
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA general, versatile and divergent synthesis of selectively deuterated amines†
Morgan
Lecomte
,
Mounsef
Lahboubi‡
,
Pierre
Thilmany‡
,
Adil
El Bouzakhi
and
Gwilherm
Evano
 *
*
Laboratoire de Chimie Organique, Service de Chimie et PhysicoChimie Organiques, Université libre de Bruxelles (ULB), Avenue F. D. Roosevelt 50, CP160/06, 1050 Brussels, Belgium. E-mail: Gwilherm.Evano@ulb.be
First published on 13th July 2021
Abstract
Deuterated organic molecules are of utmost importance in many areas of science and have been recently intensively investigated in medicinal chemistry due to their enhanced metabolic stability. The development of efficient and broadly applicable methods for the selective incorporation of deuterium atoms into organic molecules from readily available starting materials and reagents is therefore of extreme importance. Such methods however often lack generality and selectivity, notably in the nitrogen series. With nitrogen-containing molecules being indeed ubiquitous in medicinal chemistry, there is a strong need for efficient methods enabling the selective synthesis of deuterated amines. In this perspective, we report herein a general, versatile, divergent and metal-free synthesis of amines selectively deuterated at their α and/or β positions. Upon simple treatment of readily available ynamides with a mixture of triflic acid and triethylsilane, either deuterated or not, a range of amines can be smoothly obtained with high levels of deuterium incorporation by a unique sequence involving a domino keteniminium/iminium activation.
Introduction
Since its discovery in 1931 by Harold C. Urey,1 deuterium D, or 2H, one of the three isotopes of hydrogen together with protium 1H and tritium T, or 3H, having a natural abundance of 0.015%, has found many applications in various areas of science, notably when incorporated into organic molecules.2 While a carbon–deuterium bond indeed closely resembles a carbon–hydrogen3 one, the main difference lies in the higher stability of the former compared to the latter due to a smaller vibrational frequency and hence a lower zero-point energy. This subtle difference has a dramatic effect when replacing a hydrogen atom by a deuterium in a carbon chain.2 Deuterated organic molecules, in addition to their use as internal standards for mass spectrometry or NMR spectroscopy, have been indeed utilized to measure minute amounts of a specific chemical in the environment or in the body by means of the stable isotope dilution technique,2,4 to study reaction mechanisms as well as their kinetics – the use of deuterium as a tracer and kinetic isotope effects being especially useful in this area –2,5 for the elucidation of biosynthetic pathways,6 to alter the selectivity of some reactions2,7 and even to improve the properties of polymers8 and chemical stability of materials such as OLEDs.9In addition to their use in metabolism and toxicology studies,10 the increased stability of deuterated molecules has moreover been extensively studied recently in medicinal chemistry, notably to increase the metabolic stability of some drugs and/or to avoid the formation of toxic metabolites.11 Representative examples of such a strategy include deuterated Efavirenz 1d, an isotopologue of Bristol-Myers Squibb's Efavirenz utilized for the treatment of AIDS, in which the incorporation of a deuterium atom at the propargylic position inhibits its enzymatic oxidation, resulting in the formation of nephrotoxic metabolites (Fig. 1),12 or deuterated Telaprevir 2d, a deuterated analogue of Johnson&Johnson/Vertex Pharmaceuticals' drug for the treatment of hepatitis C with improved stability due to a reduced epimerization rate.13 While the deuteration of drugs has been mostly studied in academia for years and was clearly recognized as a subtle, yet powerful, medicinal chemistry tool to retain potency and selectivity together with improving the overall pharmacological profile of a drug, several companies have started to focus on this area.11,14 It can indeed breathe new life into old drugs, with Teva's Austedo® (deutetrabenazine) 3d6, the first FDA-approved deuterated drug displaying a better safety profile than its non-deuterated analogue (tetrabenazine, readily demethylated by cytochrome CYP2D6 to less active metabolites 4 and 4′) and commercialized since 2017 for the treatment of chorea associated with Huntington's disease, being certainly the most striking example.15 Other deuterated drugs in the pipeline include Avanir's AVP-786 (hexadeuterated dextromorphan 5d6) in phase III clinical trials for the treatment of agitation in Alzheimer's disease patients16 and phase II trials in major depressive disorder and schizophrenia,17 Vertex's VX-561 6d9, a deuterated form of ivacaftor in phase II trials for cystic fibrosis,18 or Concert's deuterated Paroxetine CTP-347 7d2 in phase II clinical trials for non-hormonal treatment of vasomotor symptoms. In addition to bringing new life to old drugs, deuteration is now starting to be envisioned from the early stages of drug development as exemplified with Bristol-Myers Squibb's de novo deuterated BMS-986165 8d3 developed for the treatment of psoriasis which recently succeeded in phase II trials.19 Many more deuterated small molecule drug candidates are currently in the pipeline20 and a growing number of companies are now actively pursuing the deuterated drug market which was recently estimated to represent a total value of transaction of $5 billion.11k
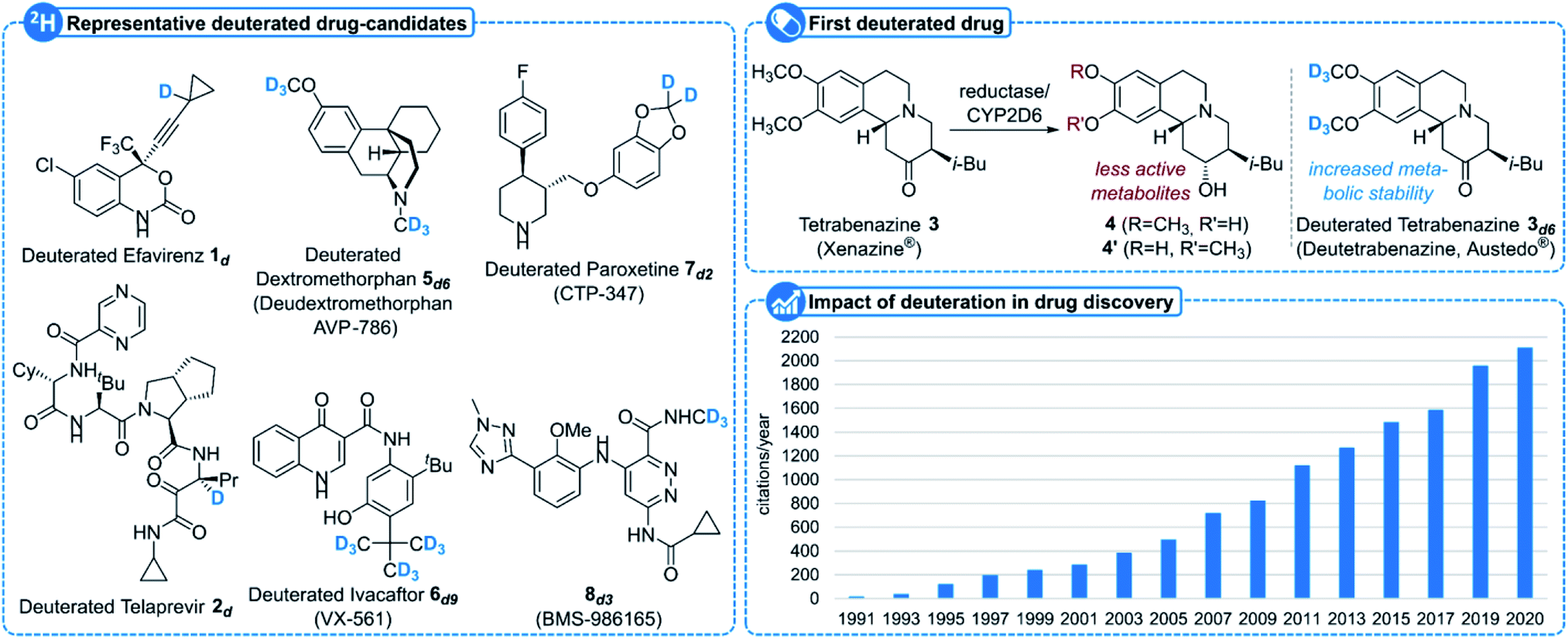 | ||
| Fig. 1 Impact of deuterated molecules in drug discovery. | ||
These recent tremendous developments obviously strongly depend on the synthesis of deuterated molecules. An analysis of the processes developed for the synthesis of all deuterated drugs/drug candidates reported to date actually reveals that they mostly rely on commercially available, simple deuterated precursors such as D2O, CD3I, CD3OD, DCO2D, CD2O, CD3S(O)CD3 or CD3NH2 which are incorporated into organic molecules using standard and classical techniques.21 Further developments in this area will clearly require the use of more advanced processes either based on the use of a broader variety of deuterated reagents or for the selective introduction of deuterium atoms. A parallel can actually be drawn with fluorinated drugs: since the first approval by the FDA of such a drug, fludrocortisone, in 1955, nearly 150 fluorinated molecules have succeeded in reaching the market and a recent analysis estimates that about 30% of drugs will soon contain at least one fluorine atom,22 this dramatic increase being mainly associated with the design of novel methods for the synthesis of fluorinated organic molecules. With deuterium being considered as the potential “next fluorine” in medicinal chemistry, a strong need for new processes streamlining the synthesis of deuterated molecules is logically emerging, such methods being expected to have an important impact in drug discovery.
