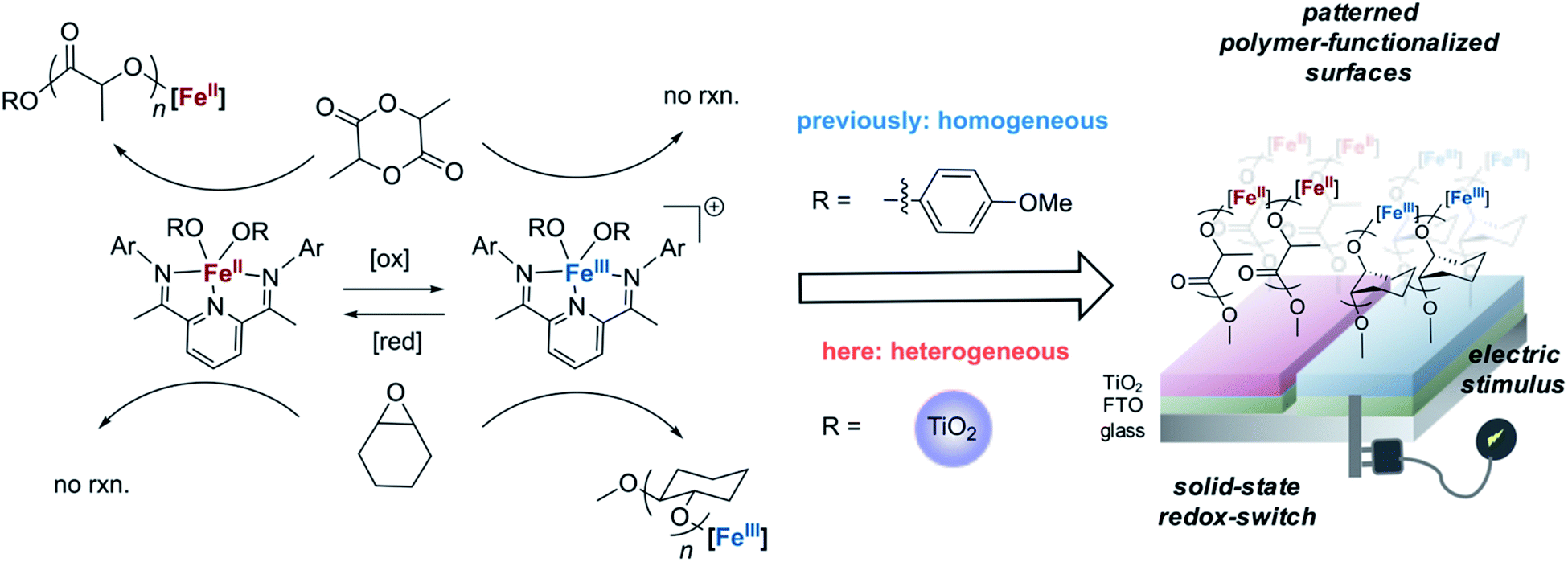
Towards the goal of developing a redox-switchable surface-initiated polymerization process, we first attempted to anchor the catalyst onto a surface. Previously, our group has found that protonolysis reactions between the bis(imino)pyridine iron(II) bisalkyl precursor 1 and various organic alcohols have proven reliable in synthesizing molecular, monomeric iron alkoxide complexes.37,44–46 Recognizing that hydroxyl groups are abundant on metal oxide surfaces, we hypothesized that surface hydroxyl groups would be good surrogates for alcohols, providing a convenient way to anchor the iron complexes directly to metal oxide surfaces. In these initial proof-of-principle investigations, we chose the semiconducting titanium dioxide as the metal oxide to support the catalyst so as to take advantage of its known surface chemistry,47 in particular the ability to introduce high densities of surface hydroxyl groups.48 To maximize surface hydroxyl densities, we treated commercially available P25 TiO2 nanoparticles with UV light, and then applied heat at 150 °C for 16 hours under reduced pressure (10−5 torr) to remove residual water without disrupting surface hydroxyl groups.49 Thermogravimetric analysis (TGA) demonstrated the efficacy of removing water with this procedure and provided an estimate of the surface hydroxyl concentration of 0.46 mmol g−1 (Fig. S2†), which is consistent with literature reports.49 Next, we exposed the nanoparticles to a diethyl ether solution of 1 to anchor the iron complex to the nanoparticles (Fig. 1a). The resulting iron-containing nanoparticles were light purple, the characteristic color of the molecular iron(II) alkoxide complexes used for lactide polymerization.37 Inductively coupled plasma optical emission spectroscopic (ICP-OES) analysis of the particles revealed an iron content that was 2.1 wt%. This high loading is consistent with nearly all of the surface hydroxyl groups being modified with an iron complex, assuming that one molecule of 1 reacts with two surface hydroxyl groups (Fig. 1a).37 This stoichiometry is consistent with the molecular iron complex bridging two consecutive hydroxyl groups on the surface of TiO2.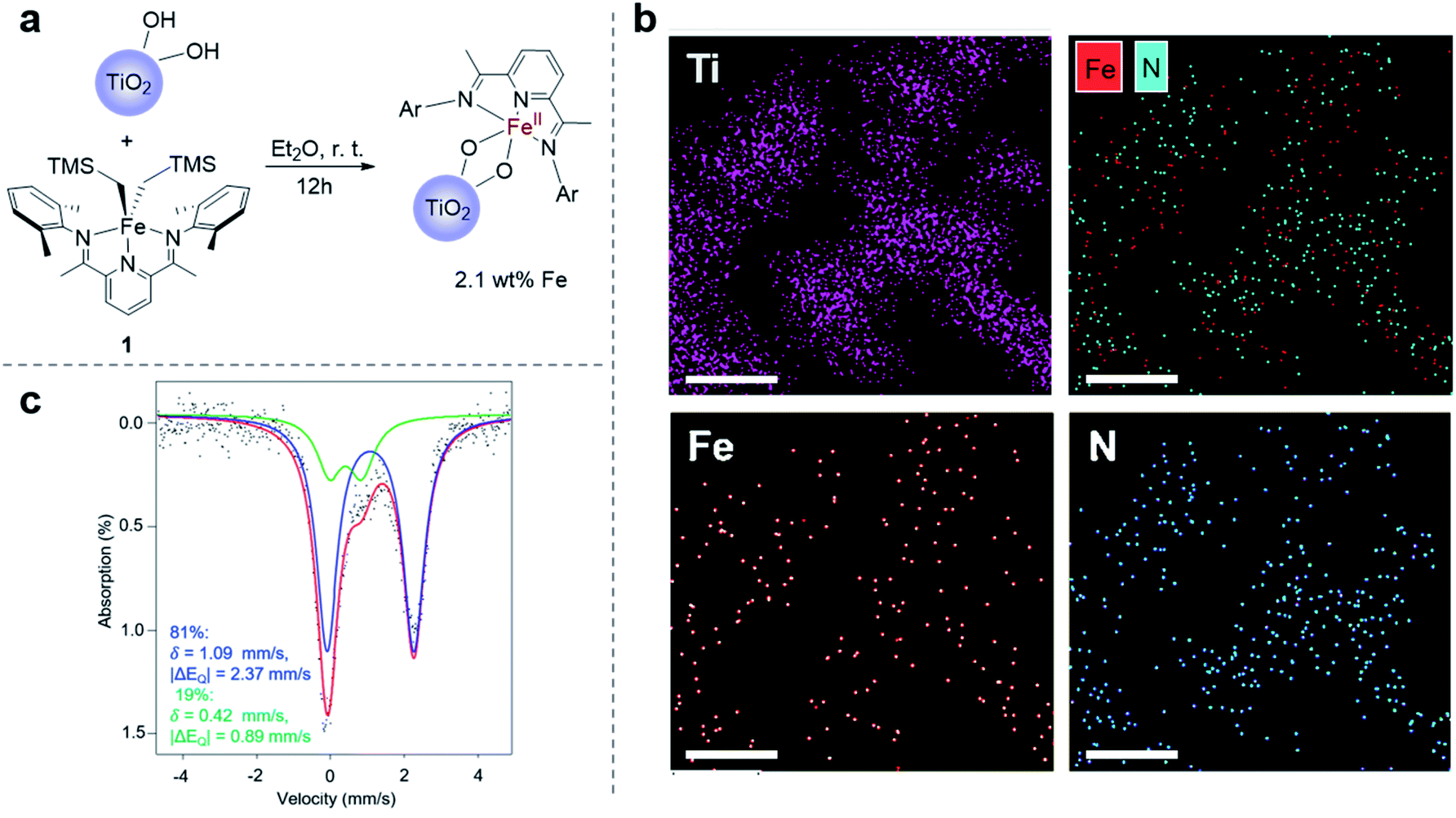 | ||
| Fig. 1 (a) Reaction scheme for anchoring the molecular iron complex onto P25 TiO2 nanoparticle (Ar = 2,6-dimethylphenyl); (b) STEM elemental mapping of the Fe(II)–TiO2 powder (scale bar: 50 nm); (c) Mössbauer spectroscopy of Fe(II)–TiO2 powder. | ||
A suite of analytical methods was used to characterize the iron-functionalized nanoparticles. That the iron complexes are evenly distributed on the surface of TiO2 is strongly supported by elemental mapping using a scanning transmission electron microscope (STEM) (Fig. 1b and S3†).50 Moreover, the spatial correlation between iron and nitrogen in the STEM is consistent with the metal complexes being deposited on the surface rather than the complexes serving as a precursor for deposition of elemental iron. Mössbauer spectroscopy provided further evidence that the molecular identity of the iron complex was maintained when supported on the TiO2 surface (Fig. 1c). The Mössbauer spectrum revealed the presence of two iron-containing species. The major species (81%) had an isomer shift of δ = 1.09 mm s−1 and a quadrupole splitting of |ΔEQ| = 2.37 mm s−1, while the minor species (19%) had δ = 0.42 mm s−1 and |ΔEQ| = 0.89 mm s−1. The molecular bis(imino)pyridine iron(II) bisphenoxide complex has Mössbauer parameters (δ = 0.94 mm s−1, |ΔEQ| = 2.19 mm s−1)51 similar to the major species observed on the functionalized nanoparticle. Importantly, the isomer shift of the precursor 1 is 0.25 mm s−1,52 which is too low to be either species observed in the functionalized nanoparticle.
Mössbauer parameters of a complex that approximates the surface bound species was calculating using DFT B3LYP/631G triple-ζ-quality basis sets TZVP.53–55 This level of theory has previously been used successfully to describe other molecular iron complexes containing the bis(imino)pyridine ligand.45,56 To model the coordination environment predicted from the stoichiometry of the chemical reaction between 1 and TiO2, an iron(II) bistitanoxide complex bridging two titanium centers was constructed (Table S1†). This model complex S2 had computed Mössbauer parameters (δ = 1.04 mm s−1, |ΔEQ| = 2.11 mm s−1) similar to the major species found in the iron-functionalized nanoparticle. Therefore, we conclude that the protonolysis reaction between 1 and the TiO2 nanoparticles yielded primarily a surface bound iron species that has a similar oxidation state, spin state and coordination environment as the molecular iron(II) alkoxide complex previously used for lactide polymerization. While we cannot yet definitively assign the structure of the minor species observed in the Mössbauer spectrum, it has similar parameters as the oxidized iron(III) complex discussed below.
