
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePeptide and protein assays using customizable bio-affinity arrays combined with ambient ionization mass spectrometry†
Xuemeng
Zhang‡
a,
Wei
Wang‡
a,
Richard N.
Zare
 *b and
Qianhao
Min
*a
*b and
Qianhao
Min
*a
aState Key Laboratory of Analytical Chemistry for Life Science, Chemistry and Biomedicine Innovation Center, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, P. R. China. E-mail: minqianhao@nju.edu.cn
bDepartment of Chemistry, Stanford University, Stanford, California 94305, USA. E-mail: rnz@stanford.edu
First published on 14th July 2021
Abstract
High-throughput identification and quantification of protein/peptide biomarkers from biofluids in a label-free manner is achieved by interfacing bio-affinity arrays (BAAs) with nano-electrospray desorption electrospray ionization mass spectrometry (nano-DESI-MS). A wide spectrum of proteins and peptides ranging from phosphopeptides to cis-diol biomolecules as well as thrombin can be rapidly extracted via arbitrarily predefined affinity interactions including coordination chemistry, covalent bonding, and biological recognition. An integrated MS platform allows continuous interrogation. Profiling and quantitation of dysregulated phosphopeptides from small-volume (∼5 μL) serum samples has been successfully demonstrated. As a front-end device adapted to any mass spectrometer, this MS platform might hold much promise in protein/peptide analysis in point-of-care (POC) diagnostics and clinical applications.
Introduction
Rapid and high-throughput determination of proteins/peptides of pathological significance from a biological specimen is highly desirable for disease diagnosis and prognostic analysis.1,2 Mass spectrometry (MS) can directly profile target proteins/peptides without the necessity of signal tags, offering a label-free acquisition modality for assaying proteins/peptides for point-of-care (POC) diagnostics.3–5 Electrospray ionization mass spectrometry (ESI-MS)6 and matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS)7 have long been regarded as two typical types of soft ionization for the MS measurement of proteins and peptides. However, the coupling of ESI-MS with liquid chromatography brings about a loss in throughput,8–10 and MALDI-MS takes tens of minutes for substantial off-line pretreatments prior to sample loading.11,12 Consequently, neither of these popular techniques allow direct translation of MS into clinical tests of proteins/peptides in analyzing a large number of samples.Ambient ionization mass spectrometry (AIMS)13,14 enables direct sampling and ionization of analytes under atmospheric pressure and room temperature with minimal or no sample preparation, creating the possibility of rapid collection of molecular information in POC bioassays. However, the most prevalent AIMS techniques, direct analysis in real time (DART) and desorption electrospray ionization (DESI), both suffer from poor extraction efficiency of macromolecules.15–19 More effective desorption/ionization and MS readout of peptides and proteins can be achieved by delivering a continuous low-flow solvent stream in a liquid microjunction surface sampling probe (LMJ-SSP),20,21 nanospray desorption electrospray ionization (nano-DESI),22–24 and swan probe.25,26 Among them, nano-DESI proposed by Laskin's group22 has demonstrated its advantages in high-efficiency extraction and quantitative determination of small molecules including phospholipids,27,28 neurotransmitters29 and prostaglandins.30 By virtue of the excellent desorption capability, this ionization technique also allowed direct detection and MS imaging of typical proteins in tissue sections.31–33 Despite these advances, selectively measuring low-abundance proteins/peptides of pathological significance from the complexity of biosamples by AIMS in a sensitive and quantitative fashion remains challenging.
Separation media with chemical affinity or biological recognition ability represent an ideal interface compatible to AIMS for interrogating biomolecules of interest in biosystems. Solid-phase microextraction (SPME) coupled to AIMS in forms of coated blade,34,35 miniaturized fiber,36 and polymer coating transfer enrichment (PCTE)37 have shown enormous benefits in the extraction and determination of metabolites, drugs, and lipids in biofluids. Yet, few efforts have been devoted to gaining the inherent ion signals of protein/peptide biomarkers under ambient conditions, owing to concerns about low efficiency in desorption/ionization. One solution is the utilization of a secondary probe to convert the biorecognition events to ion signals of mass tags with ease of ionization, but causing losses of innate molecular information.38–40 In recent years, a couple of attempts that combined AIMS with SPME interfaces or affinity materials for harvesting and MS characterization of phosphorylated and glycosylated peptides have been documented,41–43 highlighting the feasibility of AIMS determination of target proteins/peptides in complex samples. Nevertheless, the versatility, detection throughput, and quantitation capability of these approaches are still far from meeting clinical demands.
Herein, we present a versatile strategy for high-throughput and direct MS detection of targeted protein/peptide biomarkers under ambient conditions by interfacing customizable bio-affinity arrays with nanospray desorption electrospray ionization MS (BAAs-nano-DESI-MS) (Fig. 1a). This integrated AIMS platform allows the selective capturing of phosphopeptides and cis-diol biomolecules or target proteins from complex matrices on the arbitrarily pre-designed BAAs via affinity interactions ranging from coordination chemistry, covalent bonding to biological recognition, followed by successively extraction, ionization, and MS readout of the captured target species within seconds after minimum sample pretreatment. Unlike the conventional SPME-MS, in which one SPME tip (fiber,44 paper,45,46 Al foil47 or coated blade34,35) only affords the extraction and spray of a single sample, the BAAs integrated in one slide augment the throughput of biological sample handling by continuous manipulation of tens of samples. In this sense, with the arsenal of affinity probes in hand, the BAAs can be easily customized to implement the MS analysis of desired target proteins/peptides. Significantly, the BAAs-nano-DESI-MS enables the profiling and quantitation of target proteins/peptides from small-volume (∼5 μL) serum specimens, suggesting the great potential of this technique for advanced POC tests and clinical applications.