Among all deuterated functional groups, amines occupy a central place due to the vast majority of drugs containing at least one nitrogen atom and due to their easy metabolization. The selective introduction of deuterium atoms close to the nitrogen in amines could indeed represent a quite subtle strategy to increase the stability of nitrogen-containing drug candidates while bringing minor structural changes, a concept already validated with AVP-786 5d6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16,17 and BMS-986165 8d3.19 The state-of-the-art methods available for the synthesis of deuterated amines actually reveal the limited number of possibilities as well as an important number of limitations. Incorporation of deuterium α to the nitrogen atom indeed mostly relies on the alkylation of amines or surrogates with deuterated alkyl halides or synthetic equivalents and on the reduction of nitriles, imines and amides with LiAlD4, a reagent whose commercial availability greatly varies over time. Limitations met with these classical methods, mostly in terms of efficiency, generality and functional group tolerance, have stimulated the development of alternative processes relying on the use of readily available deuterated reducing agents such as PhSiD3 in the presence of a nickel catalyst23 or on the use of D2O or EtOD under reductive conditions.24 Other methods recently reported are essentially based on the use of metal catalysts25 or on a cooperative acid/base catalytic system26 for the direct deuteration of amines α and/or β to the nitrogen atom. While these methods are clearly attractive since they enable the synthesis of α- or α,β-deuterated amines from the corresponding non-deuterated ones using readily available sources of deuterium such as D2O, D2, CD3S(O)CD3, CD3COCD3 or deuterated alcohols and can be performed in a stereospecific manner with some chiral amines, they however suffer from important limitations in terms of substrate scope, regioselectivity and, more importantly, with respect to deuterium incorporation, which is far from quantitative in most cases.
16,17 and BMS-986165 8d3.19 The state-of-the-art methods available for the synthesis of deuterated amines actually reveal the limited number of possibilities as well as an important number of limitations. Incorporation of deuterium α to the nitrogen atom indeed mostly relies on the alkylation of amines or surrogates with deuterated alkyl halides or synthetic equivalents and on the reduction of nitriles, imines and amides with LiAlD4, a reagent whose commercial availability greatly varies over time. Limitations met with these classical methods, mostly in terms of efficiency, generality and functional group tolerance, have stimulated the development of alternative processes relying on the use of readily available deuterated reducing agents such as PhSiD3 in the presence of a nickel catalyst23 or on the use of D2O or EtOD under reductive conditions.24 Other methods recently reported are essentially based on the use of metal catalysts25 or on a cooperative acid/base catalytic system26 for the direct deuteration of amines α and/or β to the nitrogen atom. While these methods are clearly attractive since they enable the synthesis of α- or α,β-deuterated amines from the corresponding non-deuterated ones using readily available sources of deuterium such as D2O, D2, CD3S(O)CD3, CD3COCD3 or deuterated alcohols and can be performed in a stereospecific manner with some chiral amines, they however suffer from important limitations in terms of substrate scope, regioselectivity and, more importantly, with respect to deuterium incorporation, which is far from quantitative in most cases.
Based on our long-standing interest in the chemistry of ynamides,27 stable nitrogen-substituted alkynes,28 and in the unique reactivity of activated keteniminium ions29 derived from these reagents, we envisioned that these building blocks – that are either commercially available or readily prepared by a range of methods30 – and intermediates might provide a straightforward, versatile and divergent entry to α- and/or β-deuterated amines, which should overcome most of the limitations met with other methods. Our strategy, depicted in Fig. 2, is based on a simple, yet unexplored, and unique keteniminium/iminium activation sequence: upon reaction with an acid (deuterated or not), the starting ynamide 9 would afford a transient activated keteniminium ion 10 whose trapping with a hydride or a deuteride would yield an intermediate enamide 11. Further reaction of this enamide with an acid would then enable the generation of an iminium ion 12 whose final reduction should provide the desired amine 13.31,32 Depending on the nature of the acid and the hydride (either deuterated or not), this reaction should enable an easy and divergent access to a range of amines with selective deuteration at the α- and/or β-positions. We report in this manuscript the development of a broadly applicable versatile, and divergent synthesis of selectively deuterated amines based on this working hypothesis.
 | ||
| Fig. 2 Working hypothesis for the divergent and selective synthesis of deuterated amines. | ||
Results and discussion
Optimization of the reduction of ynamides to amines
We initiated our studies by assessing the feasibility of our working hypothesis and the identification of the optimal conditions for the reduction of ynamides to amines first using non-deuterated reagents. The main problems associated with our strategy lie in the fact that the acid and the hydride should both react faster with all intermediates than together and that the conjugated base of the acid should not competitively trap the activated keteniminium 10 and iminium 12 ions.With this goal in mind, a series of acids and hydrides, known to be compatible and typically used in ionic reductions and deoxygenations, was first evaluated using at this stage an excess of these reagents, with ynamide 9a as a model substrate in dichloromethane at 0 °C for 15 minutes. Results from these studies, shown in Fig. 3, reveal the feasibility of our approach as well as the superiority of triflic acid for the activation of both the starting ynamide 9a and the transient enamide 11a. Among the hydrides evaluated,33 triethylsilane was found to be the most efficient one: indeed, a combination of triflic acid and triethylsilane resulted in the formation of the desired amine 13a (84% 1H NMR yield) together with amide 14a (14% 1H NMR yield) resulting from the trapping of the intermediate keteniminium ion 10a by the triflate anion followed by further hydrolysis upon workup.28c Having demonstrated the feasibility of our approach and identified the best combination of the acid and reducing agent, we next focused our attention on reducing the amount of both these reagents, a critical point in the deuterated series. A brief screening of different stoichiometries, temperatures and reaction times (not shown) revealed that the use of 2.5 equivalents of triflic acid in combination with 5 equivalents of triethylsilane at room temperature for 18 hours was optimal, resulting in the formation of the reduced product 13a in 84% 1H NMR yield and 72% isolated yield.
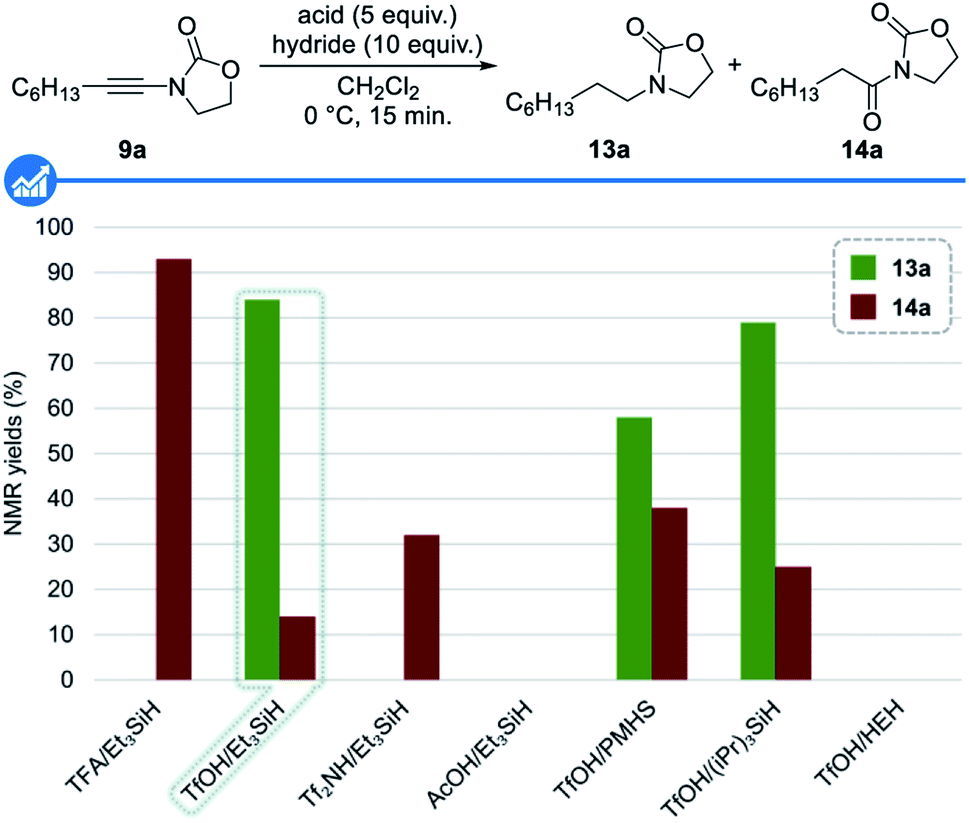 | ||
| Fig. 3 Screening of acids and hydrides for the reduction of ynamides to amines. 1H NMR yields determined with 1,1,2,2-tetrachloroethane as the internal standard; HEH: Hantzsh ester, 4-dihydro-2,6-dimethyl-3,5-pyridinedicarboxylate. | ||
With these optimized conditions in hand, we then evaluated their extension to the deuterated series: as shown in Fig. 4, the use of deuterated triethylsilane, deuterated triflic acid, or both, resulted in a smooth reduction to the corresponding α-, β- or α,β-deuterated products 13adα2, 13adß2 and 13adα2β2, in good yields, excellent and good levels of deuteration at the α and β positions being respectively attained. Importantly, a complete regioselectivity was observed in these three trials, which is in sharp contrast with previously reported techniques.
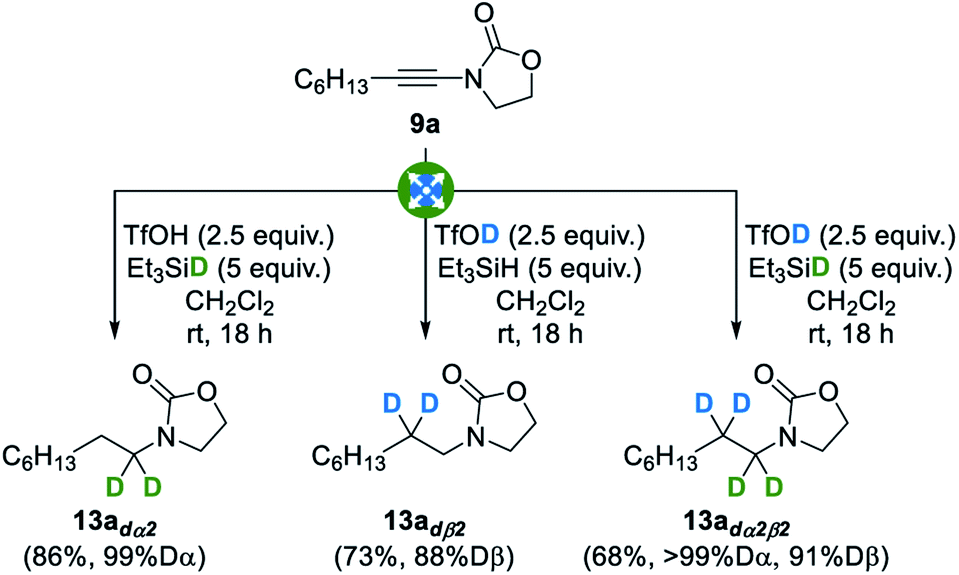 | ||
| Fig. 4 Divergent and selective deuterative reduction of ynamides to amines. | ||
Scope of the divergent synthesis of deuterated amines
We then moved to the study of the scope and limitations of this divergent synthesis of selectively deuterated amines, first focusing on α-deuterated ones 13dα2 resulting from the reduction of the corresponding ynamides 9 with deuterium-labelled triethylsilane (Fig. 5). A set of ynamides 9 with representative substitution patterns was therefore reacted under our optimized conditions: in all cases, the reduction was shown to be fully regioselective, with deuterium being selectively incorporated at the α position only, and remarkably high levels of deuteration (96 to >99% D) were systematically attained. The α-deuterated amines 13dα2 could be obtained in fair to good yields, with the major byproducts that could be detected and easily separated being the amides resulting from a competing trapping of the transient activated keteniminium ions by the triflate followed by hydrolysis of the resulting ketene N,O-acetals.28c Oxazolidinone- and pyrrolidinone-derived ynamides were found to be suitable substrates, providing the corresponding deuterated N-alkyl oxazolidinones 13a–cdα2 and lactam 13ddα2, respectively. Interestingly, N-tosyl-ynamines were also found to smoothly undergo the reductive deuteration to afford the corresponding deprotectable N-tosyl-amines 13e–mdα2 in modest to good yields. As for the substitution pattern and electronic properties of the starting ynamides, both aryl (13b–ddα2, 13f–hdα2 and 13l,mdα2) and alkyl (13adα2, 13edα2 and 13idα2) groups were found to be tolerated and even terminal and trifluoromethyl-substituted ynamides could be transformed into the corresponding selectively α-deuterated amines 13jdα2 and 13kdα2, respectively, although with reduced efficiency yet excellent levels of deuteration. Interestingly, and which nicely highlights the efficacy and selectivity of our method, an alkyne (13cdα2) was found to be compatible with the reaction conditions and no competing reduction was observed in this case. As for the substituent on the nitrogen atom in the starting ynamides 9, cleavable benzyl (13e–kdα2) and allyl (13ldα2) groups were tolerated and no products resulting from a competing Pictet–Spengler-type trapping of the transient keteniminium ions by the benzyl group, a reaction that is known to be fast and that would afford isoindolines,34 were observed. Finally, even an acid-sensitive cyclopropyl group (13mdα2) was found to survive the reductive deuteration without competing ring-opening.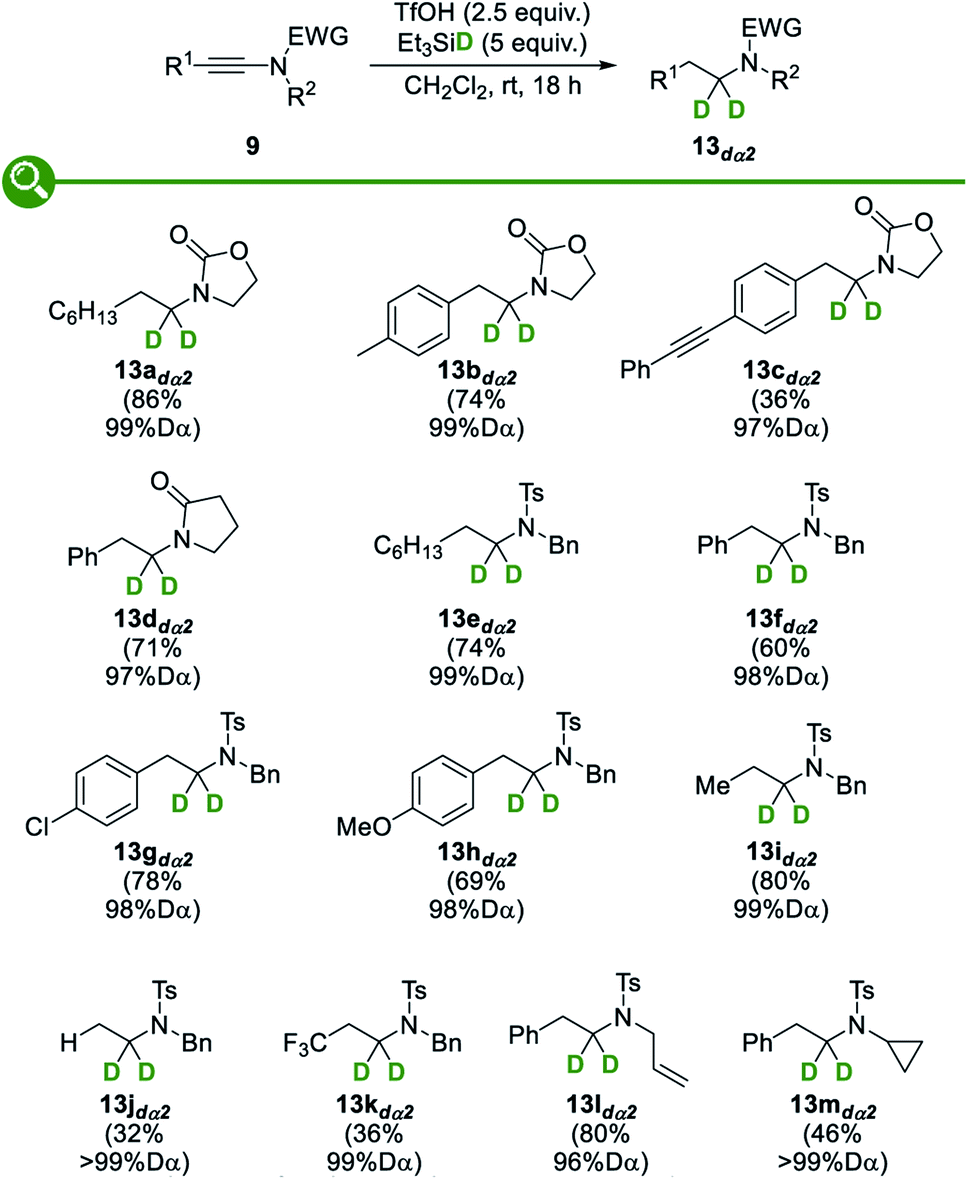 | ||
| Fig. 5 Synthesis of selectively α-deuterated amines. | ||
Having demonstrated the broad scope and the generality of our reductive deuteration for the synthesis of a range of amines selectively deuterated at the α position, we next moved to the study of the synthesis of their analogues selectively deuterated at the β position 13dß2. The exact same set of ynamides 9 was therefore reacted under our optimized conditions but now using deuterated triflic acid instead of deuterated triethylsilane (Fig. 6). This simple switch in reagents enabled the synthesis of a variety of β-deuterated amines 13dß2 that could be obtained with similar efficiencies and good levels of deuteration (82–93% D), levels that were however somehow a bit lower than the ones obtained for the deuteration at the β-position but still in the high range and highly competitive with levels of deuteration that can be obtained with other methods. As for the yields, some are somehow lower than when using triflic acid, which might be attributed to the lower acidity of TfOD vs. TfOH. As a practical note, higher levels of deuteration were attained with freshly prepared deuterated triflic acid. Importantly, the reductive deuteration was not affected by the presence of aromatic substituents that could be competitively protonated by TfOD, resulting, after deprotonation, in the release of TfOH and therefore reduced levels of deuteration at the β-position, as observed only with an especially electron-rich anisole substituent that was partially deuterated in the course of the reaction and resulted in a reduced (85% D) level of β-deuteration (13hdß2). In a similar vein, no competing deuteration of the alkene (13ldß2) or cyclopropane (13mdß2) was noted, the deuteration being selective for the β position.
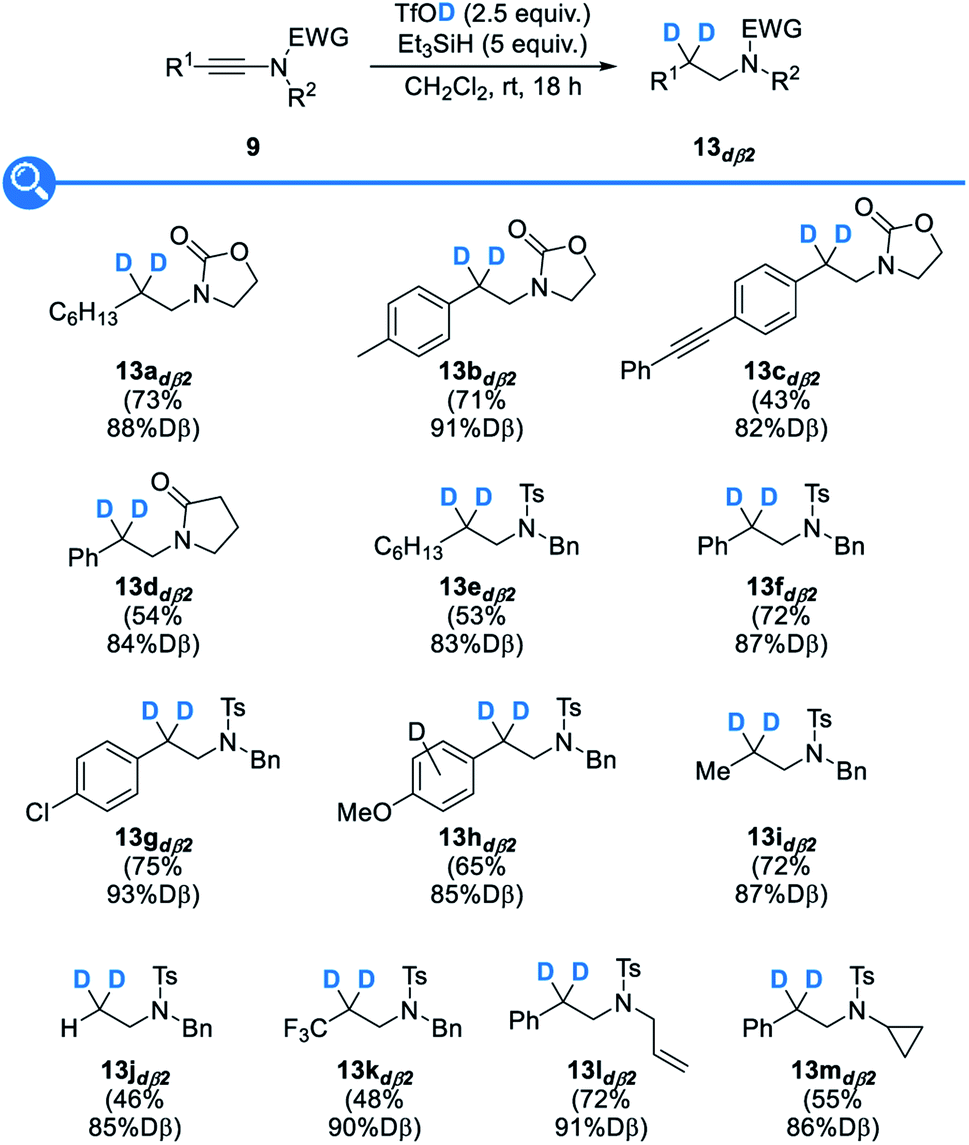 | ||
| Fig. 6 Synthesis of selectively β-deuterated amines. | ||
We finally focused our efforts on the synthesis of amines selectively deuterated at both their α and β positions, compounds that could be obtained using a combination of deuterated triflic acid and deuterated triethylsilane under our standard reaction conditions. Results summarized in Fig. 7 show the generality of this process that enables the preparation of a broad range of selectively deuterated amines – compounds that can be hardly prepared otherwise – with various protecting groups, substitution patterns and substituents. As for the trends in reactivity and deuteration levels, they logically follow the ones discussed above for the synthesis of α- and β-deuterated amines.
