
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkynyl triazenes enable divergent syntheses of 2-pyrones†
Jin-Fay
Tan
a,
Carl Thomas
Bormann
b,
Kay
Severin
 b and
Nicolai
Cramer
*a
b and
Nicolai
Cramer
*a
aLaboratory of Asymmetric Catalysis and Synthesis, EPFL SB ISIC LCSA, BCH 4305, CH-1015 Lausanne, Switzerland. E-mail: nicolai.cramer@epfl.ch
bLaboratory of Supramolecular Chemistry, EPFL SB ISIC LCS, BCH 3307, CH-1015 Lausanne, Switzerland. E-mail: kay.severin@epfl.ch
First published on 4th June 2021
Abstract
The 2-pyrone motif occurs frequently in bioactive natural products and is appreciated as synthetic intermediates. However, only few methods allow for diversifying functional group modifications on this relevant heterocycle. The distinct properties of 1-alkynyl triazenes promote a smooth addition of propiolic acids across the triple bond. Addition of catalytic amounts of silver salt induces cyclization to 2-pyrones. Depending on the reaction temperature, either 6-triazenyl or 5-triazenyl 2-pyrones are selectively formed. The triazenyl unit is subsequently replaced by a variety of valuable groups in a one-pot process yielding for instance 2-fluoro pyrones. The substitution occurs with an intriguing 1,5-carbonyl transposition. Moreover, the triazenyl group serves as traceless activating group for subsequent Diels–Alder cycloadditions and as a constituting unit for rare fused aminopyrazole pyrone heterocycles.
Introduction
2-Pyrones represent a frequently encountered framework in diverse bioactive natural products, exhibiting a broad range of antifungal, antibiotic, cytotoxic and phytotoxic activities (Fig. 1).1 These occurrences and properties sparked interests in the scaffold and motivated the development of 2-pyrone syntheses. To date, the repertoire of synthetic methods2 to access 2-pyrones include cyclizations,3 ring expansions,4 cyclotrimerizations5 and transition metal-catalyzed vinylic C–H annulations.6 However, despite these diverse strategies, the reachable functional group variability on 2-pyrones remains relatively limited compared to that of the related benzannulated congeners, the isocoumarins. For instance, reported substituents at the 6-position are largely restricted to aryl or alkyl groups, while halo or other heteroatom substituents are scarce (Fig. 2a).7–9 This might be attributed to tedious or non-viable routes to elaborate starting materials required for their construction. The limited substituent flexibility constitutes a bottleneck for medicinal chemistry studies involving these heterocycles.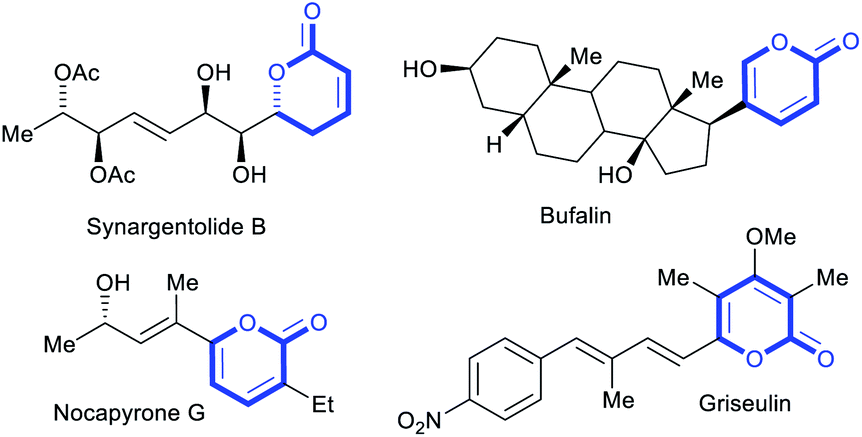 | ||
| Fig. 1 Bioactive 2-pyrone containing natural products. | ||
 | ||
| Fig. 2 Exploitation of the triazenyl group as a multi-leveraging reactivity and selectivity handle for the 2-pyrone synthesis and functionalization. | ||
Besides their bioactive properties, 2-pyrones are attractive dienes in Diels–Alder reactions delivering highly elaborate arenes after expulsion of CO2.10 However, installations of additional electron-donating substituents such as amino or methoxyphenyl groups are often required to enhance their reactivity (Fig. 2b).11 Subsequent removal of these substituents, or difficulties in subjecting them to functional group manipulations, render the normal electron-demand Diels–Alder reaction of 2-pyrones far less adopted in total synthesis than its inverse electron-demand counterpart.10a
Over the past years, the use of alkynyl triazenes has gained momentum in organic synthesis.12,13 They possess a unique ynamide-like reactivity,14 and are compatible with transition metals15 paired with versatile transformability into numerous functional groups. These values drew our interest to access triazenyl pyrones, which would in turn enable a divergent access to various functionalized pyrones. Inspired by Cui's synthesis of 2-pyrones from ynamides (Fig. 2c),3a we hereby report a smooth addition of propiolic acids to alkynyl triazenes, followed by a silver-catalyzed cyclization to triazenyl 2-pyrones (Fig. 2d). Divergent elaboration of the triazenyl groups into various valuable groups occurs under an unusual 1,5-carbonyl transposition delivering 3,4-substituted isomers. The triazene unit also serves as a traceless activating group for subsequent Diels–Alder cycloadditions and as a constituting unit for fused aminopyrazoles.
