
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesymmetrization of pibrentasvir for efficient prodrug synthesis†
Eric A.
Voight
 *a,
Stephen N.
Greszler
a,
John
Hartung
b,
Jianguo
Ji
b,
Russell C.
Klix
b,
John T.
Randolph
a,
Bhadra H.
Shelat
b,
Jan E.
Waters
a and
David A.
DeGoey
a
*a,
Stephen N.
Greszler
a,
John
Hartung
b,
Jianguo
Ji
b,
Russell C.
Klix
b,
John T.
Randolph
a,
Bhadra H.
Shelat
b,
Jan E.
Waters
a and
David A.
DeGoey
a
aDrug Discovery Science & Technology, AbbVie, Inc., 1 North Waukegan Road, North Chicago, Illinois 60064-1802, USA. E-mail: eric.a.voight@abbvie.com
bProcess Research and Development, AbbVie, Inc., 1 North Waukegan Road, North Chicago, Illinois 60064-1802, USA
First published on 29th June 2021
Abstract
A novel and practical desymmetrization tactic is described to access a new class of pibrentasvir prodrugs. The homotopic benzimidazoles of pibrentasvir (PIB) are differentiated via a one-pot di-Boc/mono-de-Boc selective N-Boc protection and formaldehyde adduct formation sequence, both enabled by crystallization-induced selectivity. The first step represents the only known application of the Horeau principle of statistical amplification for C2-symmetric polyheterocycle regioselective functionalization. The resulting versatile intermediate is employed in the high-yielding preparation of several pibrentasvir prodrug candidates.
Introduction
Mavyret® is a pan-genotype treatment for hepatitis C virus (HCV) containing glecaprevir, an NS3/4A protease inhibitor, and pibrentasvir (1, PIB), an NS5A inhibitor (Fig. 1).1 As the first pan-genotypic 8 week cure for people suffering from HCV, Mavyret® was approved by the U.S. Food and Drug Administration (FDA) in 2017 for the treatment of genotype 1-6 chronic HCV after a 98% 8 week cure rate was reported for treatment-naive patients without cirrhosis or with compensated cirrhosis.2 Enabling formulations were required for further improving bioavailability due to challenging physicochemical properties,3 so a prodrug approach was pursued, resulting in the discovery of phosphates 2, 3, and 4.4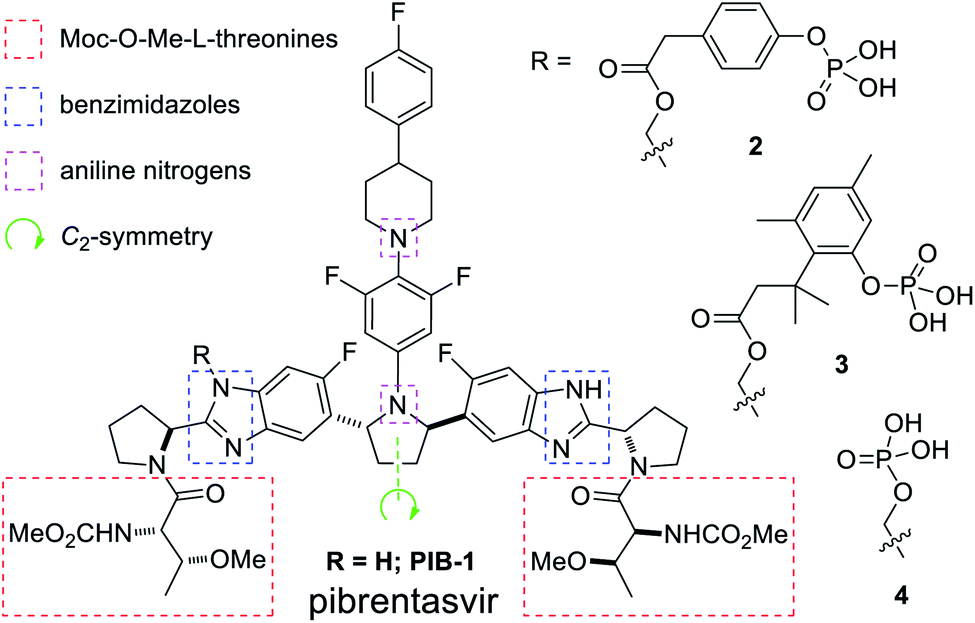 | ||
| Fig. 1 Pibrentasvir (1) structure, challenging features, and PIB prodrugs 2–4. | ||
Pibrentasvir (1, PIB) is a large (MW 1113 g mol−1) C2-symmetric drug molecule which presents several uniquely challenging structural features when considering a solubility-enhancing prodrug approach (Fig. 1). The end-cap amino acid fragments, methoxycarbonyl (Moc)-protected O-Me-L-threonines, are prone to epimerization, β-elimination, and facile Moc cleavage with nucleophiles and bases. For these reasons, preliminary attempts to attach cleavable prodrug moieties to the end-caps proved futile.4 The four PIB benzimidazole nitrogen atoms initially appeared too similar in reactivity to be functionalized selectively, complicated by the two tertiary aniline nitrogens and the fact that PIB exists as a mixture of tautomers and rotamers in solution.5 Finally, and most significantly when considering synthetic efficiency challenges, the C2-symmetric nature of PIB renders each end homotopic. This feature facilitated efficient two-directional chain synthesis in the preparation of PIB6 with stereochemical purity enhancement via the Horeau principle.7 However, without a readily apparent internal functionalization or steric proximity effect to avoid bis(functionalization),8 a statistical mixture of products was anticipated and observed in early syntheses of 2–4.4
Since the seminal reviews by Schreiber9 and Magnus8 describing two-directional synthesis and terminus differentiation, this strategy has been used successfully in a number of additional complex molecule syntheses (Fig. 2). For example, in Hoye's synthesis of the annonaceous acetogenin (+)-parviflorin, bis(epoxide) 5 was desymmetrized via reaction with a limited quantity of a lithium acetylide, giving alcohol 6 (29%) along with recovered 5 (53%).10 Two syntheses from the Burke group utilized this approach, including a related annonaceous acetogenin, uvaricin (not shown), using a dihydroxylation,11 and the C(37)-C(54) halichondrin B subunit, using an olefination/hydroboration/oxidation sequence to convert bis(lactone) 7 to desymmetrized primary alcohol 8 in 40% yield.12 In a remarkably brief route to (+)-roxaticin that showcases this approach, the Krische group desymmetrized diol 9via mono-selenide 10 formation in 50% yield.13 Our initial synthesis of prodrug 3 used a similar strategy, producing intermediate di-Bn-3 in only 19% yield from pibrentasvir. In each example, a maximum yield of 50% was expected and observed due to the standard statistical mixtures of starting material/mono/di-functionalized products (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 ratio) obtained in homotopic termini differentiation, requiring starting material recovery and resubjection to improve material throughput. In contrast, this work describes a rare example of highly controlled mono-functionalization of a complex C2-symmetric molecule via Horeau amplified di-Boc protection/crystallization-induced selective mono-Boc deprotection (de-Boc) with n-BuNH2, giving mono-N-Boc-PIB 11 in 94% yield from PIB, a 5-fold increase in desymmetrization yield versus the original route. An overall yield of ∼50% from PIB was a requirement for consideration of a pibrentasvir prodrug as a development candidate due to the high value of PIB, so this desymmetrization tactic was critical.