Surface-initiated polymerization of lactide
When exposing the iron(II) functionalized TiO2 nanoparticles to a dichloromethane solution of lactide overnight, 67% lactide consumption was observed relative to an internal standard by proton nuclear magnetic resonance spectroscopy (1H NMR). Importantly, while consumption of monomer was measurable, no evidence for poly(lactic acid) (PLA) was detectable in the supernatant by 1H NMR. This observation suggested that polymerization occurred on the surface of the nanoparticles. TGA of the nanoparticles exposed to lactide showed a 61% weight loss at 283 °C (Fig. S4a†), which is the known decomposition temperature for poly(lactic acid).57 Attenuated total reflection-Fourier transform infrared spectroscopy (ATR-FTIR) of the Fe(II)–TiO2 powder with lactide polymerization also indicated the presence of PLA on the surface of the nanoparticles (Fig. S5a†). To provide further evidence that a surface-initiated polymerization reaction occurred, the surface-grown polymers could be cleaved from the nanoparticles by treating them with iodomethane (Fig. 2a). IR of the iodomethane-treated TiO2 powder revealed no evidence for remaining poly(lactic acid), which suggested that the polymer was quantitatively cleaved from the particles (Fig. S5a†). 1H NMR analysis of the polymer cleaved from the surface revealed that methylation occurred exclusively at the carboxylate terminus, as evidenced by the singlet at 3.77 ppm (Fig. S6†). The hydroxyl chain end, which is presumed to be ligated to iron during lactide polymerization, remained unreactive as is evidenced by the resonance at 4.23 ppm. This end group has previously been assigned to the methine proton adjacent to a hydroxyl terminus of poly(lactic acid).37 That the ester end group was preferentially alkylated instead of the alcoholic end group is consistent with the greater nucleophilicity of the titanium ester chain end relative to the alcohol chain end.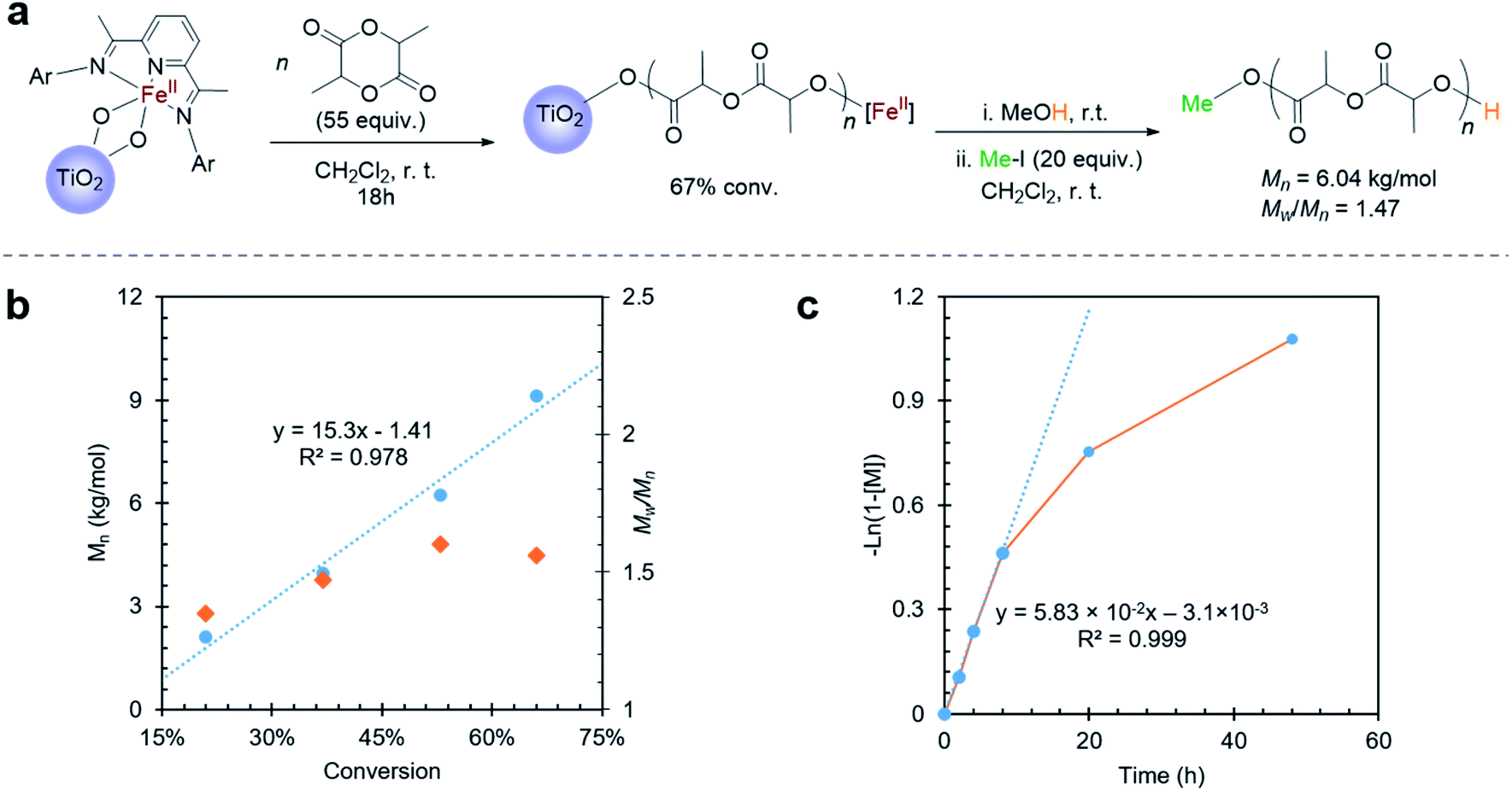 | ||
| Fig. 2 (a) Reaction scheme for lactide polymerization on Fe(II)–TiO2 particles; kinetic analysis of the lactide polymerization on Fe(II)–TiO2 particles: (b) evolution of Mnversus conversion, Mw/Mn was shown in the second y axis on the right, and (c) first order kinetic plot of lactide polymerization. | ||
Cleaving the polymer from the surface also provided a facile way to evaluate molecular weight of the polymer produced using gel permeation chromatography (GPC). GPC analysis of the cleaved poly(lactic acid) revealed a polymer with molecular weight of Mn = 6.04 kg mol−1 and a dispersity of Mw/Mn = 1.47. The polymer molecular weight agreed with the predicted molecular weight of 5.31 kg mol−1, which was calculated from the conversion of the reaction, the iron loading, and the assumption that only one polymer chain is initiated per iron center. This assumption is based on our previous results for reactions initiated from iron alkoxides derived from alcohols with acidity (pKa < 10)37 similar to Ti–OH.58 The good match between the measured and predicted molecular weights suggested that most of the iron centers anchored on TiO2 are active for lactide polymerization.
To further illustrate the benefit of the cleavage method, a time course investigation of the polymerization reaction was undertaken. In this experiment, liquid aliquots were removed at various time points, and they were analyzed by 1H NMR spectroscopy to obtain lactide conversion. Solid aliquots removed simultaneously could be subjected to methyl iodide treatment so as to obtain polymer molecular weight information by GPC. Using this combination of techniques, molecular weight, molecular weight distribution, and conversion could be monitored over time (Fig. 2b and c). This study revealed a linear increase of molecular weight with conversion, which suggested that the surface-initiated lactide polymerization had living characteristics (Fig. 2b). This behavior was similar to lactide polymerization catalyzed by the homogeneous molecular iron complex.37 Different from the homogeneous reactions, however, were slower reaction rates and broader molecular weight distributions. Examining the conversion versus time plots revealed a possible explanation (Fig. 2c). At low conversion (<40%), the reaction rate was fast and followed first order reaction kinetics. Molecular weight distributions were also narrower than observed at the end of the reaction. However, at higher conversion, the reaction deviated from first order kinetics reaching an ultimate conversion of 65%. Coincidentally, the molecular weight distribution became broader as the reaction proceeded. The slower reaction rates and higher dispersity at high conversions is consistent with mass transport becoming more prominent as the polymerization proceeds. To rule out possible contributions for the TiO2 surface, control experiments were carried out using the TiO2 particles that were not functionalized with the iron complex. These control experiments revealed no reaction, indicating that the molecular iron complex was needed to initiate lactide polymerization.