Results and discussion
Our studies were initiated by investigating the reaction between phenyl propiolic acid (1a) and alkynyl triazene 2a (Table 1). At 60 °C in toluene, addition of 1a to 2a formed acyloxy alkene 3aa in 95% yield and complete regioselectivity (entry 1). The sequential addition of catalytic AgSbF6 initiated cyclization to 6-triazenyl pyrone 4aa along with minor amounts of isomer 4aa′ (entry 2). The structure of 4aa was confirmed by X-ray crystallography.164aa′ is presumably formed from the thermodynamic regioisomer 3aa′. Indeed, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 3aa and 3aa′ was observed at 100 °C in the absence of AgSbF6 (entry 3). Performing the process at ambient temperature in CH2Cl2 led to quantitative and exclusive formation of 4aa (entries 4–6). The direct addition of AgSbF6 at the start of reaction, or using a 5 mol% loading, resulted in lower yields (entries 7 and 8). Very notably, conducting the reaction at 100 °C strongly favored the formation of isomer 4aa′ in good yields (entries 10–12). In summary, this is a simple temperature switch of the reaction outcome.
:1 mixture of 3aa and 3aa′ was observed at 100 °C in the absence of AgSbF6 (entry 3). Performing the process at ambient temperature in CH2Cl2 led to quantitative and exclusive formation of 4aa (entries 4–6). The direct addition of AgSbF6 at the start of reaction, or using a 5 mol% loading, resulted in lower yields (entries 7 and 8). Very notably, conducting the reaction at 100 °C strongly favored the formation of isomer 4aa′ in good yields (entries 10–12). In summary, this is a simple temperature switch of the reaction outcome.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | T (°C) | Solvent | % yield 3aa (3aa:3aa′) |
% yield 4aa | % yield 4aa′ |
4aa:4aa′ |
| a Conditions: (i) 0.11 mmol 1a, 0.10 mmol 2a, 0.2 M in the indicated solvent and temperature for 12 h, (ii) 10 μmol AgSbF6, 12 h, yield and ratio determined by 1H-NMR with an internal standard. b 12 h for (ii), no AgSbF6. c (i) 0.11 mmol 1a, 0.10 mmol 2a in CH2Cl2 for 2 h, (ii) 10 μmol AgSbF6, 23 °C for 1 h. d 0.11 mmol 1a, 0.10 mmol 2a, 10 μmol AgSbF6 in CH2Cl2 at 23 °C for 24 h. e 5 μmol AgSbF6. f With 0.15 mmol 1a. g With 0.18 mmol 1a. h Isolated yields. Ortep structure of 4aa with thermal ellipsoids are at 50% probability. Hydrogen atoms are omitted for clarity. | ||||||
| 1b | 60 | PhMe | 95 (>20:1)h |
Trace | — | — |
| 2 | 60 | PhMe | — | 75 | 8 | 10:1 |
| 3b | 100 | PhMe | 46 (1:1)h |
— | — | — |
| 4 | 23 | PhMe | — | 92 | — | >20:1 |
| 5 | 23 | CH2Cl2 | — | 99 | — | >20:1 |
| 6 | 23 | CH 2 Cl 2 | — | 98 | — |
>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
| 7d | 23 | CH2Cl2 | 38 (>20:1)h |
49 | — | >20:1 |
| 8e | 23 | CH2Cl2 | — | 68 | — | >20:1 |
| 9 | 100 | PhMe | — | 13 | 58 | 1:4.5 |
| 10f | 100 | PhMe | — | 12 | 72h | 1:6 |
| 11g | 100 | PhMe | — | 13 | 65 | 1:5 |
| 12f | 120 | PhMe | — | 13 | 70 | 1:5.4 |

Despite the highly-valued fluorine effect in heterocycles in drug discovery,17 fluorinated 2-pyrones remain the rarest and least reported among functionalized 2-pyrones.18 The complete lack of suitable methods to access 6-fluoro 2-pyrones underscores the formidable challenges of regioselective fluorination of 2-pyrones. To address this gap in synthetic methodology, we opted to transform the triazene unit into a fluoride substituent by the Wallach reaction.19 In a one-pot fashion, HF·py was added to the crude reaction mixture containing 4aa (Scheme 1). Surprisingly, the expected 4,5-disubstituted pyrone 5aa′ was not detected. Instead, 3,4-disubstituted isomer 5aa was isolated in 88% yield, confirmed by X-ray crystallography.16 This unexpected phenomenon was attributed to a 1,5-carbonyl transposition (vide infra). Despite the facile acidic cleavage of 4aa, its regioisomer 4aa′ was found to be unreactive or unproductive towards further transformations (see ESI† for details).