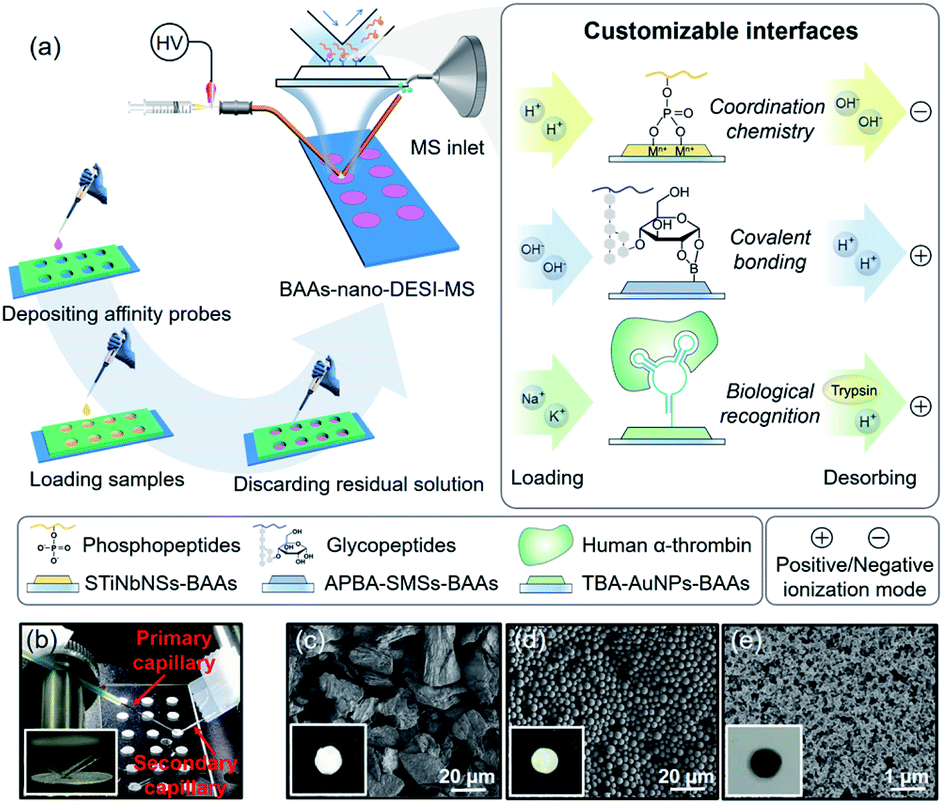 | ||
| Fig. 1 (a) Schematic of rapid extraction and direct MS determination of phosphopeptides, glycopeptides, and target proteins from biofluid samples by BAAs-nano-DESI-MS. (b) Photograph of the BAAs-nano-DESI-MS setup. SEM images of BAAs respectively constituted by (c) stacked titanoniobate nanosheets (STiNbNSs), (d) aminophenylboronic acid grafted silica microspheres (APBA-SMSs), and (e) thrombin binding aptamer modified gold nanoparticles (TBA-AuNPs). Insets show photographs of individual spots. | ||
Results and discussion
The BAAs were fabricated by recruiting a polydimethylsiloxane (PDMS) mold covered glass slide as substructure, and the slurry of bio-affinity nanomaterials were deposited and dried in the arrayed wells (3 mm in diameter, 3 mm in depth) (Fig. 1a and S1†). As a proof of concept, we first focused on characterizing phosphopeptides from complex biosamples, which are informative in the activation of kinase cascades in physiological and pathological states.48,49 Stacked titanoniobate nanosheets (STiNbNSs), which have been reported as the separation probes for enrichment of phosphopeptides,50 were deposited at the bottoms of arrayed wells to form the STiNbNSs-BAAs. The STiNbNSs were characterized as micrometer-sized particles assembled by 2D nanosheets (Fig. 1c). Nano-DESI-MS constitutes the analytical terminal by interfacing the liquid microjunction with the biomolecule-preloaded BAAs, where continuous solvent infusion extracts and mobilizes compounds from the surface (Fig. 1b, parameters of the setup are shown in Fig. S1†).51 For the measurement of phosphopeptides in the tryptic digests of β-casein (Fig. S2a†), acidic loading buffer was adopted to guarantee the coordination interaction between phosphate groups and unsaturated metal ions on the STiNbNSs-BAAs, while alkaline solvent (1% NH3·H2O in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 methanol/water) was optimized to facilitate the detachment of phosphopeptides from the STiNbNSs-BAAs as well as the deprotonation of acidic peptides in negative ionization mode (Fig. S3†). Four phosphopeptides trapped by STiNbNSs-BAAs were subsequently desorbed with alkaline solvent and generated distinct ion signals from a clean background (Fig. 2b, S3c and Table S1†).
:1 methanol/water) was optimized to facilitate the detachment of phosphopeptides from the STiNbNSs-BAAs as well as the deprotonation of acidic peptides in negative ionization mode (Fig. S3†). Four phosphopeptides trapped by STiNbNSs-BAAs were subsequently desorbed with alkaline solvent and generated distinct ion signals from a clean background (Fig. 2b, S3c and Table S1†).
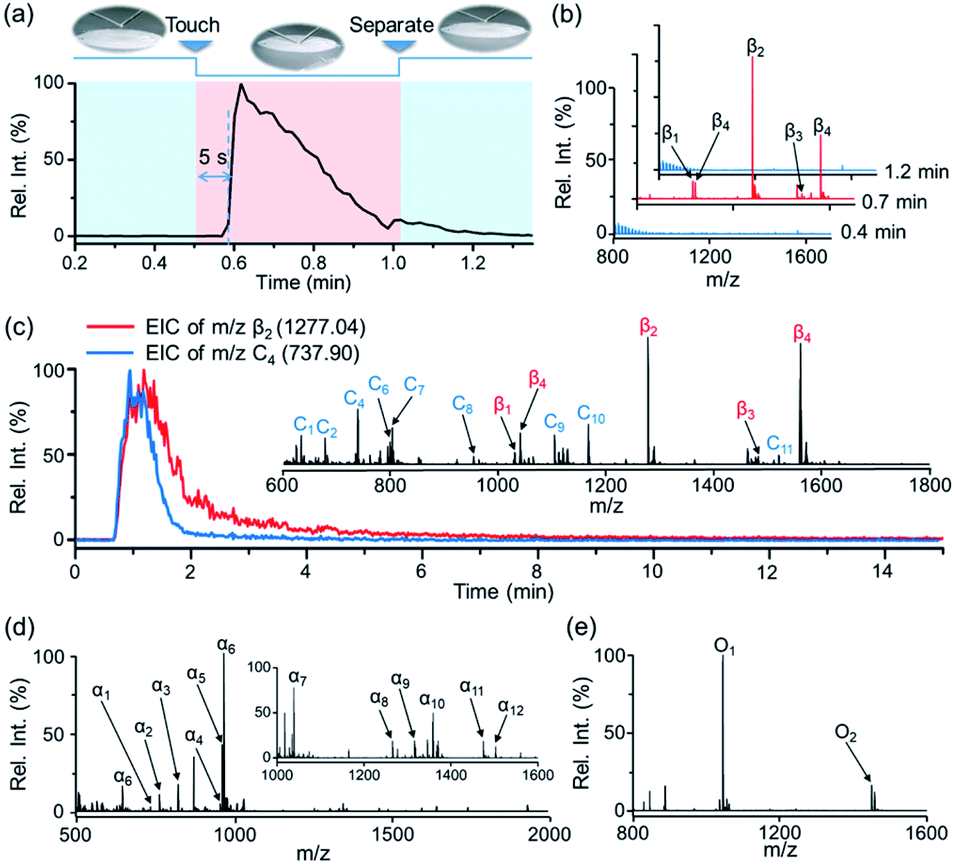 | ||
| Fig. 2 (a) EIC of m/z 1277.03 ([β2-2H]2−) corresponding to the phosphopeptide β2 from tryptic digests of β-casein (200 pmol) acquired by STiNbNSs-BAAs-nano-DESI-MS in the touch-and-desorb mode. (b) Negative ion mode mass spectra of tryptic digests of β-casein over the course of sampling process. (c) EIC of the ions respectively corresponding to the trapped phosphopeptide β2 (m/z 1277.04, red) and peptide C3 (m/z 737.90, blue) from tryptic digests of Cyt c. After rinsing the phosphopeptides-preloaded BAAs, digests of Cyt c (80 pmol) were additionally dropped onto the spot and dried at room temperature, followed by nano-DESI sampling and MS analysis. Insets show the negative ion mode mass spectrum of tryptic digests of Cyt c and phosphopeptides from tryptic digests of β-casein. (d and e) Negative ion mode mass spectra of phosphopeptides from tryptic digests of (d) α-casein (200 pmol) and (e) ovalbumin (200 pmol) obtained by STiNbNSs-BAAs-nano-DESI-MS. | ||
Notably, by using high-speed nebulizing gas to accelerate microdroplets in sampling, DESI was unable to output any signal of phosphopeptides (even from 200 pmol β-casein), but this approach caused physical destruction of sample spots (Fig. S4a and b†). In addition, matrix-free laser desorption/ionization mass spectrometry (LDI-MS) analysis also exported no observable signal of phosphopeptides due to the poor performance on transferring laser energy, and the loosely attached affinity materials may splatter while pumping vacuum and applying laser (Fig. S4c and d†). In contrast, nano-DESI-MS exhibited much higher desorption/ionization efficiency for peptides and proteins due to the continuous solvent stream for sampling, with the limit of detection as low as 5 pmol (Fig. S5†). The extraction kinetics was monitored by recording extracted ion chromatography (EIC) of