Redox-switching and surface-initiated polymerization of cyclohexene oxide
Convinced that the supported iron(II) complexes were capable of carrying out lactide polymerization in a similar way as they did in the solution state, we next explored the oxidation of the iron(II)-functionalized titanium nanoparticles with the goal of observing the switch in chemoselectivity, as previously observed.38,40 Reacting the nanoparticles with an excess of ferrocenium hexafluorophosphate (FcPF6) produced a light brown solid (Fig. 3a). Analysis of these particles by Mössbauer spectroscopy revealed once again the presence of two species, but this time the major signal (75%) was a new iron species with δ = 0.49 mm s−1 and |ΔEQ| = 0.85 mm s−1 (Fig. 3b). These parameters are similar to those obtained for the major species observed in the Mössbauer spectrum of the molecular cationic Fe(III) bisalkoxide complex 3, which exhibit δ = 0.45 mm s−1 and |ΔEQ| = 0.85 mm s−1 (Fig. S8†). Further corroborating the experimental findings were Mössbauer parameters computed for a cationic iron(III) bistitanoxide model complex S3 (Table S1,†δ = 0.46 mm s−1, |ΔEQ| = 0.98 mm s−1), which are similar to the major species found in the iron(III)-functionalized nanoparticles. Thus, we conclude that oxidation of the iron(II)-containing particles occurred successfully with minimal complex decomposition. Interestingly, the minor species (25%) in the oxidized powder had Mössbauer parameters (δ = 1.28 mm s−1, |ΔEQ| = 2.86 mm s−1) whose large isomer shift indicated a high spin Fe(II) compound. These parameters are similar but different compared to the reduced iron(II) species supported on TiO2 (Fig. 1c), which suggests the formation of a new high spin iron(II) complex. At the current stage of investigation, we cannot assign this unknown iron(II) complex, but we suspect that it may be a solvated iron(II) alkoxide complex. | ||
| Fig. 3 (a) Reaction scheme for cyclohexene oxide polymerization on Fe(III)–TiO2 particles; (b) Mössbauer spectroscopy of Fe(III)–TiO2 powder. | ||
Consistent with the homogeneous complexes,38,40 the oxidized nanoparticles were completely inactive when exposed to lactide. Moreover, unlike the iron(II)-functionalized nanoparticles, which were found to be unreactive when exposed to cyclohexene oxide, the iron(III)-functionalized nanoparticles resulted in consumption of cyclohexene oxide, giving 33% conversion after stirring overnight. Once again, conversion of the monomer occurred without evidence for polyether being formed in the supernatant, and TGA analysis of the resulting particles was consistent with surface-initiated polymerization reactions (Fig. S4b†). FTIR of the Fe(III)–TiO2 powder with cyclohexene oxide polymerization also indicated the presence of poly(cyclohexene oxide) (PCHO) on the surface of the nanoparticles (Fig. S5b†). As was the case with surface-initiated poly(lactic acid), the polymer formed in the epoxide polymerization reaction could be cleaved from the surface by treating the nanoparticles with iodomethane (Fig. S7†). GPC analysis of the PCHO product revealed a polymer molecular weight of Mn = 11.6 kg mol−1 and a molecular weight distribution of Mw/Mn = 3.08 (Fig. 3a). The molecular weight obtained was higher than the theoretical molecular weight (Mn(theor) = 5.43 kg mol−1) and, as was the case for poly(lactic acid), the molecular weight distributions were broader than observed in the solution state. Notably, the polymerization of cyclohexene oxide catalyzed by the molecular cationic iron(III) complex does not display living characteristics.38,40,41 Kinetic analysis was not carried out for the cyclohexene oxide polymerization as was done for the lactide polymerization because the kinetics for the epoxide polymerization with the homogeneous complex does not follow simple first-order kinetics. Nevertheless, we attribute the broad molecular weight distribution to mass transport limitations, which are a consequence of the iron-based complex being supported directly off the TiO2 surface. The possibility that the TiO2 surface was also initiating epoxide polymerization was ruled out with control experiments carried out on the unfunctionalized TiO2 nanoparticles.