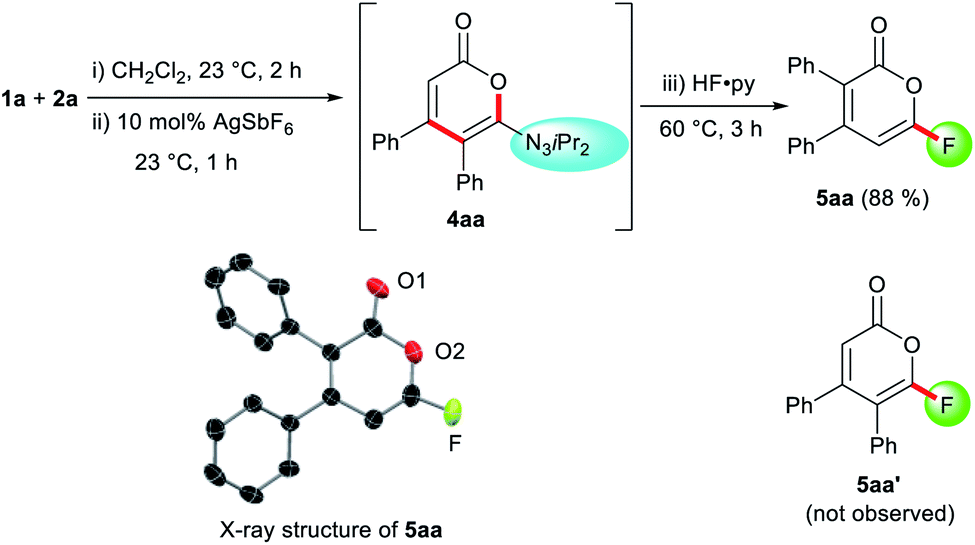 | ||
| Scheme 1 1,5-Carbonyl transposition in the one-pot formation of fluoropyrone 5aa. Ortep structure of 5aa with thermal ellipsoids are at 50% probability. Hydrogen atoms are omitted for clarity. | ||
The scope for the one-pot 6-fluoropyrone formation was subsequently evaluated (Scheme 2). Both, electron-rich and electron-poor aryl groups R1 were well tolerated, furnishing fluoropyrones 5ba–5ha in good yields, with full regioselectivity and complete 1,5-transposition. 1-Naphthyl- (5ia) and a thienyl-substituted products (5ja) were as well accessed. Alkyl groups like cyclopropyl (5ka) and propyl (5la) were well accepted. The use of parent propiolic acid 1m provided 3-phenyl-6-fluoropyrone 5ma. Concerning the alkynyl triazenes, various aryl and alkyl substituents consistently reacted well and afforded pyrones in good yields and full regioselectivity (5ab–5af). X-Ray crystallographic analysis of 5da and 5ac confirmed assignment of the substituent R1 and R2 on the pyrone core.16
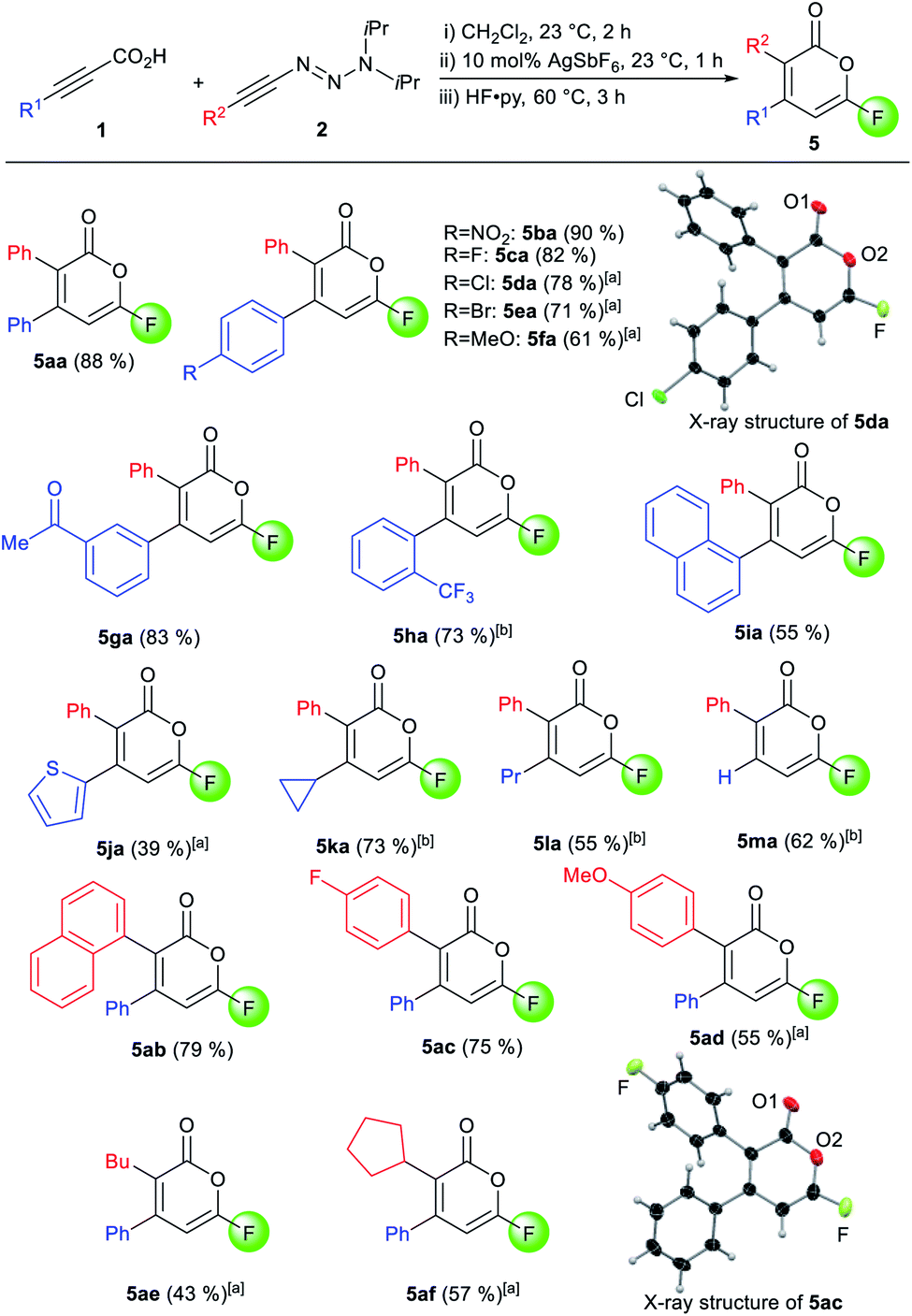 | ||
| Scheme 2 Scope for fluoropyrone formation. Conditions: (i) 0.11 mmol 1a, 0.10 mmol 2a in CH2Cl2 (0.2 M) at 23 °C for 2 h, (ii) 10 μmol AgSbF6 at 23 °C for 1 h, (iii) 1.0 mL HF·py at 60 °C for 3 h; [a] 0.5 mL HF·py at 23 °C for 48 h for step (iii); [b] 60 °C for 12 h for step (ii). | ||
During our investigations, very notable reactivity profiles were encountered with specific alkynyl triazenes. In particular, cyclopropyl-substituted substrate 2g provided pyrone-fused N-aminopyrazole 6ag in 76% yield whereas the expected fluoropyrone was not observed at all (Scheme 3).15g This highly rare fused double heterocyclic system was as well obtained with tethered 1-diynyl triazene 2h. The structures of 6ag and 6ah were unambiguously confirmed by single crystal X-ray diffraction16 We reasoned that the N-aminopyrazoles arose from triazenyl pyrone intermediates 4ag and 4ahvia the depicted mechanisms.20 For 4ag, a cyclopropane ring opening assisted by triazene or the conjugated oxygen leads to X and Y. An intramolecular cyclization and a subsequent rearomatization then give rise to 6ag. In a similar fashion, displacement of the tosylamide group in 4ah leads to 6ah, further evidenced by the isolation of tosylamide 7. These unique products underscore once again triazenes possess an untapped potential to forge novel and complex fused heterocyclic systems.