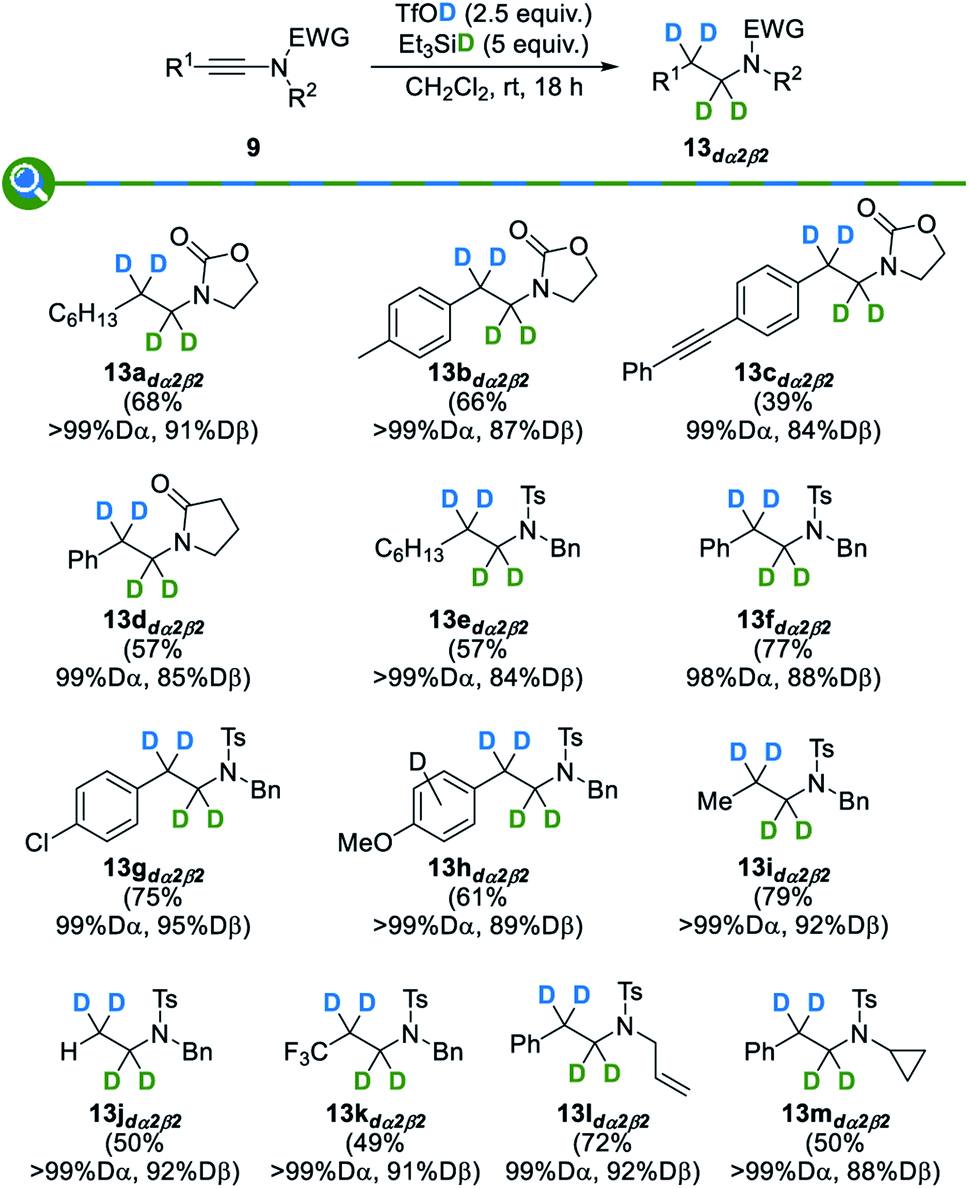 | ||
| Fig. 7 Synthesis of selectively α,β-deuterated amines. | ||
Results from these studies highlight the efficiency and selectivity of our process for the divergent preparation of amines selectively deuterated at the α position, β position, or both. By simply selecting the appropriate deuterated reagent(s), all these selectively labelled molecules can be prepared in a fully divergent manner and with high functional group tolerance and levels of deuterium incorporation. These processes could have a significant impact in medicinal chemistry, a field in which amines are ubiquitous, and we therefore decided to apply them to the synthesis of representative deuterated biologically relevant amines. The corresponding results will be described in the next section.
Application to the synthesis of deuterated biologically relevant amines
In an effort to further highlight the synthetic potential of our divergent and selective synthesis of deuterated amines, we indeed next envisioned, as a proof of concept, its use for the synthesis of simple biologically relevant amines selectively deuterated at the α- and/or β-position(s) with respect to the nitrogen atom: phenethylamine 17 and dopamine 19 were selected for this study. Phenethylamine 17 is indeed a monoamine alkaloid found in low concentrations in the central nervous system of mammals. Along with other so-called “trace amines”, phenethylamine is a major one and is known to act as a neuromodulator for structurally related classical neurotransmitters such as dopamine 19, norephedrine and serotonin.35 Therefore, trace amines are known to affect the release and uptake of the latter neurotransmitters and have thus been linked to psychiatric and neurological pathologies such as depression, schizophrenia, Tourette's syndrome and Parkinson's disease. Accordingly, their use for the development of new therapeutics has been suggested and highlighted on multiple occasions.36 Interestingly, the very first example of deuterium substitution in biologically relevant compounds, dating back to 1961, already described that the metabolism of two trace amines, p-tyramine and tryptamine, was significantly reduced when replacing α-hydrogens with deuterium atoms.37 With this in mind, we envisioned the synthesis of phenethylamine 17 deuterated at the α- and/or β-position(s) from ynamide 9n, itself readily synthesized by copper-catalyzed cross-coupling of bromoalkyne 15 with sulfonamide 16 (Fig. 8). Upon exposure of 9n to combinations of triflic acid and triethylsilane, deuterated or not, and subsequent deprotection of the anisylsulfonyl and benzyl moieties, selectively deuterated phenethylamine 17dα2, 17dß2 and 17dα2β2 could be obtained in good overall yields and with excellent deuteration levels of 97 to >99% at the α-position, and 84 to 95% at the β-position, further demonstrating the usefulness and versatility of our process for the synthesis of deuterated amines. | ||
| Fig. 8 Divergent synthesis of selectively α-, β- and α,β-deuterated phenethylamine. | ||
In an effort to go one step further, we next targeted a second biologically relevant amine, dopamine 19, a major neurotransmitter that also plays a central role in various pathologies. It is notably involved in Parkinson's disease which is characterized by a progressive loss of dopaminergic neurons, causing decreasing dopamine levels and therefore a degeneration of motor skills in affected patients. To date, the most efficient symptomatic therapy for Parkinson's disease relies on the oral administration of L-DOPA (L-3,4-dihydroxyphenylalanine) as a precursor to dopamine.38 Interestingly, and which further showcases the importance of selectively deuterated drugs, the introduction of deuterium atoms at the α- and β-positions of L-DOPA significantly slows down the metabolic breakdown of deuterated dopamine, thus improving its potency.39 Indeed, and as mentioned in the introduction, the breaking of C–D bonds during metabolic oxidation is often more difficult than that of C–H bonds, meaning that the selective deuteration of bioactive compounds might allow for smaller or less frequent dosing. In this perspective, we decided to apply our method to the divergent synthesis of α- and/or β-deuterated dopamine from readily available dichlorinated ynamide 9o that was chosen as the starting substrate to avoid a competing deuteration of its electron-rich dimethoxylated analogue with deuterated triflic acid. As shown in Fig. 9, this ynamide 9o was readily converted into selectively deuterated amines 13odα2, 13odß2 and 13odα2β2 with good yields and deuteration levels of ≥99% at the α-position and ranging from 87 to 94% at the β-position. Installation of the methyl-protected catechol moiety by a palladium-catalyzed Buchwald–Hartwig cross-coupling40 and subsequent deprotection of the anisylsulfonyl, benzyl and methyl ether moieties next afforded selectively deuterated dopamine 19dα2, 19dß2 and 19dα2β2 without affecting deuteration levels, further highlighting the versatility and efficiency of our modular synthesis of deuterated amines.
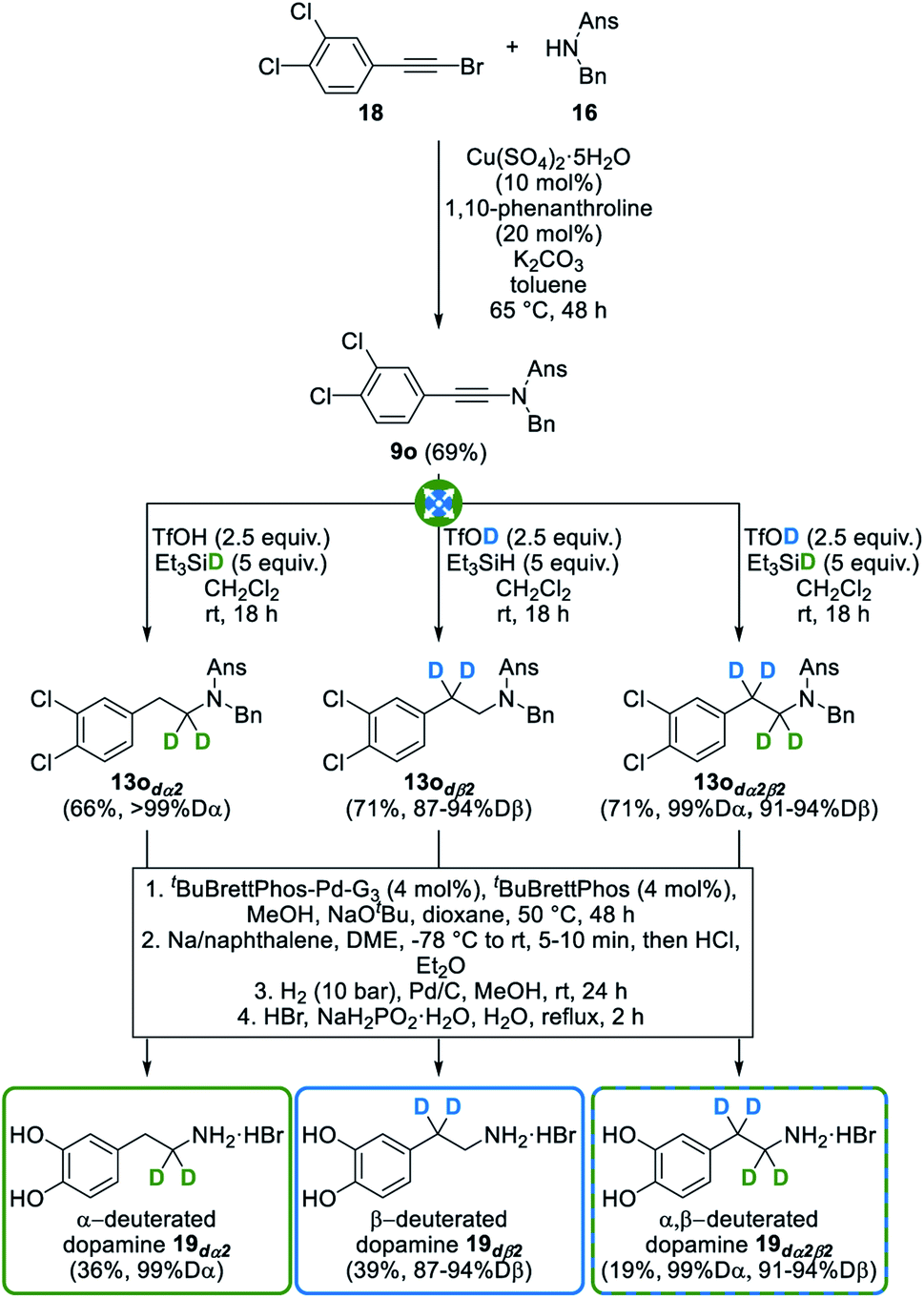 | ||
| Fig. 9 Divergent synthesis of selectively α-, β- and α,β-deuterated dopamine. | ||
After having successfully developed a straightforward and divergent access to α- and/or β-deuterated amines and applied it, as a proof of concept, to biologically relevant amines including phenethylamine and dopamine, we finally wanted to briefly evaluate the possibility of extending this strategy to the synthesis of selectively deuterated alcohols and ethers.
Extension to the synthesis of selectively deuterated ethers
In order to illustrate the generality of our method, we indeed envisioned that our strategy could be extended to other heteroatomic molecules and building blocks starting from other heterosubstituted alkynes. With this goal in mind, ynol ether 21, readily obtained in a single step from commercially available ethyl ethynyl ether 20,41 was subjected to our standard conditions and gratifyingly gave the corresponding selectively deuterated ethers 22dα2, 22dß2 and 22dα2β2 with good to excellent levels of deuteration (Fig. 10). In this case, lower yields were however obtained due to the formation of an ester side-product resulting from the hydrolysis of the intermediate ketenium ion. This side-product could however be easily removed by simply subjecting the crude reaction mixture to saponification conditions and subsequent acid–base wash. Despite lower yields, these examples again still highlight both the strong selectivity, as no residual deuteration was observed at untargeted positions, and versatility of this method. Other heterosubstituted alkynes such as thioynol ethers and bromoalkynes were however found to be reluctant substrates for this transformation.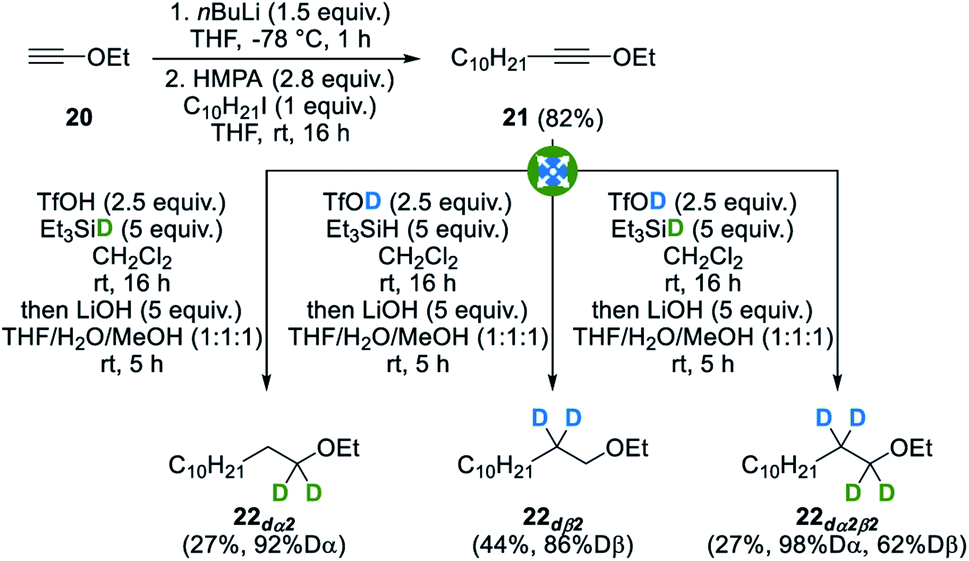 | ||
| Fig. 10 Divergent synthesis of α-, β- and α,β-selectively deuterated ethers. | ||
Conclusions
In conclusion, we have developed a simple, user-friendly and general synthesis of amines selectively deuterated at their α and/or β positions. Upon simple treatment of readily available ynamides with a mixture of triflic acid and triethylsilane, either deuterated or not, a range of amines can be smoothly obtained with high levels of deuterium incorporation. Notable features of this process, for which we envision great acceptance due to the major and growing importance of selectively deuterated amines, particularly in medicinal chemistry, include its versatility, its generality and its user-friendliness. The extension of this process to the synthesis of selectively deuterated ethers and its use for the preparation of biologically relevant amines such as phenethylamine and dopamine, deuterated with surgical precision at their α and/or β positions, further highlight the potential of this rather unique method involving moreover an underrated novel domino keteniminium/iminium activation.Data availability
All experimental procedures, characterization, copies of NMR spectra for all new compounds related to this study can be found in the ESI.Author contributions
G. E. designed the study. Experimental work and data analysis were performed by M. Le., M. La., P. T., and A. E. B.; P. T. and G. E. co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Our work was supported by the Université libre de Bruxelles (ULB), the Fédération Wallonie-Bruxelles (ARC Consolidator 2014–2019) and the FNRS (CDR 2017 J.0058.17). M. Le. and P. T. acknowledge the Fonds pour la formation à la Recherche dans l'Industrie et dans l'Agriculture (F.R.I.A.) for graduate fellowships. We kindly acknowledge Pr. Michel Luhmer (CIREM, ULB) for his help and advice for NMR analysis of deuterated molecules as well as Dr Samuel Oger (LCO, ULB) for his precious assistance during the revision of this manuscript.Notes and references
- (a) J. S. Rigden and J. O'Connell, Am. J. Phys., 2003, 71, 189 CrossRef; (b) H. C. Urey, Science, 1933, 78, 566–571 CrossRef CAS PubMed.
- J. Yang, Deuterium: Discovery and Applications in Organic Chemistry, Elsevier, 2016 Search PubMed.
- “Hydrogen” is commonly used in place of “protium” 1H, the major isotope of hydrogen. For clarity, we have utilized “hydrogen” everywhere in this manuscript but it refers to “protium” in most cases.
- For a representative example, see: R. R. Gerona, T. J. Woodruff, C. A. Dickenson, J. Pan, J. M. Schwartz, S. Sen, M. W. Friesen, V. Y. Fujimoto and P. A. Hunt, Environ. Sci. Technol., 2013, 47, 12477–12485 CrossRef CAS PubMed.
- (a) N. S. Isaacs, Physical Organic Chemistry, Longman Scientific & Technical, Burnt Mill, Harlow, Essex, England, 1995 Search PubMed; (b) K. B. Wiberg, Chem. Rev., 1955, 55, 713–743 CrossRef CAS.
- J. Rinkel and J. S. Dickschat, Beilstein J. Org. Chem., 2015, 11, 2493–2508 CrossRef CAS PubMed.
- (a) M. Miyashita, M. Sasaki, I. Hattori, M. Sakai and K. Tanino, Science, 2004, 305, 495–499 CrossRef CAS PubMed; (b) K. W. Quasdorf, A. D. Huters, M. W. Lodewyk, D. J. Tantillo and N. K. Garg, J. Am. Chem. Soc., 2012, 134, 1396–1399 CrossRef CAS PubMed.
- T. Kaino, K. Jinguji and S. Nara, Appl. Phys. Lett., 1983, 42, 567–569 CrossRef CAS.
- (a) H. Tsuji, C. Mitsui and E. Nakamura, Chem. Commun., 2014, 50, 14870–14872 RSC; (b) C. C. Tong and K. C. Hwang, J. Phys. Chem. C, 2007, 111, 3490–3494 CrossRef CAS.
- A. E. Mutlib, Chem. Res. Toxicol., 2008, 21, 1672–1689 Search PubMed.
- (a) R. D. Tung, Innovations Pharm. Technol., 2010, 32, 24–28 Search PubMed; (b) S. L. Harbeson and R. D. Tung, in Annual Reports in Medicinal Chemistry, ed. J. E. Macor, Elsevier, 2011, vol. 46, ch. 24, pp. 403–417 Search PubMed; (c) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529–2591 CrossRef CAS PubMed; (d) S. L. Harbeson and R. D. Tung, MedChem News, 2014, 24, 8–22 Search PubMed; (e) G. S. Timmins, Expert Opin. Ther. Pat., 2014, 24, 1067–1075 CrossRef CAS PubMed; (f) T. G. Gant, J. Med. Chem., 2014, 57, 3595–3611 CrossRef CAS PubMed; (g) T. Pirali, M. Serafini, S. Cargnin and A. A. Genazzani, J. Med. Chem., 2019, 62, 5276–5297 CrossRef CAS PubMed; (h) R. D. Tung, Future Med. Chem., 2016, 8, 491–494 CrossRef CAS PubMed; (i) A. Mullard, Nat. Rev. Drug Discovery, 2016, 15, 219–221 CrossRef CAS PubMed; (j) B. Halford, Chem. Eng. News, 2016, 94, 32–36 Search PubMed; (k) G. S. Timmins, Expert Opin. Ther. Pat., 2017, 27, 1353–1361 CrossRef CAS PubMed.
- (a) A. E. Mutlib, R. J. Gerson, P. C. Meunier, P. J. Haley, H. Chen, L. S. Gan, M. H. Davies, B. Gemzik, D. D. Christ, D. F. Krahn, J. A. Markwalder, S. P. Seitz, R. T. Robertson and G. T. Miwa, Toxicol. Appl. Pharmacol., 2000, 169, 102–113 CrossRef CAS PubMed; (b) A. W. Czarnik, US Pat., US 2009/0075991 A1, 2009 Search PubMed.
- F. Maltais, Y. C. Jung, M. Chen, J. Tanoury, R. B. Perni, N. Mani, L. Laitinen, H. Huang, S. Liao, H. Gao, H. Tsao, E. Block, C. Ma, R. S. Shawgo, C. Town, C. L. Brummel, D. Howe, S. Pazhanisamy, S. Raybuck, M. Namchuk and Y. L. Bennani, J. Med. Chem., 2009, 52, 7993–8001 CrossRef CAS PubMed.
- K. Sanderson, Nature, 2009, 458, 269 CAS.
- A. Sommer, C. Zhang, J. Carter, J. Arthur, M. Bradbury, T. Gant and M. Shahbaz, US Pat., US 2016/0222008 A1, 2016 Search PubMed.
- (a) J. Siffert, WO Pat., WO2016/040930 A1, 2016 Search PubMed; (b) R. P. Garay and G. T. Grossberg, Expert Opin. Invest. Drugs, 2017, 26, 121–132 CrossRef CAS PubMed.
- (a) U.S. National Library of Medicine, https://clinicaltrials.gov/ct2/show/NCT02153502, accessed May 11, 2021; (b) R. P. Garay, C. A. Zarate, T. Charpeaud, L. Citrome, C. U. Correll, A. Hameg and P.-M. Llorca, Expert Rev. Neurother., 2017, 17, 593–609 CrossRef CAS PubMed.
- V. Braman, US Pat., US 2018/0353500 A1, 2018 Search PubMed.
- R. M. Moslin, D. S. Weinstein, S. T. Wrobleski, S. J. Tokarski and A. Kumar, WO Pat., WO 2014/074661 A1, 2014 Search PubMed.
- C. Schmidt, Nat. Biotechnol., 2017, 35, 493–494 CrossRef CAS PubMed.
- J. F. Liu, S. L. Harbeson, C. L. Brummel, R. Tung, R. Silverman and D. Doller, in Annual Reports in Medicinal Chemistry, ed. R. A. Goodnow, Elsevier, 2017, ch. 14, vol. 50, pp. 519–542 Search PubMed.
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed.
- B. J. Simmons, M. Hoffmann, J. Hwang, M. K. Jackl and N. K. Garg, Org. Lett., 2017, 19, 1910–1913 CrossRef CAS PubMed.
- (a) B. Zhang, H. Li, Y. Ding, Y. Yan and J. An, J. Org. Chem., 2018, 83, 6006–6014 CrossRef CAS PubMed; (b) Y. Ding, S. Luo, A. Adijiang, H. Zhao and J. An, J. Org. Chem., 2018, 83, 12269–12274 CrossRef CAS PubMed; (c) Y. Ding, S. Luo, C. Weng and J. An, J. Org. Chem., 2019, 84, 15098–15105 CrossRef CAS PubMed; (d) R. Mészáros, B.-J. Peng, S. B. Ötvös, S.-C. Yang and F. Fülöp, ChemPlusChem, 2019, 84, 1508–1511 CrossRef PubMed; (e) A. Kurimoto, R. S. Sherbo, Y. Cao, N. W. X. Loo and C. P. Berlinguette, Nat. Catal., 2020, 3, 719–726 CrossRef CAS.
- (a) For a review, see: A. Michelotti and M. Roche, Synthesis, 2019, 51, 1319–1328 CrossRef CAS; (b) M. Maeda, O. Ogawa and Y. Kawazoe, Chem. Pharm. Bull., 1977, 25, 3329–3333 CrossRef CAS; (c) M. Takahashi, K. Oshima and S. Matsubara, Chem. Lett., 2005, 34, 192–193 CrossRef CAS; (d) E. Alexakis, M. J. Hickey, J. R. Jones, L. P. Kingston, W. J. S. Lockley, A. N. Mather, T. Smith and D. J. Wilkinson, Tetrahedron Lett., 2005, 46, 4291–4293 CrossRef CAS; (e) L. Neubert, D. Michalik, S. Bähn, S. Imm, H. Neumann, J. Atzrodt, V. Derdau, W. Holla and M. Beller, J. Am. Chem. Soc., 2012, 134, 12239–12244 CrossRef CAS PubMed; (f) G. Pieters, C. Taglang, E. Bonnefille, T. Gutmann, C. Puente, J.-C. Berthet, C. Dugave, B. Chaudret and B. Rousseau, Angew. Chem., Int. Ed., 2014, 53, 230–234 CrossRef CAS PubMed; (g) T. Maegawa, A. Akashi, H. Esaki, F. Aoki, H. Sajiki and K. Hirota, Synlett, 2005, 2005, 0845–0847 Search PubMed; (h) F. T. Jere, D. J. Miller and J. E. Jackson, Org. Lett., 2003, 5, 527–530 CrossRef CAS PubMed; (i) C. Taglang, L. M. Martínez-Prieto, I. del Rosal, L. Maron, R. Poteau, K. Philippot, B. Chaudret, S. Perato, A. Sam Lone, C. Puente, C. Dugave, B. Rousseau and G. Pieters, Angew. Chem., Int. Ed., 2015, 54, 10474–10477 CrossRef CAS PubMed; (j) K. Moozeh, S. M. So and J. Chin, Angew. Chem., Int. Ed., 2015, 54, 9381–9385 CrossRef CAS PubMed; (k) L. V. A. Hale and N. K. Szymczak, J. Am. Chem. Soc., 2016, 138, 13489–13492 CrossRef CAS PubMed; (l) S. Bhatia, G. Spahlinger, N. Boukhumseen, Q. Boll, Z. Li and J. E. Jackson, Eur. J. Org. Chem., 2016, 2016, 4230–4235 CrossRef CAS; (m) A. Michelotti, F. Rodrigues and M. Roche, Org. Process Res. Dev., 2017, 21, 1741–1744 CrossRef CAS; (n) C. Taglang, D. E. Korenchan, C. von Morze, J. Yu, C. Najac, S. Wang, J. E. Blecha, S. Subramaniam, R. Bok, H. F. VanBrocklin, D. B. Vigneron, S. M. Ronen, R. Sriram, J. Kurhanewicz, D. M. Wilson and R. R. Flavell, Chem. Commun., 2018, 54, 5233–5236 RSC; (o) B. Chatterjee, V. Krishnakumar and C. Gunanathan, Org. Lett., 2016, 18, 5892–5895 CrossRef CAS PubMed; (p) Y. Y. Loh, K. Nagao, A. J. Hoover, D. Hesk, N. R. Rivera, S. L. Colletti, I. W. Davies and D. W. C. MacMillan, Science, 2017, 358, 1182 CrossRef CAS PubMed; (q) Y. Hu, L. Liang, W.-t. Wei, X. Sun, X.-j. Zhang and M. Yan, Tetrahedron, 2015, 71, 1425–1430 CrossRef CAS.
