
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed asymmetric allylic alkylation (AAA) with alkyl sulfones as nucleophiles†
Barry M.
Trost
 *b,
Zhiwei
Jiao
*a and
Hadi
Gholami
b
*b,
Zhiwei
Jiao
*a and
Hadi
Gholami
b
aSchool of Chemistry, Sun Yat-Sen University, Guangzhou 510275, China. E-mail: jiaozhw@mail.sysu.edu.cn
bDepartmentof Chemistry, Stanford University, Stanford, CA 94305-5080, USA. E-mail: bmtrost@stanford.edu
First published on 2nd July 2021
Abstract
An efficient palladium-catalyzed AAA reaction with a simple α-sulfonyl carbon anion as nucleophiles is presented for the first time. Allyl fluorides are used as superior precursors for the generation of π-allyl complexes that upon ionization liberate fluoride anions for activation of silylated nucleophiles. With the unique bidentate diamidophosphite ligand ligated palladium as catalyst, the in situ generated α-sulfonyl carbon anion was quickly captured by the allylic intermediates, affording a series of chiral homo-allylic sulfones with high efficiency and selectivity. This work provides a mild in situ desilylation strategy to reveal nucleophilic carbon centers that could be used to overcome the pKa limitation of “hard” nucleophiles in enantioselective transformations.
Introduction
Transition-metal-catalyzed asymmetric allylic alkylation (AAA) is a powerful tool for the enantioselective construction of stereogenic centers, enabling the elaboration of complex organic molecules and synthesis of pharmaceutical intermediates and bioactive natural products.1 A variety of “soft” carbon nucleophiles (Nu-H with pKa <25) and heteroatoms have been used in AAA reactions, generating the corresponding stereogenic centers with good to excellent selectivity.2 However, transition-metal-catalyzed allylic substitution reactions with “hard” nucleophiles (Nu-H with pKa > 25) is mainly limited to non-enantioselective transformations.3 During the past decades, our group and many other research groups have made tremendous efforts to engage “unstable” nucleophiles, such as ketones, acyclic amides and nitrogen-contained heteroarenes, etc. in the palladium-catalyzed AAA reactions.4,5 Despite these achievements, a great number of “hard” carbon nucleophiles are still not compatible in palladium-catalyzed AAA reactions. One important example of such unexplored carbon based nucleophiles for AAA reactions is α-sulfonyl carbon anion (pKa = 29 for (methylsulfonyl)benzene, Fig. 1a).6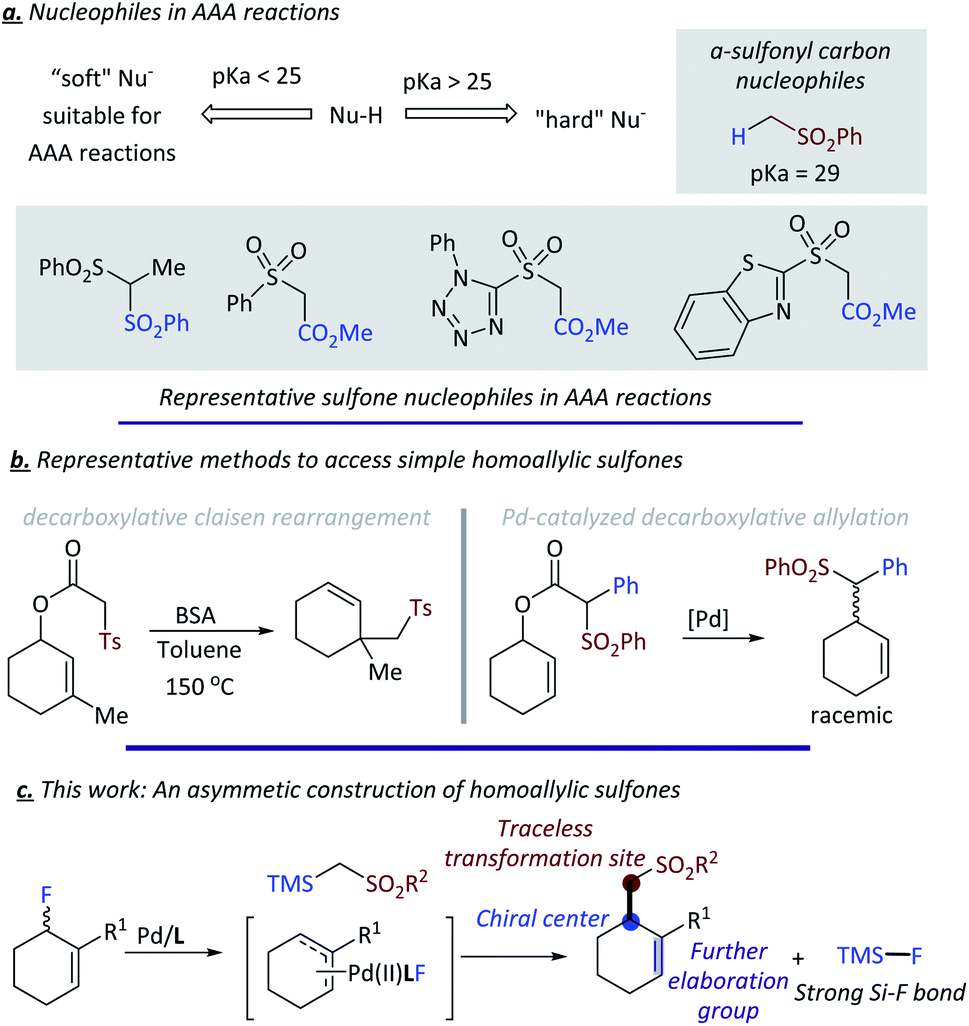 | ||
| Fig. 1 Representative methods for homo-allylic sulfones. | ||
Sulfone represents an important moiety that is widely spread in many biologically active compounds and pharmaceutical intermediates.7 Moreover; sulfones can be converted into a wide range of other groups at the α position via traceless transformations.8 Efficient utilization of α-sulfonyl carbon anion in the palladium-catalyzed AAA reaction would lead to chiral homo-allylic sulfones, which would provide promising opportunities for the exploration of chiral sulfone containing compounds (Fig. 1c). Previous exploration to build chiral homo-allylic sulfones via AAA reactions were limited to special sulfone reagents, an additional electron-withdrawing group was usually needed to enhance the acidity of α proton (Fig. 1a).9 For example, the use of the ester group allowed removal after the reaction of Krapcho demethoxycarbonylation;10 however, besides the moderate yield (60–80%), the harsh reaction conditions for demethoxycarbonylation would rule out many useful functional groups. Therefore, exploration of efficient method to realize the direct asymmetric allylic reaction of simple alkyl sulfones under mild reaction conditions is highly in needed.