:2:1 ratio) obtained in homotopic termini differentiation, requiring starting material recovery and resubjection to improve material throughput. In contrast, this work describes a rare example of highly controlled mono-functionalization of a complex C2-symmetric molecule via Horeau amplified di-Boc protection/crystallization-induced selective mono-Boc deprotection (de-Boc) with n-BuNH2, giving mono-N-Boc-PIB 11 in 94% yield from PIB, a 5-fold increase in desymmetrization yield versus the original route. An overall yield of ∼50% from PIB was a requirement for consideration of a pibrentasvir prodrug as a development candidate due to the high value of PIB, so this desymmetrization tactic was critical.
 | ||
| Fig. 2 Examples of desymmetrization reactions of complex C2-symmetric molecules. | ||
Results and Discussion
In addition to homotopic terminus differentiation challenges, potential strategies to selectively functionalize PIB had to simultaneously address benzimidazole regioisomer selectivity (Fig. 3). Since two equivalent PIB nitrogens are para-fluoro (“down” red) and two are meta-fluoro (“up” – blue), some electronic bias was anticipated. However, early routes to PIB prodrugs demonstrated poor alkylation selectivities under a variety of conditions with challenging isomer separations further complicating the aforementioned statistical terminus differentiation issue (Fig. 2).4 Since no direct PIB alkylation appeared to provide any useful levels of selectivity or clear opportunities for efficient end differentiation, a Boc protection strategy was envisioned to simplify the problem to a single benzimidazole regioselective alkylation. All reaction conditions would need to be mild enough to not degrade PIB and efficient enough to enable the 50% overall yield target. While considering the potential for biocatalytic or Miller peptide-based catalyst approaches for desymmetrization,14 approaches using simple reagents which could benefit from inherent substrate bias and/or solubility properties were prioritized.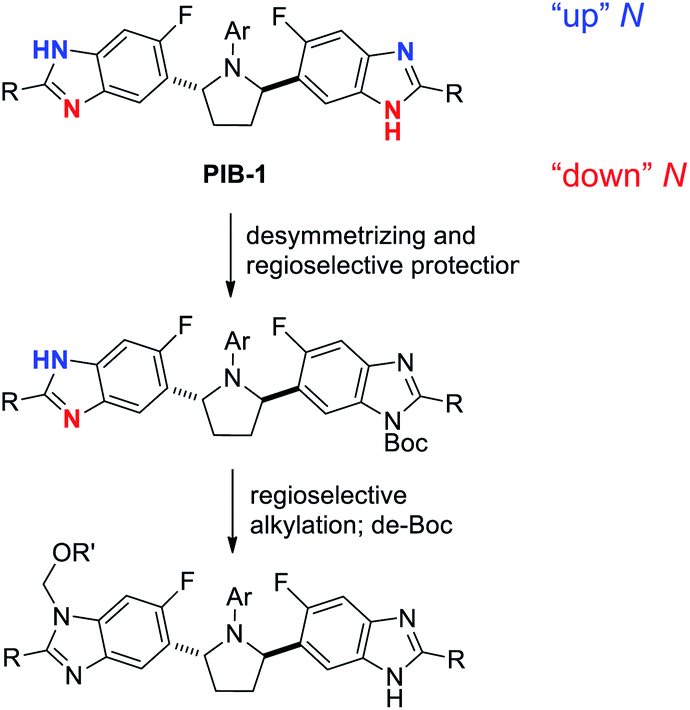 | ||
| Fig. 3 Benzimidazole nitrogen desymmetrizing and regioselective functionalization strategy. | ||
Not surprisingly, initial attempts to mono-Boc protect PIB gave a mixture of six compounds (Scheme 1), reflecting a 1:2:1 statistical ratio of starting material 1, mono-Boc (11 + 12), and di-Boc (13 + 14 + 15) isomers with a 3:1 ratio of down-mono-Boc 11 (Boc para to fluoro) to up-mono-Boc 12 (Boc meta to fluoro). This ratio favoring 11 was presumably influenced by the more nucleophilic nature of the nitrogens para to the fluorine. Following tedious chromatography, a 48% yield of mono-Boc isomers 11 and 12 was obtained. Slurrying the mixture in 3:1 MTBE/EtOAc gave a crystalline solid of 11 containing <1% 12 (3:1 12/11 in the filtrate), indicating a significant solubility difference between mono-Boc benzimidazole regiosiomers. This solubility difference would prove to be important in optimizing the formation of down-mono-Boc PIB 11.