- Y. Chang, A. Yesilcimen, M. Cao, Y. Zhang, B. Zhang, J. Z. Chan and M. Wasa, J. Am. Chem. Soc., 2019, 141, 14570–14575 CrossRef CAS PubMed.
- For review articles, see: (a) K. A. DeKorver, H. Li, A. G. Lohse, R. Hayashi, Z. Lu, Y. Zhang and R. P. Hsung, Chem. Rev., 2010, 110, 5064–5106 CrossRef CAS PubMed; (b) G. Evano, A. Coste and K. Jouvin, Angew. Chem., Int. Ed., 2010, 49, 2840–2859 CrossRef CAS PubMed; (c) G. Evano, C. Theunissen and M. Lecomte, Aldrichimica Acta, 2015, 48, 59–70 CAS.
- For representative contributions from our group, see: (a) W. Gati, M. M. Rammah, M. B. Rammah, F. Couty and G. Evano, J. Am. Chem. Soc., 2012, 134, 9078–9081 CrossRef CAS PubMed; (b) W. Gati, F. Couty, T. Boubaker, M. M. Rammah, M. B. Rammah and G. Evano, Org. Lett., 2013, 15, 3122–3125 CrossRef CAS PubMed; (c) C. Theunissen, B. Métayer, N. Henry, G. Compain, J. Marrot, A. Martin-Mingot, S. Thibaudeau and G. Evano, J. Am. Chem. Soc., 2014, 136, 12528–12531 CrossRef CAS PubMed; (d) H. A. Laub, G. Evano and H. Mayr, Angew. Chem., Int. Ed., 2014, 53, 4968–4971 CrossRef CAS PubMed; (e) M. Lecomte and G. Evano, Angew. Chem., Int. Ed., 2016, 55, 4547–4551 CrossRef CAS PubMed; (f) P. Thilmany and G. Evano, Angew. Chem., Int. Ed., 2020, 59, 242–246 CrossRef CAS PubMed.
- For review articles, see: (a) C. Madelaine, V. Valerio and N. Maulide, Chem.–Asian J., 2011, 6, 2224–2239 CrossRef CAS PubMed; (b) G. Evano, M. Lecomte, P. Thilmany and C. Theunissen, Synthesis, 2017, 49, 3183–3214 CrossRef CAS.
- (a) G. Evano, K. Jouvin and A. Coste, Synthesis, 2013, 45, 17–26 CrossRef CAS; (b) Y. Zhang, R. P. Hsung, M. R. Tracey, K. C. M. Kurtz and E. L. Vera, Org. Lett., 2004, 6, 1151–1154 CrossRef CAS PubMed; (c) T. Hamada, X. Ye and S. S. Stahl, J. Am. Chem. Soc., 2008, 130, 833–835 CrossRef CAS PubMed; (d) A. Coste, G. Karthikeyan, F. Couty and G. Evano, Angew. Chem., Int. Ed., 2009, 48, 4381–4385 CrossRef CAS PubMed; (e) K. Jouvin, F. Couty and G. Evano, Org. Lett., 2010, 12, 3272–3275 CrossRef CAS PubMed; (f) K. Jouvin, J. Heimburger and G. Evano, Chem. Sci., 2012, 3, 756–760 RSC.
- X. Zeng, Chem. Rev., 2013, 113, 6864–6900 CrossRef CAS PubMed.
- For examples of ionic reduction of enamides, see: (a) M. M. Hinman and C. H. Heathcock, J. Org. Chem., 2001, 66, 7751–7756 CrossRef CAS PubMed; (b) M. N. Masuno and T. F. Molinski, Tetrahedron Lett., 2001, 42, 8263–8266 CrossRef CAS.
- For the partial reduction of alkynes with silanes, see: (a) M. Yan, T. Jin, Y. Ishikawa, T. Minato, T. Fujita, L.-Y. Chen, M. Bao, N. Asao, M.-W. Chen and Y. Yamamoto, J. Am. Chem. Soc., 2012, 134, 17536–17542 CrossRef CAS PubMed; (b) J. Chen, X. Shen and Z. Lu, J. Am. Chem. Soc., 2020, 142, 14455–14460 CrossRef CAS PubMed.
- Y. Zhang, R. P. Hsung, X. Zhang, J. Huang, B. W. Slafer and A. Davis, Org. Lett., 2005, 7, 1047–1050 CrossRef CAS PubMed.
- (a) M. D. Berry, J. Neurochem., 2004, 90, 257–271 CrossRef CAS PubMed; (b) A. H. Lewin, AAPS J., 2006, 8, E138–E145 CrossRef CAS PubMed; (c) L. Lindemann and M. C. Hoener, Trends Pharmacol. Sci., 2005, 26, 274–281 CrossRef CAS PubMed.
- (a) M. D. Berry, Rev. Recent Clin. Trials, 2007, 2, 3–19 CrossRef CAS PubMed; (b) T. A. Branchek and T. P. Blackburn, Curr. Opin. Pharmacol., 2003, 3, 90–97 CrossRef CAS PubMed.
- B. Belleau, J. Burba, M. Pindell and J. Reiffenstein, Science, 1961, 133, 102–104 CrossRef CAS PubMed.
- W. G. Meissner, M. Frasier, T. Gasser, C. G. Goetz, A. Lozano, P. Piccini, J. A. Obeso, O. Rascol, A. Schapira, V. Voon, D. M. Weiner, F. Tison and E. Bezard, Nat. Rev. Drug Discovery, 2011, 10, 377–393 CrossRef CAS PubMed.
- (a) T. Malmlöf, D. Rylander, R.-G. Alken, F. Schneider, T. H. Svensson, M. A. Cenci and B. Schilström, Exp. Neurol., 2010, 225, 408–415 CrossRef PubMed; (b) F. Schneider, L. Erisson, H. Beygi, M. Bradbury, O. Cohen-Barak, I. D. Grachev, S. Guzy, P. S. Loupe, M. Levi, M. McDonald, J.-M. Savola, S. Papapetropoulos, W. G. Tracewell, M. Velinova and O. Spiegelstein, Br. J. Clin. Pharmacol., 2018, 84, 2422–2432 CrossRef CAS PubMed.
- C. W. Cheung and S. L. Buchwald, Org. Lett., 2013, 15, 3998–4001 CrossRef CAS PubMed.
- P. J. Kocieński, B. Pelotier, J.-M. Pons and H. Prideaux, J. Chem. Soc., Perkin Trans. 1, 1998, 1373–1382 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization and copies of NMR spectra. See DOI: 10.1039/d1sc02622d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |