
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRE-SELEX: restriction enzyme-based evolution of structure-switching aptamer biosensors†
Aimee A.
Sanford
 a,
Alexandra E.
Rangel
b,
Trevor A.
Feagin
b,
Robert G.
Lowery
c,
Hector S.
Argueta-Gonzalez
a and
Jennifer M.
Heemstra
*a
a,
Alexandra E.
Rangel
b,
Trevor A.
Feagin
b,
Robert G.
Lowery
c,
Hector S.
Argueta-Gonzalez
a and
Jennifer M.
Heemstra
*a
aDepartment of Chemistry, Emory University, Atlanta, Georgia 30322, USA. E-mail: jen.heemstra@emory.edu
bDepartment of Chemistry, Center for Cell and Genome Science, University of Utah, Salt Lake City, Utah 84112, USA
cBellBrook Labs, LLC, Madison, Wisconsin 53711, USA
First published on 24th July 2021
Abstract
Aptamers are widely employed as recognition elements in small molecule biosensors due to their ability to recognize small molecule targets with high affinity and selectivity. Structure-switching aptamers are particularly promising for biosensing applications because target-induced conformational change can be directly linked to a functional output. However, traditional evolution methods do not select for the significant conformational change needed to create structure-switching biosensors. Modified selection methods have been described to select for structure-switching architectures, but these remain limited by the need for immobilization. Herein we describe the first homogenous, structure-switching aptamer selection that directly reports on biosensor capacity for the target. We exploit the activity of restriction enzymes to isolate aptamer candidates that undergo target-induced displacement of a short complementary strand. As an initial demonstration of the utility of this approach, we performed selection against kanamycin A. Four enriched candidate sequences were successfully characterized as structure-switching biosensors for detection of kanamycin A. Optimization of biosensor conditions afforded facile detection of kanamycin A (90 μM to 10 mM) with high selectivity over three other aminoglycosides. This research demonstrates a general method to directly select for structure-switching biosensors and can be applied to a broad range of small-molecule targets.
1. Introduction
Small molecules play an important role in human health. A prominent example is their use in therapeutics, including antibiotics such as ampicillin and kanamycin or chemotherapeutics such as taxol. While these examples generally benefit overall human health, naturally occurring and synthetic small molecules can also pose detrimental effects depending on exposure levels.1 For instance, overuse of antibiotics in medicine and agriculture has led to the emergence of multi-drug resistant bacteria. This highlights the need for methods to rapidly and selectively detect these and other small-molecule analytes. Conventional detection techniques are limited in that they require expensive experimentation or user-specific expertise,1,2 and recent efforts have focused on novel approaches that are cost-effective and user-friendly without sacrificing sensitivity. This is a major challenge for small molecules due to their size and reduced number of binding epitopes compared to larger biomolecules such as proteins.Nucleic acids are well suited to address these shortcomings due to their ability to “recognize” other biomolecules through non-covalent interactions. This molecular recognition is the key feature of aptamers, which are single stranded nucleic acids that can be evolved in vitro through the Systematic Evolution of Ligands by Exponential Enrichment (SELEX) to bind to a target of interest with high affinity.3,4 Aptamers can also demonstrate impressively high selectivity by distinguishing between molecules with only subtle differences in structure.5 For these reasons, aptamer-based sensors have shown great promise for biosensing applications, and they are especially well-suited for the development of low-cost, user-friendly methods due to their stability, ease of synthesis, and re-useability.1,2 The platforms reported for aptamer-based detection of small molecules are vast, but the structure-switching biosensor format is among the most promising because the biorecognition event can be directly transduced into a readable output.6,7 One common type of structure-switching biosensor is that in which a fluorophore-labeled aptamer is hybridized to a quencher-labeled short complementary capture strand. Upon introduction of the target, the capture strand is displaced, which results in a dose-dependent increase in fluorescence.6,7 The biggest challenge with this method is that few aptamers generated using conventional SELEX inherently demonstrate the significant conformational change needed to be incorporated into structure-switching biosensors.6 As a result, the majority of biosensors reported in the literature rely on a small handful of aptamers that have inherent structure-switching capabilities.
Several approaches have been described to engineer or select structure-switching biosensors. The most common method is to first select for binding, and then test and optimize potential complementary strands to achieve structure-switching displacement behavior.8–11 However, this approach has a high failure rate as most aptamers do not undergo a significant enough change in conformation or signal response upon target recognition. More recent approaches have focused on universal designs that do not require the aptamer itself to be structure-switching.12,13 One example utilizes a mixture of exonucleases to digest sequence components that have less stable folded structures, which is typically those not bound to the target of interest.12,14 This approach has shown great promise for engineering truncations and modulating affinity of aptamers for subsequent use as biosensors.14,15 The other notable approach takes inspiration from the constant and variable regions of antibodies in that a constant G-quadruplex region is incorporated to serve as the structure-switching element coupled to an aptamer.13
An alternative strategy is to directly select for structure-switching aptamers using SELEX.16–19 Initial efforts in this area took advantage of beads to immobilize the capture strand either before or after library hybridization.16,17 Library members containing a complementary “docking” stretch will hybridize to the immobilized capture strand, then washing is performed to remove non-hybridized members. Upon addition of the target, sequences that are displaced from the capture strand can be eluted and recovered. However, a key limitation that we have found when using this method in our lab is that the equilibrium for binding of the library to the beads is being constantly re-established with each wash, and thus a significant number of non-functional sequences are inevitably recovered during the target elution step. One benefit of this method is that it does not require target immobilization, which avoids issues associated with small-molecule conjugation.17,20 However, the selection step is still considered heterogenous because it requires immobilization of the capture strand and incubation of the library with beads. This and the challenge of non-selective background elution described above is likely why this method has not yet found widespread adoption, and recent reports have questioned the utility of capture SELEX methods for downstream structure-switching biosensor applications.20,21
Seeking to overcome the aforementioned challenges, we designed a new structure-switching SELEX method that utilizes a homogenous isolation step to distinguish between active and inactive library members without the need to immobilize target or capture strand. We envisioned that restriction enzymes could be exploited for this task because they are highly selective for their cognate dsDNA palindromic recognition sites.22 Specifically, construction of a library having the restriction site encoded in the duplex region where the aptamer and capture strand hybridize would allow for library members that are displaced in response to target to evade digestion, while members that remain hybridized to be digested. Further, because this restriction site would also overlap with a primer binding site on the library, only functional and non-digested members would be capable of exponential amplification in PCR. Herein, we describe the development of our restriction enzyme SELEX (RE-SELEX) method and its validation and benchmarking using kanamycin A, a small molecule target that has been the focus of previous structure-switching evolution methods.17,23 In our initial selection design, we observed enrichment of sequences that evade restriction cleavage without binding to the target molecule. We subsequently incorporated a negative selection step to remove these sequences, and after eleven rounds of this re-optimized SELEX, we identified several candidates that showed biosensor activity with kanamycin A. The most promising sequence, K16-1, provided dose-dependent signal for kanamycin A concentrations of 90 μM to 10 mM in a structure-switching biosensor format. We performed two structure-based truncations that retained the same binding affinity and overall biosensor performance. The harshest truncation which results in a 32 nt aptamer denoted K16-1c was successfully implemented as a structure-switching biosensor for kanamycin A from 90 μM to 10 mM. We also demonstrate the selectivity of both K16-1 and K16-1c biosensors and their superior performance in the structure-switching format compared to previous sequences reported from Capture SELEX. Together, this research provides a new approach to the direct selection of structure-switching biosensors for small-molecule targets. We anticipate that this will accelerate the use of aptamers in biosensing applications for a wide range of drugs, toxins, and other biologically important molecules.
2. Results and discussion
2.1. Design of restriction enzyme SELEX libraries
SELEX libraries generally contain a random region flanked by two constant primer binding sites, and for our specific method, it was critical to locate the restriction enzyme digestion site within one of the primer binding sites. As shown in Fig. 1a, we designed a biosensor library in which the capture strand was complementary to a portion of the 5′ primer binding site and an N40 random region was located directly adjacent to this hybridization region to maximize the likelihood of achieving target-dependent displacement of the capture strand.6 One concern that arose when designing our library was that while restriction enzymes are most efficient with six flanking nucleotides on either side of the cut site, capture strands for structure-switching biosensors are generally <12 nt.8,10,11,21,24,25 Therefore, we first wanted to test the digestion efficiency of two restriction enzyme-based libraries with capture strands of varying lengths to determine the minimum number of flanking nucleotides that would still support efficient restriction cleavage. We chose BamHI-HF and EcoRI-HF as our initial restriction enzyme candidates because they are both reported to produce quantitative cleavage (ESI Fig. 1†). We found EcoRI-HF conditions to be more efficient than BamHI-HF for all capture strand lengths ranging from 8–14 nt (ESI Fig. 3†), and thus EcoRI-HF was used in all subsequent selection experiments. To explore the utility of this method in selections requiring varying buffer conditions, we also tested digestion in the absence of CutSmart Buffer and in other common SELEX buffers (ESI Fig. 4a and b†).10 We found that effective cleavage was achieved in each buffer as long as 2 mM Mg2+ was present. This was not entirely surprisingly given that EcoRI cleavage is magnesium dependent.26 This demonstrates that our method can be applied in a variety of buffer conditions and that CutSmart Buffer is not necessary.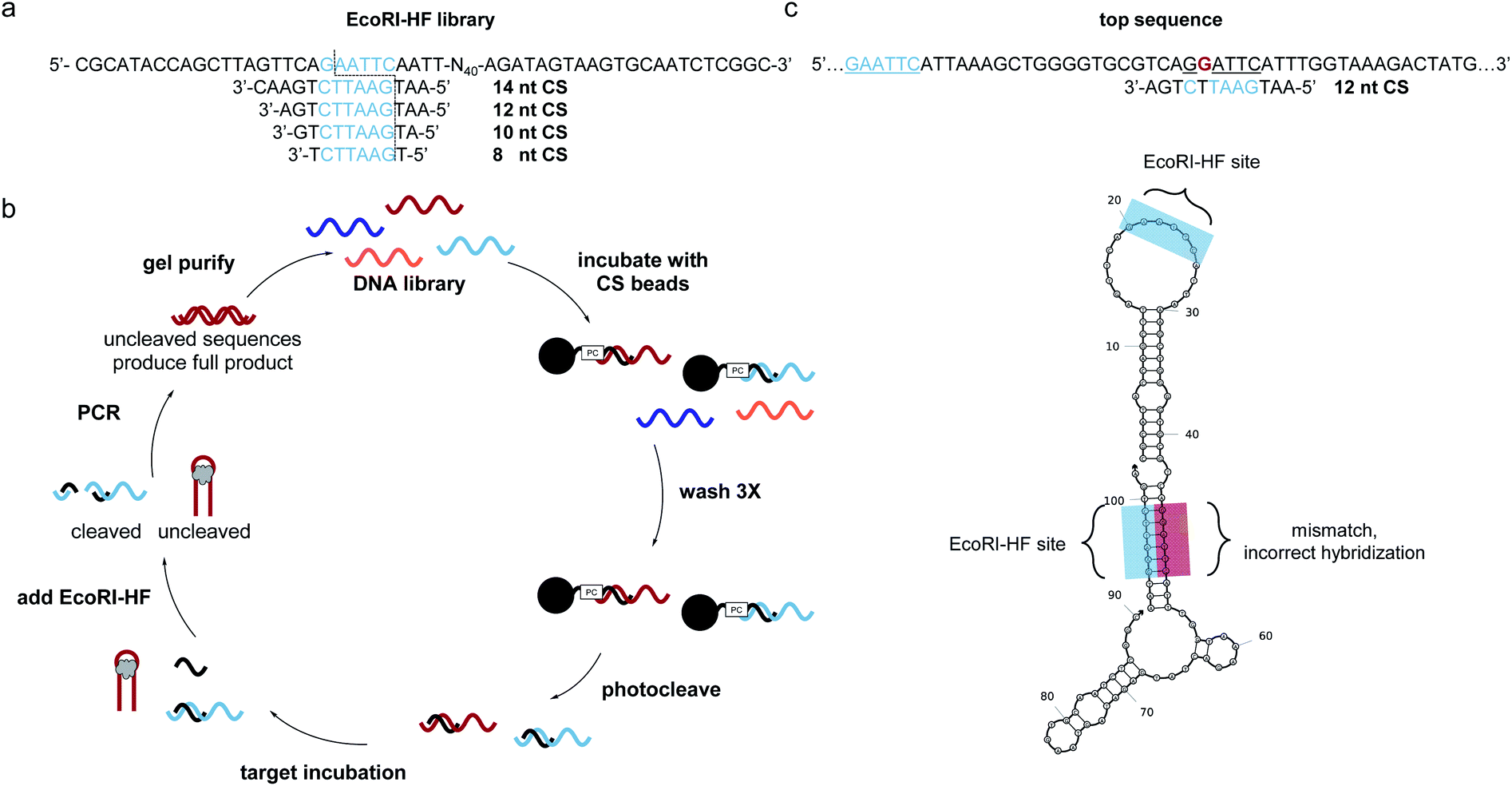 | ||
| Fig. 1 Bead assisted EcoRI-HF SELEX to generate structure-switching aptamers. (a) EcoRI-HF library and capture strand sequences with recognition sites in blue. Cut site is illustrated by dotted line. (b) FAM-labeled DNA library having an N40 random region was hybridized to immobilized capture strand and the complex washed and photocleaved. After target incubation, bound sequences were recovered following EcoRI-HF digestion and PCR amplification. Gel purified library was then carried on to subsequent rounds. ssDNA library was purified by 10% denaturing PAGE and carried on to subsequent selection rounds. (c) Highly enriched sequences did not correctly hybridize the capture strand through the built in EcoRI-HF recognition site (underlined blue) sequences containing partial recognition sites in the N40 region (underlined black) with one or more mismatches (red, bold) were present in predicted structures by NUPACK. | ||
2.2. Initial RE-SELEX design
With our library constructed and validated for restriction digest, we set about developing our selection method. We recognized that while the restriction digest step was highly efficient, some library members can adopt stable folding patterns that make hybridization to the capture strand thermodynamically impossible. These sequences would be enriched in the selection, leading to a significant non-functional background. To circumvent this challenge, we included an initial selection step to remove these stably folded sequences. This step is akin to that used in the well-known capture SELEX method, but in our approach the capture strand was synthesized directly on polystyrene beads with a photocleavable linker to ensure maximum bead loading.17 To perform the negative selection step, library members were hybridized to the immobilized capture strand (Fig. 1b). After several washes, intact biosensor sequences were collected we collected intact biosensors through UV irradiation. This reduces the chance for non-specific de-hybridization and allows for exploitation of the restriction enzyme handle to directly select for biosensor activity. Target was introduced and then EcoRI-HF added to digest non-functional sequences. To ensure that cleavage prevents subsequent amplification, we performed PCR on the cleavage products (70 nt, 20 nt) and uncleaved library (90 nt) and observed no detectable amplification for the cleaved sample (ESI Fig. 5†). Thus, the surviving library members were amplified using PCR, purified using PAGE, and subjected to a subsequent round of selection.After seven rounds of SELEX, we observed the anticipated increase in non-cleaved sequences, but upon performing a quality check experiment, found that the resistance to digestion was not target-dependent (ESI Fig. 6a and b†). To explore the source of this challenge, we performed next-generation sequencing. The data revealed an interesting pattern where many of the enriched sequences had a segment of the N40 random region that was able to hybridize to the capture strand but with a single mismatch (ESI Fig. 6c†). Thus, instead of generating aptamers that survive EcoRI-HF digestion through target induced displacement, highly enriched sequences evaded complete digestion through the single mismatch restriction enzyme cleavage (Fig. 1c). While the probability of this occurring is statistically low, selection pressure can exponentially enrich and over-populate the pool with these false positive sequences within a few rounds. Furthermore, this is exacerbated in selections with small-molecule targets, which typically require more rounds for tangible enrichment.27,28 We probed this hypothesis further by ordering a highly enriched sequence which contained a cleavage site with single mismatch in the N40 region (ESI Fig. 7†). As we suspected, this sequence showed overall less efficient cleavage of 80%, despite containing the built-in recognition site in the primer binding site. Upon removal of the primer binding site, the cleavage was only slightly higher than background at 4%. This recapitulates the ability of these false positive sequences to survive in the selection, and thus we sought to redesign our selection in order to prevent enrichment of such constructs.
2.3. Optimized EcoRI-HF SELEX
Identifying the source of the false positive sequences in our initial SELEX protocol enabled us to redesign our method to eliminate these sequences. We recognized that while the immobilized capture strand was isolating sequences that are capable of hybridization, it did not distinguish between those where the location of the complementary stretch was within the library rather than in the primer binding site as intended. As highlighted in Fig. 1, this can lead to enrichment of library:capture strand duplexes that are not efficiently digested by EcoRI-HF, regardless of the presence of target. We envisioned that these sequences could be removed by replacing the initial bead hybridization step with an initial cleavage and re-ligation step, as this would ensure that all sequences carried forward were capable of both hybridizing to the capture strand and being cleaved in the absence of target. As an added benefit, this modified protocol completely removed all bead binding steps, resulting in a fully homogeneous method that best mimics the conditions under which the biosensors will be ultimately used. As shown in Fig. 2, library was hybridized with capture strand then digested by EcoRI-HF and the cleaved sequences recovered by PAGE purification. The full-length biosensor library was then regained through ligation using a cut forward primer (5′-/FAM/CGCATACCAGCTTAGTTCAG-3′) and splint (5′-AATGAATTCTGAACTAAGCTGGTATGCG-3′) (ESI Fig. 2†). The ligation reaction was monitored using 10% denaturing PAGE for each round of SELEX and subjected to ethanol precipitation to purify and concentrate the resulting full-length library. We note that even if ligation is not quantitative, unligated sequences will just serve as spectators during the ensuing selection step and will not be PCR amplified. Although the ligation occurs within the constant primer binding site, we wanted to ensure that this step did not unnecessarily bias the library. Bulk library and re-ligated samples were subjected to high-throughput sequencing, and we were encouraged to find that 98% of the ligated sample contained unique sequences (ESI Fig. 9†). Additionally, there was no apparent change in the distribution of nucleotides in the random region before and after cleavage and ligation, suggesting that this step does not introduce measurable bias in the library.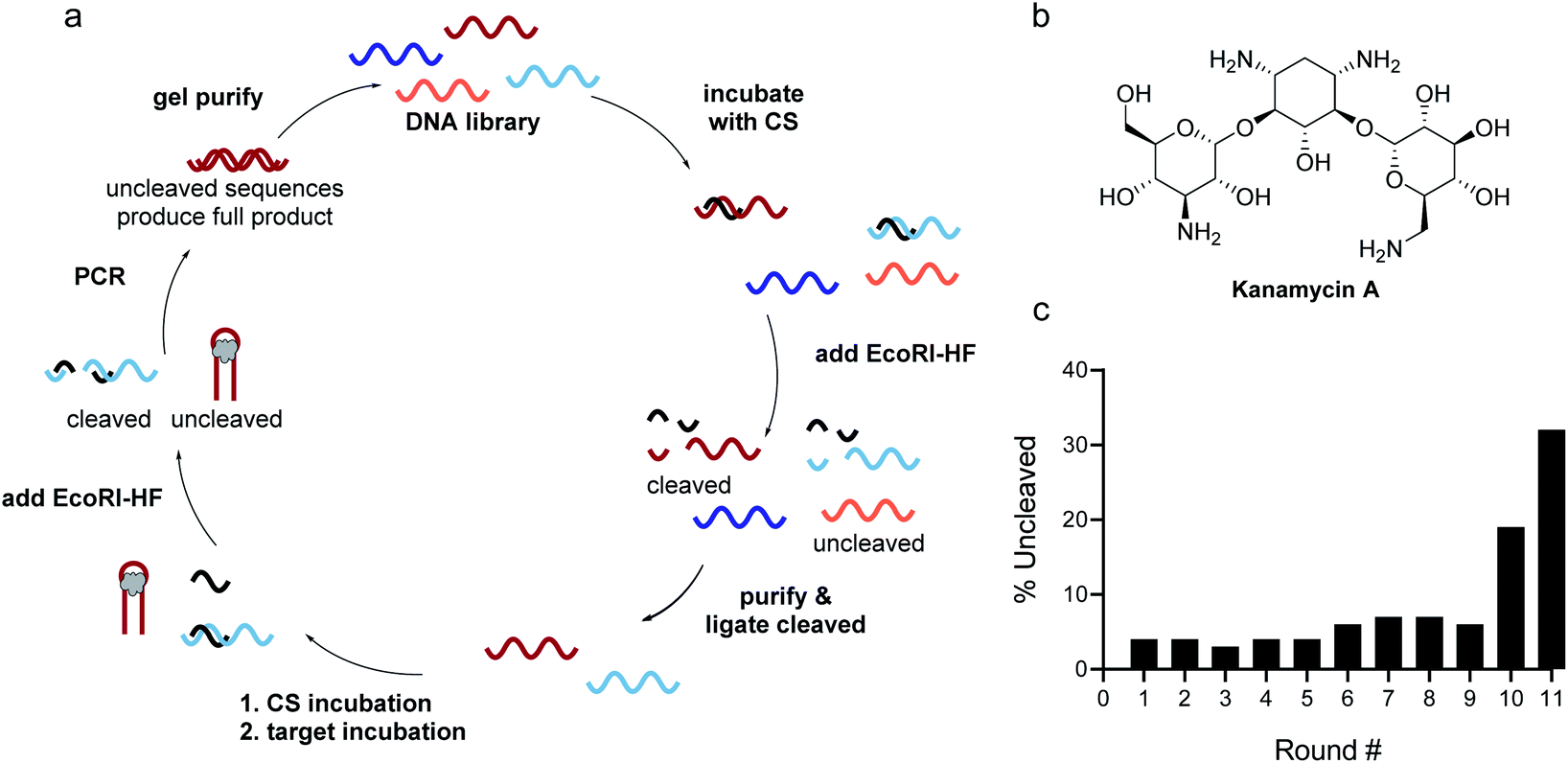 | ||
| Fig. 2 Homogenous EcoRI-HF SELEX to generate structure-switching aptamers. (a) FAM-labeled DNA library having an N40 random region was hybridized to capture strand and the complex digested with EcoRI-HF. The cleaved product (70 nt) was gel purified and full-length product (90 nt) was recovered through split ligation by T4 DNA ligase. The re-ligated library members were hybridized to free capture strand and incubated with the target. Active biosensor sequences were enriched by EcoRI-HF digestion followed by PCR amplification. ssDNA library was purified by 10% denaturing PAGE and carried on to subsequent selection rounds. (b) Chemical structure of kanamycin A. (c) Progression of EcoRI-HF SELEX to generate structure-switching aptamers to kanamycin A. Following digestion, cleavage products were monitored by 10% denaturing PAGE. Band intensity was used to quantify percent uncleaved. | ||
The selection was then carried out as described above, in that the library was hybridized to unfunctionalized capture strand in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio and then incubated with target for 1 h at 25 °C. EcoRI-HF digestion followed by PCR amplification enabled the enrichment of sequences that exhibit the desired target-dependent conformational change. To validate our method for structure-switching aptamer selection, we chose kanamycin A (Fig. 2b) as an initial target, as this small molecule has been the target used in previous structure-switching aptamer evolution methods, enabling us to benchmark our approach against these methods.17 To perform our selection, we started round one with 1 nmol of fluorescein (FAM) labeled N40 DNA library and subsequent rounds with >100 pmol. After the initial digestion and ligation, we formed the biosensor complex with 1 μM library and 2 μM capture strand through slow cooling hybridization. We then incubated the biosensor with 10 mM kanamycin A. Despite using a high concentration of target, we were somewhat concerned when we observed 4% uncleaved library in the initial round (Fig. 2c). Given the ability of our cleavage-ligation step to remove uncleavable library members, we attributed the unexpectedly high percent of uncleaved library to slightly inefficient EcoRI-HF digestion due to insufficient flanking nucleotides. However, compared to the sequences that evaded cleavage in our initial selection protocol, we recognized that in this case the surviving non-functional sequences would not have a selection advantage in the subsequent round, and thus were far less problematic. To ensure that this was the case, we incorporated a quality control step for several subsequent rounds in which a small aliquot of the enriched biosensor library was incubated with EcoRI-HF in the presence and absence of kanamycin A (ESI Fig. 10†). Our observation of increased digestion in the presence of kanamycin A compared to buffer alone provided reassurance that the selection was generating sequences having the desired structure-switching aptamer function. Given that some ligands may interfere with restriction enzyme activity, testing library cleavage in the presence of the target molecule is an important validation step. Significant enrichment above the background appeared in round 9 and increased further in round 10. While round 11 did increase as expected, there was also an increase in the background signal, which we attributed to potential contamination carried over from the initial digestion. At this point, we investigated the bulk biosensor activity of the enriched library from rounds 10 and 11 to determine whether we were ready to proceed to sequencing. For these experiments 5′-FAM-labeled library (1 μM) was hybridized to capture strand functionalized with 3′-BHQ1 (2 μM), then incubated with 1 mM kanamycin A for 1 h at 25 °C. After normalization to the fluorescence of aptamer alone, the percent displacement was calculated according to eqn (1):10
:2 ratio and then incubated with target for 1 h at 25 °C. EcoRI-HF digestion followed by PCR amplification enabled the enrichment of sequences that exhibit the desired target-dependent conformational change. To validate our method for structure-switching aptamer selection, we chose kanamycin A (Fig. 2b) as an initial target, as this small molecule has been the target used in previous structure-switching aptamer evolution methods, enabling us to benchmark our approach against these methods.17 To perform our selection, we started round one with 1 nmol of fluorescein (FAM) labeled N40 DNA library and subsequent rounds with >100 pmol. After the initial digestion and ligation, we formed the biosensor complex with 1 μM library and 2 μM capture strand through slow cooling hybridization. We then incubated the biosensor with 10 mM kanamycin A. Despite using a high concentration of target, we were somewhat concerned when we observed 4% uncleaved library in the initial round (Fig. 2c). Given the ability of our cleavage-ligation step to remove uncleavable library members, we attributed the unexpectedly high percent of uncleaved library to slightly inefficient EcoRI-HF digestion due to insufficient flanking nucleotides. However, compared to the sequences that evaded cleavage in our initial selection protocol, we recognized that in this case the surviving non-functional sequences would not have a selection advantage in the subsequent round, and thus were far less problematic. To ensure that this was the case, we incorporated a quality control step for several subsequent rounds in which a small aliquot of the enriched biosensor library was incubated with EcoRI-HF in the presence and absence of kanamycin A (ESI Fig. 10†). Our observation of increased digestion in the presence of kanamycin A compared to buffer alone provided reassurance that the selection was generating sequences having the desired structure-switching aptamer function. Given that some ligands may interfere with restriction enzyme activity, testing library cleavage in the presence of the target molecule is an important validation step. Significant enrichment above the background appeared in round 9 and increased further in round 10. While round 11 did increase as expected, there was also an increase in the background signal, which we attributed to potential contamination carried over from the initial digestion. At this point, we investigated the bulk biosensor activity of the enriched library from rounds 10 and 11 to determine whether we were ready to proceed to sequencing. For these experiments 5′-FAM-labeled library (1 μM) was hybridized to capture strand functionalized with 3′-BHQ1 (2 μM), then incubated with 1 mM kanamycin A for 1 h at 25 °C. After normalization to the fluorescence of aptamer alone, the percent displacement was calculated according to eqn (1):10
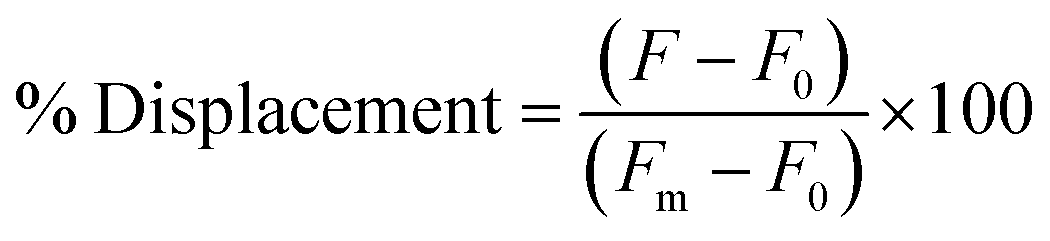 | (1) |
2.4. Next-generation sequencing to identify top candidates
The raw sequencing files were subjected to bioinformatic analysis using Emory's Galaxy server. After quality sorting and isolation of sequences that contained the intact forward primer sequence, we searched for sequences or sequence clusters that were conserved across all rounds. We utilized the FASTAptamer-count and FASTAptamer-cluster functions to analyze abundance and similarity and monitor how these had changed across each round.29 As expected, we observed a broader sequence distribution with subsequent rounds that occurs with genotypic frequency changes from enrichment (ESI Fig. 12c and d†).29 Indeed, this was also supported by the decrease in percentage of unique sequences in later rounds, as the unique sequences dropped from 99.9% in round 4 to 13.7% in round 11. We then used the FASTAptamer-compare and FASTAptamer-enrich functions to monitor changes in sequence distribution.29 The FASTAptamer-compare function is used to investigate the genotypic frequency changes during increasing rounds of SELEX by plotting the reads per minute for conserved sequences in two rounds. We found that the sequence distribution between replicate runs followed a y = x relationship but plotting round 9 and round 10 (x) with round 11 (y) resulted a broader distribution as sequences are being enriched or depleted. Round 9 to round 10 resulted in the broadest distribution, so we weighted our candidate selection with this in mind. We identified ten candidate sequences from five unique clusters with overall enrichment ≥2 from round 9 to 11 (Table 1, ESI Fig. 12 and 13†). Of these, cluster 16 and 1 were the most abundant and candidates were named using the format of [K-cluster]-[sequence #], e.g. K16-1.| Name K-cluster-# | Sequence 5′-3′ | Enrichment R9 → R10 | Enrichment R10 → R11 |
|---|---|---|---|
| K16-1 |
|
19 | 2 |
| K16-3 |
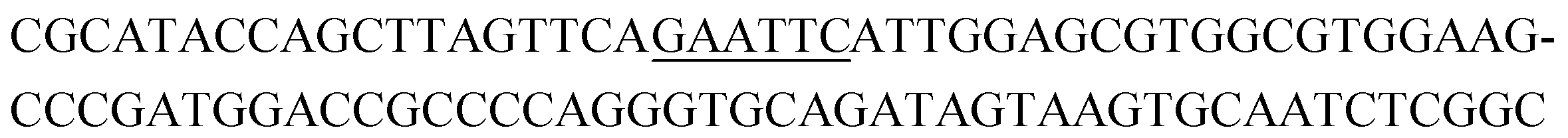
|
8 | 2 |
| K4-1 |
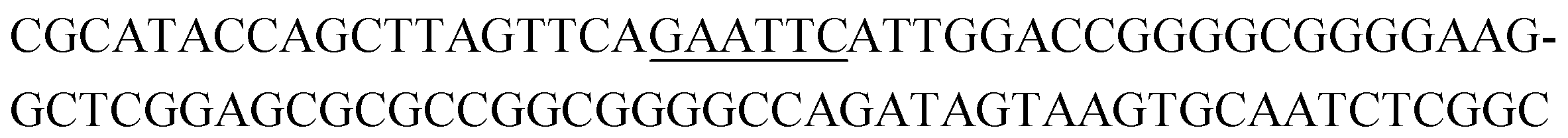
|
17 | 1 |
| K1-2 |
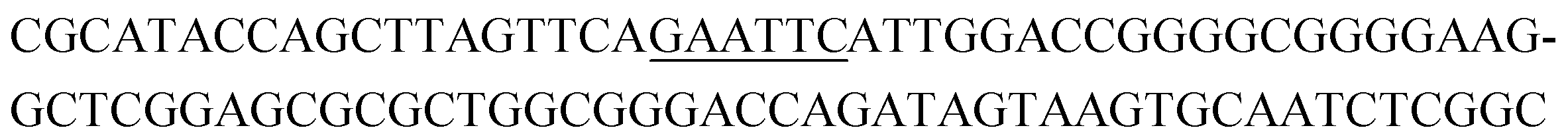
|
9 | 2 |
| K1-3 |
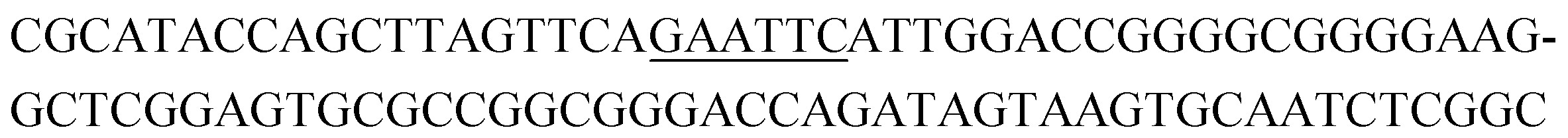
|
9 | 2 |
| K8-1 |

|
12 | 0.9 |
| K1-1 |

|
10 | 11 |
| K16-2 |

|
11 | 0.7 |
| K4-2 |
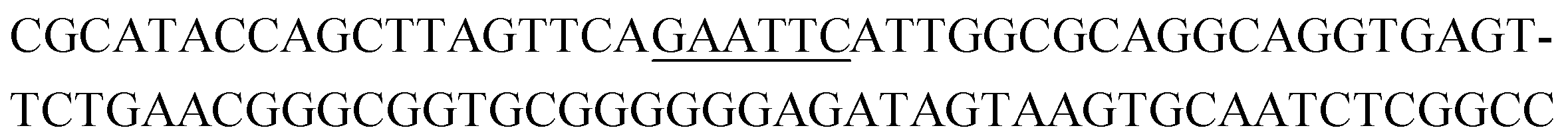
|
13 | 0.3 |
| K2-1 |
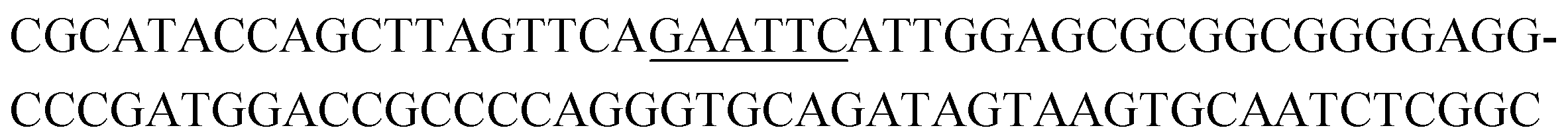
|
8 | 0.3 |
As a final bioinformatic quality control step, we wanted to ensure that there were no instances of sites within the N40 region that could hybridize to the capture strand, as had been the challenge in our initial selection protocol. To check for this, we input both the sequences and unfunctionalized capture strand in a 1:2 ratio into NUPACK software and estimated the structure of each complex at 25 °C.30 We were excited to see that all of our candidate sequences hybridized to the capture strand in the desired location in the primer binding site. Thus, we proceeded to order each candidate sequence having a 5′-FAM label, and we used a preliminary binding assay to identify top candidates to carry forward into biosensor optimization (ESI Fig. 14†). To measure target binding, excess kanamycin A was immobilized to Pierce™ NHS activated magnetic beads and each candidate sequence was incubated with the beads and the bound fraction heat eluted and quantified using a plate reader. Binding of candidate sequences was normalized to the known aptamer #3–19 from Capture SELEX.17,31 From this assay, we identified K16-1, K1-1, K1-2, and K2-1 as the top candidates to carry forward into biosensor characterization.
2.5. Characterization of candidates as kanamycin A structure-switching biosensors
Our lab has previously found that structure-switching biosensors display optimal performance at concentrations where the majority of aptamer is hybridized to the capture strand, but hybridization is not complete. This is likely because at these concentrations, the enthalpic gain from target binding is sufficient to shift equilibrium toward disassembly of the biosensor.10 Thus, we first screened varying concentrations of biosensor complex by hybridizing aptamer and capture strand in 1:1 ratio in concentrations varying from 10 nM to 1 μM. We chose K16-1 for this demonstration as it had the highest enrichment from round 9 to round 11 and was one of the most promising sequences in the initial binding screen. We monitored fluorescence using a plater reader and quantified the percent quenched using eqn (2).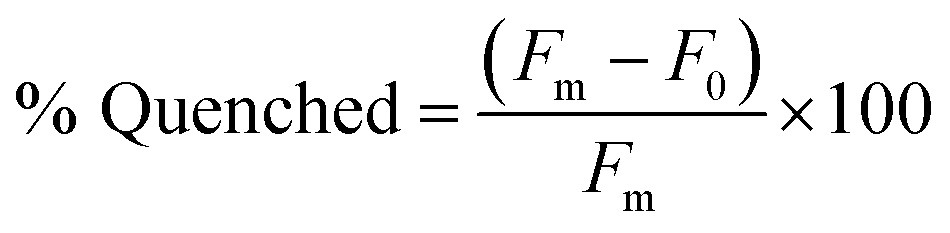 | (2) |
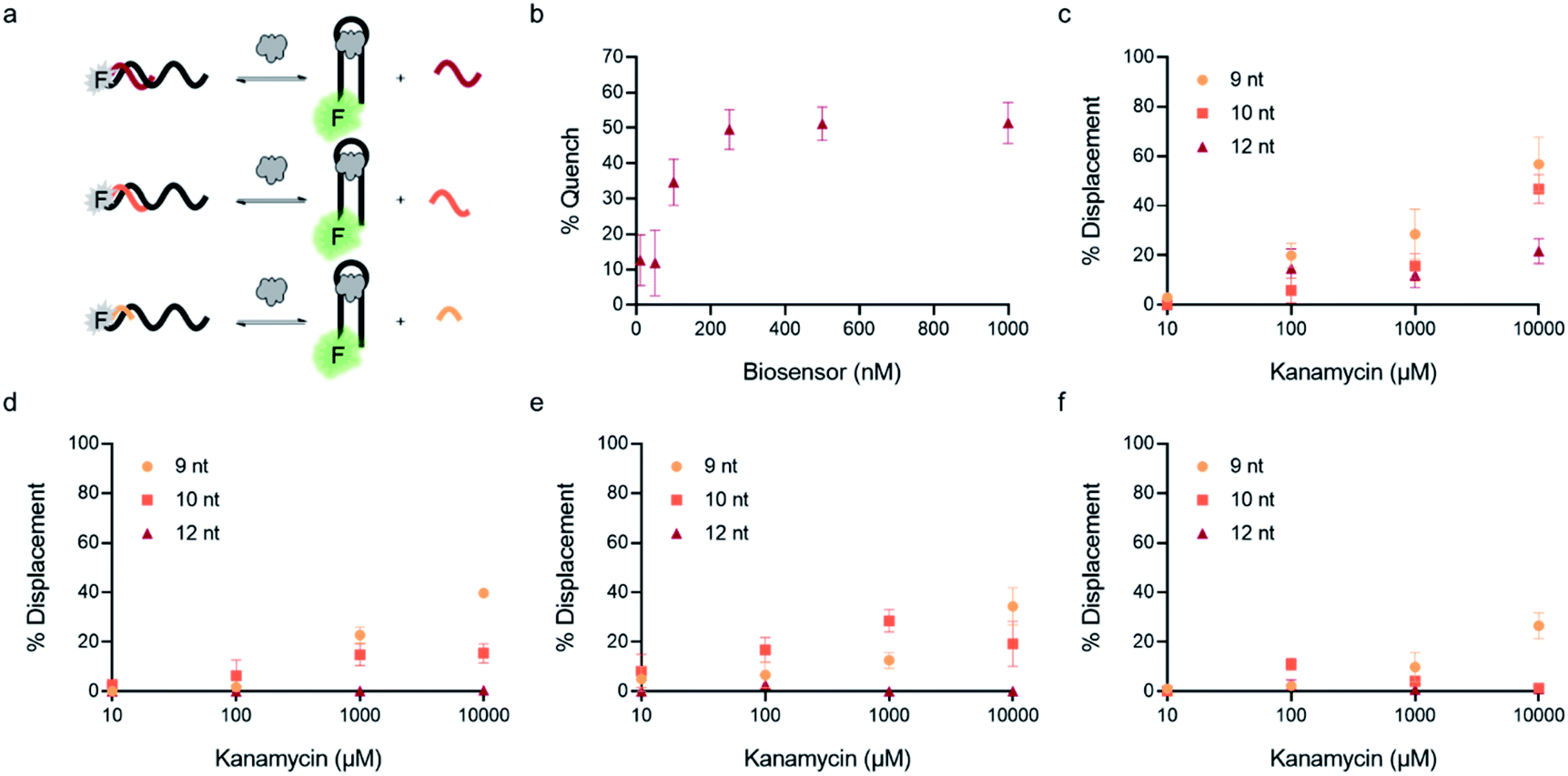 | ||
| Fig. 3 Optimization of biosensor conditions of top candidates. (a) Fluorescent structure-switching aptamer biosensors were tested using capture strands having varying lengths (9 nt, 10 nt, 12 nt). The signal is quenched in the absence of target, but addition of target results in dose-dependent displacement of the capture strand. (b) Biosensor concentration was optimized by monitoring concentration-dependent hybridization for K16-1 with 12 nt capture strand. Capture strand displacement as a function of kanamycin A concentration using varying capture stand lengths for (c) K16-1, (d) K1-1, (e) K1-2, (f) K2-1. Samples resulting in negative displacement were denoted 0% displacement. Error bars represent standard error (n = 3). | ||
In addition to overall biosensor concentration, capture strand length can also greatly affect displacement.6,10,11,16 Thus, in addition to the 12 nt capture strand analogous to that used in our selection, we also ordered 9 nt 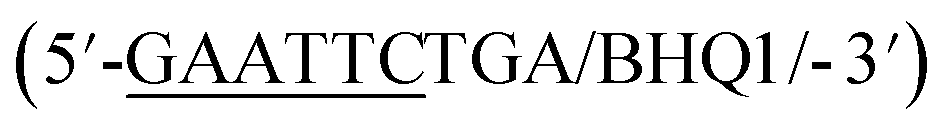 and 10 nt
and 10 nt 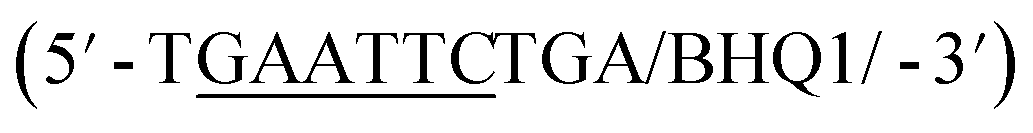 capture strands. Using our four candidate aptamer sequences, we generated solutions of 100 nM biosensor with each of the three capture strands and then tested target-dependent displacement by incubating each biosensor with kanamycin A at concentrations ranging from 10 μM to 10 mM. Most sequences resulted in low to moderate displacement for biosensors having the 9 and 10 nt capture strands and no observable displacement with the 12 nt capture strand. However, we were excited to observe that the K16-1 biosensor displayed significant displacement between 100 μM and 10 mM kanamycin A with all three capture strand lengths (Fig. 3c). As expected, the biosensor having the 9 nt capture strand provided the highest displacement, ranging from 20 ± 5% to 60 ± 10%. We attempted to further optimize the biosensor by increasing the ratio of aptamer:capture strand to 1:2 and 1:3, but we did not observe a significant difference in biosensor stability or target response (ESI Fig. 15†). Finally, to further validate our success in generating a structure-switching aptamer, we tested K16-1 using our optimized conditions (100 nM aptamer, 1:1 ratio, 9 nt capture strand) and an increased number of kanamycin A concentrations. To ensure our results were reproducible, we ran each sample in triplicate in three separate experiments, totalling nine individual replicates (Fig. 4b). Excitingly, as shown in Fig. 4b, we observed robust dose-dependent displacement from 90 μM to 10 mM kanamycin A.
capture strands. Using our four candidate aptamer sequences, we generated solutions of 100 nM biosensor with each of the three capture strands and then tested target-dependent displacement by incubating each biosensor with kanamycin A at concentrations ranging from 10 μM to 10 mM. Most sequences resulted in low to moderate displacement for biosensors having the 9 and 10 nt capture strands and no observable displacement with the 12 nt capture strand. However, we were excited to observe that the K16-1 biosensor displayed significant displacement between 100 μM and 10 mM kanamycin A with all three capture strand lengths (Fig. 3c). As expected, the biosensor having the 9 nt capture strand provided the highest displacement, ranging from 20 ± 5% to 60 ± 10%. We attempted to further optimize the biosensor by increasing the ratio of aptamer:capture strand to 1:2 and 1:3, but we did not observe a significant difference in biosensor stability or target response (ESI Fig. 15†). Finally, to further validate our success in generating a structure-switching aptamer, we tested K16-1 using our optimized conditions (100 nM aptamer, 1:1 ratio, 9 nt capture strand) and an increased number of kanamycin A concentrations. To ensure our results were reproducible, we ran each sample in triplicate in three separate experiments, totalling nine individual replicates (Fig. 4b). Excitingly, as shown in Fig. 4b, we observed robust dose-dependent displacement from 90 μM to 10 mM kanamycin A.
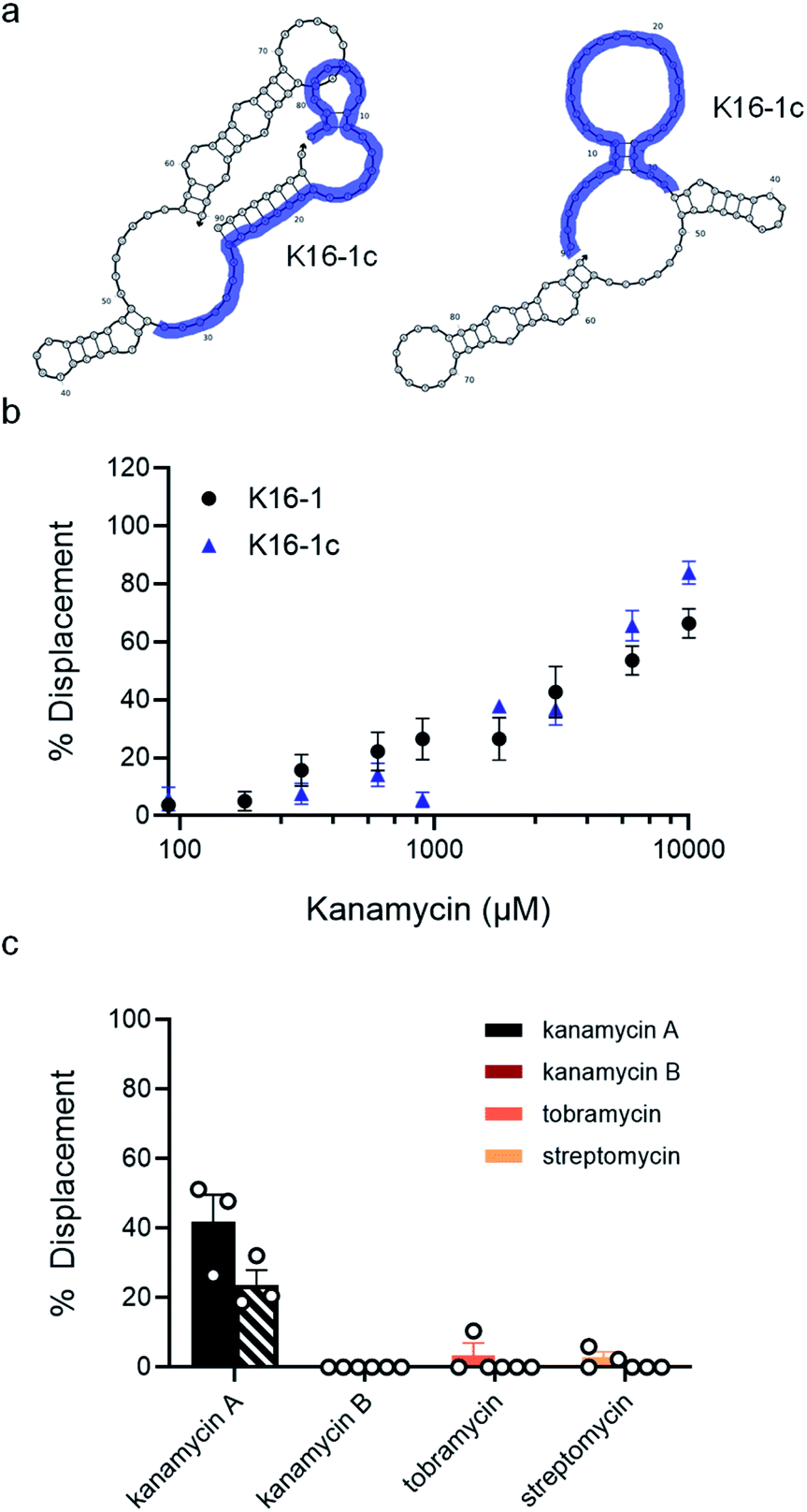 | ||
| Fig. 4 K16-1 and K16-1c function as structure-switching aptamer sensors and are specific to kanamycin A. (a) Secondary structure of K16-1 with (left) and without (right) the 9 nt capture strand calculated using NUPACK. The K16-1c truncation is highlighted in purple with (left) and without (right) the 9 nt capture strand calculated using NUPACK. (b) Capture strand displacement as a function of kanamycin A concentration with 100 nM K16-1 and 500 nM K16-1c with 100 nM and 125 nM 9 nt capture strand, respectively. Samples resulting in negative displacement were denoted 0% displacement. Error bars denote standard error (n = 9). (c) Displacement of K16-1 (solid) and K16-1c (dashed) biosensors with 1 mM kanamycin A, kanamycin B, streptomycin, or tobramycin. Error bars denote standard error (n = 3). | ||
Considering that the primary goal of structure-switching SELEX methods is to generate functional biosensors, we were interested to compare the performance of our K16-1 biosensor to a biosensor constructed from the best kanamycin aptamer generated using Capture SELEX.17,23 To do so, we optimized hybridization for the #3–19 aptamer with its cognate capture strand and in its reported binding buffer (100 mM sodium chloride, 20 mM Tris–Hydrochloride, 2 mM magnesium chloride, 5 mM potassium chloride, and 1 mM calcium chloride (pH 7.6)). We found that similar to K16-1, ideal hybridization was observed at 100 nM (ESI Fig. 16b†). At a 1:1 ratio, no change in fluorescence intensity was seen with a 9, 10, or 12 nt capture strand (ESI Fig. 16c†). This highlights that aptamers generated though selections that do not directly select for structure-switching biosensor activity cannot be easily adapted to this format. While there are several reports binding to kanamycin A, the capture SELEX aptamer does not appear to show structure-switching biosensor activity.17,20,21,23
For the next characterization, we determined the KD of K16-1 using microscale thermophoresis (MST) (ESI Fig. 17†).32 We chose this method as it does not require target immobilization and has become widely used for quantifying aptamer–small molecule binding interactions.33 Encouragingly, the observed KD of 340 ± 70 μM for K16-1 was similar to that of #3–19, which was found by MST to have a KD of 260 ± 50 μM.17,23 We note that this is weaker affinity than previously reported for the #3–19 sequence (3.9 μM), but previous characterization was carried out by elution rather than an equilibrium method such as MST and it has been established that KD can vary widely depending on the characterization method employed.5,27 Thus, we were excited that K16-1 had comparable binding affinity to #3–19 while also benefitting from inherent biosensor activity that the latter sequence does not possess.
We then sought to identify the minimal region of K16-1 required for binding to kanamycin A. Some aptamer selection methods involve minimization by removal of the constant regions. We removed both primer binding sites to give K16-1a (ESI Fig. 18a†). We were not entirely surprised that there was no detectable binding of K16-1a to kanamycin A (ESI Fig. 18b†), as our experience has shown that the constant regions are often incorporated into the binding region of optimized aptamers and thus a better approach is to pursue truncation based on structural components.33,34 We performed two truncations based on the NUPACK structures generated with and without capture strand to give 50 nt K16-1b and 32 nt K16-1c (Fig. 4a). Excitingly, MST analysis demonstrated that both of these sequences bind to kanamycin A with KD values of 400 ± 100 μM (ESI Fig. 18†). We note that our observed affinities are lower than those of some other aptamers for kanamycin A. However, this is not surprising as these other aptamers were selected only for binding and aptamers selected in the structure-switching format are known for having weaker binding affinities compared to traditional aptamers.35 We reason that for novel targets requiring high-affinity structure-switching aptamers, suitable candidates could be identified by decreasing the target concentration in later selection rounds in order to increase the selection pressure and enable isolation of candidates having increased affinity.
Since the K16-1c truncation was so drastic, we were interested to see if it still retained structure-switching biosensor capability. Given the structural changes introduced by truncation, we re-optimized biosensor concentration and stoichiometry and found that using 500 nM aptamer with 125 nM of the 9 nt capture strand provided robust target-dependent displacement (ESI Fig. 20c†). Excitingly, the dose-dependent displacement with K16-1c under these conditions closely matches that of the full aptamer from 90 μM to 10 mM kanamycin A but using a dramatically shortened sequence (Fig. 4b).
Although varying biosensor conditions are known to alter the extent and dynamic range of displacement, we found several reports also highlighting that the KD of a structure-switching aptamer to its cognate capture strand is directly proportional to the Ksens of a structure-switching aptamer biosensor (eqn (3)).36,37 Thus, the equilibrium position of hybridization affects the stability of the biosensor, in turn impacting response to ligand (ref. 10). Interestingly, this model also offers an alternative approach to measuring KD value of the aptamer for the target, enabling us to validate our MST results. Using the optimized aptamer concentrations (100 nM, 500 nm), we monitored the hybridization as a function of fluorescence quenching from 0.01 μM to 5 μM for K16-1 and 0.05 μM to 50 μM for K16-1c and found KD values of 1.0 ± 0.4 μM and 0.06 ± 0.09 μM, respectively, for biosensor hybridization (ESI Fig. 21a and b†). Dose-dependent displacement data from Fig. 4 were also graphed using GraphPad Prism and fit according to the law of mass action to determine Ksens values, which were found to be 3 ± 1 mM and 8 ± 2 mM for K16-1 and K16-1c, respectively (ESI Fig. 21c and d†). Using these values with eqn (3) provided aptamer–target KD values of 400 ± 100 μM for K16-1 and 90 ± 30 μM for K16-1c, which are in agreement with our MST data.
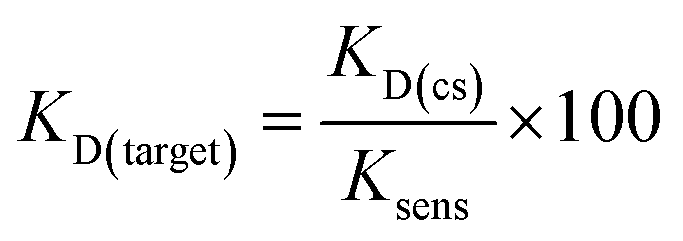 | (3) |
As a final characterization, we investigated the selectivity of our optimized biosensors for kanamycin A compared to structurally similar aminoglycosides. Using our optimized conditions with K16-1 and K16-1c, we incubated the biosensor with 1 mM and 10 mM each of kanamycin A, kanamycin B, streptomycin, and tobramycin. We observed that at 10 mM, kanamycin B, streptomycin, and tobramycin led to non-specific quenching of aptamer fluorescence (ESI Fig. 22†). However, this effect was relatively minor at 1 mM and we were able to normalize for it in the biosensor experiments. Excitingly, we found that neither kanamycin B, tobramycin, or streptomycin resulted in significant displacement, while the displacement observed for kanamycin A at 1 mM was similar to that in previous experiments at 42 ± 8% for K16-1 and 24 ± 4% for K16-1c (Fig. 4c). These data together demonstrate that K16-1 and K16-1c function as selective structure-switching biosensors for kanamycin A.
3. Conclusions
In summary, we report the first homogenous method for in vitro evolution of structure-switching DNA aptamers. By utilizing the native activity of restriction enzymes, we are able to cleave non-functional sequences and enable functional structure-switching candidates to be selectively amplified by PCR. This obviates the need to immobilize the target or capture strand on beads, which offers greater convenience and allows for selection to proceed under conditions that most closely replicate those in which the resulting biosensors will be deployed. Excitingly, after eleven rounds of selection, we identified four candidates that bind to kanamycin A and show biosensor activity. The most enriched sequence (K16-1) also showed the most robust biosensor activity, and this was achieved with minimal post-selection optimization. We were able to perform two successful truncations that resulted in an active structure-switching aptamer of 32 nt (K16-1c). While we chose kanamycin A as a model target in order to benchmark our method relative to previous structure-switching selections, our approach is anticipated to be broadly useful for other small molecule targets. Further, this is not limited to EcoRI-HF based libraries, as this general approach could be applied using other combinations of nuclease and recognition sequence. A potentially interesting future line of method development could involve adapting this approach to RNA or XNA aptamer selections. While these sequences would not serve as substrates for restriction enzymes, we envision that incorporating a short DNA restriction site within the constant region of the RNA or XNA libraries would still allow for structure-switching isolation, and it may be possible to engineer this small sequence block into RNA or XNA post-selection. Nevertheless, the relative dearth of aptamers that demonstrate structure-switching activity has represented a significant limitation in the aptamer biosensor field, and we envision that our method will overcome this limitation and significantly accelerate the development of aptamer biosensors for a wide range of biologically and environmentally important small-molecule targets.4. Experimental
4.1. Materials
All materials were purchased from commercial vendors and used without further purification. All DNA sequences were purchased from the University of Utah DNA/Peptide Synthesis Core Facility or Integrated DNA Technologies. A list of oligonucleotides is provided in ESI Fig. 2.† All oligonucleotides were purified by 10% denaturing polyacrylamide gel electrophoresis prior to use. Gel bands were excised and incubated in crush and soak buffer at 95 °C for 2 h. The DNA was then separated from the gel pieces using Cellulose Acetate Membrane Filters (ThermoFisher) and concentrated using Amicon Ultra-0.5 Centrifugal Unit with Ultracel 10 membrane (EMD Millipore).4.2. EcoRI-based SELEX
 was hybridized to unfunctionalized capture sequence
was hybridized to unfunctionalized capture sequence  in a 1:2 ratio in selection buffer to a final concentration of 1 μM library and 2 μM capture sequence (round 1, 1 nmol; round 2+, 100–200 pmol). The composition of the selection buffer was 50% 100 mM sodium chloride, 20 mM Tris–Hydrochloride, 2 mM magnesium chloride, 5 mM potassium chloride, and 1 mM calcium chloride (pH 7.6); 50% 1× Cut Smart Buffer (New England Biolabs) 50 mM potassium acetate, 20 mM Tris–Acetate, 10 mM magnesium acetate, 100 μg mL−1 bovine serum albumin (pH 7.9).
in a 1:2 ratio in selection buffer to a final concentration of 1 μM library and 2 μM capture sequence (round 1, 1 nmol; round 2+, 100–200 pmol). The composition of the selection buffer was 50% 100 mM sodium chloride, 20 mM Tris–Hydrochloride, 2 mM magnesium chloride, 5 mM potassium chloride, and 1 mM calcium chloride (pH 7.6); 50% 1× Cut Smart Buffer (New England Biolabs) 50 mM potassium acetate, 20 mM Tris–Acetate, 10 mM magnesium acetate, 100 μg mL−1 bovine serum albumin (pH 7.9).
The hybridized complex was then divided into portions with a 10:1 ratio of complex to EcoRI-HF (100000 U mL−1) (New England Biolabs). The solutions were incubated for 2 h at 37 °C, followed by 20 min at 65 °C to denature the EcoRI. The samples were then run on a 10% denaturing PAGE gel at 270 V for 30–45 minutes. The gel was stained with SYBR Gold for 20 min in 1× Tris/Borate/EDTA (TBE) buffer. The desired gel band (70 nt) was visualized using a UV-transilluminator, excised, and incubated in crush and soak buffer at 37 °C for 16 h. The DNA was then separated from the gel pieces using Cellulose Acetate Membrane Filters (ThermoFisher), ethanol precipitated, and resuspended in water (<5 μL). The remaining DNA was quantified via Nanodrop.
:1.1:1 ratio, respectively. Hybridization was performed in 1× phosphate buffered saline (PBS) with a final concentration ≥1 μM using slow cooling from 95 °C to 25 °C over 30 min. This complex was then added to the ligation reaction with final conditions: complex, 0.1 μM; T4 DNA ligase (50000 units per mL); 1× T4 DNA ligase reaction buffer (New England Biolabs). The solution was incubated for 1 h at 25 °C, followed by 20 min at 65 °C to denature T4 DNA ligase. A small portion was run on a 10% PAGE gel to determine ligation efficiency. This was calculated using percent ligated (fluorescence band volume for the ligated 90 nt band/total lane volume). The remaining DNA was purified using a MinElute PCR cleanup column and then ethanol precipitated to reduce sample volume. The remaining DNA was resuspended in selection buffer and quantified via Nanodrop.
:2 ratio in selection buffer to a final concentration of 1 μM library and 2 μM unfunctionalized capture sequence. This solution was hybridized using slow cooling from 95 °C to 25 °C over 30 min. Buffer (for negative control) or 10 mM target (for selection) were then added and the solutions incubated for 1 h at 25 °C.
4.3. EcoRI digest of biosensor complex
The biosensor complex was divided into 10 μL portions and 1 μL (5 units) EcoRI (New England Biolabs) was added to each aliquot. The solutions were incubated for 2 h at 37 °C, followed by 20 min at 65 °C to denature EcoRI. The library was recovered using a MinElute PCR cleanup column (Qiagen). Samples were analyzed by denaturing 10% PAGE to monitor digestion of the biosensor. The gels were imaged on a GE Amersham Typhoon RGB scanner using a 488 nm excitation laser and the Cy2 525BP20 emission filter. Digestion efficiency was determined by the percent cleaved (fluorescence band volume for the cleaved product/total lane volume) using ImageJ.4.4. PCR amplification of library after digest
The recovered DNA library was amplified in 50 μL PCR reactions containing 0.2 μM template, 0.5 μM primers (forward primer, 5′-/FAM/CGCATACCAGCTTAGTTCAGAATTCATT-3′; reverse primer, 5′-TTTTTTTTTTTTTTTTTTTT/Sp9/GCCGAGATTGCACTTACTATCT-3′) and 1× Hot Start Master Mix (10 mM Tris–HCl, 50 mM KCl, 1.5 mM MgCl2, 0.2 mM dNTPs, 5% Glycerol, 25 units per mL Hot Start Taq DNA polymerase, pH 8.3, New England Biolabs). The template was amplified with an initial denaturation at 95 °C for 3 min, 25 cycles of (95 °C for 30 s, 55 °C for 30 s, and 72 °C for 20 s), and a final extension 72 °C for 2 min. The amplified double stranded DNA was purified using a MinElute PCR cleanup column (Qiagen) and the labeled strand was separated on a denaturing 10% polyacrylamide gel. The desired gel band was excised and >100 picomoles recovered as described previously.4.5. Next-generation sequencing
Round 7 (bead-assisted) and rounds 4, 9, 10, and 11 were amplified in 50 μL PCR reactions containing 0.2 μM template, 0.5 μM primers (forward primer, 5′-CGCATACCAGCTTAGTTCAGAATTCATT-3′; reverse primer, 5′-GCCGAGATTGCACTTACTATCT-3′) and 1× Hot Start Master Mix (10 mM Tris–HCl, 50 mM KCl, 1.5 mM MgCl2, 0.2 mM dNTPs, 5% glycerol, 25 units per mL Hot Start Taq DNA polymerase, pH 8.3) (New England Biolabs). The template was amplified with an initial denaturation at 95 °C for 3 min, 25 cycles of (95 °C for 30 s, 55 °C for 30 s, and 72 °C for 20 s), and a final extension 72 °C for 2 min. The amplified double stranded DNA was purified using a MinElute PCR cleanup column (Qiagen). PCR products were quantified via Nanodrop and normalized to 20 ng μL−1 in nuclease free water. The samples were sent to Genewiz (South Plainfield, NJ) for Amplicon-EZ (150–500 bp) sequencing.4.6. Bioinformatics of aptamer pools
Raw sequencing data were uploaded to the Emory Galaxy server. Bioinformatic analysis was carried out as described in Alam et al.29 Briefly, the reads were first trimmed to only contain the 90 nt library sequence and filtered by quality with a minimum score of 30%. Sequences were then isolated that contained the forward primer binding site through the barcode function. The FASTAptamer-count and FASTAptamer-cluster function were run to gather information about abundance and similarity within each round. These files were then used as inputs for FASTAptamer-enrich and FASTAptamer-enrich functions. We identified candidates by overall count enrichment from round 9 to round 11. The 2D structures of the candidates hybridized with the 12 nt capture strand were modelled at 25 °C using NUPACK software.4.7. Biosensor construction
DNA stock solutions were diluted in selection buffer. Biosensors were prepared by combining FAM-labeled aptamer (100 nM) and BHQ1-labeled capture strand (100 nM) in selection buffer unless otherwise noted. To hybridize, this solution was heated to 95 °C and slow-cooled to 25 °C over 30 min in a thermal cycler. Biosensor solutions were stored at 4 °C until use.4.8. Aminoglycoside measurement
The biosensor was warmed up to room temperature and 50 μL added to 96-well black plates (Corning, #3915). In triplicate, increasing concentrations of kanamycin A (50 μL) was added to the wells and the solutions incubated for 1 h at 25 °C while protected from light using foil. Displacement was quantified by measuring the fluorescence intensity on a Cytation 5 multi-mode plate reader (BioTek) using excitation at 490 nm and emission at 520 nm (bandwidth 9, read height 6.5 mm). All samples were normalized to wells containing FAM-labeled aptamer alone. Percent displacement was calculated using eqn (1) and plotted using GraphPad Prism.4.9. MST
MST experiments were performed to determine the binding affinity of K16-1 to kanamycin A. Briefly, a serial dilution of kanamycin A in the selection buffer with 0.05% tween-20 was made to provide sixteen samples ranging from 0.15 μM to 5 mM. Cy5-labeled K16-1 at 10 nM (10 μL) was added to 10 μL of each sample. Triplicate samples from three independent experiments were run on a Monolith NT.115 Pico at 25 °C, with 5% LED power and high laser power. Data were fitted using GraphPad Prism analysis software to determine KD.Data availability
Raw next-generation sequencing reads are deposited as Sequence Read Archive files on NCBI's BioProject ID PRJNA728693.38Author contributions
T. A. F., A. E. R., and J. M. H. conceived the initial project idea. A. A. S. led the design and implementation of the reported experiments with assistance from R. G. L. and H. S. A. G. All authors provided critical feedback and helped shape the research, analysis, and manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Mike Hanson and the oligonucleotide and peptide synthesis facility at the University of Utah for oligonucleotide materials. This work was supported by the Defense Threat Reduction Agency [HDTRA118-1-0029 to J. M. H] and the National Science Foundation [CHE 1904885 to J. M. H.]. Any opinions, findings and conclusions or recommendations expressed in this publication are those of the authors and do not necessarily reflect the views of DTRA.Notes and references
- V. T. Nguyen, Y. S. Kwon and M. B. Gu, Curr. Opin. Biotechnol., 2017, 45, 15–23 CrossRef CAS.
- E. M. McConnell, J. Nguyen and Y. Li, Front. Chem., 2020, 8, 434 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CrossRef CAS.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS.
- A. Ruscito and M. C. DeRosa, Front. Chem., 2016, 4, 14 Search PubMed.
- R. Nutiu and Y. Li, J. Am. Chem. Soc., 2003, 125, 4771–4778 CrossRef CAS.
- S. D. Jhaveri, R. Kirby, R. Conrad, E. J. Maglott, M. Bowser, R. T. Kennedy, G. Glick and A. D. Ellington, J. Am. Chem. Soc., 2000, 122, 2469–2473 CrossRef CAS.
- J. Chen, Z. Fang, J. Liu and L. Zeng, Food Control, 2012, 25, 555–560 CrossRef CAS.
- F. Liu, A. Ding, J. Zheng, J. Chen and B. Wang, Sensors, 2018, 18, 1769 CrossRef.
- T. A. Feagin, D. P. V. Olsen, Z. C. Headman and J. M. Heemstra, J. Am. Chem. Soc., 2015, 137, 4198–4206 CrossRef CAS.
- Z. Zhu, T. Schmidt, M. Mahrous, V. Guieu, S. Perrier, C. Ravelet and E. Peyrin, Anal. Chim. Acta, 2011, 707, 191–196 CrossRef CAS PubMed.
- J. Canoura, Z. Wang, H. Yu, O. Alkhamis, F. Fu and Y. Xiao, J. Am. Chem. Soc., 2018, 140, 9961–9971 CrossRef CAS PubMed.
- H. Gao, J. Zhao, Y. Huang, X. Cheng, S. Wang, Y. Han, Y. Xiao and X. Lou, Anal. Chem., 2019, 91, 14514–14521 CrossRef CAS.
- J. Canoura, H. Yu, O. Alkhamis, D. Roncancio, R. Farhana and Y. Xiao, J. Am. Chem. Soc., 2021, 143, 805–816 CrossRef CAS.
- H. Yu, O. Alkhamis, J. Canoura, Y. Liu and Y. Xiao, Angew. Chem., Int. Ed., 2021, 133(31), 16938–16961 CrossRef.
- R. Nutiu and Y. Li, Angew. Chem., Int. Ed., 2005, 44, 1061–1065 CrossRef CAS PubMed.
- R. Stoltenburg, N. Nikolaus and B. Strehlitz, J. Anal. Methods Chem., 2012, 1, 14 Search PubMed.
- H. Qu, A. T. Csordas, J. Wang, S. S. Oh, M. S. Eisenstein and H. T. Soh, ACS Nano, 2016, 10, 7558–7565 CrossRef CAS PubMed.
- N. Qiao, J. Li, X. Wu, D. Diao, J. Zhao, J. Li, X. Ren, X. Ding, D. Shangguan and X. Lou, Anal. Chem., 2019, 91, 13383–13389 CrossRef CAS.
- C. Lyu, I. M. Khan and Z. Wang, Talanta, 2021, 229, 122274 CrossRef CAS PubMed.
- T. A. Feagin, N. Maganzini and H. T. Soh, ACS Sens., 2018, 3, 1611–1615 CrossRef CAS PubMed.
- S. Buckhout-White, C. Person, I. L. Medintz and E. R. Goldman, ACS Omega, 2018, 3, 495–502 CrossRef CAS.
- N. Nikolaus and B. Strehlitz, Sensors, 2014, 14, 3737–3755 CrossRef CAS.
- Z. Tan, T. A. Feagin and J. M. Heemstra, J. Am. Chem. Soc., 2016, 138, 6328–6331 CrossRef CAS PubMed.
- Z. Tan and J. M. Heemstra, ChemBioChem, 2018, 19, 1853–1857 CrossRef CAS.
- V. Pingoud, W. Wende, P. Friedhoff, M. Reuter, J. Alves, A. Jeltsch, L. Mones, M. Fuxreiter and A. Pingoud, J. Mol. Biol., 2009, 393, 140–160 CrossRef CAS.
- M. McKeague and M. C. Derosa, J. Nucleic Acids, 2012, 2012, 748913 Search PubMed.
- S. M. Shamah, J. M. Healy and S. T. Cload, Acc. Chem. Res., 2008, 41, 130–138 CrossRef CAS.
- K. K. Alam, J. L. Chang and D. H. Burke, Mol. Ther.–Nucleic Acids, 2015, 4, e230 CrossRef CAS PubMed.
- J. N. Zadeh, C. D. Steenberg, J. S. Bois, B. R. Wolfe, M. B. Pierce, A. R. Khan, R. M. Dirks and N. A. Pierce, J. Comput. Chem., 2011, 32, 170–173 CrossRef CAS PubMed.
- K. M. Song, M. Cho, H. Jo, K. Min, S. H. Jeon, T. Kim, M. S. Han, J. K. Ku and C. Ban, Anal. Biochem., 2011, 415, 175–181 CrossRef CAS.
- M. Jerabek-Willemsen, C. J. Wienken, D. Braun, P. Baaske and S. Duhr, Assay Drug Dev. Technol., 2011, 9, 342–353 CrossRef CAS.
- A. E. Rangel, Z. Chen, T. M. Ayele and J. M. Heemstra, Nucleic Acids Res., 2018, 46, 8057–8068 CrossRef CAS.
- A. D. Ortega, V. Takhaveev, S. R. Vedelaar, Y. Long, N. Mestre-Farràs, D. Incarnato, F. Ersoy, L. F. Olsen, G. Mayer and M. Heinemann, Cell Chem. Biol., 2021, 00161–00166 Search PubMed , S24519456, .
- R. Nutiu and Y. Li, Chem.–Eur. J., 2004, 10, 1868–1876 CrossRef CAS PubMed.
- N. Nakatsuka, K. A. Yang, J. M. Abendroth, K. M. Cheung, X. Xu, H. Yang, C. Zhao, B. Zhu, Y. S. Rim, Y. Yang, P. S. Weiss, M. N. Stojanović and A. M. Andrews, Science, 2018, 362, 319–324 CrossRef CAS PubMed.
- J. Hu and C. J. Easley, Analyst, 2011, 136, 3461–3468 RSC.
- Y. Kodama, M. Shumway and R. Leinonen, Nucleic Acids Res., 2012, 40, D54–D56 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02715h |
| This journal is © The Royal Society of Chemistry 2021 |