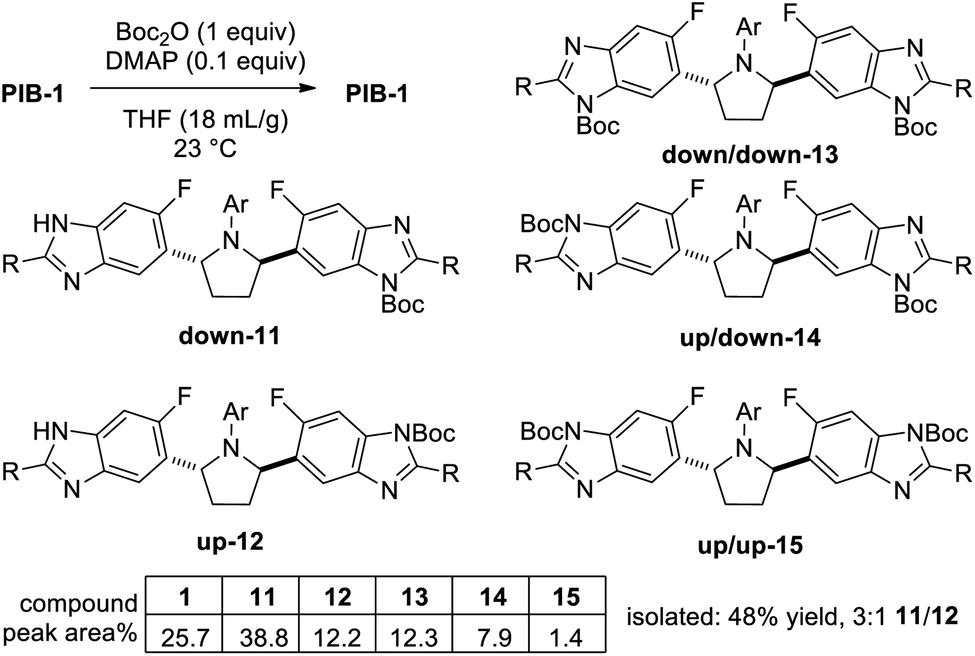 | ||
| Scheme 1 Initial attempt to mono-Boc protect PIB, 1. | ||
Reasoning that selective de-Boc of a mixture of di-Boc benzimidazoles 13, 14, and 15 may facilitate selective protection, and that both 13 and 14 could undergo Boc deprotection to give mono-down-Boc isomer 11 if relative rates of Boc cleavage were favorable, a complete reaction to a mixture of di-Boc compounds 13, 14, and 15 was carried out (Scheme 2). Following initial optimization of solvent and temperature to minimize 15,15 a 69:28:3 ratio of 13, 14, and 15, respectively, was obtained. Since (up,down)-di-Boc isomer 14 contains both an up-Boc and a down-Boc, this result represents an 83 (69 + 28/2):17 (28/2 + 3) total down/up-N-Boc ratio (5:1). When considering all species containing at least one down-Boc benzimidazole (13 and 14), this mixture constitutes a 97 (69 + 28):3 ratio (32:1), provided all undesired up-Boc can be selectively cleaved. While the first Boc protection benefits from the electronic bias imparted by the fluorine atom, the enhancement observed in the second Boc protection can be considered an example of the Horeau principle of statistical amplification.7
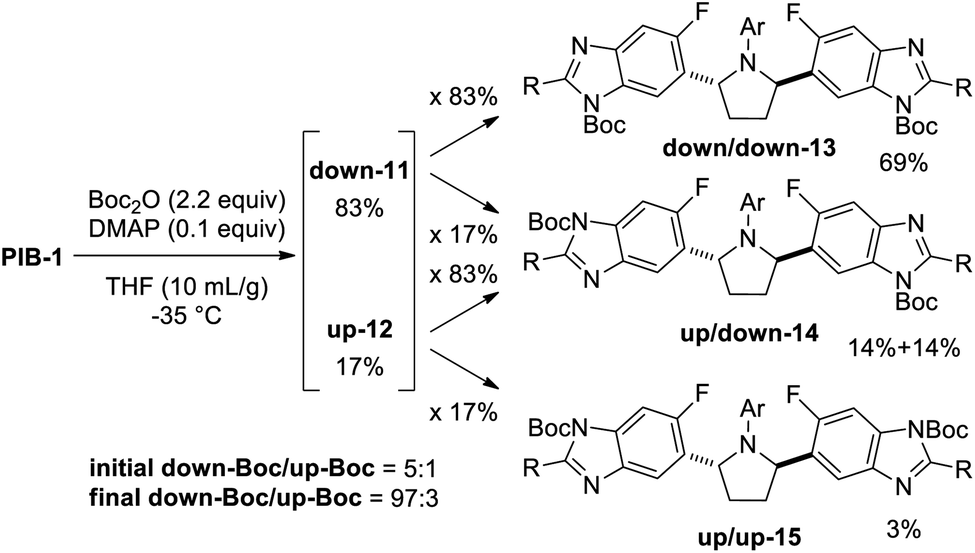 | ||
| Scheme 2 Selective di-Boc to a mixture of 13, 14, and 15. | ||
The Horeau principle is typically invoked in asymmetric synthesis to, for example, explain the upgrade in optical purity of a low-ee scalemic sample through coupling to a bifunctional linker to form a C2/meso mixture which can be more easily separated at the expense of yield. A review on this topic recently appeared.7Fig. 4 shows Horeau's first application of this principle in upgrading the enantiomeric purity of a 60% ee secondary alcohol to 87% ee by forming a mixture of C2/meso carbonates, removing the meso isomer, and cleaving the carbonate.16 In this example, since 20% (R) isomer becomes meso and 80% (S) isomer becomes meso (statistically), 32% of the carbonate mixture is readily removed, leaving a 64:4 (16:1) ratio of (R)/(S) after carbonate cleavage (87% ee). A second cycle further enhances the mixture to 96% ee at the further expense of yield. In the present case, rather than the minor/major (or equivalent major/minor) reaction product 14 being an undesirable meso isomer, the resulting up/down di-Boc 14 still contains a desired down-Boc (Scheme 2). Therefore, down/up-N regioselectivity is enhanced from 5:1 to 32:1 via the Horeau principle of statistical amplification, with 97% of the total mixture containing at least one down-N-Boc. This constitutes the first known application of this principle for the regioselective functionalization of a C2-symmetric poly-heterocycle.17 However, while both 13 and 14 contain at least one down-Boc benzimidazole, their simultaneous selective conversion to down-mono-Boc 11 without further de-Boc to PIB remained a significant hurdle to accomplishing PIB desymmetrization.
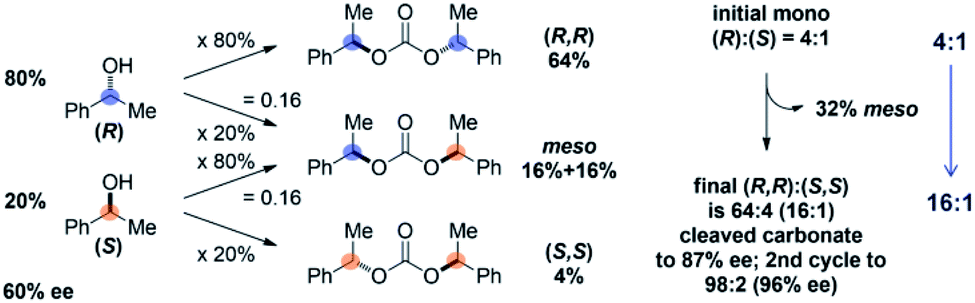 | ||
| Fig. 4 Original application of the Horeau principle of statistical amplification.16 | ||
Since mono-Boc regioisomers 11 and 12 showed favorable solubility differences in MTBE (vide supra), and it was anticipated that a primary aliphatic amine may deprotect the benzimidazoles at a slow enough rate that mono-Boc 11 could be protected from further de-Boc to PIB by crystallization,18 the reaction solvent was switched from THF to MTBE and n-butylamine was added to the reaction mixture (Scheme 3). Much to our delight, under these conditions, up-Boc cleaved significantly faster than down-Boc, with 14 converting to 11 and 15 converting to 12 quite rapidly over the first 2.5 h while 13 converted more slowly to 11. At this point, the solution was seeded with 11 (1 wt%), initiating a crystallization event. After 40 h at 23 °C, all mono-N-Boc and di-N-Boc isomers besides 13 and 11 were consumed, and a ratio of 5/84/11 for 13/11/1 was observed. Filtration gave a solid consisting of >95% 11.19 Since product solubility in acetonitrile (ACN) was low, a re-slurry in ACN gave 11 with >98% purity in 75% isolated yield.19 Here, terminus differentiation of C2-symmetric di-Boc isomer 13, the most significant component of the di-Boc mixture, was made possible through crystallization-induced protection of mono-Boc 11 against further de-Boc to PIB. Without crystallization of 11, a statistical 1:2:1 ratio of di/mono/PIB would have been generated (Fig. 2). Therefore, both Horeau amplification (di-Boc stage) and crystallization-induced protection (de-Boc stage) were required to deliver simultaneous regioselective benzimidazole functionalization and desymmetrization. To further increase the yield of the desymmetrization reaction to 94%, the conditions in Scheme 4 were employed, taking advantage of the even lower solubility of Boc-PIB 11 in ACN compared to MTBE. Overall, this di-Boc/mono-de-Boc PIB desymmetrization tactic provided a high isolated yield of a single down-mono-Boc product 11 out of the six species initially observed in attempted mono-Boc protection. While selective protection simplified the problem of PIB prodrug synthesis, regioselective functionalization of the remaining unprotected benzimidazole in 11 remained a formidable challenge (Fig. 3).
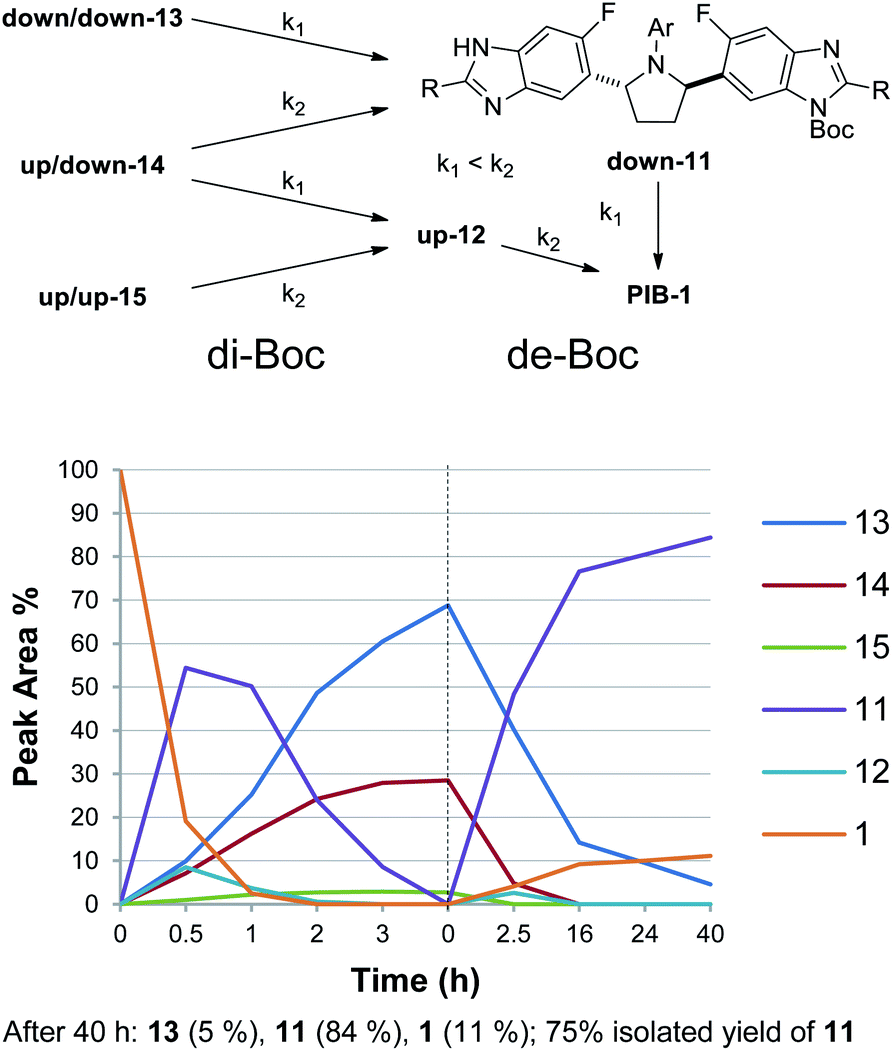 | ||
| Scheme 3 De-Boc of di-Boc mixture to give 11 selectively. De-Boc conditions: n-BuNH2 (1.5 equiv.), MTBE (8 mL g−1), 23 °C. | ||
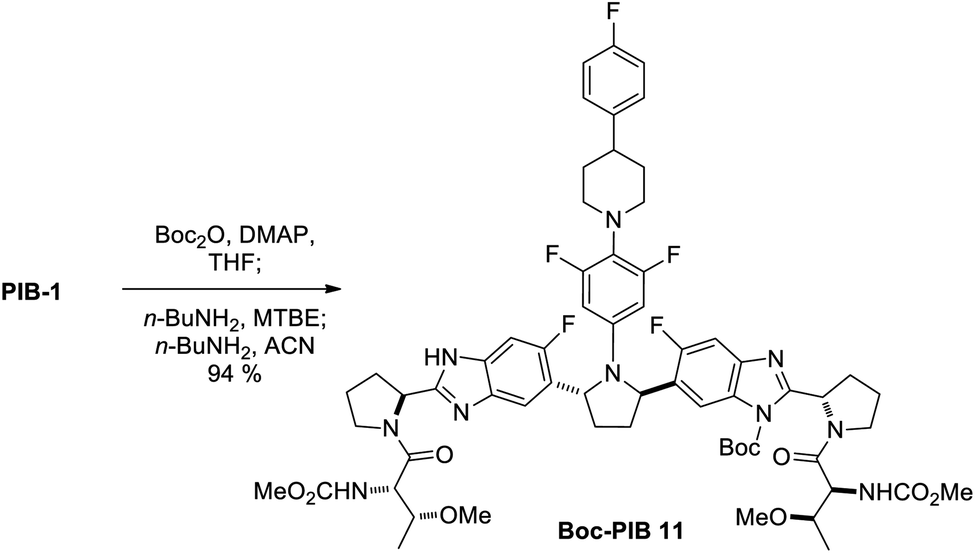 | ||
| Scheme 4 One-pot di-Boc/mono-de-Boc with ACN solvent switch. Conditions: Boc2O (1.9 equiv.), DMAP (0.1 equiv.), THF (5 mL g−1), −40 °C, 3 h; n-BuNH2 (0.8 equiv.), MTBE (10 mL g−1), 23 °C, 18 h; n-BuNH2 (1 equiv.), ACN (10 mL g−1), 23 °C, 21 h; mother liquor resubjection. | ||
Direct alkylation of Boc-PIB 11 with chloromethyl esters was feasible and used successfully for early syntheses of PIB prodrugs (Fig. 2).4 However, regioselectivities were poor and yields for the alkylation step variable after significant optimization efforts. Despite very limited precedent for regioselective benzimidazole formaldehyde adduct formation/acylation,20 the reversible nature of such an adduct offered potential benefits that could prove advantageous. To probe this strategy, Boc-PIB 11 was treated with paraformaldehyde and i-Pr2NEt in DMF (Scheme 5), then acylated with acid chloride 16.
 | ||
| Scheme 5 One-pot hydroxymethylation/acylation of 11. | ||
While good conversion to desired product was observed (85% overall yield), a disappointing 1.8:1 ratio of benzimidazole regioisomers 17 and 18 resulted, suggesting poor selectivity in the hydroxymethylation step and/or equilibration during the acylation. Undeterred by this preliminary result, Boc-PIB formaldehyde adduct 19/20 mixture was isolated by precipitation from EtOAc/MTBE/hexanes for investigation of acylation leaving group identity (Table 1).21 Standard acylating conditions (i-Pr2NEt, DMAP) were chosen as a basis for comparison between the acylating reagents, resulting in a wide range of reaction rates and product ratios. Reaction temperature was chosen for each acylating reagent to afford some conversion to product in order to measure the product isomer ratios. An increase in leaving group propensity resulted in greater conversion to the product isomer mixture as well as higher regioselectivity. With a relatively poor leaving group (OSu) in 21, the reaction reached 20% conversion after 18 h at 23 °C, and a similar product ratio was observed as in the 1-pot reaction (1.8:1). Employing the original highly electrophilic acid chloride 16 vastly improved rate (95% conversion after 1 h at −15 °C) and surprisingly improved the isomer ratio to 20:1 in favor of the desired product. Since conditions for the acylation were nearly identical to the one-pot reaction, the improved regioselectivity clearly resulted from isolation of the solid formaldehyde adduct. Therefore, the hydroxymethylation constituted a second consecutive crystallization-induced selective reaction.21
|
|
|||||
|---|---|---|---|---|---|
| Reagent | Leaving group (LG) | Temp (°C) | Time (h) | Conversion (%) |
17:18 ratio |
| 21 | OSu | 23 | 18 | 20 | 1.8:1 |
| 22 | 4-NO2PhO | 23 | 2 | 35 | 1.7:1 |
| 23 | OAt | 0 | 1 | 35 | 4.5:1 |
| 24 | C6F5O | −15 | 4 | 62 | 8:1 |
| 16 | Cl | −15 | 1 | 95 | 20:1 |
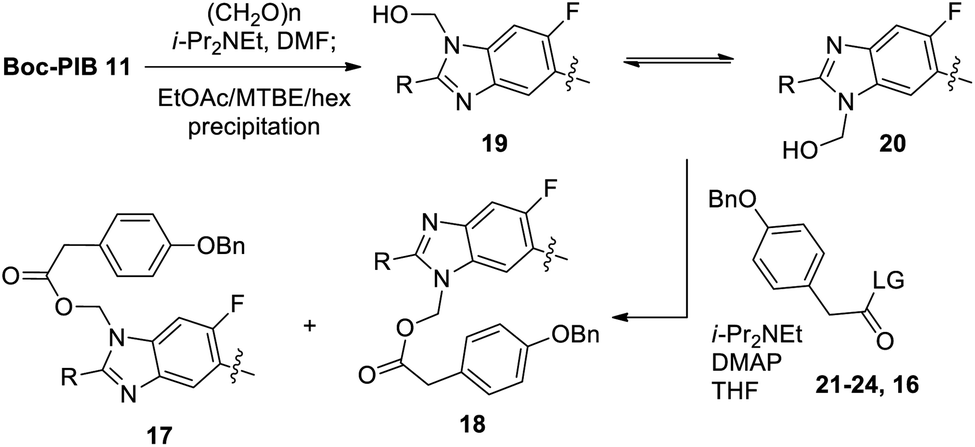
While the DMF/paraformaldehyde conditions provided adequate material for preliminary studies, a more convenient formaldehyde adduct formation was desired to avoid DMF distillation on larger scale (Scheme 6). Reaction of Boc-PIB 11 with aqueous formaldehyde in EtOAc in the absence of base was found to give a high mass balance of hydroxymethylation product (>10:1 19/11 ratio by 1H NMR in DMSO),22 providing key intermediate 19 in 99% yield.
 | ||
| Scheme 6 Preparation of key intermediate 19. | ||
Having secured a robust and scalable procedure for the synthesis of compound 19, we proceeded to investigate its use in the preparation of lead prodrugs of PIB.4 The encouraging results observed in the model system (Table 1) provided a useful starting point for the acylation of 19 with more complex structures. In order to enable a variety of potential prodrug classes, several different methods for their introduction were explored from hydroxymethyl intermediate 19. We initially examined mild acylation conditions using DMAP and Hünig's base in THF at −30 °C, which provided high regioselectivity for the desired regioisomer (25:26 = >20:1; Scheme 7, Condition A). To improve the isolation of higher purity products, however, we ultimately employed a stronger base at lower temperatures (LiHMDS, −65 °C; Scheme 7, Condition B), which offered an improved reaction profile at the expense of incomplete conversion and slightly decreased regioselectivity (25:26 = 10:1). A Boc-deprotection of the crude material preceded efficient chromatographic separation of the regioisomers, affording the penultimate dibenzylphosphate intermediate 25 in 62% yield over 3 steps from 11. To prepare the trimethyl “lock” prodrug 3,23 the cryogenic option (condition B) cleanly afforded the acylation product with high levels of selectivity when using acid chloride 27 (Scheme 7, 29:1 isomer ratio) to give an 84% isolated yield of the desired regioisomer over a similar 3-step sequence. A final hydrogenolysis with Pd/C gave the desired free phosphoric acids 2 and 3 in 83% and 64% yields, respectively.24
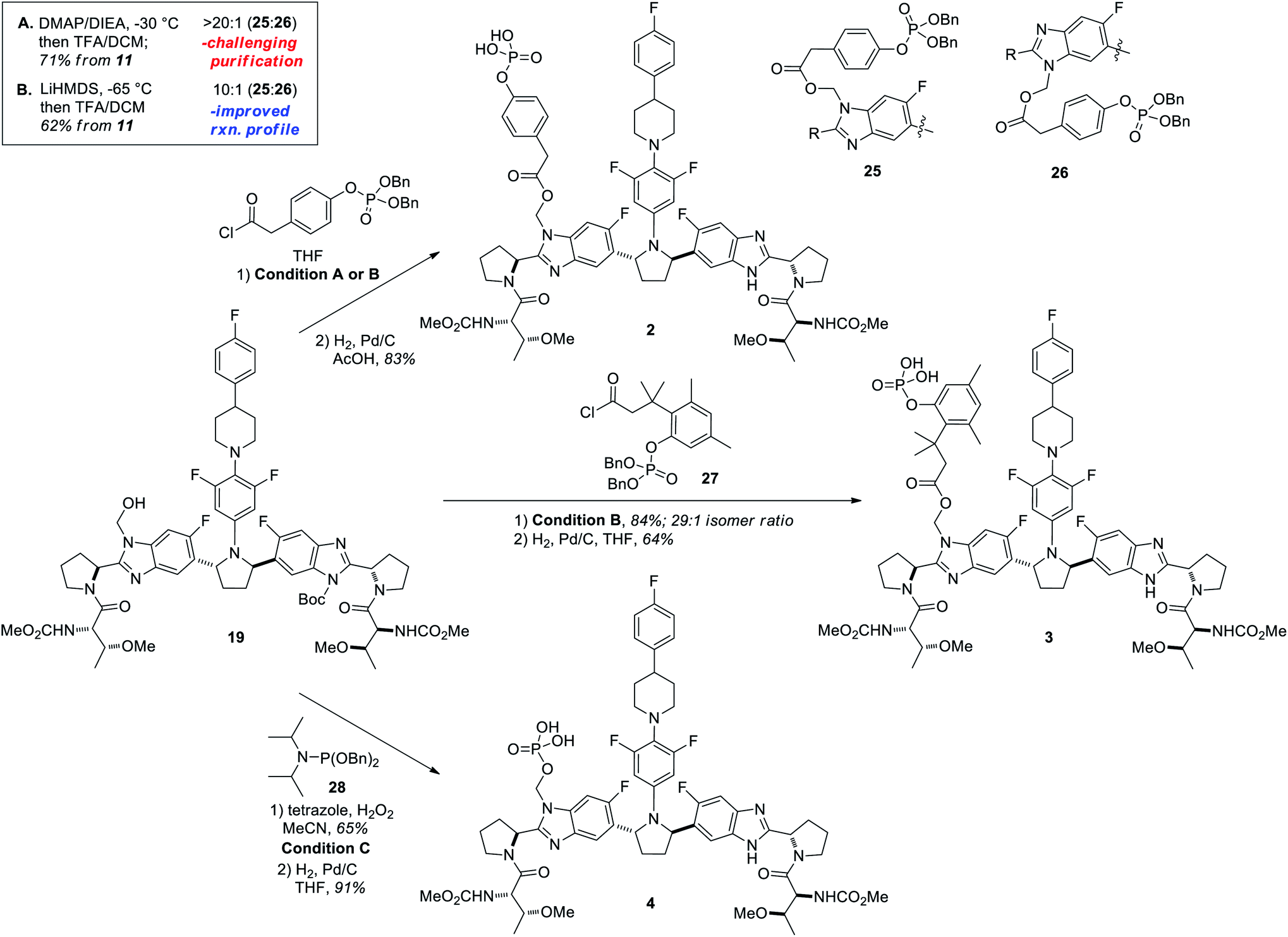 | ||
| Scheme 7 Functionalization strategies used to prepare prodrugs of PIB from 19. Condition A: (i) DMAP (5 mol%), DIEA (3.0 equiv.), acid chloride (2.5 equiv.), THF, −30 °C; (ii) 2:1 CH2Cl2:TFA, RT. Condition B: (i) LiHMDS (1.9 equiv.), THF, T < −65 °C then acid chloride (2–2.5 equiv); (ii) 2:1 CH2Cl2:TFA, RT. Condition C: (i) 28 (5.0 equiv.), tetrazole (5.0 equiv.), MeCN, −40 °C to 0 °C then H2O2 (10 equiv.); (ii) 2:1 CH2Cl2:TFA, RT. Hydrogenolyses: Pd/C (0.7 to 5 mol%), H2 (50 psi), RT, solvent (4–10 mL g−1). See ESI† for detailed procedures. | ||
To prepare phosphonoxymethyl prodrug 4 from key intermediate 19, a third functionalization strategy was required that would rapidly and selectively forge a P–O bond. After several failed attempts to directly form this bond at the phosphate oxidation state, an efficient phosphite formation was realized by employing dibenzylphosphoramidite reagent 28 with tetrazole as base (Condition C, Scheme 7).25 Following addition of hydrogen peroxide and subsequent de-Boc of the resulting crude phosphate, the dibenzylphosphonoxymethyl product was obtained in good yield (65%). Benzyl hydrogenolysis proceeded without incident, giving PIB prodrug 4 in 91% yield (Scheme 7).24
Conclusion
The initial synthetic routes to prodrugs 2–4 employed unselective direct alkylations of PIB 1 or Boc-PIB 11 with the requisite chloromethyl esters, where tedious separation of mixtures resulted in low overall yields.4 Ultimately, the development of a selective route to key intermediate 19 facilitated regioselective preparation of these lead prodrugs from PIB 1 and suitably positioned them for consideration as clinical candidates. The identification of high-yielding functionalization reactions of 19 and subsequent phosphate deprotection completed the syntheses of prodrugs 2–4, improving overall yields from 6–11% to 44–56% from PIB 1. This desymmetrization strategy could benefit the synthesis of other PIB prodrugs and other di-imidazole and di-benzimidazole analogs for HCV,4 as well as fluorescent sensors26 and ligands for organometallic complexes.27 Furthermore, extension of this concept from diheterocycles to other compound classes, for example, C2-symmetric diols,13 could also be envisioned. Finally, the desymmetrization tactic employed here to differentiate the homotopic termini of PIB 1 highlights the importance of considering classical and perhaps under-utilized strategies such as statistical amplification and exploitation of solubility differences for selectivity in even the most complex chemical settings.Data availability
Details of experimental conditions, 1H, 13C, 31P, and 19F NMR spectra for all new compounds, and HPLC/LCMS spectra for select compounds, are available in the ESI.Author contributions
E. A. V., S. N. G., J. H., R. C. K, J. T. R., and B. H. S. designed and performed the experiments; J. E. W. characterized compounds for structure confirmation; J. J. and D. A. D. supervised the project; E. A. V., S. N. G., and J. H. co-wrote the manuscript.Conflicts of interest
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. All authors are employees of AbbVie. The design, study conduct, and financial support for this research were provided by AbbVie. AbbVie participated in the interpretation of data, review, and approval of the publication.Acknowledgements
We thank the AbbVie Structural Chemistry group for compound characterization support.References
- See www.mavyret.com.
- J. Huff and R. Andersen, Glecaprevir/Pibrentasvir: The First 8-Week, Pangenotypic HCV Treatment Regimen for Patients 12 Years of Age and Older, Ann. Pharmacol., 2019, 1–15 Search PubMed.
- For a review of properties of drug molecules “beyond the rule of 5,” see: D. A. DeGoey, H.-J. Chen, P. B. Cox and M. D. Wendt, Beyond the Rule of 5: Lessons Learned from AbbVie's Drugs and Compound Collection, J. Med. Chem., 2018, 61, 2636–2651 CrossRef CAS PubMed.
- J. T. Randolph, E. A. Voight, S. N. Greszler, B. E. Uno, J. N. Newton, K. M. Gleason, D. Stolarik, C. Van Handel, D. A. J. Bow and D. A. DeGoey, Prodrug Strategies to Improve the Solubility of the HCV NS5A Inhibitor Pibrentasvir (ABT-530), J. Med. Chem., 2020, 63, 11034–11044 CrossRef CAS PubMed.
- Complex NMR spectra with significant interpretation challenges characterized PIB and most of its derivatives; see ref. 4, 6, and ESI† for details.
- R. Wagner, J. T. Randolph, S. V. Patel, L. Nelson, M. A. Matulenko, R. Keddy, J. K. Pratt, D. Liu, A. C. Krueger, P. L. Donner, D. K. Hutchinson, C. Flentge, D. Betebenner, T. Rockway, C. J. Maring, T. I. Ng, P. Krishnan, T. Pilot-Matias, C. Collins, N. Panchal, T. Reisch, T. Dekhtyar, R. Mondal, D. F. Stolarik, Y. Gao, W. Gao, D. A. Beno and W. M. Kati, Highlights of the Structure−Activity Relationships of Benzimidazole Linked Pyrrolidines Leading to the Discovery of the Hepatitis C Virus NS5A Inhibitor Pibrentasvir (ABT-530), J. Med. Chem., 2018, 61, 4052–4066 CrossRef CAS PubMed.
- For a recent review of the Horeau principle and applications, see: A. M. Harned, From determination of enantiopurity to the construction of complex molecules: The Horeau principle and its application in synthesis, Tetrahedron, 2018, 74, 3797–3841 CrossRef CAS.
