
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnsemble effects in Cu/Au ultrasmall nanoparticles control the branching point for C1 selectivity during CO2 electroreduction†
Hongyu
Shang‡
 a,
Dongjoon
Kim‡
b,
Spencer K.
Wallentine
a,
Minkyu
Kim§
b,
Daniel M.
Hofmann
c,
Runiya
Dasgupta
a,
Catherine J.
Murphy
*c,
Aravind
Asthagiri
*b and
L. Robert
Baker
*a
a,
Dongjoon
Kim‡
b,
Spencer K.
Wallentine
a,
Minkyu
Kim§
b,
Daniel M.
Hofmann
c,
Runiya
Dasgupta
a,
Catherine J.
Murphy
*c,
Aravind
Asthagiri
*b and
L. Robert
Baker
*a
aDepartment of Chemistry and Biochemistry, The Ohio State University, Columbus, Ohio, USA. E-mail: baker.2364@osu.edu
bDepartment of Chemical Engineering, The Ohio State University, Columbus, Ohio, USA. E-mail: asthagiri.1@osu.edu
cUniversity of Illinois at Urbana-Champaign, Urbana, Illinois, USA. E-mail: murphycj@illinois.edu
First published on 10th June 2021
Abstract
Bimetallic catalysts provide opportunities to overcome scaling laws governing selectivity of CO2 reduction (CO2R). Cu/Au nanoparticles show promise for CO2R, but Au surface segregation on particles with sizes ≥7 nm prevent investigation of surface atom ensembles. Here we employ ultrasmall (2 nm) Cu/Au nanoparticles as catalysts for CO2R. The high surface to volume ratio of ultrasmall particles inhibits formation of a Au shell, enabling the study of ensemble effects in Cu/Au nanoparticles with controllable composition and uniform size and shape. Electrokinetics show a nonmonotonic dependence of C1 selectivity between CO and HCOOH, with the 3Au:1Cu composition showing the highest HCOOH selectivity. Density functional theory identifies Cu2/Au(211) ensembles as unique in their ability to synthesize HCOOH by stabilizing CHOO* while preventing H2 evolution, making C1 product selectivity a sensitive function of Cu/Au surface ensemble distribution, consistent with experimental findings. These results yield important insights into C1 branching pathways and demonstrate how ultrasmall nanoparticles can circumvent traditional scaling laws to improve the selectivity of CO2R.
1 Introduction
Bimetallic alloys have shown promise towards reducing onset potentials and modifying selectivity to enhance catalyst performance for electrochemical CO2 reduction (CO2R).1,2 However, many interesting questions such as size and composition dependence in bimetallic alloys remain to be fully explored. Cu is the only pure metal that is capable of producing multiple products from CO2 under a wide range of potentials. In particular, Cu electrocatalysts have been widely studied for C–C bond formation in the production of higher order C products. From both an experimental and theoretical perspective, much work has been performed to understand the branching point between formation of C1 products and higher order C2+ products, which is believed to occur via the formation of CO dimers on the Cu surface.2–5By comparison, relatively little theoretical work has focused on understanding the branching point between formation of C1 products CO and HCOOH. This is a particularly important question since HCOOH (formic acid or formate depending on pH) is one of the few commercially viable products for CO2 utilization, if it can be produced at high selectivity.2 A number of catalysts, including CuSn3,6 SnS/Au,7 CoOx,8 oxide derived Pb,9 Bi,10 BiOx/C,11 CuBi,12 and Pd13–17 are known to selectively produce HCOOH from CO2. Recently mixed Cu/Au catalysts have been shown to also produce HCOOH with high faradaic selectivity even at the relatively low overpotential of −0.6 V vs. RHE.18 It was hypothesized that in this system Au serves as a promoter to activate Cu, which serves as the active site for selective HCOOH production. These results highlight the potential of bimetallic systems to break fundamental scaling laws, but also illustrate the need for improved understanding of C1 branching pathways. To facilitate this effort, synthesis of Cu/Au nanoparticle catalysts is desired that allows for controlled bimetallic composition with uniform size and shape.
In this work we utilize recent advances enabling the synthesis of ultrasmall (2 nm) Cu/Au bimetallic nanoparticles to investigate the effect of composition on CO2R.19 Particles used in this study represent a random alloy with no intermetallic phase or core–shell structure such that the ensemble distribution of Cu/Au surface sites is dictated primarily by particle composition. We find that at potentials ranging from −0.7 to −1.1 V vs. RHE, HCOOH production is observed with faradaic selectivity up to 16%. While pure Au particles produce only CO and H2, addition of Cu results in production of HCOOH. Kinetic measurements show that HCOOH selectivity is a nonmonotonic function of composition and is favored for particles having small amounts of Cu in Au (Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu = 3:1). Density functional theory (DFT) studies are performed to understand the branching point between CO and HCOOH. Findings show that selectivity can be described as a competition between the surface stabilization energies for CHOO*, COOH* and H* (* represents a surface adsorbed species). Cu surface sites are required to stabilize the CHOO* intermediate, which leads to production of HCOOH. However, at high Cu content the competition between CHOO* and hydrogen evolution becomes unfavorable leading to selectivity loss to H2 evolution. Specifically, it is shown that Cu3 three-fold hollow sites are required to stabilize H*, while CHOO* is effectively stabilized on Cu2 bridging sites, making selectivity a sensitive function of the ensemble distribution of Cu and Au surface sites, consistent with experimental findings.
:Cu = 3:1). Density functional theory (DFT) studies are performed to understand the branching point between CO and HCOOH. Findings show that selectivity can be described as a competition between the surface stabilization energies for CHOO*, COOH* and H* (* represents a surface adsorbed species). Cu surface sites are required to stabilize the CHOO* intermediate, which leads to production of HCOOH. However, at high Cu content the competition between CHOO* and hydrogen evolution becomes unfavorable leading to selectivity loss to H2 evolution. Specifically, it is shown that Cu3 three-fold hollow sites are required to stabilize H*, while CHOO* is effectively stabilized on Cu2 bridging sites, making selectivity a sensitive function of the ensemble distribution of Cu and Au surface sites, consistent with experimental findings.
2 Results and discussion
Kim et al. recently reported synthesis of Cu/Au bimetallic particles for CO2R, and it was shown that intermetallic Cu/Au phases increase both the turnover frequency and mass activity to CO.20 These particles had an average size of ≥7 nm and displayed a pure Au shell approximately 3 atomic layers thick, which encapsulates the intermetallic core.DFT results showed that this Au shell is consistent with the lower surface energy of Au compared to Cu leading to Au surface segregation and that higher turnover frequency to CO is the result of compressive strain on the Au shell induced by the intermetallic core.20 Although these particles are highly active for CO production, very low yields of HCOOH were observed, on the order of only a few percent faradaic selectivity, which is consistent with the absence of any Cu atoms at the catalyst surface.
However, below a certain size, bimetallic nanoparticles can no longer sustain a core–shell structure. This is the natural consequence of increasing the surface-to-bulk ratio to the point where the particle core becomes small relative to the surface area. Fig. 1a shows the calculated surface to bulk ratio for a spherical nanoparticle as a function of particle diameter. Below 3 nm the number of surface atoms becomes greater than those in the bulk. Fig. 1a also shows the theoretical threshold for spherical particles of varying Cu:Au compositions (3:1, 1:1, and 1:3) where an Au shell can no longer block Cu atoms from the particle surface. At this size the bulk becomes small relative to the surface such that it can no longer contain all the Cu atoms. This defines a maximum particle size where Cu surface sites will necessarily play an active role in CO2 reduction through the formation of Cu/Au surface ensembles. As shown, this threshold occurs at ≈6, 3, and 2 nm for Cu:Au ratios of 3:1, 1:1, and 1:3, respectively. Consequently, synthesis of monodisperse Cu/Au nanoparticles with tunable composition having sizes at or below ≈2 nm can enable systematic investigation of surface ensemble effects on reaction kinetics, which have been impossible to observe for Cu/Au bimetallic nanoparticles of larger sizes. Particularly these small sizes are required to observe the synergistic effects of Cu and Au surface sites on CO2R selectivity.
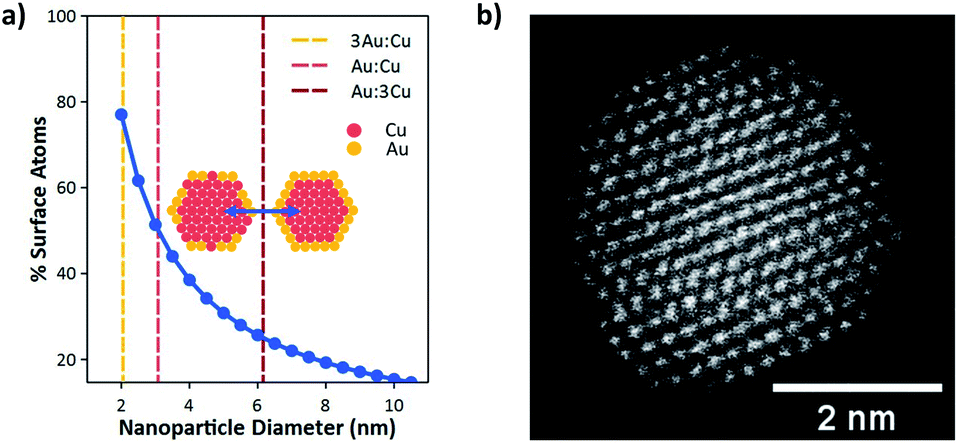 | ||
| Fig. 1 (a) Percent of surface atoms as a function of particle diameter. Vertical lines indicate the minimum diameter required to support a core–shell structure for each Cu/Au composition. (b) High-angle annular dark-field scanning transmission electron micrograph (HAADF-STEM) of a 1Au:1Cu nanoparticle. | ||
To enable these investigations, Hofmann et al.19 recently developed a method to synthesize ultrasmall Cu/Au nanoparticles with tunable composition. Here Au compositions of 0% (pure Cu), 25% (3Au:1Cu), 50% (1Au:1Cu), 75% (1Au:3Cu), and 100% (pure Au) were used to systematically study CO2R. Size histograms based on TEM images of the Cu/Au bimetallic nanoparticles have been reported previously and show that each composition has an average diameter of 2 nm.19Fig. 1b shows a high-angle annular dark-field scanning transmission electron micrograph (HAADF-STEM) of a 1Au:1Cu nanoparticle. The uniform intensity of atomic columns across the nanoparticle shows that the particle is a random alloy with no sign of intermetallic ordering or core–shell structure. Consequently, the surface composition will be a random configuration of different Au:Cu ensembles. The distribution of these ensembles will be composition dependent, assuming no preference for any cluster type, and DFT calculations show that no ensemble preference is expected (see Section S8 in the ESI†).
As synthesized, these ultrasmall nanoparticles contain a surface layer of dodecanethiol required to prevent agglomeration. As shown in Fig. S3 and S4,† these capping agents stabilize the nanoparticles, even under electrochemical reaction conditions, and there is no measurable change in the particle size distribution before and after CO2R reaction. However, as shown in Fig. S4c,† removal of the ligand does increase the particle size by about 30%. We have separately conducted a detailed study to better understand the role of this organic surface ligand on electrochemical activity of the nanoparticle catalyst.21 Although not the focus of the present study, we briefly summarize those results here. Surprisingly, we find that the dodecanethiol surface ligand acts as a permeable membrane to CO2, which has no detrimental effect on the catalytic activity. One way to understand this is that CO2 is more soluble in hydrophobic dodecane compared to water. Additionally, the absolute coverage of the dodecanethiol surface ligand in the as-synthesized particles is only a fraction of the total surface metal atoms due to steric effects between these long chain ligands. Consequently, CO2 can readily diffuse to the particle surface and react with essentially no observable transport limitations, while the presence of the surface ligand ensures the size and structure of the as-synthesized particle are carefully preserved. We find that any attempt at removal of the surface ligand results in particle agglomeration, and the ultrasmall size dispersion of the particles is quickly lost. For these reasons, we consider that the ligand plays no active role in the surface chemistry except to facilitate diffusion of reactants to and from catalytic active sites while preserving the size and structure of the catalyst. Post-reaction TEM and XPS at the S 2p edge support these conclusions (see Sections S3 and S5 of the ESI†). Additional details related to stability and deactivation of these ultrasmall particles in the presence and absence of a surface ligand can be found in that study.21
To demonstrate the unique properties of these ultrasmall bimetallic nanoparticles, CO2 reduction (CO2R) experiments were conducted from −0.7 to −1.1 V vs. RHE in 0.1 M NaHCO3 electrolyte (pH = 6.8). Post reaction transmission electron micrographs (TEM) show that the particles are stable and do not change size during reaction (see Section S3 in the ESI†). Fig. 2 shows the faradaic efficiency to CO, H2, and HCOOH as a function of composition under four potentials. We also detect trace amounts of CH4 and C2H4, but we neglect them in our analysis as these products have cumulative faradaic efficiencies less than 0.5%. We show that Au produces CO and H2, but no HCOOH while Cu produces mostly H2 with some CO and HCOOH. This result is different from larger Cu nanoparticles and polycrystalline Cu, which are selective to C2+ hydrocarbons.22,23 However, this is consistent with previous reports, which show that small Cu nanoparticles produce only trace amounts of C2H4 and CH4.24 The bimetallic compositions show a synergistic behavior towards HCOOH production. HCOOH efficiency is non-monotonic, with 3Au:1Cu showing the highest selectivity of 16% at −0.7 V vs. RHE. This is significantly higher than <5% reported previously for larger Cu/Au bimetallic nanoparticles under the same conditions.25 The faradaic selectivity to HCOOH decreases as the potential is increased, suggesting that these bimetallic nanoparticles open a pathway to HCOOH production that is uniquely selective at low overpotentials.26 Additionally, upon removal of the ligand and subsequent growth of the particle, the HCOOH selectivity for the 3Au:1Cu particles falls to zero. Re-functionalization of the enlarged particles with dodecanethiol does not restore selectivity, indicating that the thiol plays no active role in the reaction kinetics except to stabilize the ultrasmall particle size. Mass loadings, deactivation profiles, and turnover frequencies based on measured electrochemical active surface areas for each catalyst are provided in Sections S1, S2 and S4 of the ESI.†
 | ||
| Fig. 2 Faradaic efficiency of different product as a function of sample composition under (a) −0.7 V (b) −0.9 V (c) −1.0 V (d) −1.1 V vs. RHE. For clarity, the faradaic efficiency to HCOOH is shown on a different scale (right axis) than CO and H2 (left axis). | ||
To further elucidate the reaction mechanism of CO2R on these ultrasmall nanoparticles, we performed DFT calculations to evaluate the free energy of elementary proton–electron transfer steps (computational details, including solvation effects, can be found in Section S7 of the ESI†). Fig. 3 illustrates the reaction pathways for CO2 that affect HCOOH selectivity. The initial hydrogenation of CO2 can proceed via CHOO* or COOH* surface intermediates. Although both surface intermediates can hypothetically lead to HCOOH, the COOH* pathway competes with CO formation, while the CHOO* pathway selectively produces only HCOOH. DFT evaluation of the potential dependent barrier for the proton–electron transfer step on several metal surfaces has shown that C–H bond formation proceeds through a surface hydrogenation (SH) step involving transfer of a surface-bound H atom while O–H bond formation proceeds through a proton shuffling (PS) step involving transfer of a proton from solution.27–30 Based on these previous studies, we expect that the kinetics for a PS step will be fast compared to the kinetics for an SH step, when the free energy of reaction for elementary steps are similar. We note that the free energy of H* formation can affect the selectivity between PS and SH steps since the SH step needed to form CHOO* proceeds via a surface H atom transfer requiring the formation of H*.27–30 The hydrogen evolution reaction (HER) also competes directly with CO2R and will be affected by the free energy of H* formation.
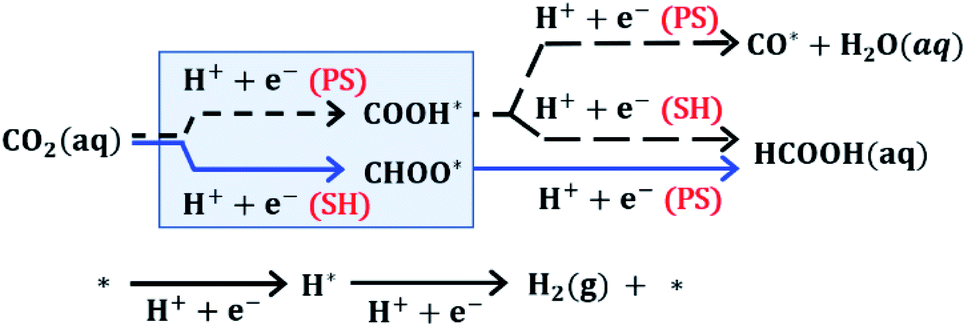 | ||
| Fig. 3 The proton–electron transfer elementary steps for CO2 electrochemical reduction. The proton–electron transfer step proceeds via water assisted proton shuffling (PS) for O–H bond formation or through water solvated surface hydrogenation (SH) for C–H bond formation. The hydrogen evolution reaction (HER) competes with CO2 electroreduction. | ||
We use DFT to evaluate the potential-dependent reaction free energy (ΔG) of the elementary steps in Fig. 3. For these calculations, we consider a (211) surface as representative of the low coordination sites found on nanoparticles as has been done previously.20,31 In this approach, we assume that quantum size effects do not contribute substantially to nanoparticle surface chemistry behavior for sizes above 1 nm as reported in several surface science studies.32–36 We find that similar trends to those reported here are also observed for the (111), (110), and (100) surfaces indicating that these effects are not strongly facet dependent (see ESI Section S11†). In the discussion of DFT results in Fig. 4 we focus on a Au(211) slab with just the surface atoms doped with contiguous Cu atoms (see Fig. S12 in the ESI† for figures of the slab with dopants). However, we have also examined models where the subsurface layers consist of Au/Cu alloys and/or Cu layers. In each system we have the top layer as Au with the introduction of contiguous Cu dopants. A comparison of the results over these models can be found in Section S9 in the ESI,† but the overall conclusions are quite similar to the results presented in Fig. 4.
 | ||
| Fig. 4 (a) ΔG of CO2R reaction pathways illustrated in Fig. 3 on Au(211) (left) and Cu(211) (right) with the relaxed adsorbates images. (b) ΔG for the reaction intermediates (COOH*, CHOO* and H*) with and without Cu surface dopants on Au(211). 0 Cu represents a pristine Au(211) surface. (c) The effect of compressive lattice strain on ΔG for the reaction intermediates (COOH*, CHOO* and H*) on a Au(211) slab. (d) The DOS for Au(211), Cu(211), and Cu dopants on Au(211). (e) The binding geometry of CHOO* (above) and COOH* (below) as a function of Cu dopants on Au(211). | ||
We initially compare Au(211) and Cu(211), which can then be used to understand the changes in selectivity for Cu/Au bimetallic nanoparticle surfaces. Fig. 4a presents the potential dependent reaction free energies of COOH* and CHOO* pathways on Au(211) and Cu(211) surfaces. Focusing on Au(211), both the COOH* and CHOO* species show nearly the same free energy of formation (0.84 eV). The reason for high CO selectivity on pure Au despite similar ΔG is that, as explained above, the PS mechanism involved in CO formation provides smaller barriers for steps with similar ΔG as shown recently by Sargent and co-workers with DFT calculations on Ag(111).29 Furthermore, the unfavorable positive free energy of formation for H* on Au surfaces will hinder HER and promote the PS over the SH mechanism (see Table S4 in the ESI†). Because the production of HCOOH requires SH for either the initial formation of CHOO* or for the subsequent conversion of COOH* to HCOOH, Au is expected to have very low selectivity to HCOOH since Au surfaces do not efficiently promote the SH steps. Accordingly, DFT calculations provide an appropriate framework for understanding the high selectivity for CO production on pure Au.
In contrast to Au, the Cu(211) surface favors CHOO* formation strongly (by nearly 0.66 eV) but also shows a favorable free energy of formation for H*. The favorable free energy of formation of H*, correlates well to the dramatic increase of HER selectivity on Cu versus Au shown in Fig. 2 and reported elsewhere. Interestingly, CHOO* formation on Cu(211) is −0.23 eV at 0 V vs. RHE, which would suggest that this intermediate forms at extremely low potentials on Cu catalysts. However, CHOO* is strongly bound and requires an uphill step to produce HCOOH (0.6 eV uphill) so actual HCOOH production in the solution would not occur until more negative potentials as observed in the experiments. At these negative potentials, the pathway to COOH* would also be opened and the selectivity between the two would be influenced by the competition between SH and PH mechanism. On Cu catalysts H* is more favorable than on Au, so we expect to see more HCOOH production as a result of the SH mechanism. This is consistent with experiments, which show both CO and HCOOH production on Cu at −0.7 V vs. RHE. Experimentally, we observe that at increasingly negative potentials the selectivity to HCOOH decreases to zero while CO production persists. Similar observations have been reported by Hori and co-workers on polycrystalline Cu,37 and this is consistent with the prediction that when all pathways are energetically accessible, PS is kinetically favored over SH.
We can use the insights gained from pure Au and Cu catalysts to understand the behavior of bimetallic nanoparticles. Focusing here on Cu doping in Au(211), Fig. 4b plots the free energy of formation of the three key species (COOH*, CHOO*, H*) for the Au(211) surface with varying number of dopants along with a pure Cu(211) surface. Examining Fig. 4b we see that the addition of Cu atoms on the Au(211) surface slightly lowers the ΔG for COOH* but has a far more dramatic effect on CHOO*. When a Cu2 ensemble is introduced, the ΔG for CHOO* becomes negative (−0.08 eV), while the ΔG for H* formation remains positive as found on Au(211) surfaces. This suggests that at low overpotential Cu2 sites are predicted to be selective toward HCOOH production over HER. In contrast, the Cu3 ensemble begins to resemble the energetics of a Cu(211) surface as ΔG for H* formation becomes negative although CHOO* is still slightly favored.
Bimetallic alloy particles can effect the activity and selectivity via three possible mechanisms: (1) the ligand effect, where alloying changes the electronic structure of the catalyst, (2) the lattice effect, where lattice strain changes the binding energy of intermediates in bimetallic systems, and (3) the ensemble effect, where mixed Cu/Au surface ensembles change surface energies of adsorbates.20,38,39 To consider which of these mechanisms are most strongly at play in the present case, Fig. 4c shows the effect of compressive lattice strain on the Au(211) surface for the three key species (COOH*, CHOO*, H*). While there are changes in ΔG with strain, these are not sufficient to affect any dramatic changes in selectivity of the CHOO* versus COOH* pathways or impact HER activity. Fig. 4d shows the electronic density of states (DOS) for the surface atoms of Au(211), Cu(211), and Cu dopants on Au(211). The electronic structure of the ensemble states for 1, 2, 3 Cu dopants are virtually identical, and have valence band maxima similar to that of copper except more narrow. The narrow energy distribution suggests that the valence states of the Cu atoms in the ensemble do not hybridize extensively with neighboring Au atoms. This narrow energy distribution does not require a single Cu site but persists even for aggregates of two or three Cu atoms in Au. Because the electronic structure does not change from 1, 2, to 3 Cu dopants, and in no case does the energy of the valence band maximum shift relative to pure Cu, the ligand effect is not sufficient to explain the changes in selectivity predicted in Fig. 4b.
Rather, the dominant effect on selectivity arises due to differences in binding induced by Cu/Au ensembles as illustrated in Fig. 4e. CHOO* has a bidentate adsorption mode on the metal surface through two M–O bonds (M = Cu or Au). In contrast, COOH* bonds through a M–C bond and a M–O bond (see Fig. 4e). The Cu–O bond is stronger than the Au–O bond and DFT calculations show that the Cu–O bond is similar in strength on Cu doped Au(211) as on pure Cu(211). This is consistent with the DOS features shown in Fig. 4d, which illustrate that the conduction band for Cu/Au(211) is similar to pure Cu and that Cu does not significantly hybridize with the surrounding Au atoms. For the Cu1 ensemble both CHOO* and COOH* benefit from the formation of a Cu–O bond, and the ΔG goes down substantially for both species (see Fig. 4b). However, as the number of Cu atoms increases from 1 to 2, the CHOO* can make two strong Cu–O bonds that leads to a negative ΔG of formation and dramatically favors CHOO* over COOH*, which has only one Cu–O bond. This leads to the sharp difference in COOH* and CHOO* when transitioning from a Cu1 to a Cu2 ensemble. Furthermore, while only two Cu atoms are required to stabilize the bidentate coordination of CHOO* to the surface, H* is best stabilized in 3-fold hollow sites of Cu. Consequently, the Cu2 ensemble is ideal for the production of HCOOH since formation of CHOO* is energetically favored over COOH*, while HER is still suppressed. This result predicts the formation of HCOOH at lower onset potentials with higher selectivity. As a higher potential is applied to the system, the COOH* pathway will become accessible, and CO will be favored over HCOOH due to the kinetic competition between PS and SH transfer mechanisms. Additionally, HER becomes increasingly favorable at more negative potentials. As such, the selectivity to HCOOH is predicted to decrease at increasing overpotential as observed experimentally.
To further support these findings we estimate the composition dependent surface ensemble distributions. As indicated by DFT calculations, there is minimal interaction between Cu and Au (Section S8). As such we consider the distribution of Cu and Au to be distributed in random ensembles, with each ensemble weighted by the nanoparticle composition. Making these assumptions, we calculate the probability distribution of Cu1, Cu2, and Cu3 ensembles as a function of composition. A detailed description of the calculation is given in the Section S6.† The Cu1 ensembles represent the majority in the probability distribution function at low Cu compositions, while Cu3 ensembles dominate at Cu compositions greater than approximately 0.4. However, there is a narrow distribution range where Cu2 ensembles represent the majority in the probability distribution function, and the calculated composition dependence is in excellent agreement with the average FE to HCOOH (Fig. 5). These results provide compelling evidence that the surface ensemble distribution is the dominate factor in determining catalytic selectivity.
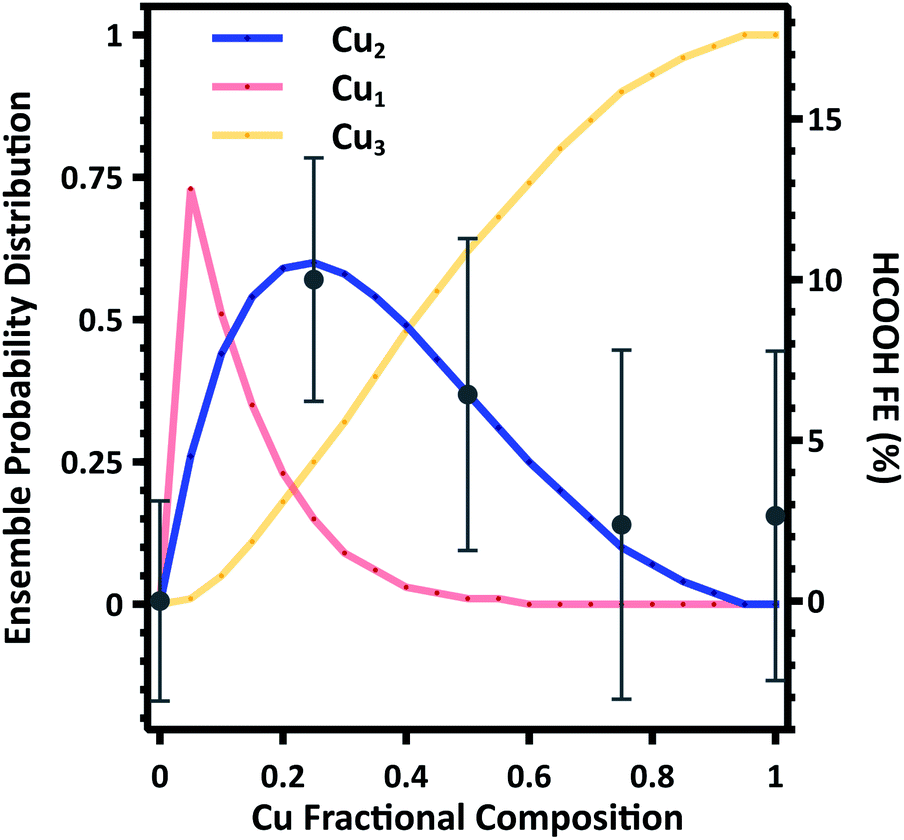 | ||
| Fig. 5 Calculated ensemble probability distribution as a function of composition. The average HCOOH Faradaic Efficiency (FE) at all potentials is plotted for comparison. Error bars represent the sum of squares error for each composition at each potential. | ||
As the number of Cu dopants is increased, the behavior begins to resemble pure Cu(211). We have also examined the other extreme where Au atoms are the minority and appear as dopants on Cu(211). While Au ensembles on Cu(211) lead to minor perturbations in the free energy calculations, each of these surfaces favor CHOO* formation over COOH*. Additionally, the binding of CHOO*, COOH*, and H* all favor adsorption on Cu sites rather than Au dopants. Thus on Cu-rich surfaces we would expect behavior similar to pure Cu. This prediction is confirmed by measurements on 1Au:3Cu samples, which display similar CO2R kinetics to pure Cu samples (see Fig. 2).
3 Conclusion
These DFT calculations indicate that certain Cu ensembles in Au catalysts provide a pathway for selective production of HCOOH at low overpotential. Consistent with DFT calculations, we find that selectivity is highest for 3Au:1Cu nanoparticles reaching 16% at −0.7 V vs. RHE, while at more negative potentials or increasing Cu content selectivity decreases due to competition with HER and CO production. These findings support the theoretical prediction that stabilization of CHOO* on Cu atom clusters provides a low overpotential window for production of HCOOH from CO2R. Experimental observations are consistent with theoretical DFT predictions and with the predicated composition dependent ensemble probability distribution in these random alloys. In fact, improved control of catalyst surface structure has potential to produce specific active sites such as Cu2 in Au, which we predict to display significantly higher selectivity compared to the ensemble average. These results provide insights into important C1 branching pathways on Cu, Au, and Cu/Au bimetallic surfaces and affirm that ultrasmall nanoparticles provide the opportunity to introduce unique metal atom ensembles that can circumvent traditional scaling laws and have a significant effect on the selectivity of CO2R.Data availability
We include all extra related data in the supporting information.Author contributions
Hongyu Shang performed kinetic and characterization measurements and was involved in writing the manuscript. Dongjoon Kim performed DFT calculations and was involved in writing the manuscript. Spencer K. Wallentine performed kinetic and characterization measurements and was involved in writing the manuscript. Minkyu Kim performed DFT calculations and was involved in writing the manuscript. Daniel M. Hofmann synthesized the nanoparticles. Runiya Dasgupta assisted with kinetic measurements. Catherine J. Murphy planned the experiments and supervised nanoparticle synthesis. Aravind Asthagiri supervised the DFT work and was involved in writing the manuscript. L. Robert Baker planned the experiments, supervised kinetic and characterization measurements and was involved in writing the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Catalyst characterization, electrokinetic measurements, and DFT calculations were supported by the National Science Foundation under NSF award number 1665280. Nanoparticle synthesis was supported by the National Science Foundation under NSF award number 1503408. HAADF-STEM and TEM imaging was performed at the Ohio State University Center for Electron Microscopy and Analysis (CEMAS). We acknowledge the Ohio Supercomputing Center for providing computational resources. Minkyu Kim received support from the National Research Foundation (NRF) of the Republic of Korea under the framework of the Priority Research Centers Program funded by the Ministry of Education (2014R1A6A1031189).Notes and references
- J. He, N. J. Johnson, A. Huang and C. P. Berlinguette, ChemSusChem, 2018, 11, 48–57 Search PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. Stephens, K. Chan and C. Hahn, et al. , Chem. Rev., 2019, 119, 7610–7672 Search PubMed.
- X. Han and V. S. Thoi, ACS Appl. Mater. Interfaces, 2020, 12, 45929–45935 Search PubMed.
- E. Pérez-Gallent, M. C. Figueiredo, F. Calle-Vallejo and M. T. Koper, Angew. Chem., 2017, 129, 3675–3678 Search PubMed.
- J. D. Goodpaster, A. T. Bell and M. Head-Gordon, J. Phys. Chem. Lett., 2016, 7, 1471–1477 Search PubMed.
- X. Zheng, Y. Ji, J. Tang, J. Wang, B. Liu, H.-G. Steinrück, K. Lim, Y. Li, M. F. Toney and K. Chan, et al. , Nat. Catal., 2019, 2, 55–61 Search PubMed.
- X. Zheng, P. De Luna, F. P. G. de Arquer, B. Zhang, N. Becknell, M. B. Ross, Y. Li, M. N. Banis, Y. Li and M. Liu, et al. , Joule, 2017, 1, 794–805 Search PubMed.
- S. Gao, Y. Lin, X. Jiao, Y. Sun, Q. Luo, W. Zhang, D. Li, J. Yang and Y. Xie, Nature, 2016, 529, 68–71 Search PubMed.
- C. H. Lee and M. W. Kanan, ACS Catal., 2015, 5, 465–469 Search PubMed.
- N. Han, Y. Wang, H. Yang, J. Deng, J. Wu, Y. Li and Y. Li, Nat. Commun., 2018, 9, 1–8 Search PubMed.
- C. W. Lee, J. S. Hong, K. D. Yang, K. Jin, J. H. Lee, H.-Y. Ahn, H. Seo, N.-E. Sung and K. T. Nam, ACS Catal., 2018, 8, 931–937 Search PubMed.
- A. Klinkova, P. De Luna, C.-T. Dinh, O. Voznyy, E. M. Larin, E. Kumacheva and E. H. Sargent, ACS Catal., 2016, 6, 8115–8120 Search PubMed.
- M. Rahaman, A. Dutta and P. Broekmann, ChemSusChem, 2017, 10, 1733–1741 Search PubMed.
- X. Bai, W. Chen, C. Zhao, S. Li, Y. Song, R. Ge, W. Wei and Y. Sun, Angew. Chem., 2017, 129, 12387–12391 Search PubMed.
- D. Gao, H. Zhou, F. Cai, D. Wang, Y. Hu, B. Jiang, W.-B. Cai, X. Chen, R. Si and F. Yang, et al. , Nano Res., 2017, 10, 2181–2191 Search PubMed.
- D. Gao, H. Zhou, F. Cai, J. Wang, G. Wang and X. Bao, ACS Catal., 2018, 8, 1510–1519 Search PubMed.
- X. Min and M. W. Kanan, J. Am. Chem. Soc., 2015, 137, 4701–4708 Search PubMed.
- Z. Tao, Z. Wu, X. Yuan, Y. Wu and H. Wang, ACS Catal., 2019, 9, 10894–10898 Search PubMed.
- D. M. Hofmann, D. H. Fairbrother, R. J. Hamers and C. J. Murphy, ACS Appl. Nano Mater., 2019, 2, 3989–3998 Search PubMed.
- D. Kim, C. Xie, N. Becknell, Y. Yu, M. Karamad, K. Chan, E. J. Crumlin, J. K. Nørskov and P. Yang, J. Am. Chem. Soc., 2017, 139, 8329–8336 Search PubMed.
- H. Shang, S. K. Wallentine, D. M. Hofmann, Q. Zhu, C. J. Murphy and L. R. Baker, Chem. Sci., 2020, 11, 12298–12306 Search PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 Search PubMed.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 Search PubMed.
- R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 Search PubMed.
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- H. Mistry, R. Reske, P. Strasser and B. R. Cuenya, Catal. Today, 2017, 288, 30–36 Search PubMed.
- X. Nie, M. R. Esopi, M. J. Janik and A. Asthagiri, Angew. Chem., Int. Ed., 2013, 52, 2611 Search PubMed.
- X. Nie, W. Luo, M. J. Janik and A. Asthagiri, J. Catal., 2014, 312, 108–122 Search PubMed.
- A. Seifitokaldani, C. M. Gabardo, T. Burdyny, C.-T. Dinh, J. P. Edwards, M. G. Kibria, O. S. Bushuyev, S. O. Kelley, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2018, 140, 3833–3837 Search PubMed.
- J. Hussain, H. Jónsson and E. Skúlason, ACS Catal., 2018, 8, 5240–5249 Search PubMed.
- X. Liu, J. Xiao, H. Peng, X. Hong, K. Chan and J. K. Nørskov, Nat. Commun., 2017, 8, 1–7 Search PubMed.
- C. Dong, C. Lian, S. Hu, Z. Deng, J. Gong, M. Li, H. Liu, M. Xing and J. Zhang, Nat. Commun., 2018, 9, 1–11 Search PubMed.
- G. Tritsaris, J. Greeley, J. Rossmeisl and J. K. Nørskov, Catal. Lett., 2011, 141, 909–913 Search PubMed.
- T. Jiang, D. Mowbray, S. Dobrin, H. Falsig, B. Hvolbæk, T. Bligaard and J. K. Nørskov, J. Phys. Chem. C, 2009, 113, 10548–10553 Search PubMed.
- I. V. Yudanov, A. Genest, S. Schauermann, H.-J. Freund and N. Rosch, Nano Lett., 2012, 12, 2134–2139 Search PubMed.
- S. Back, M. S. Yeom and Y. Jung, ACS Catal., 2015, 5, 5089–5096 Search PubMed.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 Search PubMed.
- H. Li, K. Shin and G. Henkelman, J. Chem. Phys., 2018, 149, 174705 Search PubMed.
- P. Liu and J. K. Nørskov, Phys. Chem. Chem. Phys., 2001, 3, 3814–3818 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: (1) Experimental details. (2) Catalyst cyclic voltammograms and deactivation profiles. (3) Pre-reaction and post-reaction TEM. (4) Catalyst electrochemical surface areas and turn over frequencies. (5) Pre-reaction and post-reaction XPS of the Au and 3Au:1Cu nanoparticles. (6) Ensemble distribution calculation. (7) Computational details. (8) Cu–Cu surface dopant interactions, and (9) Alternative bimetallic slab models, and (10) Configurations of adsorbates on Au(211) slab. (11) DFT calculations on low-Miller index facets of Au. See DOI: 10.1039/d1sc02602j |
| ‡ These authors contributed equally to this work. |
| § Present address: Yuengnam University, School of Chemical Engineering, Gyeongsan, Republic of Korea. |
| This journal is © The Royal Society of Chemistry 2021 |