the phosphopeptide β2 (m/z 1277.03) (Fig. 2a). After the liquid microjunction touched the surface of STiNbNSs-BAAs, termed touch-and-desorb mode, it took around 5 s to see the onset of ion signals coming from phosphopeptides (Fig. 2a and Video S1†), which peaked within seconds and then gradually declined with consumption of the pre-captured peptides. The delayed emergence of ion signals should be attributed to the dead volume (∼2 μL) in the secondary capillary. Moreover, the extraction kinetics of phosphopeptides was compared with that of nonphosphopeptides from digests of cytochrome c (Cyt c) by respectively recording the EIC of m/z 1277.03 and 737.90 (nonphosphopeptide C3, Fig. 2c inset and Table S2†). During the co-sampling process, phosphopeptides displayed an elution curve with upward and downward sections that lagged those physically adsorbed peptides (Fig. 2b). As a control, the tryptic digests from β-casein and Cyt c preloaded on the sample spot made of silica microspheres (SMSs) exhibited highly consistent elution curves (Fig. S6†), verifying the critical role of coordinatively unsaturated metal ions exposed on the STiNbNSs in the retention of phosphopeptides.
We further inspected the acquisition selectivity and capacity of STiNbNSs-BAAs-nano-DESI-MS by using tryptic digests of other phosphorylated proteins. For α-casein with 22 phosphorylation sites (UniProtKB database), only 3 phosphopeptides can be discerned in the spectrum of direct MS analysis (Fig. S2b†), whereas 12 phosphopeptides varying in length and phosphate group number were unambiguously identified by STiNbNSs-BAAs-nano-DESI-MS (Fig. 2d and Table S3†). Furthermore, 2 phosphorylated fragments could be filtered from the tryptic digests of ovalbumin (Fig. 2e and Table S4†), in comparison with the overwhelming signals of ordinary peptides by direct nano-DESI-MS analysis (Fig. S2c†). This MS approach also tolerates the interference from 10- to 100-fold nonphosphorylated peptides originated from BSA, Cyt c and lysozyme (Fig. S7† and Table S5†), manifesting the great specificity to target peptides.
As crucial factors of quantitative analysis, ion signal stability and reproducibility were assessed. As shown in Fig. 3a, the sampling site (∼200 μm × 200 μm) was directed to 9 evenly distributed micro-areas in the touch-and-desorb mode on a single spot of STiNbNSs-BAAs for peptide desorption and MS measurement (Fig. 3a). EICs of individual and combined ion signals from phosphopeptides (m/z 1029.90, 1277.03, 1482.07, 1039.75 and 1560.12) were recorded along with the nano-DESI probe hopping from one site to another (Fig. 3b and S8†), showing a great difference in the integral area of each desorption event, with a coefficient of variation higher than 30%. The nano-DESI-MS imaging also showed the biased distribution of phosphopeptides on the edge of spot (Fig. S9†), exhibiting a significant spatial heterogeneity due to the “coffee ring” effect of affinity materials during the preparation of BAAs. To overcome the signal variation, line scan sampling mode was additionally employed to average the ion signals by continuously dragging the nano-DESI probe across the arrayed sample spots in close proximity (Fig. 3c). In this sampling mode, the peak area in the EIC of total ions of phosphopeptides was elevated with the increase of loading amounts of tryptic digests of β-casein (Fig. 3d), and a good linear relationship (R2 = 0.996) in the range from 2 pmol to 200 pmol and satisfactory recoveries (from 95.6% to 109.3%) could be obtained (Fig. 3e and Table S6†), thereby laying the foundation for relative quantification of phosphopeptides in biosamples without addition of an internal standard. To pursue both signal stability and detection sensitivity, we further used the drop-and-desorb sampling strategy, in which a droplet of extraction solvent (5 μL) was preloaded onto sample spots for 30 s and subsequently aspirated into MS by nano-DESI probe. Due to the full transmission of pre-captured phosphopeptides into extraction solvent, the ion signal appeared comparable to those observed after a conventional in-solution sample pretreatment (Fig. S10†). In the drop-and-desorb mode, by plotting the intensity ratio of peptide β4 to internal standard peptide (EAIpYAAPFAKKK, [IS-2H]2−, m/z 706.82) against β-casein loading amount, a calibration curve with a good linear relationship (R2 = 0.998) was obtained (Fig. S11†). The recoveries of this sampling mode were between 102.4% and 108.4% (Table S7†), manifesting high accuracy in quantification analysis.
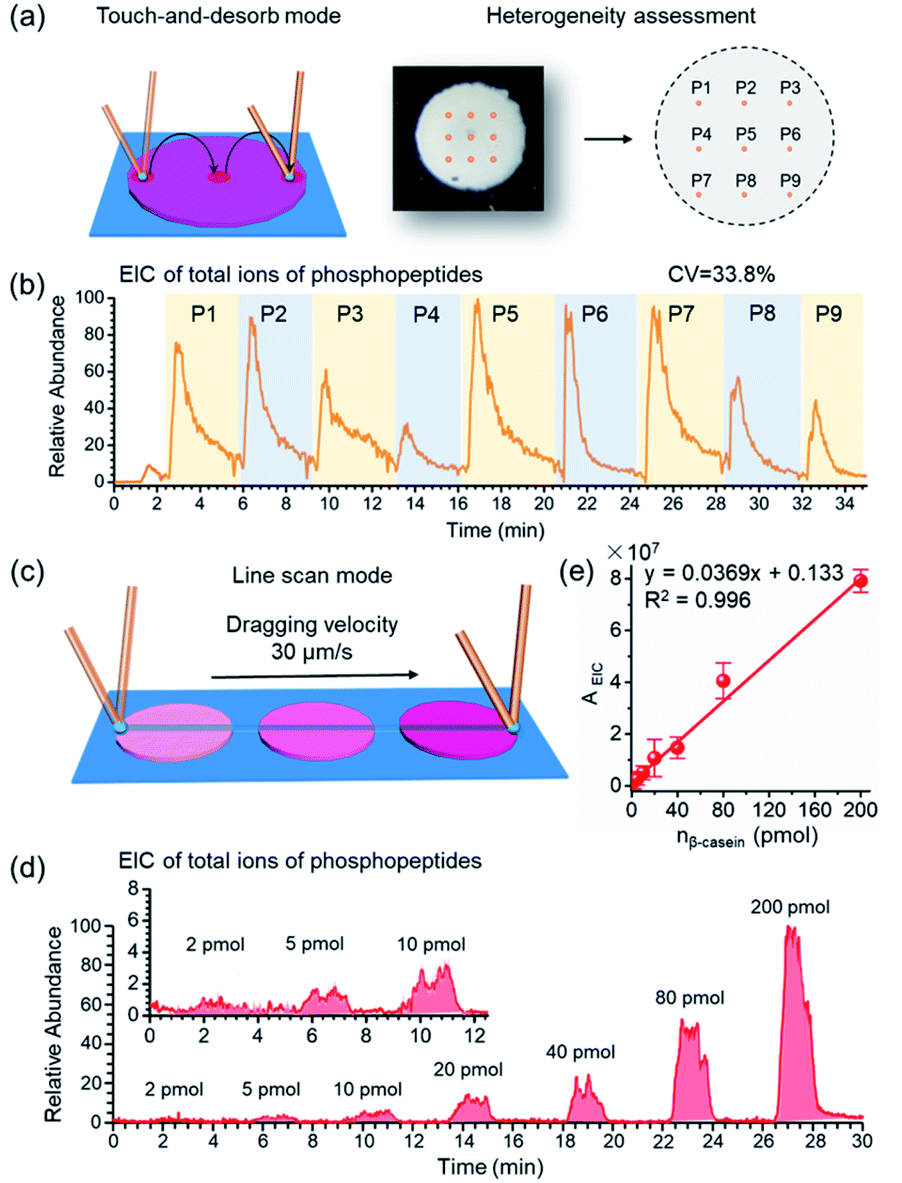 | ||
| Fig. 3 (a) Schematic illustration of the assessment of sample spot heterogeneity by nano-DESI-MS in touch-and-desorb mode. (b) EIC of total ions of phosphopeptides (m/z 1029.90, 1277.03, 1482.07, 1039.75 and 1560.12) from tryptic digests of β-casein on 9 sampling sites obtained by BAAs-nano-DESI-MS. (c) Schematic illustration of STiNbNSs-BAAs-nano-DESI-MS in line scan mode. The nano-DESI probe was dragged across the arrayed spots by moving the sample stage at speed of 30 μm s−1 for sampling and MS analysis. (d) EIC of total ions of phosphopeptides from tryptic digests of β-casein with increasing loading amount acquired by BAAs-nano-DESI-MS in line scan mode. (e) EIC peak area of total ions of phosphopeptides as a function of loading amounts of tryptic digests of β-casein. | ||
The essence of retention and desorption of biomolecules on BAAs lies in formation and breakdown of the specific interaction between bio-affinity surface and target species. Accordingly, any specific binding mechanism that contributes to biomolecule recognition and capturing can be included into the toolbox for configuring the BAAs coupled nano-DESI-MS. We then expand the objectives to other proteins/peptides of biological significance through remodeling the chemistry of BAAs.
Biomolecules with cis-diol moieties including catecholamine neuro-transmitters, nucleosides, saccharides, and glycoproteins constitute a rich pool of potential biomarkers and therapeutic targets in clinical diagnostics.52,53 Boronate affinity chemistry is a facile and efficient strategy to reversibly capture and release cis-diol compounds by harnessing the forming and breaking of covalent bonds of pH-dependent cyclic esters (Fig. 1a).54–57 To implement the capturing and MS readout of cis-diol biomolecules, aminophenylboronic acid grafted silica microspheres (APBA-SMSs) were synthesized as the separation medium for fabrication of BAAs (Fig. 1d, S12 and S13†). Alkaline loading buffer contributes to the formation of borate esters, while acidic extraction solvent facilitates the hydrolysis of the ester linkages and boosts the ionization of cis-diol species on positive ionization mode. First, dopamine (D), isoprenaline (IE) and epinephrine (E) were recruited to demonstrate the effectivity of APBA-SMSs-BAAs-nano-DESI-MS in the measurement of cis-diol biomolecules (Table S8†). Ion signals corresponding to the target cis-diol molecules were distinctly observed against 100-fold non-target substances by APBA-SMSs-BAAs-nano-DESI-MS (Fig. S14a and b†). Similarly, this MS platform also allowed the biased detection of the nucleosides containing a pentose with cis-diol moiety against nontarget counterparts (Fig. S14c and d†).
Protein glycosylation is a fundamental post-translation modifications,58,59 and methodologies that can efficiently gain the information of glycopeptides and glycoproteins from highly complicated biosamples are greatly demanded in global analysis of protein glycosylation. Due to the cis-diol groups contained in glycopeptides, the reversible covalent interaction between boronic acids and cis-diol groups can form to achieve the specific recognition of glycopeptides. After verifying the capability of the proposed AIMS platform in the interrogation of small molecules bearing cis-diols, we next investigated its performance on probing glycosylated peptides. MS peaks derived from 15 glycopeptides in the digests of horseradish peroxidase (HRP) were unambiguously picked out of the background by APBA-SMSs-BAAs-nano-DESI-MS in touch-and-desorb mode (Fig. 4a and Video S2†). Table S9† lists the information of captured glycopeptides from digests of HRP. Moreover, under ambient ionization condition, feature signal extraction of glycopeptides could overcome the influence from digests of 100-fold BSA (Fig. S15†).
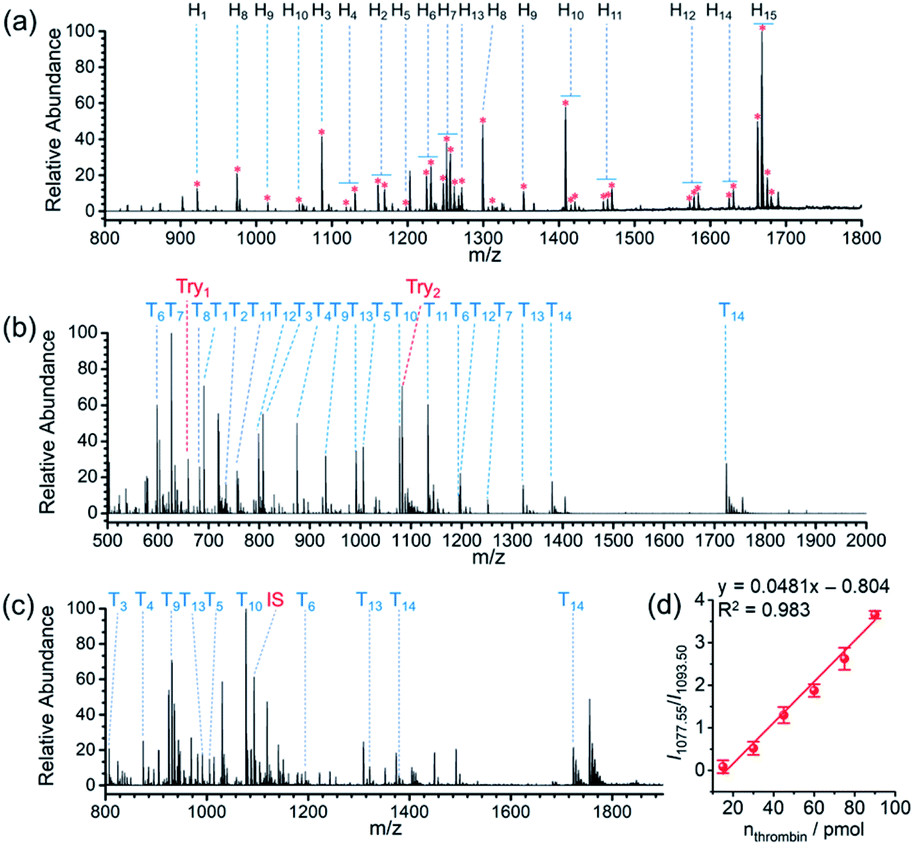 | ||
| Fig. 4 (a) Positive ion mode mass spectrum of glycopeptides from tryptic digests of HRP (200 pmol) obtained by APBA-SMSs-BAAs-nano-DESI-MS. (b) Positive ion mode mass spectra of tryptic digests of thrombin with loading amount of 80 pmol obtained by AuNPs-TBA-BAAs-nano-DESI-MS. (c) Positive ion mode mass spectrum of thrombin (45 pmol) spiked 10-fold diluted FBS after processing on TBA-AuNPs-BAAs. A standard peptide (GGDADEYLIPQQGHHHHHH) was added as an internal standard (IS, m/z 1093.50). (d) Ion signal intensity ratio of peptide T10 to IS as a function of the loading amount of thrombin in the presence of 10-fold diluted FBS. The data was obtained from three independent experiments. | ||
Because the STiNbNSs-BAAs/APBA-SMSs-BAAs hyphenated nano-DESI-MS platform restricts the scope of target biomolecules to those bearing unique chemical groups (e.g., phosphate or cis-diol groups), we explored the possibility of further upgrading its universality to general protein biomarkers by introducing biorecognition modules involving identification of protein epitopes. Nucleic acid aptamer can be regarded as an emerging artificial antibody that can bind to a specific target molecule while possessing higher synthetic availability and stability than antibody.60–62 We switched the bio-affinity materials to thrombin binding aptamer-grafted gold nanoparticles (TBA-AuNPs) (Fig. 1e), and constructed a “proof-of-concept” MS assaying platform of protein biomarkers by ambient ionization (see ESI† for the detailed Experimental procedures). After selective binding of thrombin on the BAAs, trypsin solution was deposited on site to convert the detection of intact protein to measurement of its offspring peptides with greater ease of ionization.3,7 MS peaks from 14 unique peptides of thrombin (80 pmol) were obtained by nano-DESI-MS (Fig. 4b, Table S10 and Video S3†), while no signals from thrombin but only two peaks from trypsin autolysis appeared in the mass spectrum without TBA functionalization (Fig. S16 and Table S11†). Even in the presence of 10-fold diluted fetal bovine serum (FBS) and high-abundance standard protein digests, TBA-AuNPs-BAAs were still capable of exclusively capturing thrombin and exporting its signature peptides via on-site proteolysis (Fig. 4c, S17 and S18†). Because the ion signal of peptide T10 ([T10 + 2H]2+, m/z 1077.55) showed the slowest attenuation as the loading amount decreased, it was selected as the identifiable index of thrombin in quantification. A good linear relationship (R2 = 0.994) could be obtained from the buffer with high concentration of salt by plotting the intensity ratio of peptide T10 to internal standard peptide (GGDADEYLIPQQGHHHHHH, [IS + 2H]2+, m/z 1093.50) (Fig. S19†). The identity of this ion signal was further validated by MS/MS (Fig. S20†). Even in the presence of 10-fold diluted FBS (total protein concentration is approximately 0.2–0.4 mg mL−1), a calibration curve with a good linear relationship (R2 = 0.983) was also obtained (Fig. 4d), underscoring the ability of the TBA-AuNPs-BAAs-nano-DESI-MS platform in quantitative analysis of target proteins in complex matrix.
To demonstrate the applicability of our BAAs-nano-DESI-MS method in assays of protein/peptide biomarkers in clinical biofluid samples, we further used this MS platform to measure endogenous phosphopeptides in human serum. Serum specimens were collected from healthy adults and pancreatic cancer patients, and then subjected to STiNbNSs-BAAs-nano-DESI-MS interrogation in the drop-and-desorb mode (Fig. 5a). The internal standard phosphopeptide (FQ[pS]EEQQQTEDELQDK) was introduced for relative quantification. As illustrated in Fig. 5b, 4 endogenous phosphopeptides ([HS1-2H]2−m/z 693.25, [HS2-2H]2−m/z 728.77, [HS3-2H]2−m/z 771.30 and [HS4-2H]2−m/z 806.82, see Table S12† for the peptide sequence) degraded from phosphorylated fibrinopeptide A (FPA) were specifically extracted from human serum and identified by STiNbNSs-BAAs-nano-DESI-MS. Fig. 5c summarizes the mass spectra of serum samples from 8 healthy adults and 5 cancer patients. The relative abundance of all 4 peaks of phosphopeptides varied little among samples of healthy persons, while pancreatic cancer patients showed significant individual differences in HS4 expression level. According to the statistic signal intensity ratio of phosphopeptides to IS, phosphopeptide HS1 and HS4 in cancerous samples respectively underwent 6.1-fold downregulation and 3.7-fold upregulation as compared with serum samples of healthy controls (Fig. 5d). The results reflected aberrant activation of proteases, kinases, and phosphatases implicated in tumor physiopathology, which govern the cleavage and reverse phosphorylation of fibrinogen α-chain.63,64 Previous studies also indicated the upregulated HS4 (phosphorylated FPA) as one of the most sensitive index to distinguish pancreatic cancer and cancer-free controls.65,66 The line scan mode can be alternatively operated to characterize the differential expression of phosphorylated FPA between cancer and normal samples without internal standard, merely according to the profile of EIC of corresponding ions. As the nano-DESI probe scanning across the spots of normal (N1, N2) and cancer samples (C1, C2), EIC of m/z 806.82 manifested significantly enhanced abundance of phosphopeptide HS4 in the spots of pancreatic cancer serum (C1, C2), while little difference was observed for the ion signal of phosphopeptide HS3 (m/z 771.30) between cancer sera and normal controls (Fig. 5e). The array layout of BAAs-nano-DESI-MS platform allows simultaneous processing of tens of samples, with sampling time of each spot less than 10 s (drop-and-desorb mode), thus enabling a rapid, high-throughput MS assay of disease-associated proteins/peptides in clinical samples.
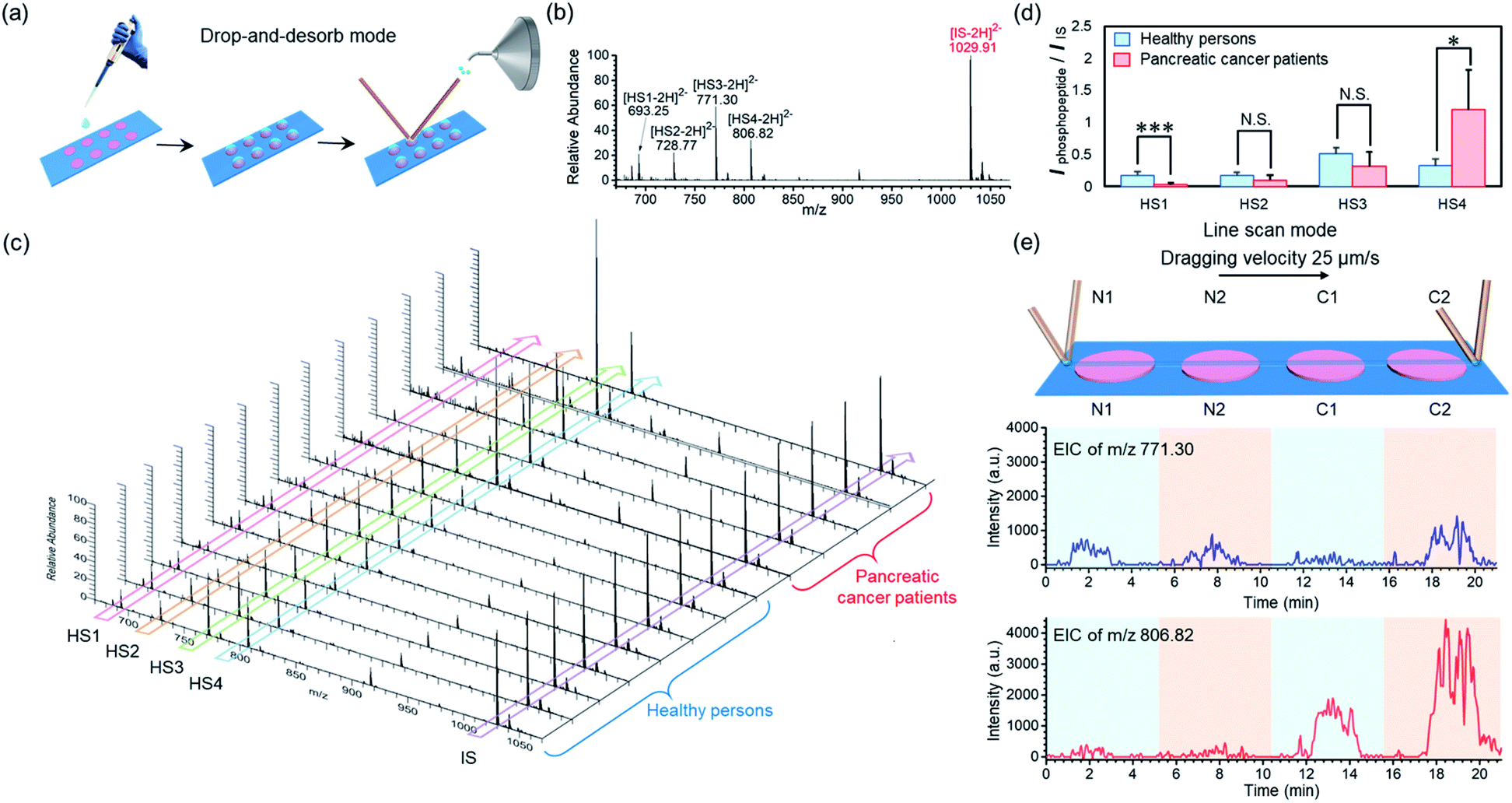 | ||
| Fig. 5 (a) Schematic illustration of a high-throughput serum phosphopeptide assay by STiNbNSs-BAA-nano-DESI-MS in the drop-and-desorb mode. (b) Typical negative ion mode mass spectrum of phosphopeptides from serum in healthy controls by STiNbNSs-BAA-nano-DESI-MS. A standard phosphopeptide was added as an internal standard (IS, m/z 1029.91). (c) Normalized negative ion mode mass spectra of serum phosphopeptides from pancreatic cancer patients and healthy persons. (d) Comparison of the MS intensity ratio of the target phosphopeptides to the internal standard phosphopeptide in pancreatic cancer patients and healthy people. Statistical analysis: two-tailed Student's t-test (*P < 0.05; ***P < 0.001; N.S., not significant). (e) EIC of the ions (HS3: m/z 771.30, HS4: m/z 806.82) from phosphopeptides in two healthy persons (N1, N2) and two cancer patients (C1, C2) obtained by STiNbNSs-BAA-nano-DESI-MS in line scan mode. | ||
Conclusions
In summary, a versatile MS approach for rapid, high-throughput assays of protein/peptide biomarkers under ambient conditions was developed by interfacing predefined BAAs with nano-DESI-MS in a highly integrated format. Having tackled the shortcomings of the conventional AIMS in desorption/ionization of large biomolecules, this BAAs-nano-DESI-MS platform allows continuous capture, extraction, and MS readout of target proteins/peptides with improved sensitivity. Benefiting from the generous toolbox of affinity materials, we have succeeded in selective MS interrogation and quantification of phosphopeptides, cis-diol biomolecules, and thrombin from complex samples by freely switching the BAAs component from titania-based material, to boronate affinity chromatographic material, and to aptamer-functionalized recognition medium. This method has shown its favorable clinical applicability in profiling serum phosphorylated FPA as potential markers for pancreatic cancer. Unlike peptide and protein assays depending on fluorescence detection, the approach we have described is able to detect many more biomarkers at one time. As progress in MS miniaturization is made, the proposed front-end device might be adapted to any mass spectrometer. This holds great promise in clinical and POC testing of protein/peptide biomarkers in biofluids, thus paving the way for advancing MS technology from bench to bedside.Author contributions
Qianhao Min and Richard N. Zare conceived and directed the project. Xuemeng Zhang and Wei Wang performed the experiment and gathered experimental data. Xuemeng Zhang analyzed and interpreted the experimental data and wrote the first draft of the article. Qianhao Min and Richard N. Zare corrected the manuscript, finalized the manuscript draft, and guided during the revision. All authors contributed to the discussions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is financially supported by the National Natural Science Foundation of China (No. 21974062, 92053102, 21622505), and the Fundamental Research Funds for the Central Universities (No. 020514380255, 020514380141).Notes and references
- L. Wu and X. Qu, Chem. Soc. Rev., 2015, 44, 2963–2997 RSC.
- X.-P. He, Y. Zang, T. D. James, J. Li and G.-R. Chen, Chem. Soc. Rev., 2015, 44, 4239–4248 RSC.
- X. Zhang, S. Zhu, Y. Xiong, C. Deng and X. Zhang, Angew. Chem., Int. Ed., 2013, 52, 6055–6058 CrossRef CAS PubMed.
- Y. Wang, W. Zhang and Z. Ouyang, Chem. Sci., 2020, 11, 10506–10516 RSC.
- C. R. Ferreira, K. E. Yannell, A. K. Jarmusch, V. Pirro, Z. Ouyang and R. G. Cooks, Clin. Chem., 2016, 62, 99–110 CrossRef CAS PubMed.
- J. B. Fenn, M. Mann, C. K. Meng, S. F. Wong and C. M. Whitehouse, Science, 1989, 246, 64–71 CrossRef CAS PubMed.
- G. Yao, C. Deng, X. Zhang and P. Yang, Angew. Chem., Int. Ed., 2010, 49, 8185–8189 CrossRef CAS PubMed.
- K. A. Brown, T. Tucholski, A. J. Alpert, C. Eken, L. Wesemann, A. Kyrvasilis, S. Jin and Y. Ge, Anal. Chem., 2020, 92, 15726–15735 CrossRef CAS PubMed.
- Y. Kuang, J. Cao, F. Xu and Y. Chen, Anal. Chem., 2019, 91, 8820–8826 CrossRef CAS PubMed.
- L. Yu, J. Ding, Y.-L. Wang, P. Liu and Y.-Q. Feng, Anal. Chem., 2016, 88, 1286–1293 CrossRef CAS PubMed.
- Q. Wu, B. Jiang, Y. Weng, J. Liu, S. Li, Y. Hu, K. Yang, Z. Liang, L. Zhang and Y. Zhang, Anal. Chem., 2018, 90, 2671–2677 CrossRef CAS PubMed.
- J. Peng, H. Niu, H. Zhang, Y. Yao, X. Zhao, X. Zhou, L. Wan, X. Kang and R. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 32613–32621 CrossRef CAS PubMed.
- R. G. Cooks, Z. Ouyang, Z. Takats and J. M. Wiseman, Science, 2006, 311, 1566–1570 CrossRef CAS PubMed.
- C. L. Feider, A. Krieger, R. J. DeHoog and L. S. Eberlin, Anal. Chem., 2019, 91, 4266–4290 CrossRef CAS PubMed.
- Z. Takats, J. M. Wiseman, B. Gologan and R. G. Cooks, Science, 2004, 306, 471–473 CrossRef CAS PubMed.
- R. B. Cody, J. A. Laramée and H. D. Durst, Anal. Chem., 2005, 77, 2297–2302 CrossRef CAS PubMed.
- S. Lendor, G. A. Gómez-Ríos, E. Boyacı, H. Vander Heide and J. Pawliszyn, Anal. Chem., 2019, 91, 10141–10148 CrossRef CAS PubMed.
- P. Khanipour, M. Loffler, A. M. Reichert, F. T. Haase, K. J. J. Mayrhofer and I. Katsounaros, Angew. Chem., Int. Ed., 2019, 58, 7273–7277 CrossRef CAS PubMed.
- J. E. Chipuk, M. H. Gelb and J. S. Brodbelt, Anal. Chem., 2010, 82, 4130–4139 CrossRef CAS PubMed.
- C. L. Feider, N. Elizondo and L. S. Eberlin, Anal. Chem., 2016, 88, 11533–11541 CrossRef CAS PubMed.
- Z. Huang, Q. Wu, H. Lu, Y. Wang and Z. Zhang, Anal. Chem., 2020, 92, 11250–11259 CrossRef CAS PubMed.
- P. J. Roach, J. Laskin and A. Laskin, Analyst, 2010, 135, 2233–2236 RSC.
- X. Li, R. Yin, H. Hu, Y. Li, X. Sun, S. K. Dey and J. Laskin, Angew. Chem., Int. Ed., 2020, 59, 22388–22391 CrossRef CAS PubMed.
- R. Yin, K. E. Burnum-Johnson, X. Sun, S. K. Dey and J. Laskin, Nat. Protoc., 2019, 14, 3445–3470 CrossRef CAS PubMed.
- D.-Q. Jin, Y. Zhu and Q. Fang, Anal. Chem., 2014, 86, 10796–10803 CrossRef CAS PubMed.
- D.-Q. Jin, S.-W. Shi, Y. Ma and Q. Fang, Anal. Chem., 2020, 92, 9214–9222 CrossRef CAS PubMed.
- I. Lanekoff, M. Thomas and J. Laskin, Anal. Chem., 2014, 86, 1872–1880 CrossRef CAS PubMed.
- H.-M. Bergman and I. Lanekoff, Analyst, 2017, 142, 3639–3647 RSC.
- H.-M. Bergman, E. Lundin, M. Andersson and I. Lanekoff, Analyst, 2016, 141, 3686–3695 RSC.
- K. D. Duncan, R. Fang, J. Yuan, R. K. Chu, S. K. Dey, K. E. Burnum-Johnson and I. Lanekoff, Anal. Chem., 2018, 90, 7246–7252 CrossRef CAS PubMed.
- C. C. Hsu, P. T. Chou and R. N. Zare, Anal. Chem., 2015, 87, 11171–11175 CrossRef CAS PubMed.
- C. C. Hsu, N. M. White, M. Hayashi, E. C. Lin, T. Poon, I. Banerjee, J. Chen, S. L. Pfaff, E. R. Macagno and P. C. Dorrestein, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14855–14860 CrossRef CAS PubMed.
- O. J. Hale and H. J. Cooper, Anal. Chem., 2021, 93, 4619–4627 CrossRef CAS PubMed.
- G. A. Gomez-Rios and J. Pawliszyn, Angew. Chem., Int. Ed., 2014, 53, 14503–14507 CrossRef CAS PubMed.
- M. Tascon, G. A. Gomez-Rios, N. Reyes-Garces, J. Poole, E. Boyaci and J. Pawliszyn, Anal. Chem., 2017, 89, 8421–8428 CrossRef CAS PubMed.
- H. Piri-Moghadam, F. Ahmadi, G. A. Gómez-Ríos, E. Boyacı, N. Reyes-Garcés, A. Aghakhani, B. Bojko and J. Pawliszyn, Angew. Chem., Int. Ed., 2016, 55, 7510–7514 CrossRef CAS PubMed.
- W. Zhang, S. Chiang, Z. Li, Q. Chen, Y. Xia and Z. Ouyang, Angew. Chem., Int. Ed., 2019, 58, 6064–6069 CrossRef CAS PubMed.
- S. Xu, M. Liu, Y. Bai and H. Liu, Angew. Chem., Int. Ed., 2021, 60, 1806–1812 CrossRef CAS PubMed.
- S. Chen, Q. Wan and A. K. Badu-Tawiah, J. Am. Chem. Soc., 2016, 138, 6356–6359 CrossRef CAS PubMed.
- S. Xu, W. Ma, Y. Bai and H. Liu, J. Am. Chem. Soc., 2019, 141, 72–75 CrossRef CAS PubMed.
- Y. Zhao, X. Gong, X. Si, Z. Wei, C. Yang, S. Zhang and X. Zhang, Analyst, 2015, 140, 2599–2602 RSC.
- M. Tascon, V. Singh, M. Huq and J. Pawliszyn, Anal. Chem., 2019, 91, 4762–4770 CrossRef CAS PubMed.
- S. H. Yang, E. H. Wang, J. A. Gurak, S. Bhawal, R. Deshmukh, A. B. Wijeratne, B. L. Edwards, F. W. Foss, R. B. Timmons and K. A. Schug, Langmuir, 2013, 29, 8046–8053 CrossRef CAS PubMed.
- E. C. Meurer, D. M. Tomazela, R. C. Silva, F. Augusto and M. N. Eberlin, Anal. Chem., 2002, 74, 5688–5692 CrossRef CAS PubMed.
- H. Wang, J. Liu, R. G. Cooks and Z. Ouyang, Angew. Chem., Int. Ed., 2010, 49, 877–880 CrossRef CAS PubMed.
- Y. Yang, H. Liu, Z. Chen, T. Wu, Z. Jiang, L. Tong and B. Tang, Anal. Chem., 2019, 91, 12874–12881 CrossRef CAS PubMed.
- P.-K. So, B.-C. Yang, W. Li, L. Wu and B. Hu, Anal. Chem., 2019, 91, 9430–9434 CrossRef CAS PubMed.
- I. Vivanco and C. L. Sawyers, Nat. Rev. Cancer, 2002, 2, 489–501 CrossRef CAS PubMed.
- L. Chang and M. Karin, Nature, 2001, 410, 37–40 CrossRef CAS PubMed.
- X. Chen, S. Li, X. Zhang, Q. Min and J. J. Zhu, Nanoscale, 2015, 7, 5815–5825 RSC.
- P. J. Roach, J. Laskin and A. Laskin, Analyst, 2010, 135, 2233–2236 RSC.
- M. M. Reddy, R. Wilson, J. Wilson, S. Connell, A. Gocke, L. Hynan, D. German and T. Kodadek, Cell, 2011, 144, 132–142 CrossRef CAS PubMed.
- M. A. Wolfert and G.-J. Boons, Nat. Chem. Biol., 2013, 9, 776–784 CrossRef CAS PubMed.
- D. Li, Y. Chen and Z. Liu, Chem. Soc. Rev., 2015, 44, 8097–8123 RSC.
- Z. Liu and H. He, Acc. Chem. Res., 2017, 50, 2185–2193 CrossRef CAS PubMed.
- H. Xiao, W. Chen, J. M. Smeekens and R. Wu, Nat. Commun., 2018, 9, 1692 CrossRef PubMed.
- R. Xing, Y. Ma, Y. Wang, Y. Wen and Z. Liu, Chem. Sci., 2019, 10, 1831–1835 RSC.
- K. Ohtsubo and J. D. Marth, Cell, 2006, 126, 855–867 CrossRef CAS PubMed.
- S. Gilgunn, P. J. Conroy, R. Saldova, P. M. Rudd and R. J. O'Kennedy, Nat. Rev. Urol., 2013, 10, 99–107 CrossRef CAS PubMed.
- W. Tan, M. J. Donovan and J. Jiang, Chem. Rev., 2013, 113, 2842–2862 CrossRef CAS PubMed.
- H. M. Meng, H. Liu, H. Kuai, R. Peng, L. Mo and X. B. Zhang, Chem. Soc. Rev., 2016, 45, 2583–2602 RSC.
- S. Xie, Y. Du, Y. Zhang, Z. Wang, D. Zhang, L. He, L. Qiu, J. Jiang and W. Tan, Nat. Commun., 2020, 11, 1347 CrossRef CAS PubMed.
- L. M. Matrisian, G. W. Sledge and S. Mohla, Cancer Res., 2003, 63, 6105–6109 CAS.
- C. G. Binnie, J. M. Hettasch, E. Strickland and S. T. Lord, Biochemistry, 1993, 32, 107–113 CrossRef CAS PubMed.
- B. Velstra, M. A. Vonk, B. A. Bonsing, B. J. Mertens, S. Nicolardi, A. Huijbers, H. Vasen, A. M. Deelder, W. E. Mesker, Y. E. M. van der Burgt and R. A. E. M. Tollenaar, J. Canc. Res. Clin. Oncol., 2015, 141, 531–541 CrossRef CAS PubMed.
- V. Abbasciano, L. Graziano, S. Guerra, D. Mazzotta, V. Pollinzi, G. Gilli and G. Zavagli, Oncology, 1991, 48, 377–382 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02311j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |