
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceResolving electron injection from singlet fission-borne triplets into mesoporous transparent conducting oxides†
Melissa K.
Gish
 a,
Emily K.
Raulerson
a,
Ryan T.
Pekarek
a,
Ann L.
Greenaway
a,
Karl J.
Thorley
b,
Nathan R.
Neale
a,
John E.
Anthony
b and
Justin C.
Johnson
*a
a,
Emily K.
Raulerson
a,
Ryan T.
Pekarek
a,
Ann L.
Greenaway
a,
Karl J.
Thorley
b,
Nathan R.
Neale
a,
John E.
Anthony
b and
Justin C.
Johnson
*a
aNational Renewable Energy Laboratory, 15013 Denver West Pkwy, CO 80401, USA. E-mail: justin.johnson@nrel.gov
bDepartment of Chemistry, University of Kentucky, Lexington, Kentucky 40506, USA
First published on 19th July 2021
Abstract
Photoinduced electron transfer into mesoporous oxide substrates is well-known to occur efficiently for both singlet and triplet excited states in conventional metal-to-ligand charge transfer (MLCT) dyes. However, in all-organic dyes that have the potential for producing two triplet states from one absorbed photon, called singlet fission dyes, the dynamics of electron injection from singlet vs. triplet excited states has not been elucidated. Using applied bias transient absorption spectroscopy with an anthradithiophene-based chromophore (ADT-COOH) adsorbed to mesoporous indium tin oxide (nanoITO), we modulate the driving force and observe changes in electron injection dynamics. ADT-COOH is known to undergo fast triplet pair formation in solid-state films. We find that the electronic coupling at the interface is roughly one order of magnitude weaker for triplet vs. singlet electron injection, which is potentially related to the highly localized nature of triplets without significant charge-transfer character. Through the use of applied bias on nanoITO:ADT-COOH films, we map the electron injection rate constant dependence on driving force, finding negligible injection from triplets at zero bias due to competing recombination channels. However, at driving forces greater than −0.6 eV, electron injection from the triplet accelerates and clearly produces a trend with increased applied bias that matches predictions from Marcus theory with a metallic acceptor.
Introduction
Molecular chromophores with strong and panchromatic absorption are important elements for maximizing photoelectrochemical conversion of the visible solar spectrum. Dye-sensitized solar cells (DSSCs), for example, combine wide band-gap metal-oxide semiconductors with molecular photosensitizers to generate electricity from sunlight; however, in the past 25 years, power conversion efficiencies (PCE) have increased only slightly from 10% in 1997 (ref. 1) to 13.5% in 2021.2 Utilizing chromophores with exciton multiplication potential, such as singlet fission (SF) dyes, offers an opportunity to exceed conventional PCE limits.3,4 Understanding of SF in various molecular chromophores has advanced significantly over the past decade.5–9 In the ideal scenario, each incoming photon can be converted to two electron–hole pairs, as singlet fission is a spin-allowed transformation of one singlet to two triplet excitons. A dye-sensitized solar cell based solely on SF dyes would double photocurrent but halve photovoltage of the corresponding optimized conventional DSSC, resulting in no gain in PCE.10 However, by combining a SF chromophore with a conventional red-absorbing chromophore, significant increases in DSSC efficiency could be realized.10,11 In addition to the potential for charge transfer to the oxide, the primary distinction between prior SF studies and those involving DSSCs is the unique arrangement of chromophores at the surface of TiO2, which is likely neither crystalline nor isotropic as in solution. Several initial attempts to build and characterize SF DSSCs, or DSSC-like architectures, have been reported. In some cases, evidence for enhanced photocurrent due to singlet fission was discovered.12–14 However, distinct challenges have also been realized, including competitive excimer formation and fast charge transfer from the singlet state.14–16Excited-state electron injection at metal oxide:molecule interfaces has been extensively studied for organometallic complexes, particularly for ruthenium trisbipyridine derivatives (Ru(bpy)32+, Ru) at the TiO2 interface.17–19 Intersystem crossing (ISC) for Ru occurs in 100s of femtoseconds due to strong spin–orbit coupling, meaning the majority of observable electron injection in a transient absorption experiment is due to triplets. In many organometallics, a metal-to-ligand charge transfer state (MLCT) is created upon visible light photoexcitation, where the promoted excited electron is spatially separated from the cation on the metal centre. Whereas singlet injection is typically complete within 1 ps, triplet electron injection from a Ru derivative occurs on the order of 10s to 100s of picoseconds, which remains significantly faster than competing processes and results in high injection efficiency.17,18
A major difference between organometallic complexes and organic SF molecules is the spatial separation between the electron and hole prior to electron injection. Recent work from our group showed triplet charge transfer rates between pentacene and molecular acceptors that were significantly lower than the predicted rates from the diffusion model, suggesting intrinsically slow triplet dissociation kinetics.20 We conjectured that the native localized properties of the triplet render its electronic interaction with charge acceptors much weaker than that of singlets. In addition, our past work has demonstrated that electron transfer from singlet and triplet states follow Marcus type trends in rate constant vs. driving force from SF chromophores to molecular acceptors, with the singlet and triplet each possessing its own normal and inverted region.21 Electronic delocalization differences between singlets and triplets are likely to be much less severe in MLCT dyes typically employed in DSSCs due to their low exchange splitting compared with conjugated organic systems. Additional insight into the important charge-generation processes involving triplets could be gained through systematic investigation into the interaction of purely organic triplets with different types of charge acceptors.
Tuning driving force using molecular structure (i.e., with electron withdrawing and donating substituents), as we had previously achieved, is useful in some cases but can be difficult over large potential ranges and can introduce undesired or unknown changes in donor–acceptor electronic coupling. Changes in dye–TiO2 electronic coupling related to molecular design are frequently reported to influence the charge transfer rate constant,18,22 but changes related to driving force are scarce. In the regime of relatively strong coupling, the dependence on driving force is found to be minimal, as sub-ps injection is often observed.23 However, the reverse process of back electron transfer has been reported to show a Marcus inverted region,24,25 partially due to the weaker electronic coupling between the oxidized dye and the injected electron in TiO2 that brings such rates into the ns regime. The forward electron transfer from triplet states has been investigated in an MLCT DSSC system, showing an increase followed by a saturation with increasing driving force expected for Marcus behaviour in the normal regime.26 The triplet was also found to inject much more slowly than the singlet, although this was mostly attributed to the density of states at the TiO2 band edge at the respective singlet vs. triplet energies, and not to localization properties that might influence electronic coupling.
To explore the full extent of driving force regimes with singlet fission dyes, we have chosen to employ a potential bias on nanocrystalline indium tin oxide (nanoITO) substrates decorated with dyes. ITO has distinct advantages over TiO2 in terms of the ability to modulate the Fermi level through a large voltage range.27 As we reveal through transient absorption experiments below, the photophysics of the carboxylic acid functionalized polyacene derivative anthradithiophene (ADT-COOH, Fig. 1A), a well-characterized SF system,28,29 are altered in a systematic way at different potentials, leading to an opening of the triplet charge transfer channel not available at zero bias (Fig. 1B). Unique features of this architecture enable control, though not entirely independent, of the singlet and triplet electron transfer dynamics that directly influence the photophysical outcome.
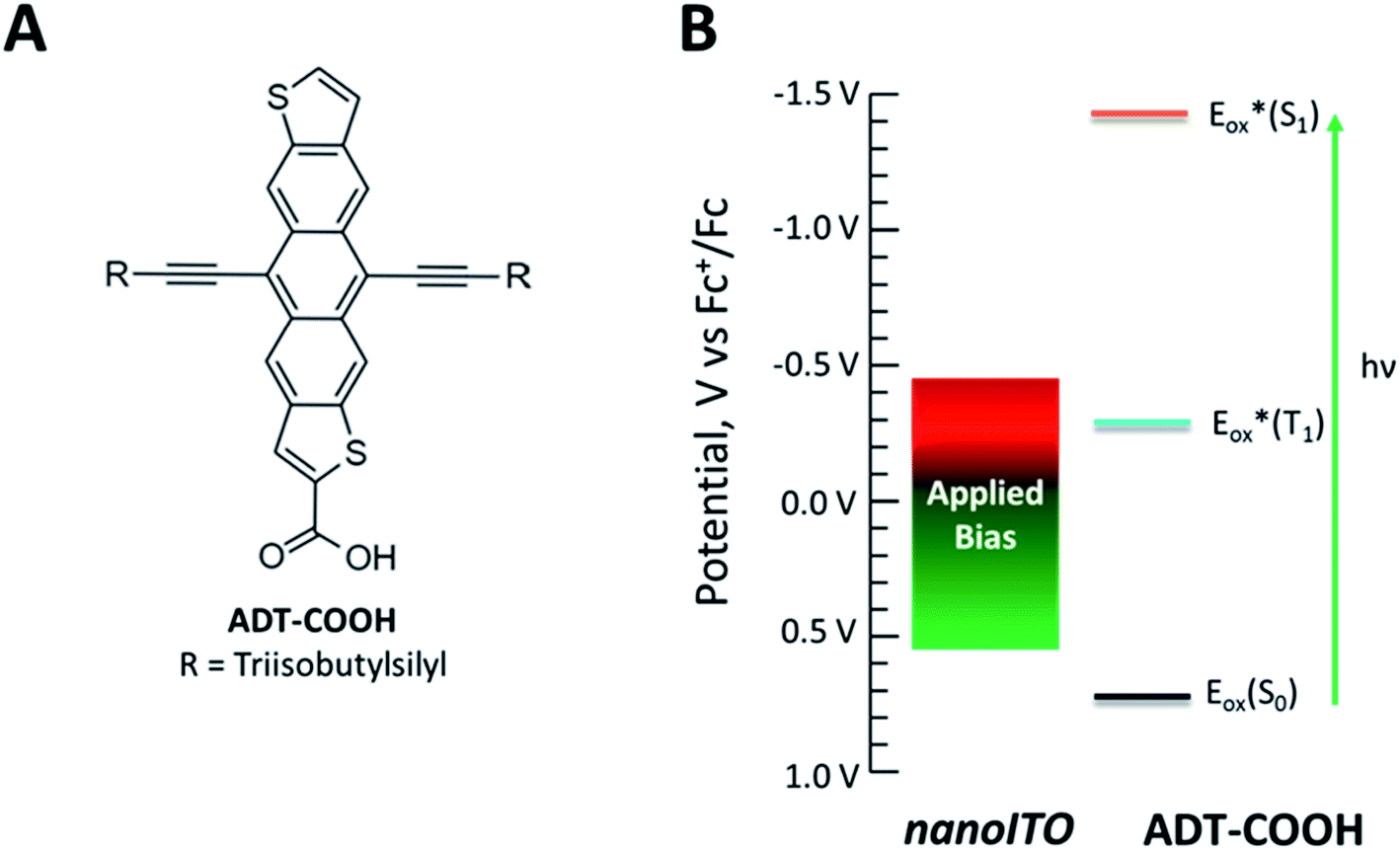 | ||
| Fig. 1 (A) Molecular structure of ADT-COOH. (B) Diagram of the range of potentials studied in this experiment and the redox properties of ADT-COOH. | ||
Experimental
Chromophore synthesis
Synthesis of ADT-COOH is described in ref. 28.Dye loading
nanoITO films were submerged in a saturated (>200 μM) solution of ADT-COOH dissolved in THF overnight. The films were rinsed with THF to remove excess unbound dye and dried under a stream of N2.Sample preparation
For dry transient absorption experiments, slides were placed in a sealed chamber in a nitrogen glove box. For triplet sensitization experiments, ADT-COOH and anthracene were dissolved in tetrahydrofuran in a 1 mm cuvette, as described in ref. 28. For applied bias transient absorption experiments, a spectroelectrochemical cell from RedoxMe (Spectro-EFC SMA 905 1.75 mL) was assembled in a nitrogen glovebox and filled with 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) in acetonitrile (MeCN) and sealed. The sample acted as the working electrode, while the reference and counter electrodes were both platinum wires. A Pine Instruments WaveNano potentiostat was used to apply a potential to the sample (working electrode) using the AfterMath software to monitor the current. Prior to starting the transient absorption experiment, the sample was allowed to stabilize for 10 min after the potential was applied.Atomic layer deposition
Atomic Layer Deposition (ALD) of aluminum oxide was performed using a Beneq TFS 200 operated at 90 °C and 5 mbar. Trimethylaluminum and water from stainless steel bubblers at 20 °C were used as precursors with nitrogen as a carrier gas. Deposition consisted of alternating 0.2 s exposures of trimethylaluminum and water separated by 5 s purge times. Seven exposure sequences (ALD 'cycles') were used to deposit ∼0.8 nm aluminum oxide (Al2O3), based on the regular calibration for this tool.Cyclic voltammetry
Cyclic voltammetry of ADT-COOH was performed in 0.1 M TBAPF6 in degassed dichloromethane. The working and counter electrodes were platinum with a Ag/AgNO3 reference electrode. Potentials were referenced to Fc+/Fc.Steady state spectroelectrochemistry
Steady state spectroelectrochemical measurements of ADT-COOH in solution were completed in an 0.1 M bis(trifluoromethane)sulfonimide lithium salt in dry acetonitrile to identify the cation spectrum. A three-electrode configuration with a Ag wire pseudoreference electrode and Pt working and counter electrodes with an internal standard of ferrocene. The potentiostat was a Princeton Applied Research Model 263 potentiostat and bias was applied in 50 or 100 mV intervals. An OceanFX spectrometer (Ocean Optics, UV-vis) and a NIRQuest 512-2.2 spectrometer (Ocean Optics, NIR) was used to collect absorption spectra. All measurements were completed in an argon-filled glovebox with electrical and fibre optic feedthroughs. The UV-vis/NIR spectrum at open circuit voltage was subtracted from applied potentials to determine the difference spectrum.Photocurrent
Photocurrent measurements were performed in 0.1 M TBAPF6 in dry acetonitrile in a standard three-electrode set up. The working electrode was nanoITO:ADT-COOH with Pt counter and reference electrodes. Potentials were referenced to Fc+/Fc. The chronoamperometry data was collected on a Biologics potentiostat using EC-Lab software. The potential was stepped at 100 mV intervals from OCV and the current was allowed to equilibrate before beginning the photocurrent measurement. After the current equilibration, a 530 nm (20 mW) LED (ThorLabs, M530F2) illuminated the working electrode from the back in 3 second on/off intervals through 10 cycles. The photocurrent was taken as the difference in the current between the LED on and LED off states.UV-visible spectroscopy
Steady state absorption spectra were collected using a UV-visible-NIR absorption spectrophotometer (Agilent Technologies, model 8453 A) with an air blank used for baseline correction.Transient absorption spectroscopy
A Coherent Libra Ti:sapphire laser (1 kHz, 800 nm output, 150 fs pulse width) was used for ultrafast transient absorption experiments. The 500 nm pump pulses were generated in a TOPAS-C optical parametric amplifier. The white light probe pulses were produced by focusing a small portion of the Libra output into a thin or thick sapphire window for visible (λprobe = 440–800 nm) or NIR (λprobe = 750–1600 nm) measurements, respectively. Pump and probe were spatially overlapped at the sample and a mechanical delay stage was used to delay the probe relative to the pump. A portion of the probe was redirected before the sample as a reference to reduce noise to <0.1 mOD. Changes in the probe were monitored through a fibre optic coupled multichannel spectrometer with a CMOS sensor. Helios software from Ultrafast Systems was used to collect the data. Data were chirp corrected and analysed with Ultrafast Systems' SurfaceXplorer software.Results
The signature vibronic peaks of ADT-COOH in solution in the UV-visible absorption spectrum are unperturbed upon adsorption to nanoITO (Fig. S1A†), indicating that the intermolecular electronic coupling between ADT-COOH molecules is weak compared with what is observed in polycrystalline films.28 The broad underlying absorption in nanoITO:ADT-COOH is representative of the optical band gap of the nanoITO with some contribution from light scattering, which is typical in mesoporous metal oxide films. The ground state oxidation potential of ADT-COOH is 0.72 V vs. Fc+/Fc as determined through cyclic voltammetry (Fig. S1B†). The excited state oxidation potentials for ADT-COOH (referred to as 1 in eqn (1a), (1b) and (2)) excited singlet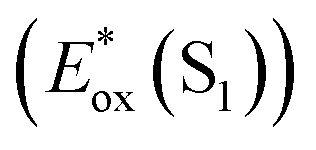 and excited triplet
and excited triplet 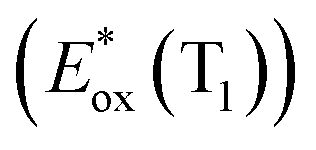 are calculated using eqn (1a) and (1b), respectively:17,30
are calculated using eqn (1a) and (1b), respectively:17,30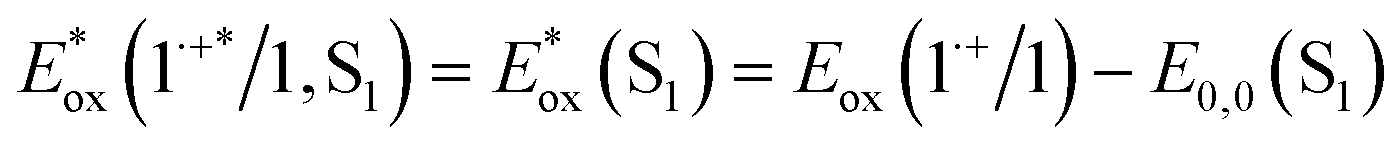 | (1a) |
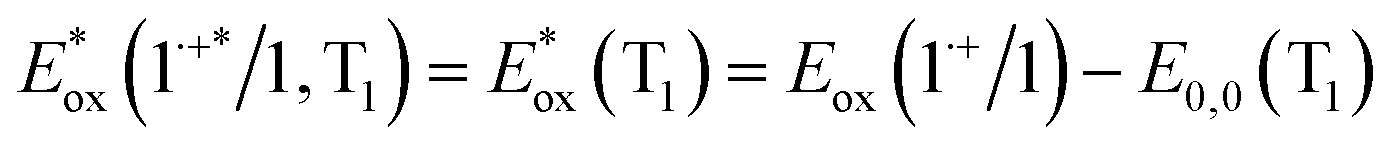 | (1b) |
Table 1 provides the relevant parameters for eqn (1a) and (1b), where the singlet energy (E0,0(S1)) is 2.14 eV, as determined in our previous work from the intersection of the UV-visible and emission spectra,28 and we estimate the triplet energy (E0,0(T1)) as roughly 1 eV from previously published studies on similar ADT derivatives.29 We note that the actual triplet energy may be slightly higher for ADT-COOH (∼1.1 eV) based on evidence for roughly isoergic singlet fission behaviour in neat films.28 For the purposes of our calculations, we use a triplet energy value of 1.05 eV. The calculated excited state oxidation potentials are −1.42 V vs. Fc+/Fc for the singlet and −0.33 V vs. Fc+/Fc for the triplet. As described by Romero et al., a single electron transfer on a per molar basis assumes a conversion factor of 1 eV/V accounting for the unit discrepancy between Eox and E0,0.30
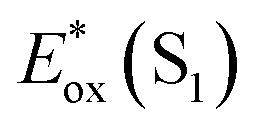
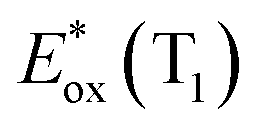
The excited state oxidation potential dictates the driving force (ΔGET) for molecular excited state electron injection into the metal oxide conduction band (ECB), as illustrated by eqn (2):
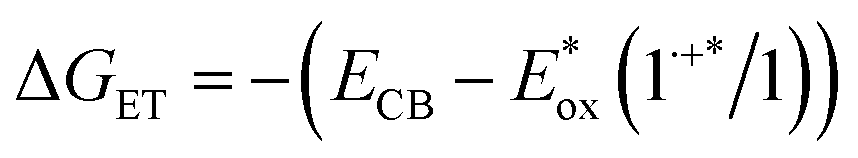 | (2) |
Based on this equation, the driving force for electron injection from triplet excited states from ADT-COOH into typical metal oxide semiconductors, is about 1 V more positive than the high driving force for electron injection from the first excited singlet.
To accurately assign features of the transient absorption data for the dye-sensitized films, we determined the spectral signatures for the singlet, the triplet, and the cation, or oxidized, ADT-COOHvia separate measurements. The singlet excited state spectrum was determined via transient absorption spectroscopy of ADT-COOH in THF solution (Fig. S1C,† purple). The singlet is characterized by a ground state bleach (GSB) at 575 nm, a stimulated emission feature at 625 nm, and two photoinduced absorption (PIAs) bands peaking at 500 nm, and 540 nm, and a broad absorption beyond the stimulated emission (650–750 nm). The spectral signature for oxidized ADT-COOH (cation, Fig. S1C,† orange) was determined through steady state spectroelectrochemistry, where the UV-visible spectra are monitored for changes as a function of applied potential. The oxidized cation spectrum consists of a GSB mirroring the ground state absorption (Fig. S1A†), as well as a broad absorption feature spanning 600 nm through 750 nm. The triplet–triplet absorption spectrum of ADT-COOH (Fig. S1C,† teal) was uncovered through sensitization with anthracene in THF solution. The triplet also contains GSB features, but they are outweighed by PIA peaks spanning 540–575 nm, and 580–625 nm. We note that there are no positive PIA features from the cation in the triplet region of interest for this work (λ = 540–575 nm).
As a benchmark comparison with prior SF-DSSC studies, we show transient absorption data collected under dry conditions. We note that pulse energies are maintained at low levels (60 nJ per pulse) to avoid nonlinear annihilation effects and dye desorption/degradation.31 To confirm the spectral assignments in our transient absorption experiments, we compared our sample of interest nanoITO:ADT-COOH (Fig. S2†) to nanoITO/Al2O3:ADT-COOH (Fig. 2), where we applied a thin insulating layer of aluminium oxide (Al2O3) on the nanoITO surface via atomic layer deposition (ALD). The alumina minimizes the kinetic competition of electron injection from the ADT-COOH singlet state by creating a physical tunnelling barrier, which allows us to isolate singlet fission dynamics at the interface.15 In Fig. 2A, the transient absorption spectra of the insulated sample are compared to the spectral signatures of the triplet excited state, singlet excited state, and the cation. The initially created excited state at 1.8 ps after photoexcitation is closely matched to the singlet excited state. Through 4.8 ns, a small peak at 560 nm appears, which matches well with the sensitized triplet spectrum. Monitoring the triplet signature at 560 nm (Fig. 2B) with this insulating layer present shows an initial rise (τ = 3.5 ps). A secondary rise in the triplet occurs with a time constant of 1195 ps. The singlet excited state decay measured at 745 nm is exponential with a time constant matching that of the secondary triplet rise (τ = 1112 ps). This behaviour is consistent with a biphasic singlet fission mechanism in nanoITO/Al2O3:ADT-COOH.
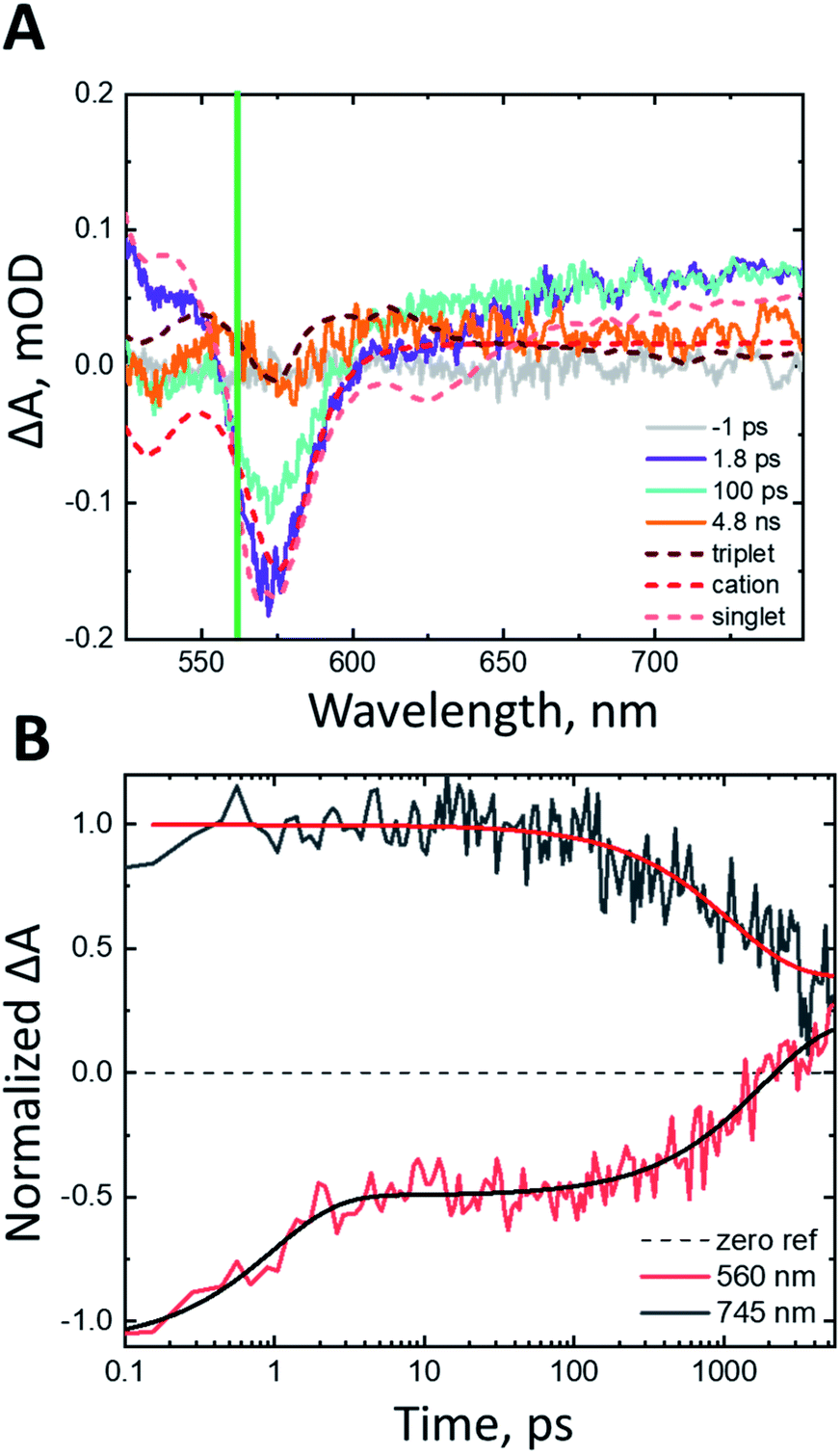 | ||
| Fig. 2 (A) Transient absorption spectra at pump-probe delays of −1 ps (grey), 1.8 ps (purple), 100 ps (teal), 4.8 ns (orange) after 500 nm photoexcitation (60 nJ per pulse) in N2 atmosphere. The spectral components of interest as shown as dashed lines. The triplet–triplet absorption spectrum as determined from anthracene sensitization in THF solution is shown in dark red, the cation as determined from steady state spectroelectrochemistry is shown in red and the singlet excited state from THF solution transient absorption measurements is shown in light red. These spectra are shown separately in Fig. S1C.† The green line denotes the wavelength of interest with kinetics shown in panel B. (B) Rise and decay kinetics at probe wavelengths of 560 nm and 745 nm of nanoITO/Al2O3:ADT-COOH after 500 nm photoexcitation (60 nJ per pulse) in N2 atmosphere. | ||
Fig. S2A† shows the transient absorption spectra of nanoITO:ADT-COOH in an N2 atmosphere. At 1.8 ps after photoexcitation, the spectrum is largely dominated by the singlet excited state, which evolves to a superposition of the cation and triplet excited state after 100 ps. Singlet injection is competitive with singlet fission, as evidenced by the lack of amplitude change in the GSB over the first 100 ps with an associated amplitude change in the broad PIA at wavelengths greater than 650 nm. The singlet decay at 745 nm is multiexponential and can be represented by three time constants: 2 ps, 18 ps, and 381 ps. The wavelength of interest for the triplet–triplet absorption spectrum is 570 nm (Fig. S2B†). A convolution of the singlet, cation, and triplet states causes the prominent triplet peak to appear slightly red-shifted from 560 nm in the sensitized spectrum (Fig. S1C†) to 570 nm in Fig. S2.† The rise is biexponential with time constants of 1.5 ps and 17 ps, which is close to the singlet decay time scale determined at 745 nm. Within the 5 ns observation window, the triplet signature decays back to baseline with a time constant of 1780 ps, which we tentatively assign to triplet–triplet or triplet-hole annihilation (vide infra).
With comparison to the insulated case in Fig. 2, the nanoITO:ADT-COOH spectra in Fig. S2† reveal two important conclusions: (1) the triplet decay in the presence of singlet injection is accelerated, most likely by the presence of holes; (2) the singlet fission kinetics overlap significantly with the singlet injection kinetics, as observed in the non-insulated case (Fig. 4, 5 and S2†), creating a nearly equal competition at zero applied bias. These observations serve as a baseline for our assignments with an applied potential, which we now describe below for uninsulated nanoITO:ADT-COOH.
Studying the behavior of these films in situ is critical to understanding the appropriate design parameters necessary for high-efficiency SF-DSSCs. The transient absorption spectra of nanoITO:ADT-COOH in 0.1 M TBAPF6 in MeCN after 500 nm photoexcitation (60 nJ per pulse) with no applied bias are shown in Fig. 3A. The spectrum initially resembles that of the singlet (Fig. S1C,† purple) at a pump-probe delay of 1.8 ps. At 150 ps, the broad PIA in the red spectral region has decayed, while the GSB at 575 nm has remained constant and a new GSB feature is uncovered at 545 nm, consistent with the cation spectrum (Fig. S1C,† orange). A peak consistent with the triplet excited state (Fig. S1C,† teal) grows in between 550 and 570 nm. This behavior is consistent with electron injection from the singlet excited state into the nanoITO occurring concurrently with singlet fission. As the pump-probe delay increases to 4.1 ns, all features decay significantly, with only a small amplitude remaining at 5 ns. With no applied bias, the charge separated state resulting from singlet injection decays within our observation window, likely through back electron transfer from the nanoITO to the cation of ADT-COOH.
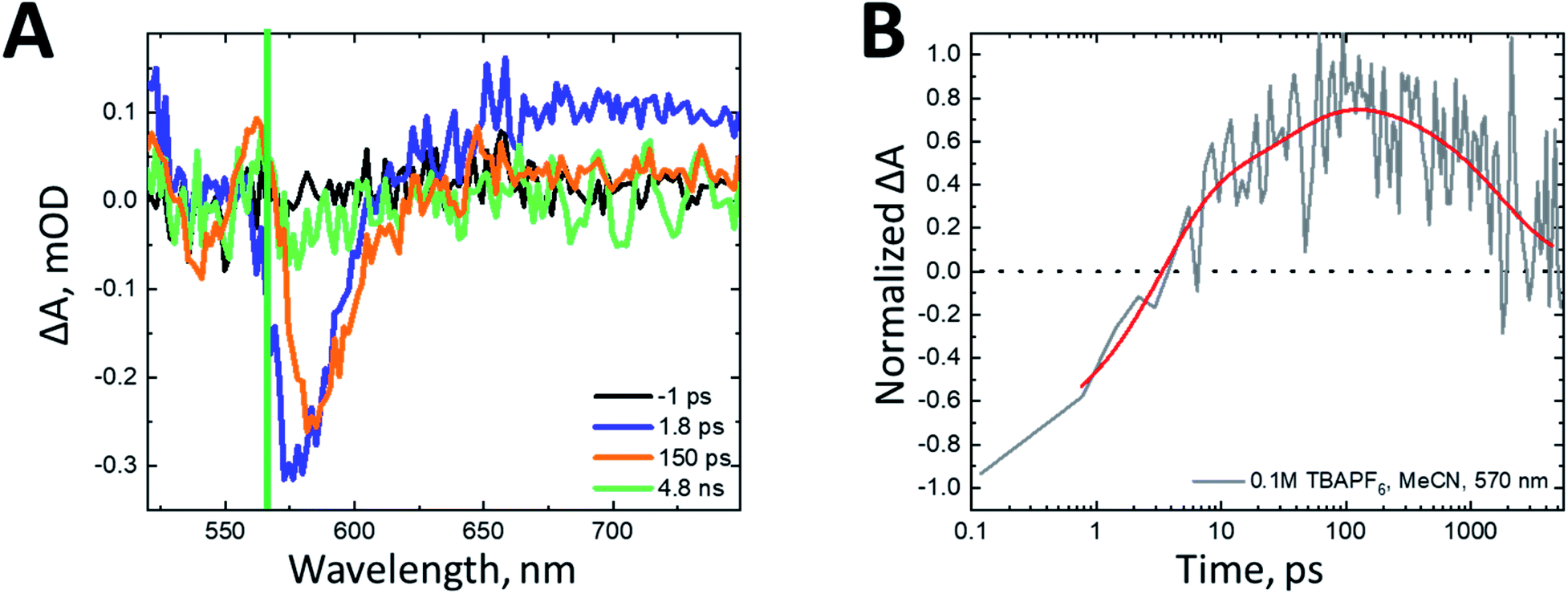 | ||
| Fig. 3 (A) Transient absorption spectra of nanoITO:ADT-COOH in 0.1 M TBAPF6 in MeCN with no applied bias after 500 nm (60 nJ per pulse) photoexcitation at pump-probe delays of −1 ps (black), 1.8 ps (blue), 150 ps (green), and 4.1 ns (orange). The green line denotes 570 nm, the wavelength probed in panel B. (B) Rise and decay kinetics at 570 nm of nanoITO:ADT-COOH in 0.1 M TBAPF6 in MeCN with no applied bias after 500 nm (60 nJ per pulse) photoexcitation. The kinetics data are normalized to the minimum of the ground state bleach and the triexponential fit is shown in red. | ||
Fig. 3B displays the kinetics of nanoITO:ADT-COOH at 570 nm, the peak of triplet absorption. The rise is biexponential with a 3.2 ps component accounting for about 70% of the increase and a 36 ps component for the remaining 30%. The decay of the triplet signal occurs with a time constant of 1782 ps. The triplet decay mechanism is likely not through electron injection into the nanoITO due to a lack of sufficient driving force at OCV conditions. Instead, triplets are probably decaying through an annihilation process, similar to that of the sample under dry conditions in N2 atmosphere (Fig. S2†), where the triplet rises biexponentially and decays with a time constant of 1780 ps.
To modulate the driving force for triplet injection, we applied a constant potential to the photoelectrode, where nanoITO:ADT-COOH is the working electrode, and the counter and reference electrodes are both Pt wires. Potentials were referenced to Fc+/Fc. We stepped the potential applied to the photoelectrode and monitored the current using chronoamperometry. The transient absorption experiment began once the current stabilized after a few minutes. Positive applied potentials lower the Fermi level in the nanoITO to increase the driving force for both singlet and triplet electron injection, whereas negative potentials increase the charge injection barrier. Fig. 4 shows a direct comparison between the dynamics of nanoITO:ADT-COOH at 0.35 V and 0.55 V vs. Fc+/Fc. At early pump-probe delays of 270 fs (Fig. 4A), the spectra are qualitatively similar, exhibiting spectral characteristics consistent with the excited singlet. The normalized spectra at 5 ns after photoexcitation exhibit many of the same features consistent with the cation, but there is a distinct difference at 570 nm, where the triplet excited state absorbs. At 0.35 V vs. Fc+/Fc, the triplet signal remains a strong PIA, while at 0.55 V, this feature has decayed into a weak GSB, representative of the cation.
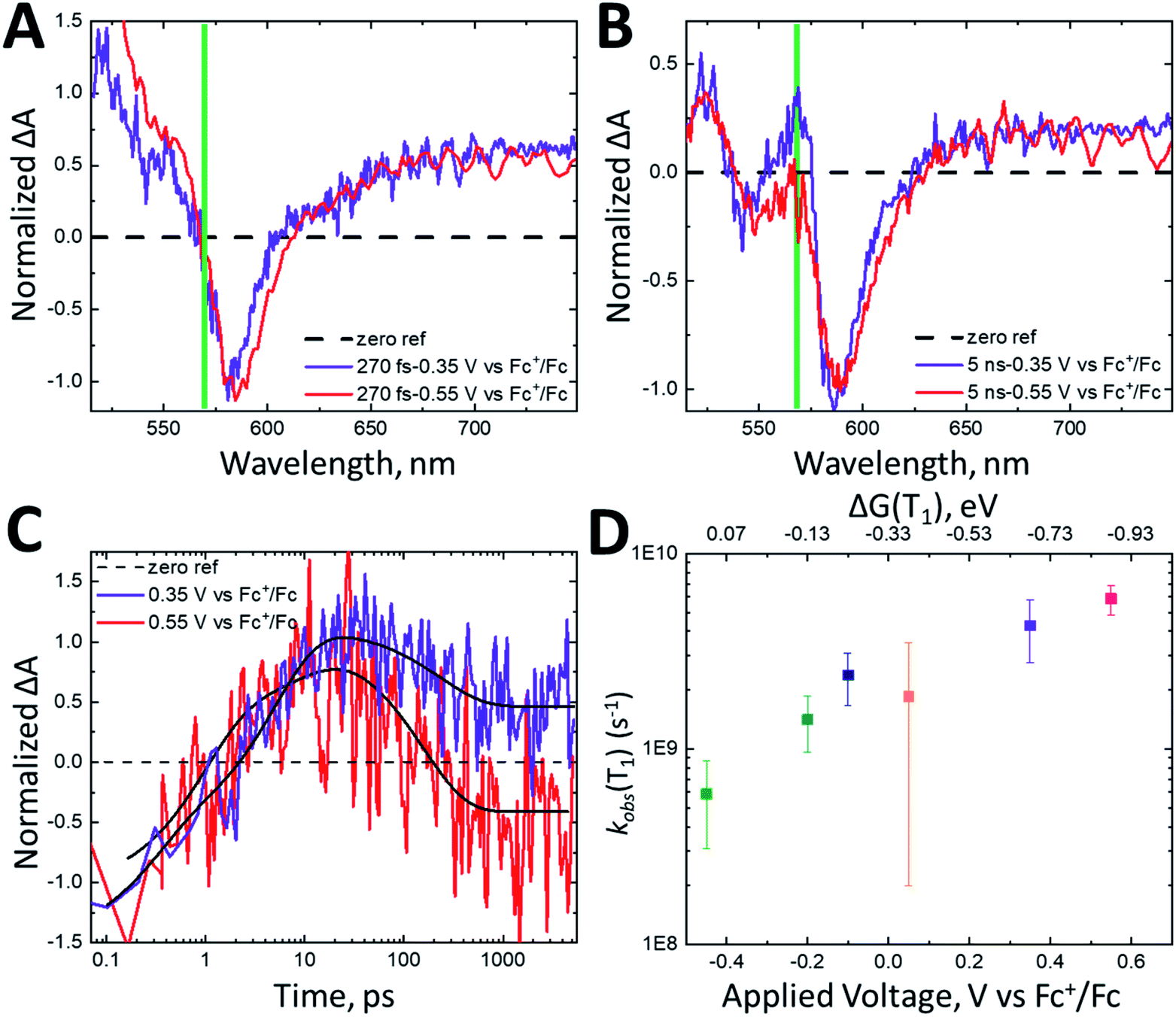 | ||
| Fig. 4 Normalized transient absorption spectra of nanoITO:ADT-COOH in 0.1 M TBAPF6 in MeCN at an applied potential of 0.35 V vs. Fc+/Fc (purple) and 0.55 V vs. Fc+/Fc (red) at (A) 270 fs and (B) 5 ns after 500 nm (60 nJ per pulse) photoexcitation. The green line in the spectra denotes the wavelength of interest at 570 nm, where the triplet absorbs. (C) Normalized kinetics monitored at 570 nm for nanoITO:ADT-COOH in 0.1 M TBAPF6 in MeCN at applied potentials of 0.35 V vs. Fc+/Fc (purple) and 0.55 V vs. Fc+/Fc (red). The kinetics are normalized to the GSB and triexponential fits are shown in black lines. (D) Observed triplet lifetimes (kobs(T1)) measured at 570 nm as a function of applied bias in V vs. Fc+/Fc. The top axis shows the driving force for triplet injection (ΔG(T1)) in eV as calculated using eqn (2). The data points here are averaged from three repeat experiments and the error bars are the averaged errors from the fits. | ||
The dynamics of the feature at 570 nm, shown in Fig. 4C, demonstrate a similar rise time scale (within 10 ps) at all biases, but starkly different decays. The decay for the 0.55 V bias is more rapid than for 0.35 V, and the positive feature decays into a GSB after about 200 ps. While it is difficult to quantify the efficiency of electron injection with the significantly superimposed spectral features of nanoITO:ADT-COOH, the increased rate of triplet decay in addition to the relative difference in triplet PIA vs. cation GSB magnitude between 0.35 V and 0.55 V at 5 ns implicates higher injection efficiencies with increased bias. This is particularly evident in Fig. 4C, where under an applied potential 0.55 V vs. Fc+/Fc, the signal at late times is negative. This is indicative of more cations present at higher applied potentials. The observed rate constants of triplet decay (kobs(T1)) as a function of applied bias (bottom axis) and driving force for triplet injection (ΔG(T1), top axis) are shown in Fig. 4D. Because of the strong spectral overlap of all features in ADT-COOH and the difficulty of isolating pure triplet excited state dynamics, we denote the observed triplet lifetimes as kobs(T1). A faster decay of kobs(T1) at an increasingly positive potential is consistent with increased electron injection from the triplet excited state.
The near-IR region of the spectrum for nanoITO:ADT-COOH has fewer overlapping spectral features than the visible spectral region, although there is no clearly observed triplet signature. Here, the singlet has a broad photoinduced absorption spanning 900–1600 nm, while there is a distinct cation feature centered at 1030 nm as determined through steady state spectroelectrochemistry (Fig. S1D†). Whereas we can probe the singlet directly at 1350 nm with no overlapping features, the cation feature exists on top of a portion of the singlet, thus we cannot observe pure cation dynamics by simply probing at 1030 nm.
Under no applied potential, the NIR spectrum (Fig. S3A†) at 230 fs has significant singlet character. The singlet excited state, monitored at 1350 nm, decays biexponentially (Fig. 5B) with time constants of 7.2 ps and 620 ps with roughly equal amplitude decays. The rise of the cation is close to the initial decay of the singlet at 10.6 ps (Fig. 5A). The cation subsequently decays significantly within the observation window with a time constant of 1319 ps. The singlet excited state decays faster at 0.35 V vs. Fc+/Fc than under no applied potential (τ = 4 ps, 167 ps). Interestingly, the singlet excited state decay remains unchanged between applied voltages of 0.35 V and 0.55 V vs. Fc+/Fc (Fig. 5B). The roughly 5 ps rise time of the cation is similar to the fast decay of the singlet for both 0.35 and 0.55 V vs. Fc+/Fc. The time scale for decay of the cation increases from 1319 ps at OCV to 1390 ps and >5000 ps at 0.35 V and 0.55 V vs. Fc+/Fc, respectively. Fitting the fast decay of the singlet at 1350 nm and plotting as a function of applied bias (Fig. 5C) shows little to no change in the rate constant.
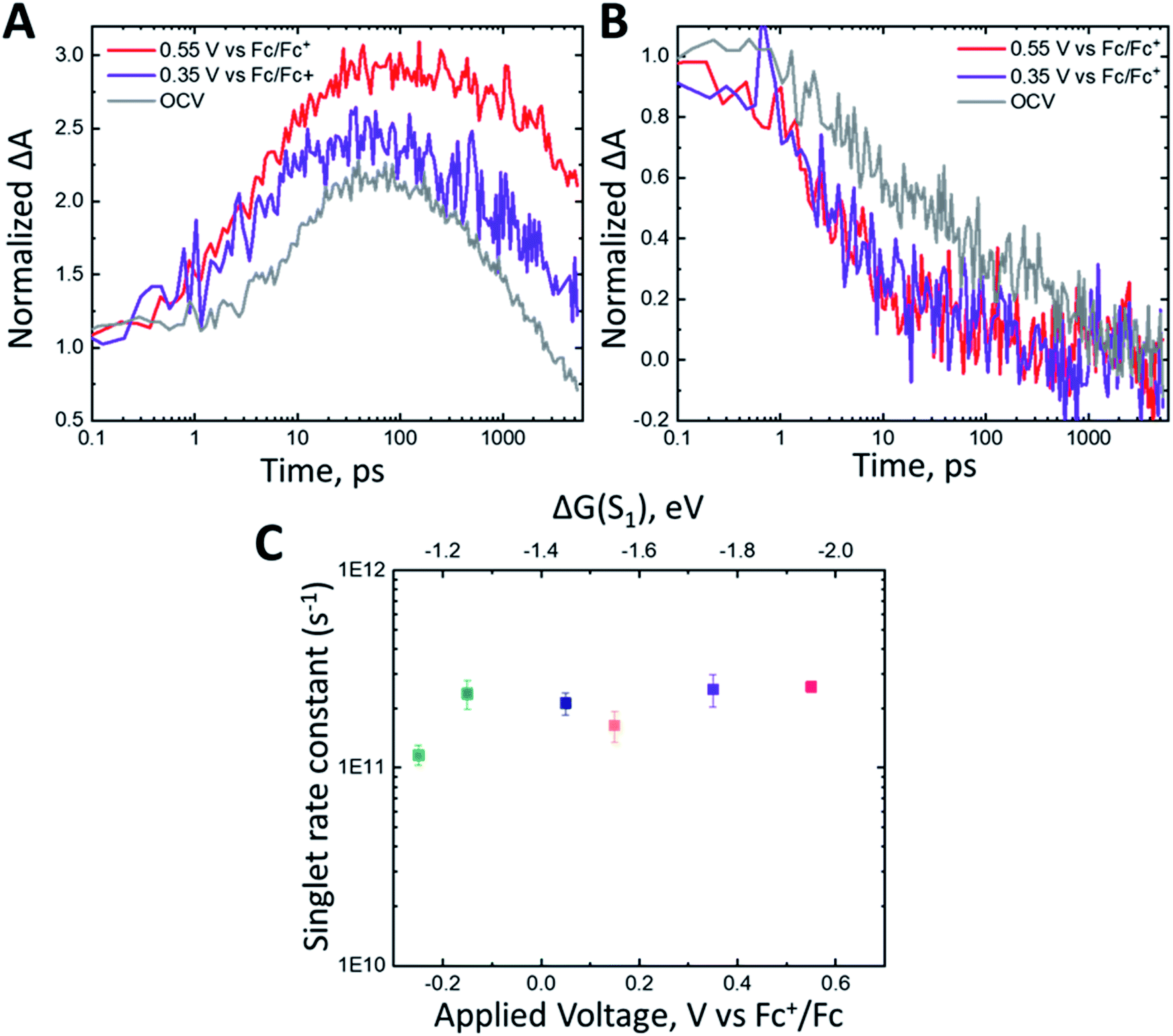 | ||
| Fig. 5 Normalized kinetics monitored at (A) 1030 nm and (B) 1350 nm for nanoITO:ADT-COOH in 0.1 M TBAPF6 in MeCN at open circuit voltage (gray), 0.35 V (purple) and 0.55 V vs. Fc+/Fc (red). The kinetics were normalized at the initial time point. (C) Singlet decay rate constant (s−1) measured at 1350 nm as a function of applied voltage (V vs. Fc+/Fc). The top axis shows the driving force for triplet injection (ΔG(S1)) in eV as calculated using eqn (2). | ||
The nearly identical singlet kinetics vs. bias are not mirrored in the normalized cation dynamics (Fig. 5A), where the magnitude of the cation rise is greater for 0.55 V than 0.35 V vs. Fc+/Fc. The spectra in Fig. S3† also show a distinct difference in the relative amplitude of the cation feature between OCV (Fig. S3A†), 0.35 V vs. Fc+/Fc (Fig. S3B†) and 0.55 V vs. Fc+/Fc (Fig. S3C†). When normalized at 100 fs, the maximum amplitude of the cation signal is 1.5× larger at 0.55 V than at OCV. This appears to be related to the reduction in observed triplet lifetimes (Fig. 4D) as a function of applied potential. Over the full potential range (Fig. 4D), the rate of triplet decay increases by about one order of magnitude. These observations are consistent with increased electron injection from the triplet excited state with increasing driving force, reaffirming the observations made in the visible spectral region. Therefore, we conjecture that the increase in relative peak amplitudes of the cation (Fig. 5A) can be correlated with the increased rate of triplet decay.
Discussion
Singlet fission
Intermolecular interactions have a significant role in dictating singlet fission dynamics,32 and the translation of prior work with ADT-COOH on planar substrates28 to the situation on nanoITO is unknown. As we previously demonstrated, the three-dimensional interactions between multiple layers of molecules in a thin film produce a delocalized singlet exciton that enables a significant component of ultrafast SF, which is absent here. Further, the roughness of mesoporous nanoITO most likely produces intermolecular ADT-COOH geometries that are distinct from those found in thin films deposited on a planar substrate. The mechanism of adsorption to a mesoporous oxide is through the carboxylic acid binding group, thus all interactions between molecules are lateral on the nanoITO surface. New parameters besides intermolecular forces become relevant on the nanoITO surface, such as surface loading, and nanoparticle size, but because our primary goal has been maximizing the rate and yield of SF, we have used saturated surface coverages,22 which likely increase favourable intermolecular interactions.Despite these distinctions between monolayer and crystal, remarkably we observe biphasic triplet growth that is consistent with fast SF from near-optimal intermolecular geometries, followed by slow singlet fission unique to the insulated nanoITO/Al2O3:ADT-COOH. The slow component is likely due to those molecular pairs that may be oriented appropriately, but not ideally for SF. This “parallel” model of SF has precedence in other disordered molecular aggregates.33 Another possible explanation for the delayed rise is diffusion to preferred sites,34 although the degree to which ps-scale diffusion plays a role in surface-bound dyes is unknown.35 While it is difficult to quantify, the transient absorption data indicate a qualitatively similar amplitude of generated triplets at all applied potentials and amongst different samples on nanoITO. This is expected as we would predict a similar distribution of favourable molecular dispositions for SF for all nanoITO substrates that would be unaffected by an applied voltage.
With no barrier to electron injection in the uninsulated nanoITO:ADT-COOH samples, the kinetic scheme becomes more complex, and triplet formation is obscured by singlet injection and cation formation. Disentangling the various mechanisms for excited state decay (i.e. electron injection, SF) requires modelling the rate and yield of the primary pathway (electron injection) vs. the variable of applied potential.
Excited state electron injection and Marcus theory
An increase in singlet injection rate constant with increased driving force is expected from Marcus theory in the “normal” regime: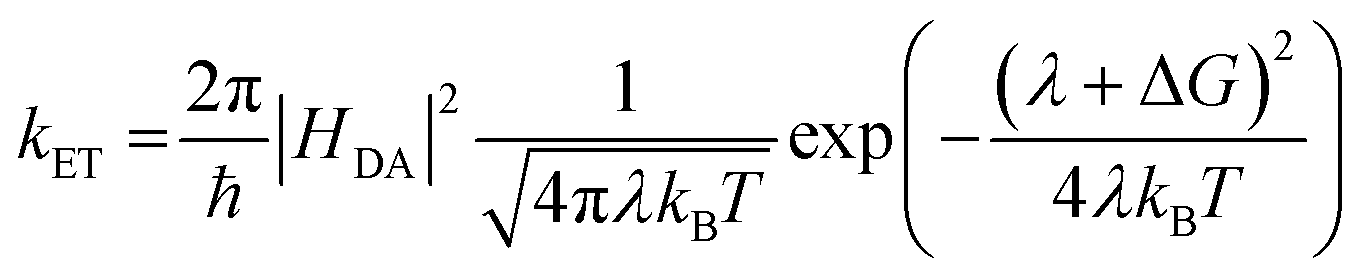 | (3) |
The lifetime dependence on driving force was analyzed using the mathematical solution derived by Farnum and coworkers.27 Briefly, we can define the injection rate constant based on the possibility of multiple electron injection events from ADT-COOH into the continuum of acceptor states in the nanoITO:
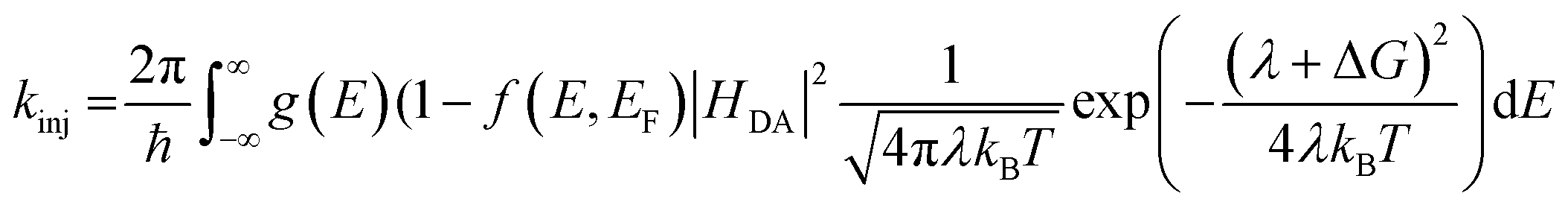 | (4) |
Eqn (4) incorporates the traditional formulation of the Marcus equation in eqn (3) with the addition of g(E) (1 − f(E, EF)) to account for the distribution of acceptor states in the nanoITO. Because this distribution is large, the assumption holds that g(E) and HDA(E) are constant with respect to energy. The Fermi–Dirac distribution function defines all levels above or equal to the Fermi level (EF) as empty, while all states below EF are filled in the low temperature limit. Within this limit eqn (4) becomes eqn (5):
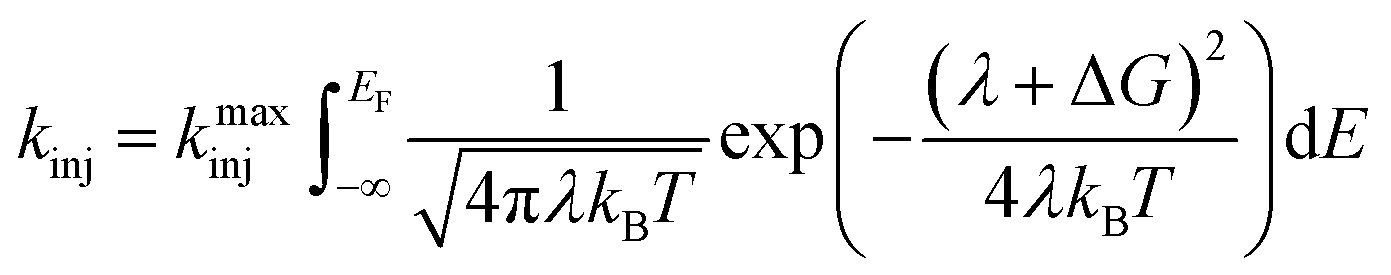 | (5) |
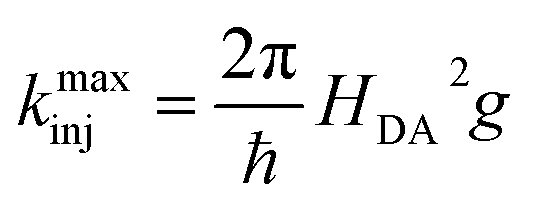 | (6) |
Eqn (5) can be solved analytically to give the cumulative distribution function,
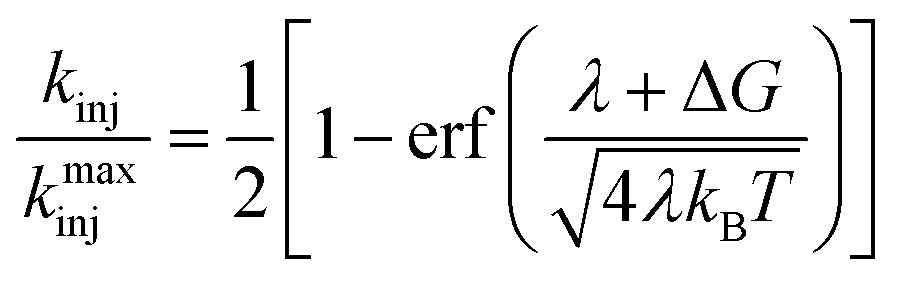 | (7) |
Using eqn (6) and (7) we can determine the electronic coupling, HDA and λ for singlet and triplet electron injection from ADT-COOH to the nanoITO. Based on similarities of film fabrication, we use the value of g calculated by Farnum and coworkers to be 0.45 eV−1 for oxidized nanoITO .27
Electron injection from the singlet excited state
The fast component of the singlet decay at 1350 nm under increasingly positive applied potentials (Fig. 5C) is roughly constant in the experiment potential range. Because the nanoITO is a continuum of acceptor states, we do not observe the expected turnover predicted for the Marcus inverted regime. Instead, we see an unchanging rate constant consistent with reaching a limit for the singlet electron injection at high driving force (ΔG(S1) = −1.2 eV). Previous studies of molecular donor-acceptor systems with a singlet fission molecular donor (pentacene) show that the optimal driving force for singlet electron transfer is approximately −0.6 eV.21Fig. 6A shows the ratio kinj(S1)/kmaxinj(S1) as a function of driving force plotted with eqn (7) using varying values for the reorganization energy λ. It is difficult to determine an accurate value of λ with these data because we are not accessing a regime where singlet injection is significantly suppressed. Applying the negative potentials necessary to access this regime was not possible due to desorption of ADT-COOH at high negative biases. There is also likely some convolution of the singlet fission rate constant in kinetics at lower driving forces that is difficult to separate from singlet injection. Calculating HDA for singlet injection using eqn (6) yields a value of 3.5 meV (28 cm-1), which is the expected order of magnitude based on prior work.18,21,27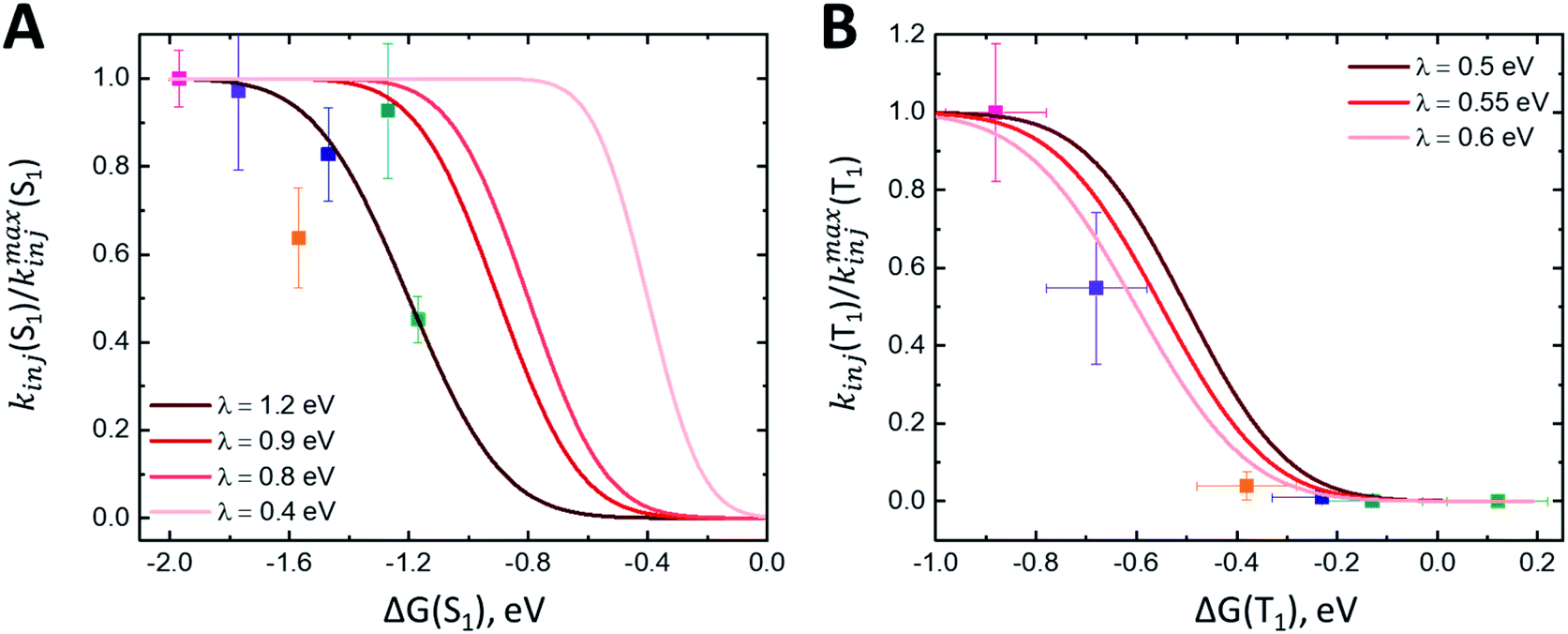 | ||
| Fig. 6 (A) Plot of kinj(S1)/kmaxinj(S1) calculated using fits at 1350 nm as a function of driving force ΔG(S1). Fits to eqn (7) with varying λ values shown in red. (B) Plot of kinj(T1)/kmaxinj(T1) calculated using fits at 570 nm as a function of driving force ΔG(T1). Fits to eqn (7) with varying λ values shown in red. | ||
Electron injection from the triplet excited state
The triplet signature in the visible spectral region is convoluted with the singlet excited state and cation spectral features, affecting the observed rate constant at 570 nm. To assess the time scale for triplet injection more accurately, we performed a correction to the triplet decay (kobs(T1)) using the cation signal in the NIR. The cation signal (Fig. 5A) continues to evolve between 0.35 V and 0.55 V vs. Fc+/Fc despite the absence of change in the singlet kinetics between these two applied potentials (Fig. 5B), and we assume that a portion of the amplitude at late times is due to injected electrons from the triplet state. To determine a relative electron injection from triplet excited state yield vs. driving force, we normalize the 1030 nm cation kinetics at 50 ps and subtract the OCV data from each applied potential dataset. This removes the contribution of electron injection from the singlet excited state from the cation signal (expected to dominate at zero bias) and isolates the triplet charge transfer portion of the dynamics. We cannot rule out that some portion of the extended lifetime at 1030 nm may also be partially due to a suppression of charge recombination from the nanoITO because of the positive applied potential. Nonetheless, dividing the kobs for triplet decay by the amplitude of the ΔΔA signal at 1000 ps gives us a proxy for the true kinj(T1).Fig. 6B shows kinj(T1)/kmaxinj(T1)as a function of ΔG(T1) and the fits to eqn (7) with varying values of λ. Error bars for ΔG(T1) account for possible triplet energies between 1.0–1.1 eV. At low driving forces (ΔG ≥ −0.2 eV), the ratio of kinj(T1)/kmaxinj(T1) suggests there is no substantial triplet injection. Triplet injection commences at ΔG < −0.3 eV and accelerates dramatically through the rest of the potential range. Importantly, the onset of triplet injection in Fig. 6B also correlates with an increase in observed photocurrent (Fig. S4A and B†). Fits to these data suggest a λ between 0.5 and 0.6 eV, which agrees with reorganization energies for charge transfer from triplet excited states observed in other studies with singlet fission molecules.21
The derived HDA for electron injection from the triplet excited state using eqn (6) is 0.53 meV (4.2 cm−1), an order of magnitude smaller than that of the singlet. We conjecture that the highly localized nature of triplets compared to singlets contributes to the stark contrast in electronic coupling values.20,21,28,36,37 This localization of the triplet excited state on ADT-COOH may lead to the difficulties in extracting charge effectively, leading to low photocurrents and ultimately poor device performance if competing pathways are present. We note that this is not an inherent problem with all triplets. With ruthenium-based organometallic complexes used in DSSCs, intersystem crossing occurs within 100 fs and the majority of the injection events that follow are from the triplet MLCT state (3MLCT), which has an intrinsically long lifetime of 100 s of ns. Injection yields for these complexes often approach 100%, in part because charge separation from the 3MLCT is effectively “built-in” to the character of the excited state. If triplet injection must occur on a sub-ns timescale to be efficient, endowing such charge-transfer character in a SF system would seem to be a contradiction, as most singlet fission molecules require a large exchange interaction (i.e., significant HOMO/LUMO orbital overlap) in order to create the requisite energy balance to enable fast triplet-pair formation. However, clever schemes in which fast evolution away from the triplet pair formation site to a secondary triplet site that includes significant charge-transfer character could be envisaged.38
Conclusion
In summary, we have demonstrated a range of electrochemical potentials for singlet fission-borne triplet injection from ADT-COOH into a mesoporous metal oxide nanoITO. A substantial difference in driving force for electron injection exists between the singlet and triplet excited states of ADT-COOH, necessitating a large applied potential for triplet electron injection to occur. The reorganization energy for electron injection from the triplet excited state is estimated to be between 0.5–0.6 eV, although the calculated electronic coupling for triplets is an order of magnitude lower than that of the singlet. Localization of the triplet state, which likely causes weak coupling with the metal oxide, will need to be overcome to reduce overpotentials and increase SF-DSSC efficiency. A strategy that could be implemented is to incorporate an organometallic chromophore with an MLCT state to act as a triplet energy acceptor to act as an intermediate towards electron injection. Similar approaches have been used for triplet–triplet upconversion devices. Overall, this study establishes that electron injection from the triplet excited state in SF chromophores is attainable and demonstrates a method for determining the optimal driving force for electron injection from SF-generated triplet states.Author contributions
M. K. G. prepared solution and film samples for and performed TA spectroscopy, performed applied bias TA spectroscopy, and analyzed the data under the guidance of J. C. J. E. K. R. prepared nanoITO films and performed electrochemistry and R. T. P. prepared nanoITO paste and films and assisted with electrochemistry and data analysis under the guidance of N. R. N. A. L. G. deposited alumina using ALD. K. J. T. synthesized ADT-COOH under the guidance of J. E. A. M. K. G. and J. C. J. wrote the paper with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge G. Michael Carroll for his assistance with steady state spectroelectrochemical and cyclic voltammetry measurements. This work was authored by Alliance for Sustainable Energy, LLC, the manager and operator of the National Renewable Energy Laboratory for the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308. Funding provided by U.S. Department of Energy, Office of Basic Energy Sciences, Division of Chemical Sciences, Biosciences, and Geosciences. The views expressed in the article do not necessarily represent the views of the DOE or the U.S. Government. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U.S. Government purposes. Dye synthesis efforts were supported by the National Science Foundation under Cooperative Agreement No. 1849213 and CHE-2003735 for ITO electrode preparation.References
- K. Sharma, V. Sharma and S. S. Sharma, Nanoscale Res. Lett., 2018, 13, 381 CrossRef PubMed.
- D. Zhang, M. Stojanovic, Y. Ren, Y. Cao, F. T. Eickemeyer, E. Socie, N. Vlachopoulos, J.-E. Moser, S. M. Zakeeruddin, A. Hagfeldt and M. Gratzel, Nat. Commun., 2021, 12, 1777 CrossRef CAS PubMed.
- M. C. Beard, J. C. Johnson, J. M. Luther and A. J. Nozik, Philos. Trans. R. Soc., A, 2015, 373 Search PubMed.
- M. K. Gish, N. A. Pace, G. Rumbles and J. C. Johnson, J. Phys. Chem. C, 2019, 123, 3923–3934 CrossRef CAS.
- J. C. Johnson, Commun. Chem., 2021, 4, 85 CrossRef.
- M. B. Smith and J. Michl, Annu. Rev. Phys. Chem., 2013, 64, 361–386 CrossRef CAS PubMed.
- D. Casanova, Chem. Rev., 2018, 118, 7164–7207 CrossRef CAS PubMed.
- N. V. Korovina, N. F. Pompetti and J. C. Johnson, J. Chem. Phys., 2020, 182, 040904 CrossRef PubMed.
- K. M. Felter and F. C. Grozema, J. Phys. Chem. Lett., 2019, 10, 7208–7214 CrossRef CAS PubMed.
- M. C. Hanna and A. J. Nozik, J. Appl. Phys., 2006, 100, 074510 CrossRef.
- I. Paci, J. C. Johnson, X. Chen, G. Rana, D. Popovic, D. E. David, A. J. Nozik, M. A. Ratner and J. Michl, J. Am. Chem. Soc., 2006, 128, 16546–16553 CrossRef CAS PubMed.
- J. N. Schrauben, Y. Zhao, C. Mercado, P. I. Dron, J. L. Ryerson, J. Michl, K. Zhu and J. C. Johnson, ACS Appl. Mater. Interfaces, 2015, 7, 2286–2293 CrossRef CAS PubMed.
- A. Kunzmann, M. Gruber, R. Casillas, J. Zirzlmeier, M. Stanzel, W. Peukert, R. R. Tykwinski and D. M. Guldi, Angew. Chem., Int. Ed. Engl., 2018, 57, 10742–10747 CrossRef CAS PubMed.
- T. Banerjee, S. P. Hill, A. Hermosilla-Palacios, B. D. Piercy, J. Haney, B. Casale, A. E. DePrince III, M. D. Losego, V. D. Kleiman and K. Hanson, J. Phys. Chem. C, 2018, 122, 28478–28490 CrossRef CAS.
- N. A. Pace, D. H. Arias, D. B. Granger, S. Christensen, J. E. Anthony and J. C. Johnson, Chem. Sci., 2018, 9, 3004–3013 RSC.
- E. Sundin, R. Ringstrom, F. Johansson, B. Kucukoz, A. Ekebergh, V. Gray, B. Albinsson, J. Martensson and M. Abrahamsson, J. Phys. Chem. C, 2020, 124, 20794–20805 CrossRef CAS.
- D. L. Ashford, M. K. Gish, A. K. Vannucci, M. K. Brennaman, J. L. Templeton, J. M. Papanikolas and T. J. Meyer, Chem. Rev., 2015, 115, 13006–13049 CrossRef CAS PubMed.
- D. F. Zigler, Z. A. Morseth, L. Wang, D. L. Ashford, M. K. Brennaman, E. M. Grumstrup, E. C. Brigham, M. K. Gish, R. J. Dillon, L. Alibabaei, G. J. Meyer, T. J. Meyer and J. M. Papanikolas, J. Am. Chem. Soc., 2016, 138, 4426–4438 CrossRef CAS PubMed.
- M. K. Brennaman, R. J. Dillon, L. Alibabaei, M. K. Gish, C. J. Dares, D. L. Ashford, R. L. House, G. J. Meyer, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 13085–13102 CrossRef CAS PubMed.
- N. A. Pace, T. T. Clikeman, S. H. Strauss, O. V. Boltalina, J. C. Johnson, G. Rumbles and O. G. Reid, J. Phys. Chem. C, 2020, 134, 26153–26164 CrossRef.
- N. A. Pace, N. V. Korovina, T. T. Clikeman, S. Holliday, D. B. Granger, G. M. Carroll, S. U. Nanayakkara, J. E. Anthony, I. McCulloch, S. H. Strauss, O. V. Boltalina, J. C. Johnson, G. Rumbles and O. G. Reid, Nat. Chem., 2020, 12, 63–70 CrossRef CAS PubMed.
- K. Hanson, M. K. Brennaman, A. Ito, H. Luo, W. Song, K. A. Parker, R. Ghosh, M. R. Norris, C. R. K. Glasson, J. J. Concepcion, R. Lopez and T. J. Meyer, J. Phys. Chem. C, 2012, 116, 14837–14847 CrossRef CAS.
- S. A. Miller, B. A. West, A. C. Curtis, J. M. Papanikolas and A. M. Moran, J. Chem. Phys., 2011, 135, 081101 CrossRef PubMed.
- X. Dang and J. T. Hupp, J. Am. Chem. Soc., 1999, 121, 8399–8400 CrossRef CAS.
- G. Ramakrishna and H. N. Ghosh, J. Phys. Chem. B, 2001, 105, 7000–7008 CrossRef CAS.
- H.-Y. Hsu, C.-W. Cheng, W.-K. Huang, Y.-P. Lee and E. W.-G. Diau, J. Phys. Chem. C, 2014, 118, 16904–16911 CrossRef CAS.
- B. H. Farnum, Z. A. Morseth, M. K. Brennaman, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 15869–15872 CrossRef CAS PubMed.
- M. K. Gish, K. J. Thorley, S. R. Parkin, J. E. Anthony and J. C. Johnson, ChemPhotoChem, 2021, 5, 68–78 CrossRef CAS.
- C.-K. Yong, A. J. Musser, S. L. Bayliss, S. Lukman, H. Tamura, O. Bubnova, R. K. Hallani, A. Meneau, R. Resel, M. Maruyama, S. Hotta, L. M. Herz, D. Beljonne, J. E. Anthony, J. Clark and H. Sirringhaus, Nat. Commun., 2017, 8, 15953 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- K. Hanson, M. K. Brennaman, H. Luo, C. R. K. Glasson, J. J. Concepcion, W. Song and T. J. Meyer, ACS Appl. Mater. Interfaces, 2012, 4, 1462–1469 CrossRef CAS PubMed.
- J. N. Schrauben, J. L. Ryerson, J. Michl and J. C. Johnson, J. Am. Chem. Soc., 2014, 136, 7363–7373 CrossRef CAS PubMed.
- R. D. Pensack, A. J. Tilley, S. R. Parkin, T. S. Lee, M. M. Payne, D. Gao, A. A. Jahnke, D. G. Oblnsky, P.-F. Li, J. E. Anthony, D. S. Seferos and G. D. Scholes, J. Am. Chem. Soc., 2015, 137, 6790–6803 CrossRef CAS PubMed.
- S. T. Roberts, R. E. McAnally, J. N. Mastron, D. H. Webber, M. T. Whited, R. L. Brutchey, M. E. Thompson and S. E. Bradforth, J. Am. Chem. Soc., 2012, 134, 6388–6400 CrossRef CAS PubMed.
- M. K. Brennaman, M. K. Gish, L. Alibabaei, M. R. Norris, R. A. Binstead, A. Nayak, A. M. Lapides, W. Song, R. J. Brown, J. J. Concepcion, J. L. Templeton, J. M. Papanikolas and T. J. Meyer, J. Phys. Chem. C, 2018, 122, 13017–13026 CrossRef CAS.
- S. L. Bayliss, K. J. Thorley, J. E. Anthony, H. Bouchiat, N. C. Greenham and A. D. Chepelianskii, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 115432 CrossRef.
- N. V. Korovina, C. H. Chang and J. C. Johnson, Nat. Chem., 2020, 12, 391–398 CrossRef CAS PubMed.
- T. Dilbeck and K. Hanson, J. Phys. Chem. Lett., 2018, 9, 5810–5821 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03253d |
| This journal is © The Royal Society of Chemistry 2021 |