To date, two elegant non-enantioselective reports on direct allylic alkylation of sulfones employed decarboxylation as the driving force. A thermodynamic decarboxylative Claisen-rearrangement reaction reported by Craig et al.11 under harsh reaction conditions (150 °C) would restrict the diversity of functional groups (Fig. 1b). Another example is a palladium-catalyzed intramolecular decarboxylative allylation of sulfonyl acetic esters with rac-BINAP as the ligated ligand (Fig. 1b).12 Notably, the conditions developed by Tunge et al. still needed high reaction temperature or microwave conditions to get acceptable results. In most cases, the key nucleophiles were stabilized by both sulfonyl and phenyl or heteroatoms (pKa = 23.4 for (benzylsulfonyl)benzene).6 Herein, we report our endeavor and initial results on the palladium-catalyzed AAA reaction with simple α-sulfonyl carbon anion as the nucleophile (pKa > 25, Fig. 1c).
Optimization of conditions
Recently, our and other groups have found that phosphoramidite and diamidophosphite ligands could facilitate transition-metal catalyzed transformations via in situ deprotonation of pro-nucleophiles.4l,13 Notably, the Sawamura group found that the chiral phosphoramidite ligated palladium catalyst can facilitate the asymmetric allylic alkylation at the “hard”α position of 2-alkyl pyridines without additives.4l The above achievements inspired us to utilize these unique ligands and exploit α-sulfonyl nucleophiles in AAA reactions. We started our research with commercial compound 1a-1 as a donor, and tert-butyl cyclohex-2-en-1-yl carbonate 2a-1 as the model counterpart (Fig. 2a). Ligand L1 (Fig. 2c) which has proven to be a suitable ligand for palladium-catalyzed transformations involving a deprotonation mechanism was selected as the ligated ligand to test different conditions.14,15 We soon realized that in the absence of additional base, the reaction gave no desired results. The tert-butoxide presumably generated in situ from Pd-mediated ionization of 2a-1 is incompetent to efficiently deprotonate the sulfone 1a-1 or perhaps the carboxylate leaving group never lost CO2 to form tert-butoxide. We surmised that addition of an external base could lead to deprotonation. Addition of LiHMDS, KHMDS and NaO-tert-Bu to facilitate the desired deprotonation failed to give acceptable results. Perhaps, these strong bases interfered with the ionization event of 2a-1 and the moisture sensitive nature of these strong bases makes the reaction hard to handle. We turned our attention to find mild conditions to generate the corresponding α-sulfonyl carbanion in a catalytic manner without additional stoichiometric base. | ||
| Fig. 2 Optimization of conditions. | ||
Recently, our group and the Hartwig group found that allylic fluoride can be used as an excellent electrophilic precursor in transition metal-catalyzed asymmetric allylic alkylation, generating the nucleophile anion by in situ fluoride induced desilylation, respectively.16 The strategy invoked a synergistic interplay of the fluoride leaving group to facilitate the generation of the electrophilic metal-allyl complex and delivery of a catalytically activated nucleophilic anion by desilylation. We sought to utilize this approach to engage α-sulfonyl 1a in palladium-catalyzed asymmetric allylic alkylation with allyl fluoride 2a (Fig. 2b). Encouragingly, using L1 gave the desired product 3a with good yield (83%) and excellent enantioselectivity (91% ee). When t-BuOMe was used as the solvent, the product 3a was obtained in lower 74% yield but better 94% ee. When L2 was used as the supporting ligand, the product 3a was obtained in 51% yield and 83% ee. Some other phosphoramidite ligands were also tested. L3 afforded the product in a moderate 72% ee with a poor 35% yield. On the other hand, L4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17 and L5, which were successful ligands in our previous palladium-catalyzed transformations, did not afford the desired results. Nevertheless, Sphos L6 afforded 3a in moderate 61% yield, thus this ligand was selected as the supporting ligand for the non-enantioselective transformations. The solvent effect was very important for this transformation and only ethereal solvents gave the desired sulfone 3a with acceptable results. Other solvents such as toluene and DCE gave trace amounts of the desired product. t-BuOMe was proved to be the optimal solvent for the enantioselective transformations. Active CpPd(η3-C3H5) was another key factor for this reaction; other palladium sources such as commonly used Pd(dba)2 gave poor results.
17 and L5, which were successful ligands in our previous palladium-catalyzed transformations, did not afford the desired results. Nevertheless, Sphos L6 afforded 3a in moderate 61% yield, thus this ligand was selected as the supporting ligand for the non-enantioselective transformations. The solvent effect was very important for this transformation and only ethereal solvents gave the desired sulfone 3a with acceptable results. Other solvents such as toluene and DCE gave trace amounts of the desired product. t-BuOMe was proved to be the optimal solvent for the enantioselective transformations. Active CpPd(η3-C3H5) was another key factor for this reaction; other palladium sources such as commonly used Pd(dba)2 gave poor results.
Results and discussion
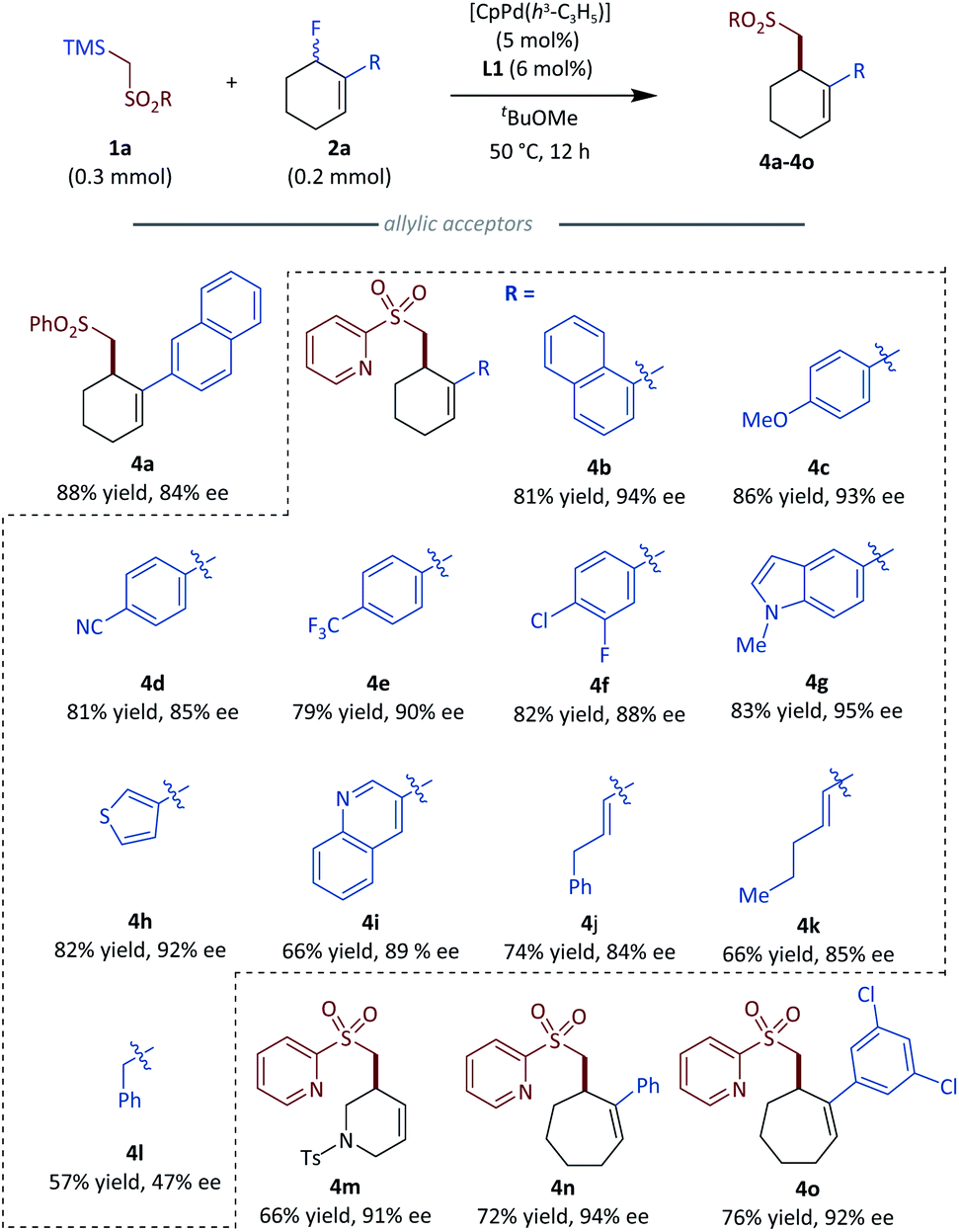
To generate more elaborate chiral homo-allylic sulfones, we first tested the scope of different sulfone donors with allyl fluoride 2a as the reaction counterpart (Fig. 3). Aryl sulfones bearing an electron-donating (3b) or an electron-withdrawing (3c) group were suitable in our system, giving good to excellent results. The substrates bearing halogen atoms, such as fluoro and chloro afforded corresponding products with good results (3e and 3f). Sterically hindered 2-naphthyl sulfone (3f) and bioactive 7-coumarinyl sulfone (3g) also gave rise to the desired products in good to excellent enantioselectivity. Additionally, different heteroaryl sulfones,18 such as 2-pyridyl sulfone (3h), 4-pyridyl sulfone (3i), 1,3-pyrimidinyl sulfone (3j) and benzothiazolyl sulfone (3k) all gave the desired products with excellent results (>94% ee). Besides the above aryl sulfones, simple alkyl sulfones were also tested with the optimized conditions, and the corresponding chiral products could be obtained with slightly diminished enantioselectivity compared to aryl sulfones (3l, 3m). Notably, sulfonamide which is a privileged functionality in modern drug discovery also could produce the corresponding homoallylic chiral sulfonamides with good results (3n, 3o).19 An interesting anion shift was observed for the reaction with benzyl sulfone, which gave the expected product 3p in a good 63% yield and excellent 92% ee with a unseparated minor regioisomer 3p′. A similar shift is not exhibited in Tunge's decarboxylative allylations of benzyl alkyl sulfone.12 Regrettably, vinyl and alkynyl sulfones did not give the desired products. The absolute configuration of 3h was determined by X-ray crystallography; the stereochemical outcome for all other homo-allylic sulfones was assigned by analogy. | ||