- S. R. Magnuson, Two-Directional Synthesis and its Use in Natural Product Synthesis, Tetrahedron, 1995, 51, 2167–2213 CrossRef CAS.
- C. S. Poss and S. L. Schreiber, Two-Directional Chain Synthesis and Terminus Differentiation, Acc. Chem. Res., 1994, 27, 9–17 CrossRef CAS.
- T. R. Hoye and Z. Ye, Highly Efficient Synthesis of the Potent Antitumor Annonaceous Acetogenin (+)-Parviflorin, J. Am. Chem. Soc., 1996, 118, 1801–1802 CrossRef CAS.
- S. D. Burke and L. Jiang, Formal Synthesis of Uvaricin via Palladium-Mediated Double Cyclization, Org. Lett., 2001, 3, 1953–1955 CrossRef CAS PubMed.
- B. C. Austad, A. C. Hart and S. D. Burke, Halichondrin B: synthesis of the C(37)-C(54) subunit, Tetrahedron, 2002, 58, 2011–2026 CrossRef CAS.
- S. B. Han, A. Hassan, I. S. Kim and M. J. Krische, Total Synthesis of (+)-Roxaticin via C-C Bond Forming Transfer Hydrogenation: A Departure from Stoichiometric Chiral Reagents, Auxiliaries, and Premetalated Nucleophiles in Polyketide Construction, J. Am. Chem. Soc., 2010, 132, 15559–15561 CrossRef CAS PubMed.
- For a recent review, see: A. J. Metrano and S. J. Miller, Peptide-Based Catalysts Reach the Outer Sphere through Remote Desymmetrization and Atroposelectivity, Acc. Chem. Res., 2019, 52, 199–215 CrossRef CAS PubMed.
- Decreasing the temperature from 23 °C to −35 °C in THF enhanced the down/up selectivity from 3:1 (Scheme 1) to 5:1 (Scheme 2) while maintaining a reasonable rate. Further decreasing the temperature led to solubility issues, while other solvents (MeTHF, MTBE, ACN, DCM, toluene) led to poor selectivity and/or solubility issues.
- J. P. Vigneron, M. Dhaenens and A. Horeau, Maximizing optical purity of a partially resolved product without using a chiral compound, Tetrahedron, 1973, 29, 1055–1059 CrossRef CAS.
- The only additional known example of the Horeau principle being invoked to rationalize selectivity outside the context of enantioselective synthesis is in Hoye’s report on alkene geometry statistical amplification: T. R. Hoye and J. C. Sudhadolnik, Stereocontrolled synthesis of 2,5-linked bistetrahydrofurans via the triepoxide cascade reaction, Tetrahedron, 1986, 42, 2855–2862 CrossRef CAS.
- For a recent example of crystallization protecting against undesired double reaction, see: T. Seo, K. Kubota and H. Ito, Selective Mechanochemical Monoarylation of Unbiased Dibromoarenes by in Situ Crystallization, J. Am. Chem. Soc., 2020, 142, 9884–9889 CrossRef CAS PubMed.
- The initially isolated solid contained a 3.4/95.3/1.3 ratio of 13/11/1, while the mother liquor was 36.7/46.7/16.6 (9% 11 loss by HPLC). Then, the reslurry in ACN (5 mL/g, slow cooling from 75 °C to 23 °C with 1 h age prior to filtration), showed a 43.1/38.9/18 ratio of 13/11/1 in the supernatant before filtration. See the ESI† for details.
- For example, see: J. C. Sih, W. B. Im, A. Robert, D. R. Graber and D. P. Blakeman, Studies on (H+-K+)-ATPase Inhibitors of Gastric Acid Secretion. Prodrugs of 2-[(2-Pyridinylmethyl)sulfinyl]-benzimidazole Proton-Pump Inhibitors, J. Med. Chem., 1991, 34, 1049–1062 CrossRef CAS PubMed.
- This selective benzimidazole hydroxymethylation can be considered an unusual example of a Crystallization-Induced Diastereomer Transformation (CIDT): K. M. J. Brands and A. J. Davies, Crystallization-Induced Diastereomer Transformations, Chem. Rev., 2006, 106, 2711–2733 CrossRef CAS PubMed.
- Interpretation of 19/20 identity was complicated in that varying degrees of hydrolysis back to 11 and solution-state interconversion were consistently observed in attempts to monitor reactions or characterize the material. Therefore, although a solid isolation of 19 was performed, its identity was assigned based on isolated acylation/de-Boc product characterization results. See the ESI† for details. These persistent byproducts appeared to be derived from the acid chloride without incorporation of PIB, which led us to propose ketene-derived products or anhydrides that were difficult to remove without reverse-phase chromatography.
- Acid chloride 27 was prepared via an abbreviated 4-step route that avoided the oxidation/reduction steps in the published route: M. G. Nicolaou, C. Yuan and R. T. Borchardt, Phosphate Prodrugs for Amines Utilizing a Fast Intramolecular Hydroxy Amide Lactonization, J. Org. Chem., 1996, 61, 8636–8641 CrossRef CAS . See the ESI† for details..
- See the ESI† for detailed reaction and purification procedures.
- W. Bannwarth and A. Trzeciak, A Simple and Effective Chemical Phosphorylation Procedure for Biomolecules, Helv. Chim. Acta, 1987, 70, 175–186 CrossRef CAS.
- Y.-C. Wu, J.-Y. You, J.-C. Xie, S.-L. Li, D. Cao and Z.-Y. Wang, Colorimetric and ratiometric fluorescent sensor for F− based on benzimidazole-naphthalene conjugate: Reversible and reusable study & design of logic gate function, Dyes Pigm., 2017, 140, 47–55 CrossRef CAS.
- C. Rajnak, J. Titis, O. Fuhr, M. Ruben and R. Boca, Low spin Fe(II) complexes formed of monosubstitued 2,6-bis(2-benzimidazolyl)pyridine ligands, Polyhedron, 2017, 123, 122–131 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02396a |
| This journal is © The Royal Society of Chemistry 2021 |