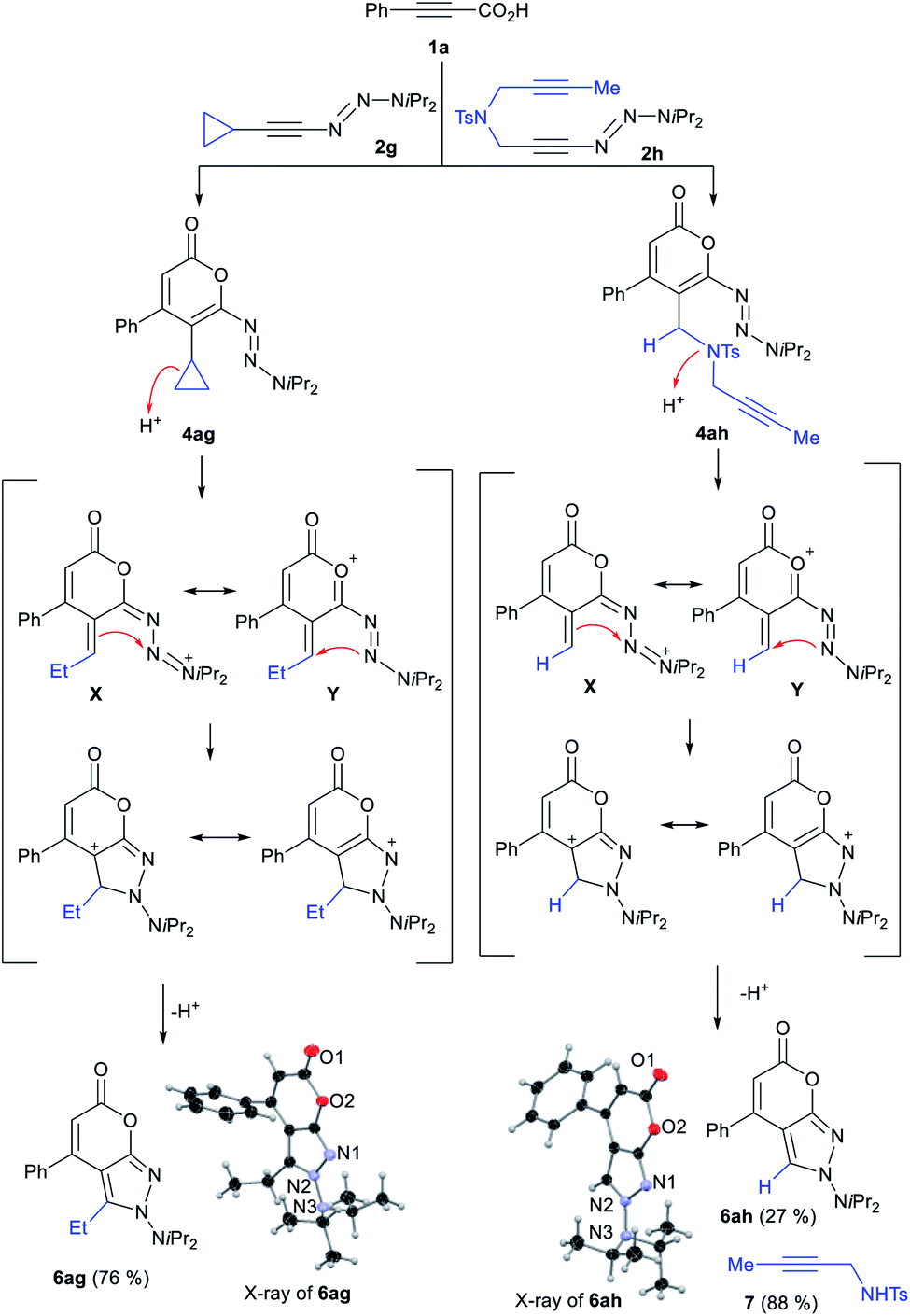 | ||
| Scheme 3 N-Aminopyrazole formation from 2g or 2h. Condition: (i) 0.11 mmol 1a, 0.10 mmol 2a in CH2Cl2 (0.2 M) at 23 °C for 2 h, (ii) 10 μmol AgSbF6 at 23 °C for 1 h, (iii) 1.0 mL HF·py at 60 °C for 3 h. | ||
Next, we investigated further one-pot divergent functionalizations of the triazenyl pyrones (Scheme 4). For instance, performing the one-pot process with 1a and 2a, chloro- (8), bromo- (9) and iodo-pyrones (10) were efficiently prepared in high yields using the corresponding trimethylsilyl halides. Again, the 1,5-carbonyl transposition was observed for these products. The use of MeOH as the nucleophile gave a 4.4:1 mixture of transposed dimethoxy orthoester 11 and its non-transposed isomer 11′. Notably, triazenyl pyrones engage in normal-electron demand Diels–Alder reactions. For instance, its reaction with acetylene dicarboxylate tetrasubstituted gave arene 12. The triazenyl group was cleaved after the cycloaddition, presumably by a thermally-induced radical mechanism,21 as evidenced by some amounts of triazenyl arene 13. Anthraquinone 14 was furnished in 69% yield after two consecutive cycloadditions on p-quinone were engaged, followed by triazenyl elimination. The use of a larger excess p-quinone gave naphthoquinone 15. Remarkably, the triazene was cleaved cleanly in all Diels–Alder examples. Contrasting to conventional strategies to activate pyrones for Diels–Alder cycloadditions, the triazenyl pyrones offer a highly efficient alternative to access densely substituted arenes leveraging the triazene moiety as a traceless activating group.
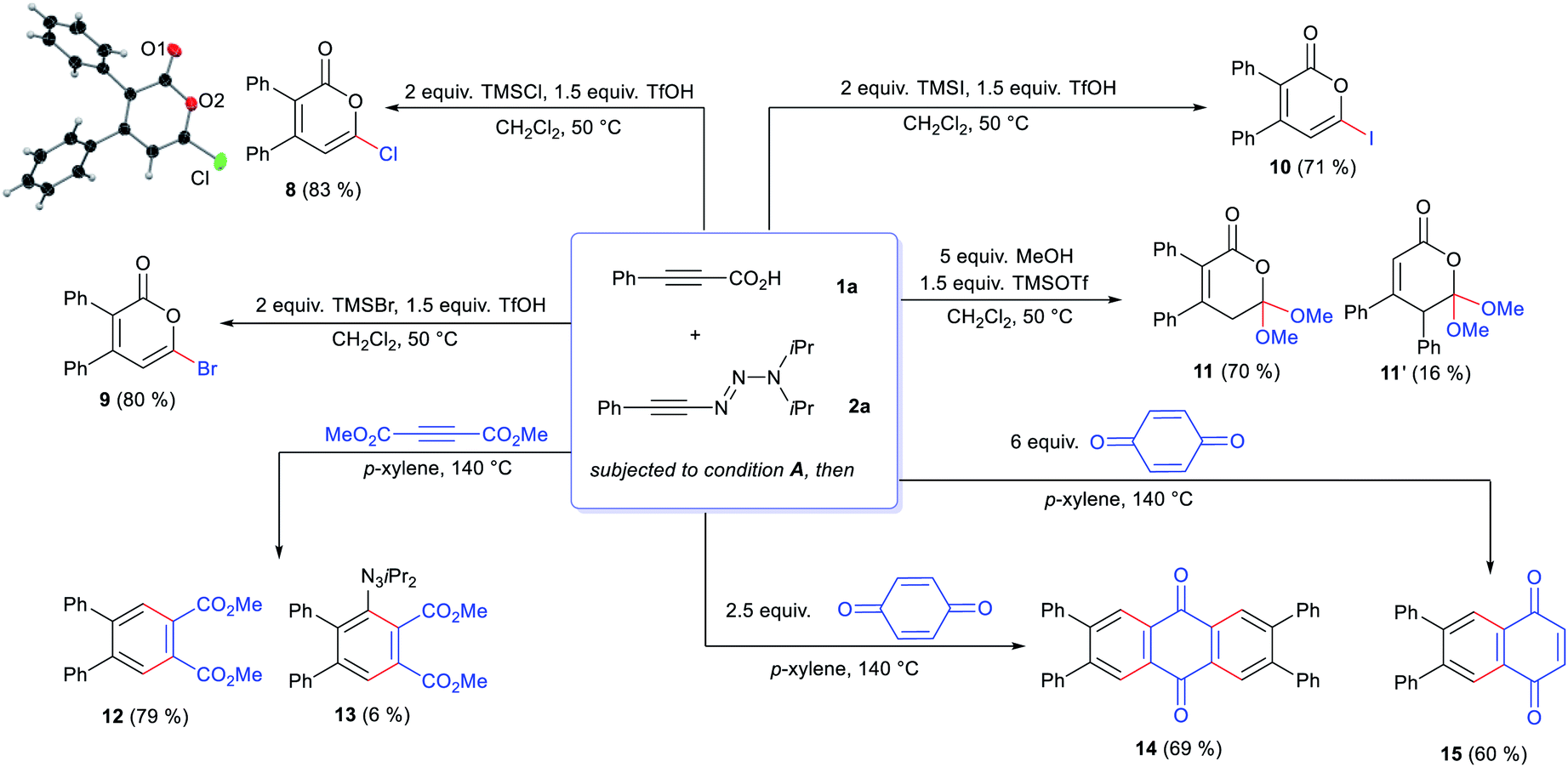 | ||
| Scheme 4 One-pot divergent formation of functionalized 2-pyrones and engagement in Diels–Alder cycloadditions. Condition A: 0.11 mmol 1a, 0.10 mmol 2a, 0.2 M CH2Cl2 at 23 °C for 2 h, then 10 μmol AgSbF6 at 23 °C for 1 h. | ||
To the best of our knowledge, the observed pyrone 1,5-carbonyl transposition has not been reported to date. Three plausible pathway for this transposition can be envisioned (Scheme 5).22–24 Initial protonation of the carbonyl oxygen atom would form pyranol A. For pathway I, a subsequent nucleophile addition leads to B.22b,22c In turn, B eventually undergoes a oxa 6π-electrocyclic ring opening23 leading to δ-hydroxy unsaturated acyl triazene C. Lactonization of the activated acyl triazene unit24 delivers transposed product 5. For pathway II, the hydroxyl group of A alternatively might undergo a substitution by the nucleophile generating cationic intermediate D and a molecule of water. Species D may be stabilized by its pyrylium mesomer E. Both are capable of providing 5 upon triazene displacement by re-addition of the previously expelled water. A third pathway involves trapping of the putative cation F by an external water molecule to give isomeric hydroxy-pyrones G and H. However, the non-transposed isomer was not observed with any halide nucleophiles. The insufficient discrimination between G and H reduces the likelihood of pathway III. Similarly, pathway II has a lower possibility as A should also be capable of generating the non-transposed isomer with halides.
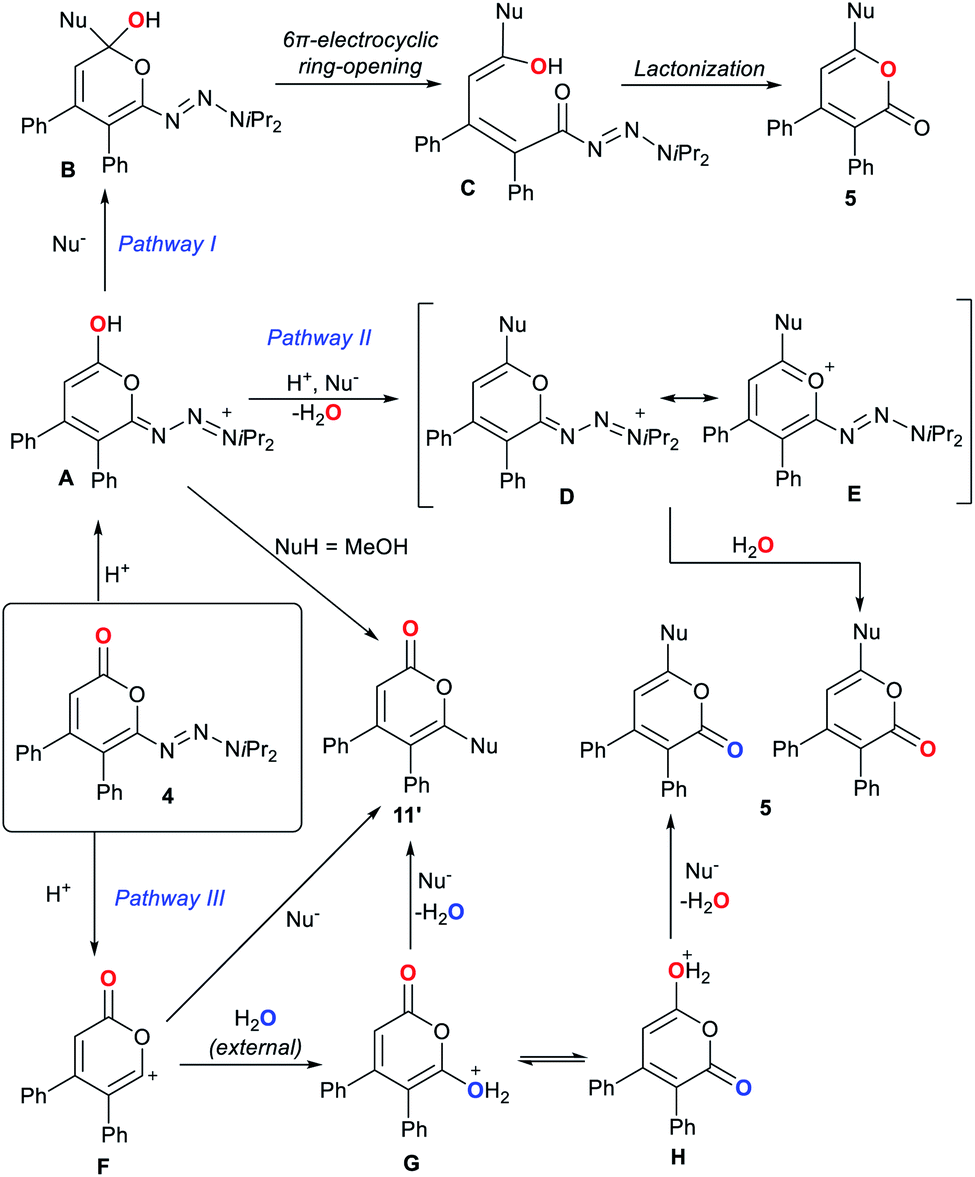 | ||
| Scheme 5 Possible mechanism pathways for the 1,5-carbonyl transposition. | ||
Experimentally, isotope spiking experiments with 50 equivalents of H218O on the fluorination and methoxylation did not provide any incorporation of 18O in 5aa and 11 based on 13C NMR and mass spectrometry (Scheme 6).25 This is hinting towards an intramolecular mechanism. Therefore, illustrated pathway I might be the most plausible mechanism for the transposition. Minor isomer 11′ would be generated from A using methanol as a nucleophile.
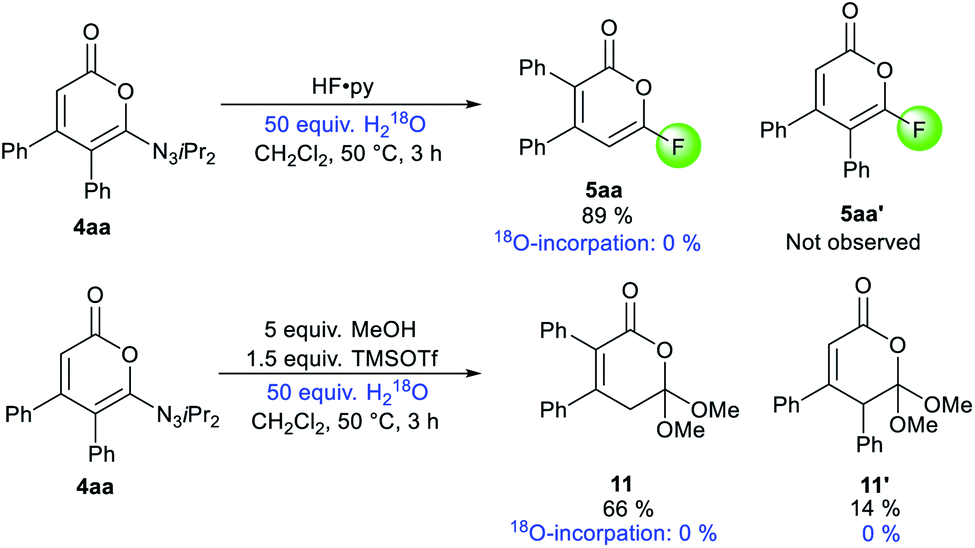 | ||
| Scheme 6 18O-Isotope labelling experiments of pyrone 4aa. | ||
Conclusions
In summary, we reported a one-pot divergent formation of a broad range of functionalized 2-pyrones by leveraging the reactivity of alkynyl triazenes on several instances. They first facilitate the addition of carboxylic acids across the alkyne triple bond. Secondly, it can be easily converted into a wide range of desirable functionalities. These comprise the synthesis of unreported 6-fluoropyrones, as well as pyrones with chloride, bromide, iodide and methoxy substituents at the 6-position. Notably, we have unraveled an unusual and mechanistically intriguing 1,5-carbonyl transposition, evidencing the non-innocent behavior of the triazenyl group. Furthermore, the triazenyl unit can be used to access rare pyrone-fused pyrazole heterocycles. Moreover, the triazene substituent acts as a traceless activating group facilitating normal electron-demand Diels–Alder cycloadditions, affording densely substituted arene products.Data availability
The datasets supporting this article have been uploaded as part of the supplementary material. Crystallographic data for 4aa, 5aa, 5ca, 5ac, 6ag, 6ah and 8 has been deposited at the CCDC under 2075639 and 2075641-2075646 and can be obtained from http://www.ccdc.cam.ac.uk.Author contributions
JFT, KS and NC conceived, designed and directed the project. JFT conducted the experiments. CTB synthesized the alkynyl triazenes. All authors wrote the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work is supported by EPFL. We thank Dr R. Scopelliti and Dr F. Fadaei-Tirani for X-ray crystallographic analysis of compounds 4aa, 5aa, 5da, 5ac, 6ag, 6ah and 8.Notes and references
- (a) A. A. Q. Al-Khdhairawi, G. A. Cordell, N. F. Thomas, N. B. S. Nagojappa and J.-F. F. Weber, Org. Biomol. Chem., 2019, 17, 8943 Search PubMed; (b) A. G. Tempone, D. D. Ferreira, M. L. Lima, T. A. C. Silva, S. E. T. Borborema, J. Q. Reimão, M. K. Galuppo, J. M. Guerra, A. J. Russell, G. M. Wynne, R. Y. L. Lai, M. M. Cadelis and B. R. Copp, Eur. J. Med. Chem., 2017, 139, 947 Search PubMed; (c) G. P. McGlacken and I. J. Fairlamb, Nat. Prod. Rep., 2005, 22, 369 Search PubMed.
- J. S. Lee, Mar. Drugs, 2015, 13, 1581 Search PubMed.
- (a) Y. Shen, C. Wang, W. Chen and S. Cui, Org. Chem. Front., 2018, 5, 3574 Search PubMed; (b) T. Luo, M. Dai, S.-L. Zheng and S. L. Schreiber, Org. Lett., 2011, 13, 2834 Search PubMed; (c) T. Luo and S. L. Schreiber, Angew. Chem., Int. Ed., 2007, 46, 8250 Search PubMed.
- (a) T. Kondo, Y. Taguchi, Y. Kaneko, M. Niimi and T. Mitsudo, Angew. Chem., Int. Ed., 2004, 43, 5369 Search PubMed; (b) P. Mingo, S. Zhang and L. S. Liebeskind, J. Org. Chem., 1999, 64, 2145 Search PubMed.
- J. Louie, J. E. Gibby, M. V. Farnworth and T. N. Tekavec, J. Am. Chem. Soc., 2002, 124, 12188 Search PubMed.
- (a) Q.-L. Yang, Y.-K. Xing, X.-Y. Wang, H.-X. Ma, X.-J. Weng, X. Yang, H.-M. Guo and T.-S. Mei, J. Am. Chem. Soc., 2019, 141, 18970 Search PubMed; (b) R. Mandal and B. Sundararaju, Org. Lett., 2017, 19, 2544 Search PubMed.
- (a) Y. Qu and G. A. Kraus, Tetrahedron Lett., 2017, 58, 892 Search PubMed; (b) W. A. Boulanger and J. A. Katzenellenbogen, J. Med. Chem., 1986, 29, 1159 Search PubMed; (c) P. Martin, E. Steiner, J. Streith, T. Winkler and D. Bellus, Tetrahedron, 1985, 41, 4057 Search PubMed.
- (a) F. Camps, J. M. Moreto, S. Ricart, J. M. Vinas, E. Molins and C. Miravitilles, J. Chem. Soc., Chem. Commun., 1989, 1560 Search PubMed; (b) S. A. Ahmed, E. Bardshiri and T. J. Simpson, Tetrahedron Lett., 1988, 29, 1595 Search PubMed; (c) S. H. Cho and L. S. Liebeskind, J. Org. Chem., 1987, 52, 2631 Search PubMed.
- D. S. Ziegler, L. Klier, N. Mgller, K. Karaghiosoff and P. Knochel, Synthesis, 2018, 50, 4383 Search PubMed.
- (a) Q. Cai, Chin. J. Chem., 2019, 37, 946 Search PubMed; (b) K. Afarinkia, V. Vinader, T. D. Nelson and G. H. Posner, Tetrahedron, 1992, 48, 9111 Search PubMed.
- A. I. Katri and S. D. Samant, Synthesis, 2015, 47, 343 Search PubMed.
- G. Kiefer, T. Riedel, P. J. Dyson, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2015, 54, 302 Search PubMed.
- A. A. Suleymanov and K. Severin, Angew. Chem., Int. Ed., 2021, 60, 6879 Search PubMed.
- F. Perrin, G. Kiefer, L. Jeanbourquin, S. Racine, D. Perrotta, J. Waser, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2015, 54, 13393 Search PubMed.
- (a) J.-F. Tan, C. T. Bormann, K. Severin and N. Cramer, ACS Catal., 2020, 10, 3790 Search PubMed; (b) C. T. Bormann, F. G. Abela, R. Scopelliti, F. Fadaei-Tirani and K. Severin, Eur. J. Org. Chem., 2020, 2130 Search PubMed; (c) J.-F. Tan, C. T. Bormann, F. G. Perrin, F. M. Chadwick, K. Severin and N. Cramer, J. Am. Chem. Soc., 2019, 141, 10372 Search PubMed; (d) T. Wezeman, R. Scopelliti, F. Fadaei Tirani and K. Severin, Adv. Synth. Catal., 2019, 361, 1383 Search PubMed; (e) A. A. Suleymanov, R. Scopelliti, F. Fadaei Tirani and K. Severin, Adv. Synth. Catal., 2018, 360, 4178 Search PubMed; (f) D. Kossler, F. G. Perrin, A. A. Suleymanov, G. Kiefer, R. Scopelliti, K. Severin and N. Cramer, Angew. Chem., Int. Ed., 2017, 56, 11490 Search PubMed; (g) L. N. Jeanbourquin, R. Scopelliti, F. Tirani Fadaei and K. Severin, Helv. Chim. Acta, 2017, 100, e1700186 Search PubMed.
- Supplementary crystallographic data of the following compounds can be found from The Cambridge Crystallographic Data Centre: 2075639 (4aa), 2075641 (5aa), 2075642 (5ca), 2075643 (5ac), 2075644 (6ag), 2075645 (6ah) and 2075646 (8).†.
- (a) X. Wang, J. Lei, Y. Liu, Y. Ye and J. Li, Org. Chem. Front., 2021, 8, 2079 Search PubMed; (b) Y. A. Serguchev, M. V. Ponomarenko and N. V. Ignat’ev, J. Fluorine Chem., 2016, 185, 1 Search PubMed; (c) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 Search PubMed; (d) V. A. Petrov, Fluorinated Heterocyclic Compounds: Synthesis, Chemistry, and Applications, ed. John Wiley & Sons, Hoboken, 2009, pp. 1–432 Search PubMed.
- (a) Y. Wang and J. D. Burton, J. Org. Chem., 2006, 71, 3859 Search PubMed; (b) V. Kvitu, Helv. Chim. Acta, 1990, 73, 411 Search PubMed; (c) D. C. England, E. A. Donald and F. J. Weigert, J. Org. Chem., 1981, 46, 144 Search PubMed.
- (a) P. J. Riss, S. Kuschel and F. I. Aigbirhio, Tetrahedron Lett., 2012, 53, 1717 Search PubMed; (b) M. N. Rosenfeld and D. A. Widdowson, J. Chem. Soc., Chem. Commun., 1979, 914 Search PubMed; (c) O. Wallach and F. Heusler, Liebigs Ann. Chem., 1888, 243, 219 Search PubMed.
- (a) W.-S. Yong, S. Park, H. Yun and P. H. Lee, Adv. Synth. Catal., 2016, 358, 1958 Search PubMed; (b) W. Yang, Z. Yang, L. Xu, L. Zhang, X. Xu, M. Miao and H. Ren, Synth. Commun., 2014, 44, 2478 Search PubMed; (c) W. Yang, Z. Yang, L. Xu, L. Zhang, X. Xu, M. Miao and H. Ren, Angew. Chem., Int. Ed., 2013, 52, 14135 Search PubMed; (d) D. B. Kimball, R. Herges and M. M. Haley, J. Am. Chem. Soc., 2002, 124, 1572 Search PubMed.
- (a) O. Nuyken, J. Stebani, T. Lippert, A. Wokaun and A. Stasko, Macromol. Chem. Phys., 1995, 196, 751 Search PubMed; (b) K. Vaughan and M. T. H. Liu, Can. J. Chem., 1981, 59, 923 Search PubMed.
- (a) X. Wu, J. Lei and Z. L. Song, Chin. Chem. Lett., 2011, 22, 306 Search PubMed; (b) A. Nangia and P. B. Rao, Tetrahedron Lett., 1993, 34, 2681 Search PubMed; (c) W. G. Dauben and D. M. Michno, J. Org. Chem., 1977, 42, 682 Search PubMed.
- A. I. Khatri and S. D. Samant, RSC Adv., 2015, 5, 2009 Search PubMed.
- I. R. Landman, E. Acuña-Bolomey, R. Scopelliti, F. Fadaei-Tirani and K. Severin, Org. Lett., 2019, 21, 6408 Search PubMed.
- (a) T. L. Mega and R. L. van Etten, J. Am. Chem. Soc., 1993, 115, 12056 Search PubMed; (b) J. Diakur, T. T. Nakashima and J. C. Vederas, Can. J. Chem., 1980, 58, 1311 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization of all new compounds. CCDC 2075639, 2075641–2075646. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02583j |
| This journal is © The Royal Society of Chemistry 2021 |