| Fig. 3 Substrate scope of different sulfone donors. | ||
Next we turned our attention to the substrate scope of allylic fluorides. To make the detection and separation of product easier, 2-pyridyl sulfone 1h was selected as the standard nucleophile for most substrates (Fig. 4). Allylic fluorides bearing 2-naphthyl (4a), more sterically hindered 1-naphthyl (4b), electron-rich aryl (4c) and electron-deficient aryl (4d, 4e) all gave a range of chiral homo allylic sulfones with good yields and good to excellent ee values. Substrate bearing a chlorine atom, which is a good handle for further functionalization, was compatible with the reaction conditions (4f). Heteroarenes are also good choices for the reaction. Electron-rich indolyl (4g), thiophenyl (4h), and electron-deficient quinolinyl (4i) were all successfully employed to give the desired products with excellent results (>89% ee). Besides the different aryls, alkenyl and alkyl substituted allylic fluorides were also subjected to the optimized conditions. Alkenyl substituted acceptors gave the desired homo-allylic sulfones in good yield with slightly lower ee values compared to aryl substituted acceptors (4j, 4k). Simple benzyl substituted acceptor gave 57% yield but a poor 47% ee (4l). Added flexibility of the benzyl substituent could account for the compromised selectivity. A nitrogen-containing heterocyclic acceptor was also tested and delivered the desired product 4m in 66% yield and an excellent 91% ee. Besides the six membered all carbon cyclic acceptors, medium-sized rings such as seven-membered cyclic acceptors also proved to be good substrates for this reaction, giving the desired products with excellent ee values (4n, 4o). Unfortunately, five-membered acceptors are not suitable for our current conditions, as the active allylic fluoride preferentially eliminated HF thereby forming cyclopentadiene spontaneously.20
 | ||
| Fig. 4 Substrates scope of different allylic acceptors. | ||
To demonstrate the synthetic application of this transformation, the scale of the reaction was increased to 1.0 mmol (Fig. 5a). The palladium loading could be reduced to 2 mol% with 3 mol% of ligand L1 whereby the product 3a was obtained in 68% yield with 92% ee. Furthermore, the sulfone group in the products provides an enabling handle for further transformations (Fig. 5b). For example the sulfone group in 3a could be reductively cleaved with Na(Hg) to access chiral allylic methyl compound 5a in 70% yield. Notably, access to compound 5a is difficult by other methods. Here the sulfone donor acts as a formal methylation reagent.21 Additionally, the sulfone group could be replaced by an ester group via a two-steps synthetic sequence in 82% yield (5b). Furthermore, the heteroaryl sulfonyl group in 3k could be used as a precursor for Julia–Kocienski olefination whereby upon reaction with benzaldehyde the skipped diene 5c forms in excellent yield and geometric selectivity (5c) for the E isomer.22 The 1,3-diene unit in 4j is a good counterpart for an intermolecular Diels–Alder reaction, which afforded fused cyclic compound 5d in 63% yield (dr = 3.5:1) upon reaction with N-methyl maleimide.23
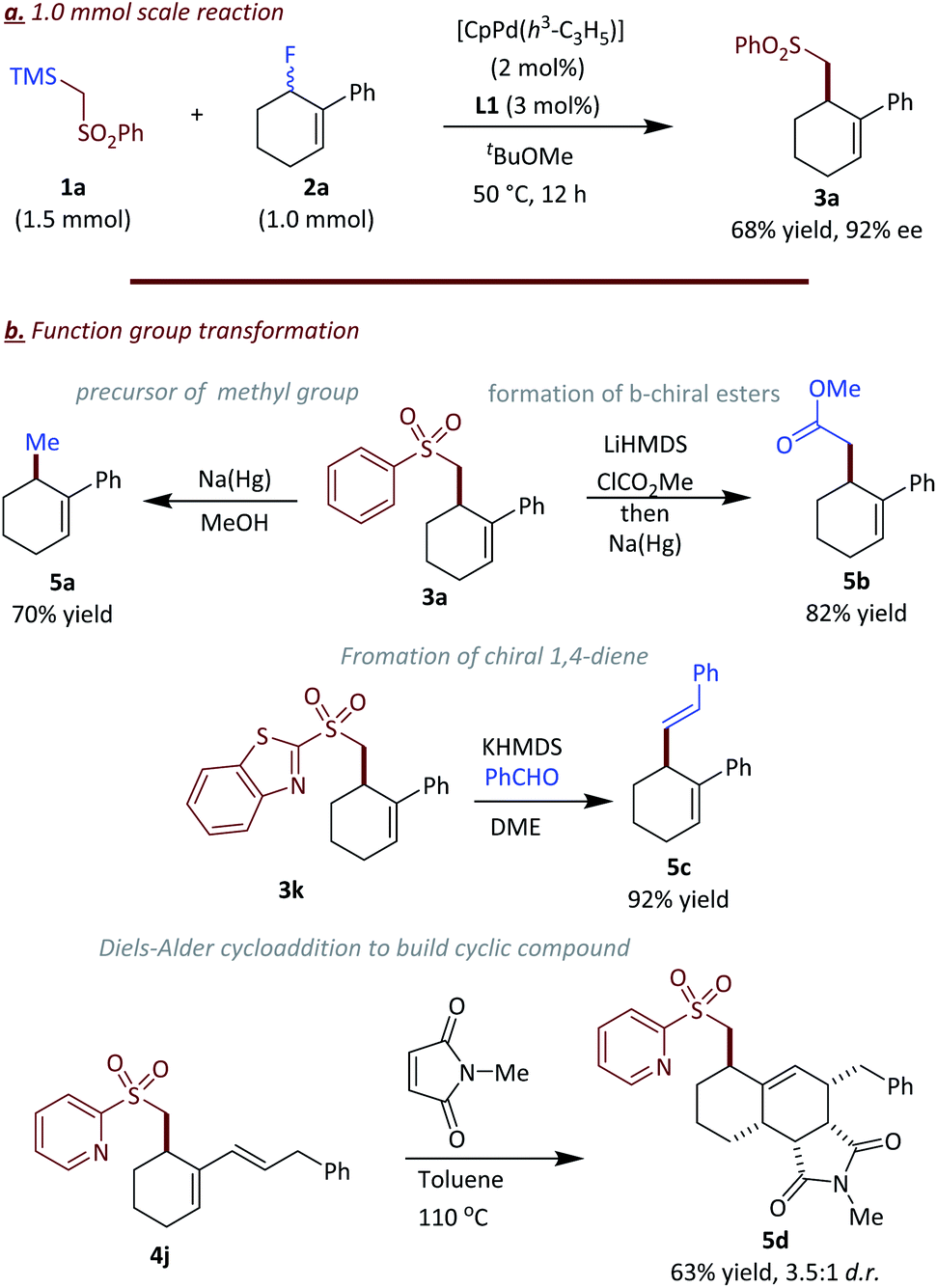 | ||
| Fig. 5 Derivatization of the products. | ||
Conclusions
In conclusion, we realized the first palladium-catalyzed AAA reaction with “hard” α-sulfonyl carbanions as the nucleophiles. This transformation provides a rapid entry to chiral homo-allylic sulfones that otherwise are challenging to obtain. The presence of a sulfone motif provides a powerful handle for subsequent structural elaborations. The “outer sphere” reaction pathway is presumed to be involved in this transformation,24 however, another possible reaction pathway involved anionic Si–F species couldn't be ruled out.25 Detailed mechanistic studies and application of this novel AAA reaction in the synthesis of biologically active compounds and analogy of natural products are ongoing.Author contributions
Z. Jiao and H. Gholami performed the synthetic experiments. B. M. Trost supervised the research. B. M. Trost, Z. Jiao and H. Gholami wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Zhijun Zuo for the measurement of optical rotation. We thank the Tamaki Foundation and Chugai Pharmaceuticals for their generous partial funding of our program. We thank the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT) of Ministry of Education of China.Notes and references
- (a) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395–422 CrossRef CAS PubMed; (b) Z. Lu and S. Ma, Angew. Chem., Int. Ed., 2008, 47, 258–297 CrossRef CAS PubMed; (c) Q. Cheng, H.-F. Tu, C. Zheng, J.-P. Qu, G. Helmchen and S.-L. You, Chem. Rev., 2019, 119, 1855–1969 CrossRef CAS PubMed; (d) L. Süsse and B. M. Stoltz, Chem. Rev., 2021, 121, 4084–4099 CrossRef PubMed.
- (a) B. M. Trost, Chem. Pharm. Bull., 2002, 50, 1–14 CrossRef CAS PubMed; (b) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2944 CrossRef CAS PubMed; (c) B. M. Trost, M. R. Machacek and A. Aponick, Acc. Chem. Res., 2006, 39, 747–760 CrossRef CAS PubMed; (d) N. Samar, Z. Ameer Fawad, A. Sajjad, S. Irum, I. Ali and F. Sadia, Curr. Org. Chem., 2019, 23, 1168–1213 CrossRef.
- (a) J. Zhang, C. Stanciu, B. Wang, M. M. Hussain, C.-S. Da, P. J. Carroll, S. D. Dreher and P. J. Walsh, J. Am. Chem. Soc., 2011, 133, 20552–20560 CrossRef CAS PubMed; (b) S.-C. Sha, J. Zhang, P. J. Carroll and P. J. Walsh, J. Am. Chem. Soc., 2013, 135, 17602–17609 CrossRef CAS PubMed; (c) S. B. Lang, K. M. O'Nele and J. A. Tunge, J. Am. Chem. Soc., 2014, 136, 13606–13609 CrossRef CAS PubMed; (d) T. Maji and J. A. Tunge, Org. Lett., 2014, 16, 5072–5075 CrossRef CAS PubMed; (e) M.-J. Tom and P. A. Evans, J. Am. Chem. Soc., 2020, 142, 11957–11961 CrossRef CAS PubMed; (f) W. Shao, C. Besnard, L. Guénée and C. Mazet, J. Am. Chem. Soc., 2020, 142, 16486–16492 CrossRef CAS PubMed; (g) D. Pal, T. B. Wright, R. O'Connor and P. A. Evans, Angew. Chem., Int. Ed., 2021, 60, 2987–2992 CrossRef CAS PubMed.
- Review: J. A. Tunge, Isr. J. Chem., 2020, 60, 351–359 CrossRef CAS Recent publications: (a) M. Braun, F. Laicher and T. Meier, Angew. Chem., Int. Ed., 2000, 39, 3494–3497 CrossRef CAS; (b) D. C. Behenna and B. M. Stoltz, J. Am. Chem. Soc., 2004, 126, 15044–15045 CrossRef CAS PubMed; (c) B. M. Trost and J. Xu, J. Am. Chem. Soc., 2005, 127, 2846–2847 CrossRef CAS PubMed; (d) B. M. Trost and J. Xu, J. Am. Chem. Soc., 2005, 127, 17180–17181 CrossRef CAS PubMed; (e) S.-L. You and L.-X. Dai, Angew. Chem. Int. Ed, 2006, 45, 5246–5248 CrossRef CAS PubMed; (f) B. M. Trost, J. Xu and M. Reichle, J. Am. Chem. Soc., 2007, 129, 282–283 CrossRef CAS PubMed; (g) K. Zhang, Q. Peng, X.-L. Hou and Y.-D. Wu, Angew. Chem., Int. Ed., 2008, 47, 1741–1744 CrossRef CAS PubMed; (h) B. M. Trost and D. A. Thaisrivongs, J. Am. Chem. Soc., 2008, 130, 14092–14093 CrossRef CAS PubMed; (i) J. Streuff, D. E. White, S. C. Virgil and B. M. Stoltz, Nat. Chem., 2010, 2, 192–196 CrossRef CAS PubMed; (j) J. Mao, J. Zhang, H. Jiang, A. Bellomo, M. Zhang, Z. Gao, S. D. Dreher and P. J. Walsh, Angew. Chem., Int. Ed., 2016, 55, 2526–2530 CrossRef CAS PubMed; (k) P. Starkov, J. T. Moore, D. C. Duquette, B. M. Stoltz and I. Marek, J. Am. Chem. Soc., 2017, 139, 9615–9620 CrossRef CAS PubMed; (l) R. Murakami, K. Sano, T. Iwai, T. Taniguchi, K. Monde and M. Sawamura, Angew. Chem., Int. Ed., 2018, 57, 9465–9469 CrossRef CAS PubMed; (m) B. M. Trost, W.-J. Bai, C. Hohn, Y. Bai and J. J. Cregg, J. Am. Chem. Soc., 2018, 140, 6710–6717 CrossRef CAS PubMed; (n) P. J. Moon, Z. Wei and R. J. Lundgren, J. Am. Chem. Soc., 2018, 140, 17418–17422 CrossRef CAS PubMed; (o) H.-H. Zhang, J.-J. Zhao and S. Yu, J. Am. Chem. Soc., 2018, 140, 16914–16919 CrossRef CAS PubMed.
- Representative enantioselective examples with other metal as catalysts: (a) Y. Makida, H. Ohmiya and M. Sawamura, Angew. Chem., Int. Ed., 2012, 51, 4122–4127 CrossRef CAS PubMed; (b) Y. Makida, Y. Takayama, H. Ohmiya and M. Sawamura, Angew. Chem., Int. Ed., 2013, 52, 5350–5354 CrossRef CAS PubMed; (c) M. Chen and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 13972–13979 CrossRef CAS PubMed; (d) X.-J. Liu, C. Zheng, Y.-H. Yang, S. Jin and S.-L. You, Angew. Chem., Int. Ed., 2019, 58, 10493–10499 CrossRef CAS PubMed; (e) C. I. Jette, Z. J. Tong, R. G. Hadt and B. M. Stoltz, Angew. Chem., Int. Ed., 2020, 59, 2033–2038 CrossRef CAS PubMed; (f) A. H. Hoveyda, Y. Zhou, Y. Shi, M. K. Brown, H. Wu and S. Torker, Angew. Chem., Int. Ed., 2020, 59, 21304–21359 CrossRef CAS PubMed.
- (a) F. G. Bordwell, N. R. Vanier, W. S. Matthews, J. B. Hendrickson and P. L. Skipper, J. Am. Chem. Soc., 1975, 97, 7160–7162 CrossRef CAS; (b) F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- (a) N. A. Tamayo, M. H. Norman, M. D. Bartberger, F.-T. Hong, Y. Bo, L. Liu, N. Nishimura, K. C. Yang, S. Tadesse, C. Fotsch, J. Chen, S. Chmait, R. Cupples, C. Hale, S. R. Jordan, D. J. Lloyd, G. Sivits, G. Van and D. J. St. Jean, J. Med. Chem., 2015, 58, 4462–4482 CrossRef CAS PubMed; (b) M. Feng, B. Tang, S. H. Liang and X. Jiang, Curr. Med. Chem., 2016, 16, 1200–1216 CrossRef CAS PubMed; (c) J. E. Pero, J. M. Matthews, D. J. Behm, E. J. Brnardic, C. Brooks, B. W. Budzik, M. H. Costell, C. A. Donatelli, S. H. Eisennagel, K. Erhard, M. C. Fischer, D. A. Holt, L. J. Jolivette, H. Li, P. Li, J. J. McAtee, B. W. McCleland, I. Pendrak, L. M. Posobiec, K. L. K. Rivera, R. A. Rivero, T. J. Roethke, M. R. Sender, A. Shu, L. R. Terrell, K. Vaidya, X. Xu and B. G. Lawhorn, J. Med. Chem., 2018, 61, 11209–11220 CrossRef CAS PubMed.
- (a) B. M. Trost and C. A. Merlic, J. Org. Chem., 1990, 55, 1127–1129 CrossRef CAS; (b) B. M. Trost and C. A. Kalnmals, Chem. –Eur. J., 2018, 24, 9066–9074 CrossRef CAS PubMed.
- (a) B. M. Trost, J. D. Chisholm, S. T. Wrobleski and M. Jung, J. Am. Chem. Soc., 2002, 124, 12420–12421 CrossRef CAS PubMed; (b) M. Gärtner, G. Satyanarayana, S. Förster and G. Helmchen, Chem. –Eur. J., 2013, 19, 400–405 CrossRef PubMed.
- (a) A. P. Krapcho, Synthesis, 1982, 805–822 CrossRef CAS; (b) A. P. Krapcho, Synthesis, 1982, 893–914 CAS.
- D. Bourgeois, D. Craig, N. P. King and D. M. Mountford, Angew. Chem., Int. Ed., 2005, 44, 618–621 CrossRef CAS PubMed.
- J. D. Weaver and J. A. Tunge, Org. Lett., 2008, 10, 4657–4660 CrossRef CAS PubMed.
- (a) B. M. Trost and G. Mata, Angew. Chem., Int. Ed., 2018, 57, 12333–12337 CrossRef CAS PubMed; (b) Y.-Z. Liu, Z. Wang, Z. Huang, X. Zheng, W.-L. Yang and W.-P. Deng, Angew. Chem., Int. Ed., 2020, 59, 1238–1242 CrossRef CAS PubMed; (c) P. Kumari, W. Liu, C.-J. Wang, J. Dai, M.-X. Wang, Q.-Q. Yang, Y.-H. Deng and Z. Shao, Chin. J. Chem., 2020, 38, 151–157 CrossRef CAS; (d) J. Liu, C.-G. Cao, H.-B. Sun, X. Zhang and D. Niu, J. Am. Chem. Soc., 2016, 138, 13103–13106 CrossRef CAS PubMed.
- (a) B. M. Trost and T. M. Lam, J. Am. Chem. Soc., 2012, 134, 11319–11321 CrossRef CAS PubMed; (b) B. M. Trost, T. M. Lam and M. A. Herbage, J. Am. Chem. Soc., 2013, 135, 2459–2461 CrossRef CAS PubMed.
- B. M. Trost, D. Zell, C. Hohn, G. Mata and A. Maruniak, Angew. Chem., Int. Ed., 2018, 57, 12916–12920 CrossRef CAS PubMed.
- (a) B. M. Trost, H. Gholami and D. Zell, J. Am. Chem. Soc., 2019, 141, 11446–11451 CrossRef CAS PubMed; (b) T. W. Butcher, J. L. Yang, W. M. Amberg, N. B. Watkins, N. D. Wilkinson and J. F. Hartwig, Nature, 2020, 583, 548–553 CrossRef CAS PubMed.
- (a) B. M. Trost, D. A. Thaisrivongs and E. J. Donckele, Angew. Chem., Int. Ed., 2013, 52, 1523–1526 CrossRef CAS PubMed; (b) B. M. Trost and Z. Jiao, J. Am. Chem. Soc., 2020, 142, 21645–21650 CrossRef CAS PubMed.
- (a) P. R. Blakemore, J. Chem. Soc. Perkin, 2002, 1, 2563–2585 RSC; (b) E. Rodrigo, I. Alonso, J. L. García Ruano and M. B. Cid, J. Org. Chem., 2016, 81, 10887–10899 CrossRef CAS PubMed; (c) J. E. Pero, J. M. Matthews, D. J. Behm, E. J. Brnardic, C. Brooks, B. W. Budzik, M. H. Costell, C. A. Donatelli, S. H. Eisennagel, K. Erhard, M. C. Fischer, D. A. Holt, L. J. Jolivette, H. Li, P. Li, J. J. McAtee, B. W. McCleland, I. Pendrak, L. M. Posobiec, K. L. K. Rivera, R. A. Rivero, T. J. Roethke, M. R. Sender, A. Shu, L. R. Terrell, K. Vaidya, X. Xu and B. G. Lawhorn, J. Med. Chem., 2018, 61, 11209–11220 CrossRef CAS PubMed.
- P. J. Mäder and L. Kattner, J. Med. Chem., 2020, 23, 14243–14275 CrossRef PubMed.
- J. J. Tufariello, A. C. Bayer and J. J. Spadaro, J. Am. Chem. Soc., 1979, 101, 3309–3315 CrossRef CAS.
- (a) E. J. Barreiro, A. E. Kümmerle and C. A. M. Fraga, Chem. Rev., 2011, 111, 5215–5246 CrossRef CAS PubMed; (b) Y. Chen, Chem. –Eur. J., 2019, 25, 3405–3439 CrossRef CAS PubMed.
- (a) P. R. Blakemore, W. J. Cole, P. J. Kocieński and A. Morley, Synlett, 1998, 1998, 26–28 CrossRef; (b) T. K. Macklin and G. C. Micalizio, Nat. Chem., 2010, 2, 638–643 CrossRef CAS PubMed.
- (a) K. Black, P. Liu, L. Xu, C. Doubleday and K. N. Houk, Proc. Nat. Acad. Sci., 2012, 109, 12860–12865 CrossRef CAS; (b) R. Hongyu and H. Gangliang, Curr. Org. Synth., 2016, 13, 847–860 CrossRef.
- J. A. Keith, D. C. Behenna, N. Sherden, J. T. Mohr, S. Ma, S. C. Marinescu, R. J. Nielsen, J. Oxgaard, B. M. Stoltz and W. A. Goddard, J. Am. Chem. Soc., 2012, 134, 19050–19060 CrossRef CAS PubMed.
- (a) Y. Hatanaka and T. Hiyama, J. Org. Chem., 1988, 53, 918–920 CrossRef CAS; (b) H. F. Sore, W. R. J. D. Galloway and D. R. Spring, Chem. Soc. Rev., 2012, 41, 1845–1866 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2082853. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02599f |
| This journal is © The Royal Society of Chemistry 2021 |