Electrochemical features of iron-complexes anchored on TiO2
With the successful anchoring of the reactive iron complexes on the TiO2 surfaces and their redox-switching capabilities demonstrated, we then moved to test the polymerization reactions on conductive surfaces so that electrochemical potential could be used to affect redox-switching.59 To maximize the yield of surface grafted polymers for the ease of characterization, we constructed an electrode with P25 TiO2 nanoparticles as the active material and fluorine doped tin oxide (FTO) as the conductive substrate. For this purpose, we modified a procedure that has worked well for the fabrication of electrodes in dye-sensitized solar cells.60 It started with applying as a film made from a slurry of P25 TiO2 nanoparticles in a mixture of Triton X-100, acetylacetone and deionized water. These films were applied either using a doctor blade or using an air brush. Annealing in air at 450 °C for 30 min gave the FTO electrode coated with TiO2 particles. After annealing, the electrode was soaked in a diethyl ether solution of 1 (0.02 M) to support the iron complex on the TiO2 surface. The surfaces contained 1.7 ± 0.5 wt% iron, which was similar to the loading obtained using the TiO2 nanoparticles and translates to 0.031 ± 0.009 mg iron per cm2 on the surface. The iron(II) functionalized electrode was then exposed to lactide in dichloromethane (0.35 M) for 12 h. Subsequent FTIR characterization of the functionalized surface after polymerization confirmed the presence of poly(lactic acid) with the appearance of an absorption band at 1710 cm−1 (Fig. 4a). In addition to this characteristic band, the surface-initiated poly(lactic acid) also contained a broad band at 1610 cm−1, which was not observed in the drop-casted polymer nor in IR taken of the iron(II) functionalized electrode prior to polymerization. We hypothesize that this broad feature is due to the interaction of surface-initiated polyester carbonyl with the Lewis acidic electrode surface.61 That these interactions are not observed for the drop-casted polymer illustrate how covalent interactions between the polymer and the surface differ from physical deposition of polymer on the surface.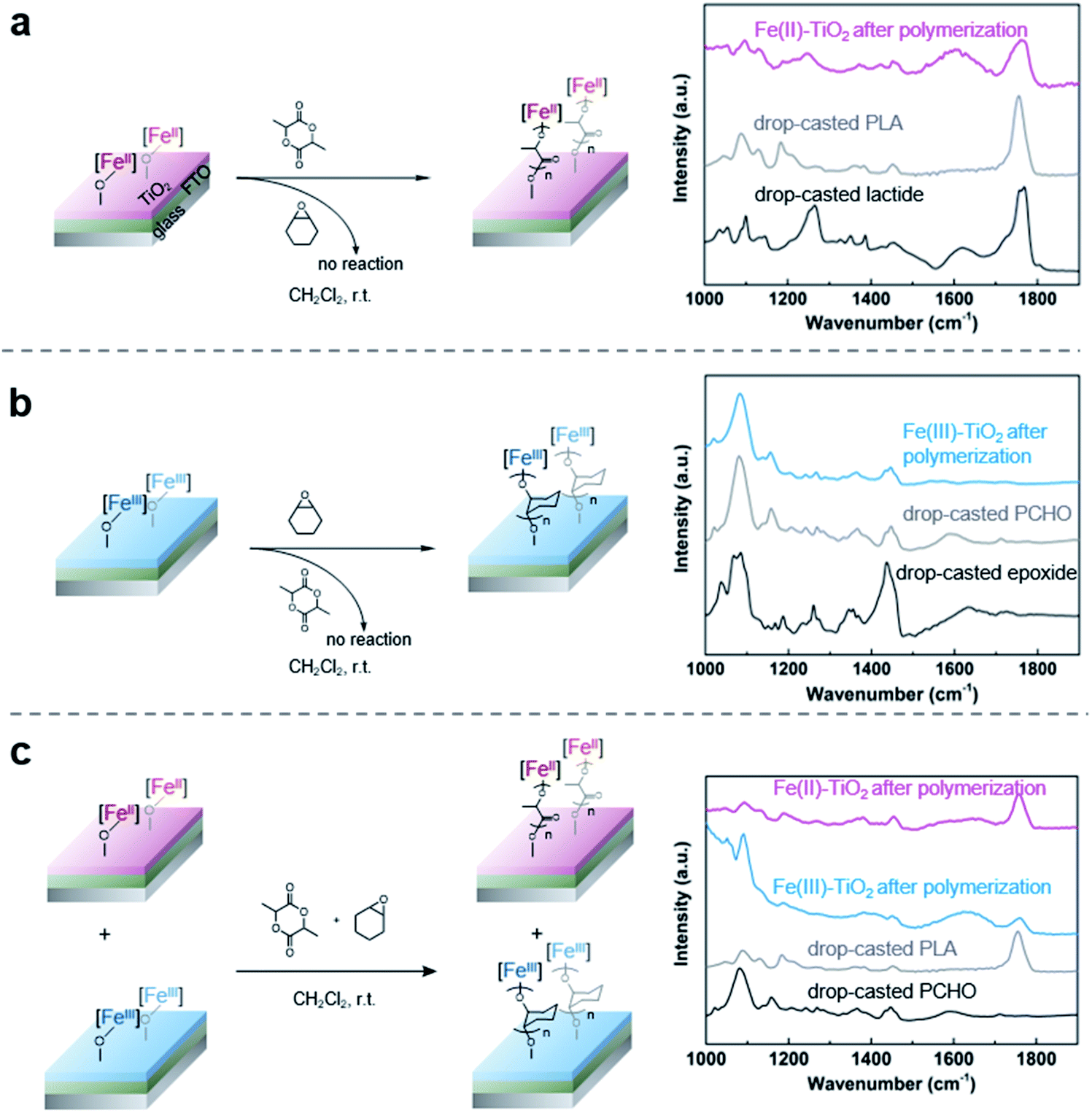 | ||
| Fig. 4 Altering the reactivities of the surface-initiated polymerization by an electrochemical switch (a) surface-initiated poly(lactic acid) can be formed from Fe(II)-modified TiO2 electrode; (b) Fe(III)-modified TiO2 electrode oxidized by potentiostatic electrolysis can trigger surface-initiated poly(cyclohexene oxide) polymerization; (c) selective surface-initiated polymerization by soaking a combination of Fe(II) and Fe(III) electrodes in a solution of mixture monomers. | ||
The electrochemical behavior of the iron-functionalized electrodes was next studied by cyclic voltammetry (CV). To minimize potential complications to the CV data due to electrochemical capacitance that is characteristic of high-surface-area materials such as P25 TiO2 nanoparticles,62 we elected to carry out the initial experiments on a TiO2 surface with relatively small surface areas. This goal was achieved by growing TiO2 directly on titanium mesh using atomic layer deposition (ALD). Specifically, 50 nm TiO2 was deposited following previously published procedures.63 The sample was then treated with a solution of 1, and CV were collected using the Fe(II)–TiO2 electrode as the working electrode and two platinum wires that served as the counter and reference electrodes (Fig. 5a). A half-wave potential of E1/2 = −0.40 V (vs. Fc+/Fc) was measured, which was ascribed to the reversible redox conversion of iron(II) to iron(III). This result represents a 0.50 V positive shift relative to the molecular iron alkoxide complex measured in solution (Fig. 5b, E1/2 = −0.90 V),37,40,41 implying that the iron center is more electron deficient when in contact with TiO2 than the dissolved molecular iron phenoxide complex.38,40 This result is consistent with the less electron donating capability of the inorganic metal-oxide ligand compared with organic phenoxide ligand, and also may explain the small amounts of iron(III) complexes observed by Mössbauer spectroscopy on the surface of TiO2 (Fig. 1c). The oxidative and reductive peaks observed in the CV were separated, but their separation remained unchanged when the scan rates were varied from 20 mV s−1 to 400 mV s−1. Plotting the peak current densities as a function of the scan rate revealed a linear relationship for the anchored complex, whereas a root-square relationship was observed for the dissolved molecular complex (Fig. 5c). In totality, these results strongly suggest that the electron transfer process is not mass-transport limited,59 which is consistent with the redox events occurring from surface-bound species.
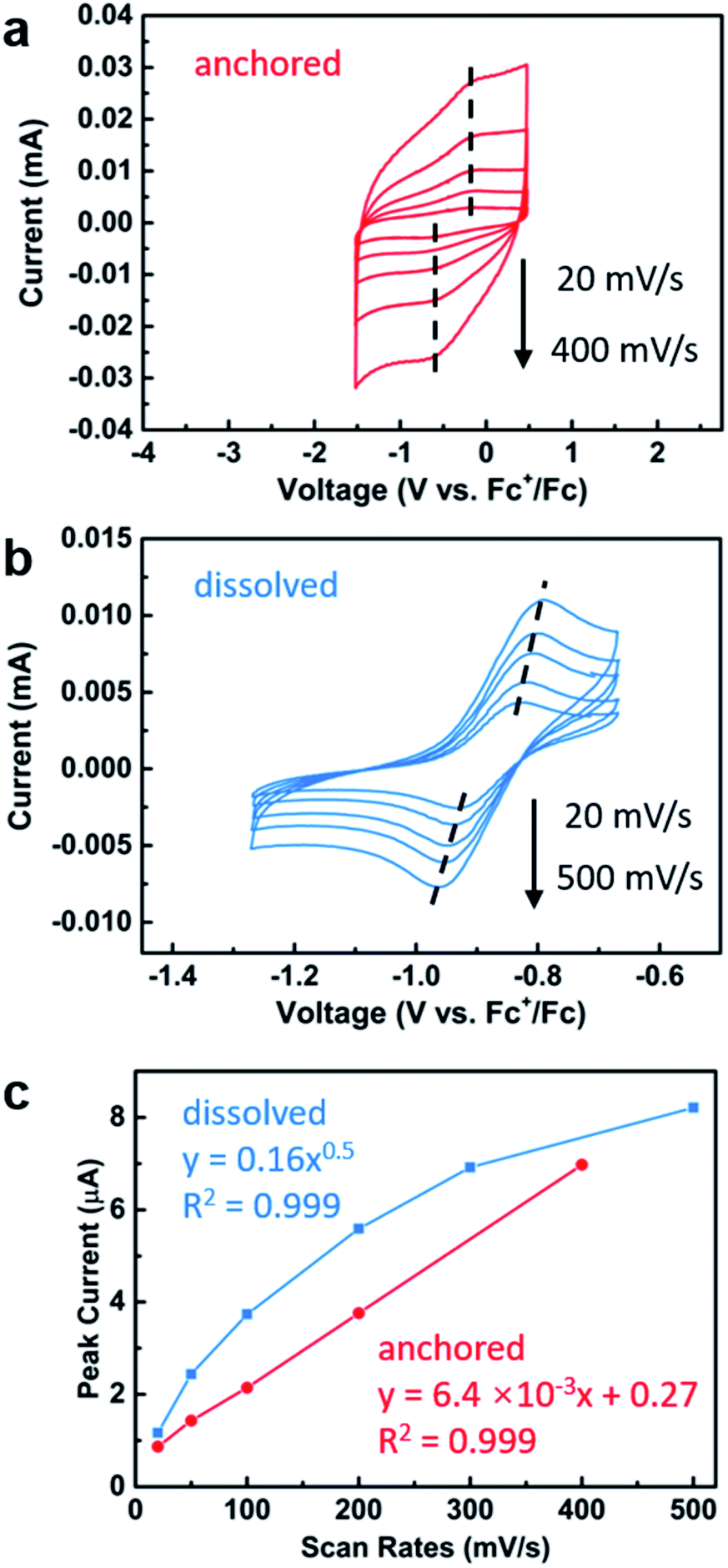 | ||
| Fig. 5 Cyclic voltammetry of (a) Fe(II)–TiO2–Ti mesh electrode and (b) molecular bis(imino)pyridine iron bisphenoxide complex with varying scan rates; (c) plot of scan rates versus cathodic peak currents. | ||
Electrochemically patterning of polyesters and polyethers on surfaces
As CV demonstrated the electrochemical reversible redox reactions are preserved for surface-anchored iron complexes, potentiostatic electrolysis was next used to oxidize the iron(II) functionalized electrodes. A divided two-electrode configuration was used for these experiments, using the iron(II) functionalized TiO2/FTO electrode as the working electrode and a lithium metal strip as the counter and reference electrodes. The counter/reference electrode was isolated from the working electrode by a fritted tube coated with a lithium ion-containing polymer membrane. We recently demonstrated that a similar electrode design was effective in carrying out bulk electrolysis of the molecular iron complexes with minimal influence from parasitic reactions occurring on the counter electrode.40 An oxidizing potential of 0.3 V vs. Fc+/Fc was applied to the working electrode for one hour, at which point the current dropped below 3 μA. The oxidized iron(III) electrode was then exposed to a stirred dichloromethane solution containing cyclohexene oxide (1.4 M). FTIR of the plate after polymerization showed characteristic absorption bands of PCHO (Fig. 4b). Importantly, exposing a combination of iron(II) and iron(III) electrodes to a mixture of cyclohexene oxide (1.4 M) and lactide (0.35 M) in dichloromethane resulted in chemoselective growth of PLA on the iron(II) electrode and PCHO on the iron(III) electrode (Fig. 4c). Noteworthy was a small amount of PLA observed on the iron(III) electrode prepared electrochemically, which may result from incomplete electrochemical oxidation. Notwithstanding this complication, this portion of experiments confirmed that electrochemistry is a reliable method to alter the oxidation state and, hence, the reactivity of surface anchored iron catalysts towards different polymerizations in a single step from a mixture of monomers.Finally, we set out to demonstrate how the surface-initiated redox-switchable polymerization reactions could be used to create surfaces selectively patterned with different polymers. As a proof-of-principle for this concept, we fabricated a substrate containing two electrically isolated zones by using reactive-ion etching to isolate two FTO strips on an insulating glass surface (Fig. 6 and S1†). A TiO2 layer was deposited on the two conducting channels, and the iron complex was then anchored onto the surface as previously described. To differentiate the two FTO strips, one strip was oxidized by exposing it to an oxidizing potential. The resulting substrate was then exposed to a dichloromethane solution containing a mixture of lactide (0.35 M) and epoxide (1.4 M) monomers and stirred for 24 h. FTIR analysis of the plate revealed that each side of the plate was functionalized differently (Fig. S11†). On the iron(II) side of the plate, characteristic bands for surface-initiated PLA was observed at 1710 cm−1 and 1610 cm−1, while no evidence for PCHO was observed. In contrast, the IR spectrum of the oxidized iron(III) side of the plate were dominated by bands from PCHO (1110 cm−1). A small band at 1710 cm−1 from PLA was also detectable, which is consistent with partial oxidation during the electrochemical oxidation as previously discussed.
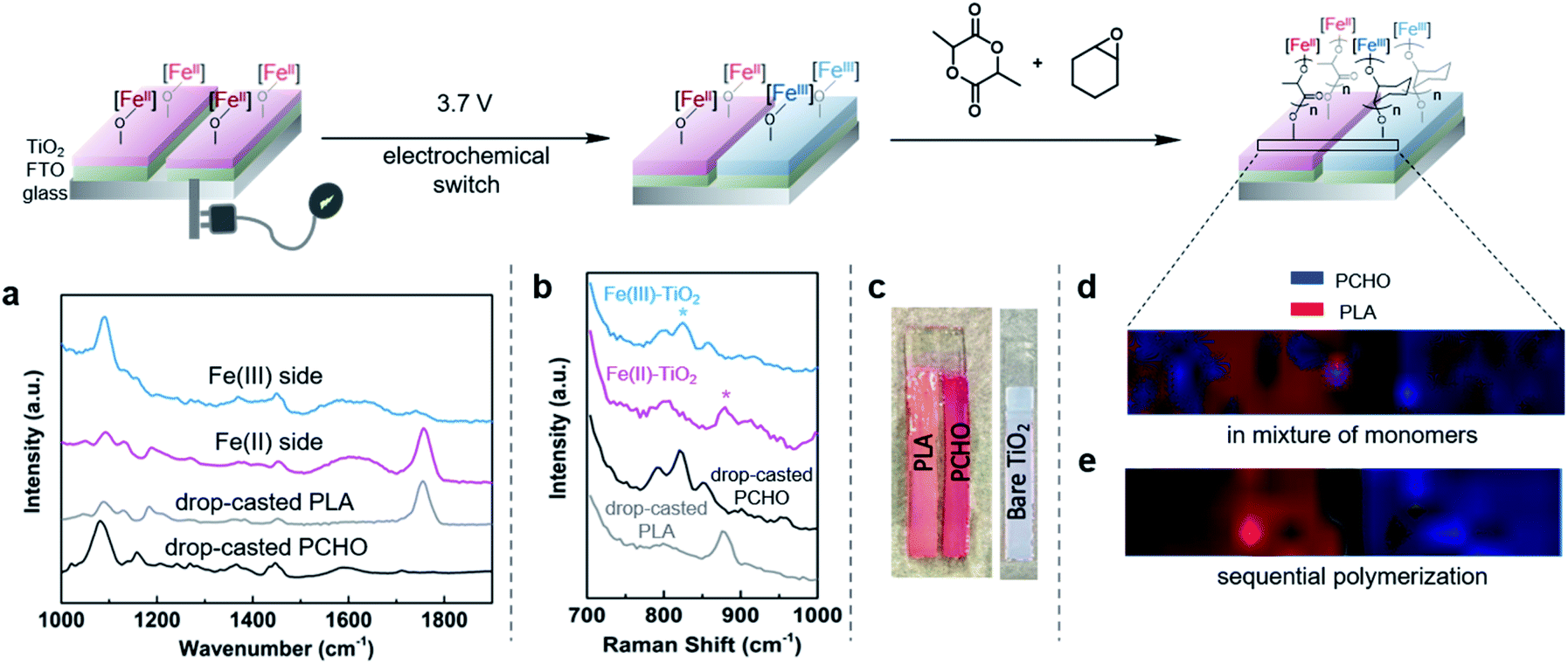 | ||
| Fig. 6 Spatial control over chemoselectivity for the surface-initiated polymerization through application of electrochemical potential. (a) IR analysis of the binarily functionalized electrically discriminated plate obtained by soaking the plate in cyclohexene oxide and lactide solution sequentially. (b) Representative Raman spectroscopy of Fe(II)-modified TiO2 glass electrode and Fe(III)-modified TiO2 glass electrode after polymerization; (c) dyeing the binarily functionalized electrically discriminated plate with rhodamine 6G solution; Raman mapping of the surface of the electrode (d) after polymerization in a mixture of monomers and (e) after sequential polymerization, PLA (red), PCHO (blue) and glass (grey). | ||
To further investigate the spatial distribution of the PLA and PCHO on functionalized substrates, we explored Raman spectroscopy as a potential characterization technique. Similar to the above investigations using IR spectroscopy, we first examined surface-grown PLA on an Fe(II)–TiO2 plate and surface-grown PCHO on an Fe(III)–TiO2 plate. As shown in Fig. 6b, bands at 820 cm−1 and 880 cm−1 can be utilized to characterize PLA and PCHO, respectively. Next, the automatic stage of the Raman spectrometer permitted us to visualize the chemical composition on the surface of the substrate. A sample area of 9600 × 1600 μm2 across the substrate was scanned, and the resulting two-dimensional map is shown in Fig. 6d. Results illustrated that PLA and PCHO were primarily segregated to the reduced and oxidized sides of the plate, respectively. This observation is consistent with our expected reactivity of the iron-based complexes and illustrates the power of the technique to form differentially functionalized surfaces from mixture of monomers. It is notable that the amount of PLA and PCHO was not evenly distributed over each side of the plate. Some areas of the reduced electrode, and especially the oxidized electrode, demonstrated evidence for deposition of PCHO and PLA, respectively. This observation is likely due to a combination of factors. First, the poor resolution and low signal to noise of the bands in the Raman spectra used to distinguish PLA from PCHO likely contribute to false positive indication of PCHO on the iron(II) side and PLA on the iron(III) side. Second, the observed signals from PLA on the iron(III) side could be from incomplete electrochemical oxidation of Fe(II)-complex as discussed earlier in this article. A third reason for observing this “crossover” reactivity is due to mobility of the electrochemical current due to imperfections incurred during the fabrication of the divided plate. Lastly, the uneven thickness of TiO2 layers may also lead to the non-uniform mapping result.
To circumvent the complications associated with the simultaneous polymerization of lactide and epoxide, the electrode was exposed to the two monomers sequentially. As described above, one of the two electroactive strips on the electrode was oxidized by exposing it to an oxidizing potential. The plate was then treated with a cyclohexene oxide solution (1.4 M) followed by a lactide solution (0.35 M). As expected, the polymerization reactions proceeded more efficiently by exposing the plate to the monomers sequentially rather than concurrently. The different outcomes for these two scenarios can be explained by considering the reaction kinetics. In the homogeneous reactions, lactide and epoxide polymerizations are significantly slower when carried out in mixtures of both monomers compared to in pure monomers.37,38 This observation suggests that the epoxide and lactide monomers likely serve as competitive inhibitors for the iron(III) and iron(II) sites, respectively. By carrying out the reactions sequentially, this competitive inhibition is avoided and reactions occur with minimal interference. Consequently, sequential addition of monomers to the plate containing iron(II) and iron(III) zones resulted in a more well defined patterned surface. FTIR of the bifunctional plate after sequential addition of lactide and epoxide monomers shows minimal PLA signal (1710 cm−1) observed on the Fe(III)-functionalized side (Fig. 6a). Moreover, Raman mapping of the resulting product also confirmed better chemoselectivity with minimal crossover of lactide polymerization on the iron(III) side and epoxide polymerization on the iron(II) side of the plate (Fig. 6e). These results indicated that better compositional homogeneity could be achieved through sequential rather than simultaneous cyclohexene oxide and lactide polymerization.
Highlighting the benefits of the method for obtaining patterned functionalized surfaces, the bulk properties of the functionalized surfaces reflect the compositional differences between the two regions of the bifunctional plate. For example, water contact-angle measurements of the two surfaces revealed a larger angle for the iron(II) side (19.3°) compared to the iron(III) side (14.7°), which is consistent with the more hydrophobic polyester compared to the polyether (Fig. S12†). The contact angles measured were significantly smaller than the contact angles for drop casted polymer. These results reflect a more hydrophilic surface that is likely a consequence of the low Ti–OH density on the nanoparticles. To better visualize the differences between the surfaces, the polymer-modified electrode was exposed to a solution containing rhodamine 6G dye. Significant differences were revealed between bare TiO2, the polymer brushes formed on the iron(II) functionalized side, and the polymer brushes formed on the iron(III) functionalized side of the bifunctional plate (Fig. 6c). Whereas the bare TiO2 plate did not adsorb R6G, by comparison, the iron(II) functionalized side containing mostly polyester was dyed a light pink color and the iron(III) functionalized side containing mostly polyether was bright red. This outcome reflects the high propensity for polyether to bind cationic dyes and illustrates how application of electrochemical potential can alter the properties of surfaces through chemoselective polymerization reactions.
Conclusions
While immobilization of molecular catalysts onto solid supports has long been recognized as an opportunity to combine the advantages of homogeneous and heterogeneous catalysis, utilizing supported catalysts to synthesize well-defined but compositionally heterogeneous patterned surfaces remains largely unexplored. Here, we describe how redox-switchable polymerization catalysis can be used to efficiently produce patterned polymer surfaces by taking advantage of molecular iron complexes supported onto semiconducting surfaces. The anchored complexes catalyze surface-initiated ring-opening polymerization reactions, and responded to applied electrochemical potentials so as to alter their reactivity for two different monomers. As a result, binary surfaces containing polymer patterns are obtained wherein the spatial resolution of the pattern is dictated by electrically isolated zones imprinted on the electrode surface. While the spatial resolution obtained in this proof-of-concept system is not as well resolved as other more established methods,19–21 future innovations in catalyst design and substrate fabrication will likely address this limitation. Motivating this endeavor is the relative simplistic synthetic procedures required for patterning surfaces with multiple polymer brushes, and the ability to access to different monomers compared to those historically used to obtain patterned surfaces with multiple polymer brushes.28–30 Consequently, the method can be applied for the rapid generation of sophisticated patterned surfaces with multiple regions containing different chemical compositions. Such materials will likely be valuable for many applications, including anti-fouling coatings, sensors, and applications in the electronics industry.Data Availabilty
All data for this article is included in the supplementary material.Author contributions
M. Q., H. Z. and Q. D. contributed equally to this work. M. Q., H. Z. and Q. D. designed and performed the experiment; J. L. collected the IR spectra; R. M. conducted Mössbauer measurements; Y. Z. carried out electron microscopy characterizations; N. D. prepared electrode materials; D. W. and J. A. B. directed and supervised the research; all authors discussed and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank support by NSF as part of the Center for Integrated Catalysis (CHE-2023955). R. A. M. is grateful for postdoctoral support from the European Union's Horizon 2020 Research and Innovation Programme under grant agreement No. 752684. The contact angle test was performed at the Center for Nanoscale Systems at Harvard University, which is supported by the NSF award No. 1541959. We thank Dr Hongyu Sun, Mr Stephen Shepard, and Prof. Matthias Waegele for their technical assistance and insightful discussions.References
- C. Copéret, M. Chabanas, R. P. Saint-Arroman and J.-M. Basset, Angew. Chem., Int. Ed., 2003, 42, 156–181 Search PubMed.
- J. D. A. Pelletier and J.-M. Basset, Acc. Chem. Res., 2016, 49, 664–677 Search PubMed.
- C. Copéret and J.-M. Basset, Adv. Synth. Catal., 2007, 349, 78–92 Search PubMed.
- D. T. Genna, A. G. Wong-Foy, A. J. Matzger and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 10586–10589 Search PubMed.
- R. Ye, A. V. Zhukhovitskiy, C. V. Deraedt, F. D. Toste and G. A. Somorjai, Acc. Chem. Res., 2017, 50, 1894–1901 Search PubMed.
- R. M. Bullock, A. K. Das and A. M. Appel, Chem.–Eur. J., 2017, 23, 7626–7641 Search PubMed.
- J. Shen, R. Kortlever, R. Kas, Y. Y. Birdja, O. Diaz-Morales, Y. Kwon, I. Ledezma-Yanez, K. J. P. Schouten, G. Mul and M. T. M. Koper, Nat. Commun., 2015, 6, 8177 Search PubMed.
- A. Maurin and M. Robert, J. Am. Chem. Soc., 2016, 138, 2492–2495 Search PubMed.
- S. W. Sheehan, J. M. Thomsen, U. Hintermair, R. H. Crabtree, G. W. Brudvig and C. A. Schmuttenmaer, Nat. Commun., 2015, 6, 6469 Search PubMed.
- W. Li, S. W. Sheehan, D. He, Y. He, X. Yao, R. L. Grimm, G. W. Brudvig and D. Wang, Angew. Chem., Int. Ed., 2015, 127, 11590–11594 Search PubMed.
- H.-Y. Liu, C. C. Cody, J. A. Jacob-Dolan, R. H. Crabtree and G. W. Brudvig, ACS Energy Lett., 2020, 5, 3195–3202 Search PubMed.
- J. D. Blakemore, A. Gupta, J. J. Warren, B. S. Brunschwig and H. B. Gray, J. Am. Chem. Soc., 2013, 135, 18288–18291 Search PubMed.
- M. K. Brennaman, R. J. Dillon, L. Alibabaei, M. K. Gish, C. J. Dares, D. L. Ashford, R. L. House, G. J. Meyer, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 13085–13102 Search PubMed.
- A. M. Oertel, V. Ritleng and M. J. Chetcuti, Organometallics, 2012, 31, 2829–2840 Search PubMed.
- M. A. Lebedeva, T. W. Chamberlain, M. Schröder and A. N. Khlobystov, Chem. Mater., 2014, 26, 6461–6466 Search PubMed.
- S. Edmondson, V. L. Osborne and W. T. S. Huck, Chem. Soc. Rev., 2004, 33, 14–22 Search PubMed.
- J. O. Zoppe, N. C. Ataman, P. Mocny, J. Wang, J. Moraes and H.-A. Klok, Chem. Rev., 2017, 117, 1105–1318 Search PubMed.
- W.-L. Chen, R. Cordero, H. Tran and C. K. Ober, Macromolecules, 2017, 50, 4089–4113 Search PubMed.
- J. E. Poelma, B. P. Fors, G. F. Meyers, J. W. Kramer and C. J. Hawker, Angew. Chem., Int. Ed., 2013, 125, 6982–6986 Search PubMed.
- Z. Xie, C. Chen, X. Zhou, T. Gao, D. Liu, Q. Miao and Z. Zheng, ACS Appl. Mater. Interfaces, 2014, 6, 11955–11964 Search PubMed.
- N. D. Dolinski, Z. A. Page, E. B. Callaway, F. Eisenreich, R. V. Garcia, R. Chavez, D. P. Bothman, S. Hecht, F. W. Zok and C. J. Hawker, Adv. Mater., 2018, 30, 1800364 Search PubMed.
- S.-J. Park, T. A. Taton and C. A. Mirkin, Science, 2002, 295, 1503–1506 Search PubMed.
- N. Schüwer and H.-A. Klok, Adv. Mater., 2010, 22, 3251–3255 Search PubMed.
- W. Zhao, Q. Ye, H. Hu, X. Wang and F. Zhou, J. Mater. Chem. B, 2014, 2, 5352–5357 Search PubMed.
- S. J. Yuan, F. J. Xu, S. O. Pehkonen, Y. P. Ting, K. G. Neoh and E. T. Kang, Biotechnol. Bioeng., 2009, 103, 268–281 Search PubMed.
- D. B. Chrisey, A. Pique, R. Modi, H. D. Wu, R. C. Y. Auyeung and H. D. Young, Appl. Surf. Sci., 2000, 168, 345–352 Search PubMed.
- H. Moon, H. Seong, W. C. Shin, W.-T. Park, M. Kim, S. Lee, J. H. Bong, Y.-Y. Noh, B. J. Cho, S. Yoo and S. G. Im, Nat. Mater., 2015, 14, 628–635 Search PubMed.
- M. C. R. Tria and R. C. Advincula, Macromol. Rapid Commun., 2011, 32, 966–971 Search PubMed.
- Y. Liu, V. Klep and I. Luzinov, J. Am. Chem. Soc., 2006, 128, 8106–8107 Search PubMed.
- F. Zhou, Z. Zheng, B. Yu, W. Liu and W. T. S. Huck, J. Am. Chem. Soc., 2006, 128, 16253–16258 Search PubMed.
- U. Lüning, Angew. Chem., Int. Ed., 2012, 51, 8163–8165 Search PubMed.
- A. J. Teator, D. N. Lastovickova and C. W. Bielawski, Chem. Rev., 2016, 116, 1969–1992 Search PubMed.
- C. K. A. Gregson, V. C. Gibson, N. J. Long, E. L. Marshall, P. J. Oxford and A. J. P. White, J. Am. Chem. Soc., 2006, 128, 7410–7411 Search PubMed.
- E. M. Broderick, N. Guo, T. Wu, C. S. Vogel, C. Xu, J. Sutter, J. T. Miller, K. Meyer, T. Cantat and P. L. Diaconescu, Chem. Commun., 2011, 47, 9897–9899 Search PubMed.
- S. M. Quan, J. Wei and P. L. Diaconescu, Organometallics, 2017, 36, 4451–4457 Search PubMed.
- D. N. Lastovickova, H. Shao, G. Lu, P. Liu and C. W. Bielawski, Chem.–Eur. J., 2017, 23, 5994–6000 Search PubMed.
- A. B. Biernesser, B. Li and J. A. Byers, J. Am. Chem. Soc., 2013, 135, 16553–16560 Search PubMed.
- A. B. Biernesser, K. R. Delle Chiaie, J. B. Curley and J. A. Byers, Angew. Chem., Int. Ed., 2016, 55, 5251–5254 Search PubMed.
- A. M. Doerr, J. M. Burroughs, S. R. Gitter, X. Yang, A. J. Boydston and B. K. Long, ACS Catal., 2020, 10, 14457–14515 Search PubMed.
- M. Qi, Q. Dong, D. Wang and J. A. Byers, J. Am. Chem. Soc., 2018, 140, 5686–5690 Search PubMed.
- K. R. D. Chiaie, L. M. Yablon, A. B. Biernesser, G. R. Michalowski, A. W. Sudyn and J. A. Byers, Polym. Chem., 2016, 7, 4675–4681 Search PubMed.
- J. Lahann and R. Langer, Macromol. Rapid Commun., 2001, 22, 968–971 Search PubMed.
- G. Carrot, D. Rutot-Houzé, A. Pottier, P. Degée, J. Hilborn and P. Dubois, Macromolecules, 2002, 35, 8400–8404 Search PubMed.
- C. M. Manna, A. Kaur, L. M. Yablon, F. Haeffner, B. Li and J. A. Byers, J. Am. Chem. Soc., 2015, 137, 14232–14235 Search PubMed.
- K. R. D. Chiaie, A. B. Biernesser, M. A. Ortuño, B. Dereli, D. A. Iovan, M. J. T. Wilding, B. Li, C. J. Cramer and J. A. Byers, Dalton Trans., 2017, 46, 12971–12980 Search PubMed.
- M. A. Ortuño, B. Dereli, K. R. D. Chiaie, A. B. Biernesser, M. Qi, J. A. Byers and C. J. Cramer, Inorg. Chem., 2018, 57, 2064–2071 Search PubMed.
- Z.-S. Wang, H. Kawauchi, T. Kashima and H. Arakawa, Coord. Chem. Rev., 2004, 248, 1381–1389 Search PubMed.
- C. Arrouvel, M. Digne, M. Breysse, H. Toulhoat and P. Raybaud, J. Catal., 2004, 222, 152–166 Search PubMed.
- C.-Y. Wu, K.-J. Tu, J.-P. Deng, Y.-S. Lo and C.-H. Wu, Materials, 2017, 10, 566 Search PubMed.
- Y. Zhao, X. Yan, K. R. Yang, S. Cao, Q. Dong, J. E. Thorne, K. L. Materna, S. Zhu, X. Pan, M. Flytzani-Stephanopoulos, G. W. Brudvig, V. S. Batista and D. Wang, ACS Cent. Sci., 2018, 4, 1166–1172 Search PubMed.
- A. B. Biernesser, Synthesis of Diverse Degradable Polymers by Redox-Switchable Iron-Based Catalysis, Boston College, 2017.
- A. M. Tondreau, C. C. H. Atienza, J. M. Darmon, C. Milsmann, H. M. Hoyt, K. J. Weller, S. A. Nye, K. M. Lewis, J. Boyer, J. G. P. Delis, E. Lobkovsky and P. J. Chirik, Organometallics, 2012, 31, 4886–4893 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 Search PubMed.
- M. Römelt, S. Ye and F. Neese, Inorg. Chem., 2009, 48, 784–785 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 Search PubMed.
- S. F. McWilliams, E. Brennan-Wydra, K. C. MacLeod and P. L. Holland, ACS Omega, 2017, 2, 2594–2606 Search PubMed.
- S. Yang, Z.-H. Wu, W. Yang and M.-B. Yang, Polym. Test., 2008, 27, 957–963 Search PubMed.
- J. Cheng and M. Sprik, J. Chem. Theory Comput., 2010, 6, 880–889 Search PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, 2nd edn, 2001 Search PubMed.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Mueller, P. Liska, N. Vlachopoulos and M. Graetzel, J. Am. Chem. Soc., 1993, 115, 6382–6390 Search PubMed.
- A. Nan, R. Turcu, I. Craciunescu, C. Leostean, I. Bratu and J. Liebscher, J. Phys.: Conf. Ser., 2009, 182, 012070 Search PubMed.
- H. Zhou and Y. Zhang, J. Phys. Chem. C, 2014, 118, 5626–5636 Search PubMed.
- Y. Lin, S. Zhou, X. Liu, S. Sheehan and D. Wang, J. Am. Chem. Soc., 2009, 131, 2772–2773 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02163j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |