
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA modular approach to mechanically gated photoswitching with color-tunable molecular force probes†
Ross W.
Barber
 and
Maxwell J.
Robb
*
and
Maxwell J.
Robb
*
Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, USA. E-mail: mrobb@caltech.edu
First published on 22nd July 2021
Abstract
Molecular force probes conveniently report on mechanical stress and/or strain in polymers through straightforward visual cues. Unlike conventional mechanochromic mechanophores, the mechanically gated photoswitching strategy decouples mechanochemical activation from the ultimate chromogenic response, enabling the mechanical history of a material to be recorded and read on-demand using light. Here we report a completely redesigned, highly modular mechanophore platform for mechanically gated photoswitching that offers a robust, accessible synthesis and late stage diversification through Pd-catalyzed cross-coupling reactions to precisely tune the photophysical properties of the masked diarylethene (DAE) photoswitch. Using solution-phase ultrasonication, the reactivity of a small library of functionally diverse mechanophores is demonstrated to be exceptionally selective, producing a chromogenic response under UV irradiation only after mechanochemical activation, revealing colored DAE isomers with absorption spectra that span the visible region of the electromagnetic spectrum. Notably, mechanically gated photoswitching is successfully translated to solid polymeric materials for the first time, demonstrating the potential of the masked diarylethene mechanophore for a variety of applications in force-responsive polymeric materials.
Introduction
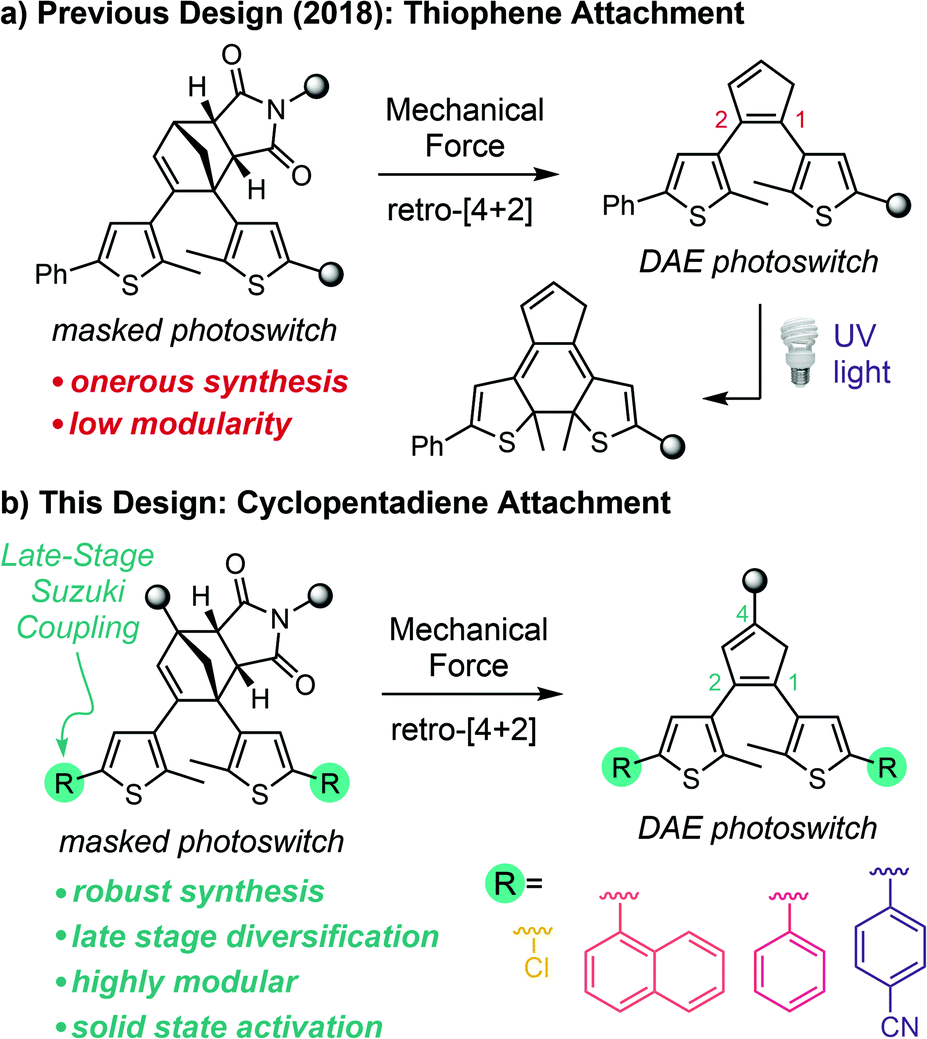
The development of stress–sensitive molecules called mechanophores has advanced the scope and functionality of stimuli-responsive polymers.1,2 Mechanical stress typically leads to nonspecific homolytic scission of polymer chains; however, mechanophores covalently incorporated into polymers harness these normally destructive forces to effect productive chemical transformations.3 Mechanical force is both ubiquitous and straightforward to apply using a range of different methods that provide temporal and spatial control,4–9 making it an appealing stimulus for responsive materials. A wide variety of mechanophores have been developed that produce functional changes in polymers subjected to mechanical force including catalyst activation,10,11 conductivity switching,12–14 small molecule release,15–17 fluorescence changes,18–21 and chemiluminescence,22 among others. Mechanophores that generate a change in color are particularly useful due to their ability to act as molecular force beacons, providing straightforward visual identification of the locations of stress and/or strain in polymeric materials.23 Nevertheless, the reversibility of typical mechanochromic mechanophores like spiropyran,6 naphthopyran,24 rhodamine,25 and diarylbibenzofuranone,26 for example, limits the mechanical history of a material from being recorded for future detection.27 Moreover, many mechanochromic mechanophores also lack mechanochemical specificity, as the same reactions that produce the chromogenic response can be promoted by alternative stimuli such as heat, light, or chemical reagents in the absence of mechanical force.6,24,25,28Inspired by the work of Craig and Boulatov on mechanical gating,29 our group introduced the concept of mechanically gated photoswitching to address, in part, some of the limitations of mechanochromic mechanophores in stress reporting materials.30 In our original design, mechanical force induces the formal retro-[4 + 2] cycloaddition reaction of a cyclopentadiene–maleimide Diels–Alder adduct to reveal a diarylethene (DAE) photoswitch, which is subsequently transformed to the conjugated colored isomer under UV light via a 6π electrocyclic ring-closing reaction (Scheme 1a). In this case, color generation only occurs under UV irradiation if mechanical force has first unmasked the DAE photoswitch, and thus the photochemical ring-closing reaction is gated by the mechanochemical retro-[4 + 2] cycloaddition reaction. An important feature of this general molecular design strategy is that mechanochemical activation of the mechanophore is decoupled from the ultimate functional response of the masked intermediate. In principle, this approach enables independent control over each reaction through structural variation, imparting a high degree of modularity to the system. Additionally, the DAE photoswitch generates color reversibly once unmasked by mechanical force, generally with excellent fatigue resistance.31 While the ring-closed isomer reverts to its colorless form under visible light, irradiation with UV light regenerates the colored isomer and the process can be repeated over many cycles, ensuring that evidence of mechanical activation is preserved.30
 | ||
| Scheme 1 Molecular design strategies for mechanically gated photoswitching. | ||
Despite the promising molecular design strategy, our initial mechanophore is hampered by an inefficient and onerous synthesis and a lack of chemical modularity, which restrict its application and further development. Moreover, unresolved issues of selectivity that manifest as slight coloration under UV light prior to mechanical activation30 and unsuccessful attempts to realize mechanically gated photoswitching in solid polymeric materials have prompted us to completely redesign the mechanophore. Here we present a second-generation platform for mechanically gated photoswitching that relies on a robust synthesis and, critically, enables late-stage diversification to provide a library of color-tunable, masked DAE mechanophores (Scheme 1b). The reactivity of the mechanophores is demonstrated to be incredibly selective, with no detectable coloration under UV light prior to mechanical activation. Notably, mechanically gated photoswitching is also successfully translated to solid polymeric materials, opening the door to promising potential applications in stress sensing, encryption, and patterning.
Results and discussion
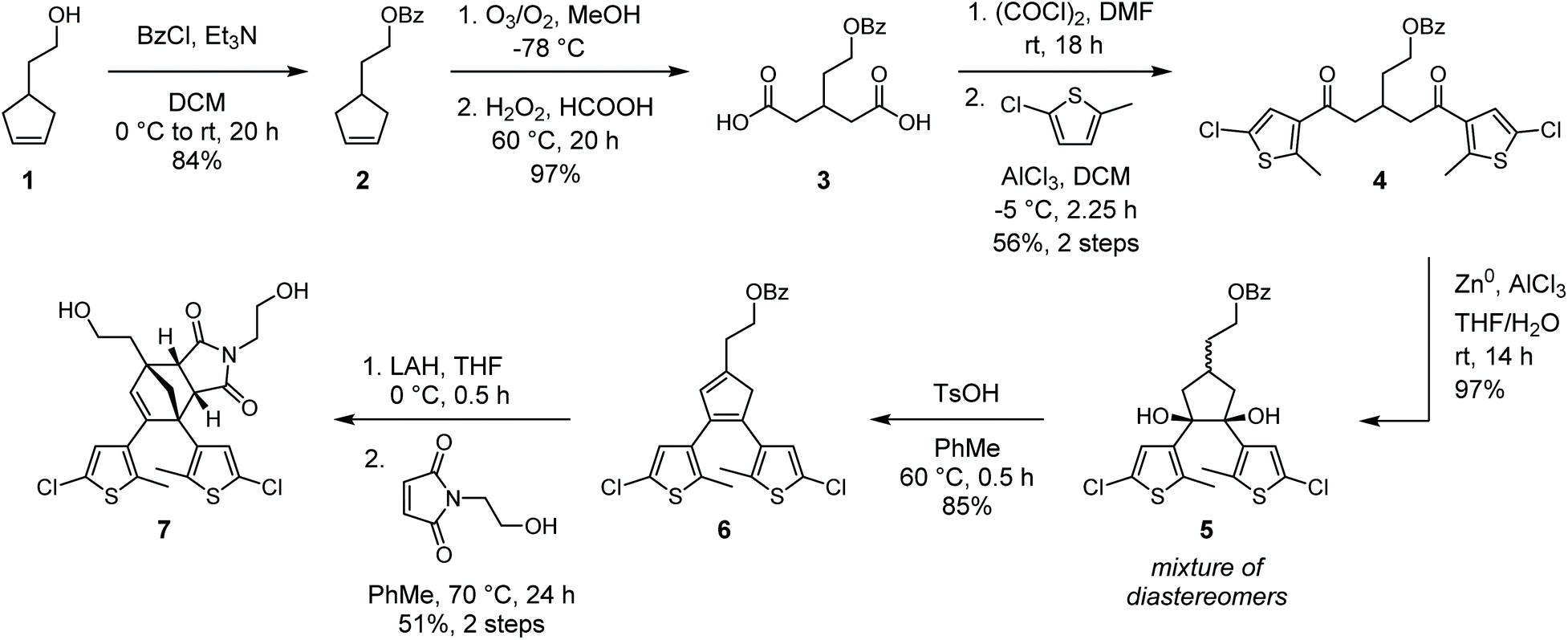
In our original synthesis of the masked DAE mechanophore, a key step involved the Diels–Alder reaction between a maleimide dienophile and an asymmetric 1,2-disubstituted cyclopentadiene that exists as a mixture of three inseparable tautomers. The reaction produces six constitutional isomers from which the desired Diels–Alder adduct, with endo stereochemistry and polymer attachment located on one of the thiophene rings proximal to the cyclopentadiene–maleimide junction, was isolated in just 20% yield (see Scheme 1a).30 This particular isomer was targeted on the basis of investigations by Stevenson and De Bo on analogous furan–maleimide Diels–Alder adducts that demonstrated the greatest mechanochemical activity for this combination of regiochemistry and stereochemistry.32 Furthermore, separation of the target cyclopentadiene–maleimide Diels–Alder adduct was only achieved using supercritical fluid chromatography, which limits the accessibility of the chemistry. Finally, the lack of modularity inherent to the original synthesis restricts the ease with which the mechanochemical properties of the mechanophore and the photochemical properties of the masked DAE photoswitch are modified.We hypothesized that relocating the polymer attachment position from one of the thiophene substituents to the 4-position of the cyclopentadiene ring would not only enable access to a Diels–Alder adduct with the desired proximal regiochemistry, but the increased symmetry of the 1,2,4-trisubstitution pattern would obviate the synthetic challenges encountered previously (see Scheme 1b). Trisubstituted cyclopentadienes exist preferentially as the two tautomers with maximal substitution of the double bonds;33 in this case, at the 1- and 3-positions. If the identity of the thienyl substituents at the 1- and 2-positions of the cyclopentadiene is the same, the two tautomers are identical. Thus, reaction with the maleimide dienophile is expected to produce only two Diels–Alder adducts possessing either endo or exo stereochemistry, the distribution of which can be controlled by varying the reaction conditions. Moreover, substitution of the thiophene rings with chlorine at the 5-positions would provide functional handles for substrate diversification by Pd-catalyzed cross-coupling reactions later in the synthesis, allowing the photophysical properties of the photoswitch to be conveniently modulated. The predisposition of this structural design toward mechanochemical activation was validated with density functional theory (DFT) calculations using the constrained geometries simulate external force (CoGEF) method,34,35 which predict the desired formal retro-[4 + 2] cycloaddition reaction upon molecular elongation to successfully unmask the cyclopentadiene-based DAE photoswitch (Fig. S1†). The mechanochemical reaction is predicted to occur with a maximum force (Fmax) of 4.9 nN, which is in the same range as the predicted Fmax value of 4.6 nN calculated for the first-generation mechanophore.30
With our new molecular design in sight, we set out to synthesize the cyclopentadiene–maleimide Diels–Alder adduct (Scheme 2). Protection of 4-(2-hydroxyethyl)cyclopentene 1 by esterification with benzoyl chloride yielded cyclopentene 2, which was then cleaved by ozonolysis followed by oxidative workup to provide dicarboxylic acid 3 in excellent yield. Next, conversion to the diacyl chloride was enacted with oxalyl chloride and catalytic DMF. Reaction times longer than 12 h were necessary to ensure good conversion, as shorter times yielded a significant amount of the corresponding cyclic anhydride. Reaction of the crude diacyl chloride with 2-chloro-5-methyl thiophene and AlCl3 afforded dithienyl diketone 4 in 56% yield over the two steps. We note that similar reactions performed on an alternative substrate containing a methylene linker between the benzoate ester and glutaric acid backbone instead of the ethylene tether in 3 resulted in the formation of a significant amount of a dihydropyran rearrangement side product. With 4 in hand, our attention turned to the formation of the 5-membered ring that would eventually become the cyclopentadiene core. DAE photoswitches are commonly formed from 1,2-disubstituted cyclopentenes by the reductive coupling of 1,5-diaryl diketones under McMurry conditions.36 We expected that subjecting 4 to these conditions would afford a 1,2,4-trisubstituted cyclopentene that could be oxidized to the corresponding cyclopentadiene (6) with molecular bromine, as reported by Branda37 and successfully employed in the synthesis of our first-generation mechanophore.30 While McMurry coupling afforded the desired cyclopentene in approximately 75% yield, oxidation to the cyclopentadiene proved fruitless with molecular bromine as well as several other oxidants tested under a variety of conditions. Instead, we ultimately explored the use of a reductive pinacol coupling to produce cyclopentanediol 5 with a total oxidation state identical to that of the target cyclopentadiene 6, conveniently avoiding additional redox manipulations necessitated by the McMurry sequence. The pinacol coupling proceeded in nearly quantitative conversion to cyclopentanediol 5, which was isolated as a mixture of diastereomers. This mixture was then dehydrated under acidic conditions to generate cyclopentadiene 6 in 85% yield. Analysis by 1H NMR spectroscopy confirmed that cyclopentadiene 6 exists as a single tautomer, which DFT calculations predict to be the most energetically favorable by 5.6 kcal mol−1 (see the ESI† for details). Removal of the benzoate protecting group with LAH produced the transiently stable alcohol, which was treated in rapid succession with N-(2-hydroxyethyl)maleimide and heated at 70 °C for 24 h to provide endo Diels–Alder adduct diol 7 in 51% yield over the two steps. Diol 7 is readily separated from the exo stereoisomer by reverse phase column chromatography, or alternatively, simply by recrystallization. Importantly, refluxing a diester derivative of cyclopentadiene–maleimide adduct 7 in toluene for 24 h results in negligible retro-Diels–Alder reaction, confirming the thermal stability of the core structure (Fig. S2†).
 | ||
| Scheme 2 Synthesis of cyclopentadiene–maleimide Diels–Alder adduct diol 7. | ||
Diversification of Diels–Alder adduct diol 7 into several different masked DAE photoswitches with varied photophysical properties was achieved by Suzuki–Miyaura cross-coupling using several commercially available aryl boronic acids to demonstrate the modularity of the scaffold (Scheme 3a). Despite the generally low reactivity of unactivated aryl chlorides toward oxidative addition, we found that transformation of 7 could be successfully executed to produce derivatives 8a–c in fair to very good yields with a variety of aryl boronic acids using the Buchwald third generation XPhos precatalyst.38 The specific aryl boronic acids employed were chosen to provide ring-closed DAE isomers that exhibit absorption maxima across a wide range of the visible spectrum, in addition to the parent 5-chlorothiophene substrate that was expected to produce a yellow-colored ring-closed DAE isomer upon mechanochemical unmasking and subsequent irradiation with UV light.36,39,40 Next, esterification of each of the four diols with α-bromoisobutyryl bromide afforded 9a–d, which were employed as bis-initiators in the controlled radical polymerization of methyl acrylate to yield poly(methyl acrylate) (PMA) polymers centrally functionalized with each Diels–Alder adduct mechanophore. Characterization of polymers 10a–d by gel permeation chromatography (GPC) monitored with multi-angle light scattering and refractive index (RI) detectors confirmed that the polymers have similar molecular weight and narrow dispersity, with Mn = 96–112 kg mol−1 and Đ = 1.05–1.07 (see the ESI† for details). Overall, the preparation of chain-centered polymers incorporating a diverse range of functional masked DAE mechanophores requires no more than three steps from diol 7. Late stage mechanophore diversification by Pd-catalyzed cross-coupling reactions, supported by the ready availability of a wide variety of aryl boronic acids, makes 7 a highly modular intermediate for the implementation of mechanically gated photoswitching.
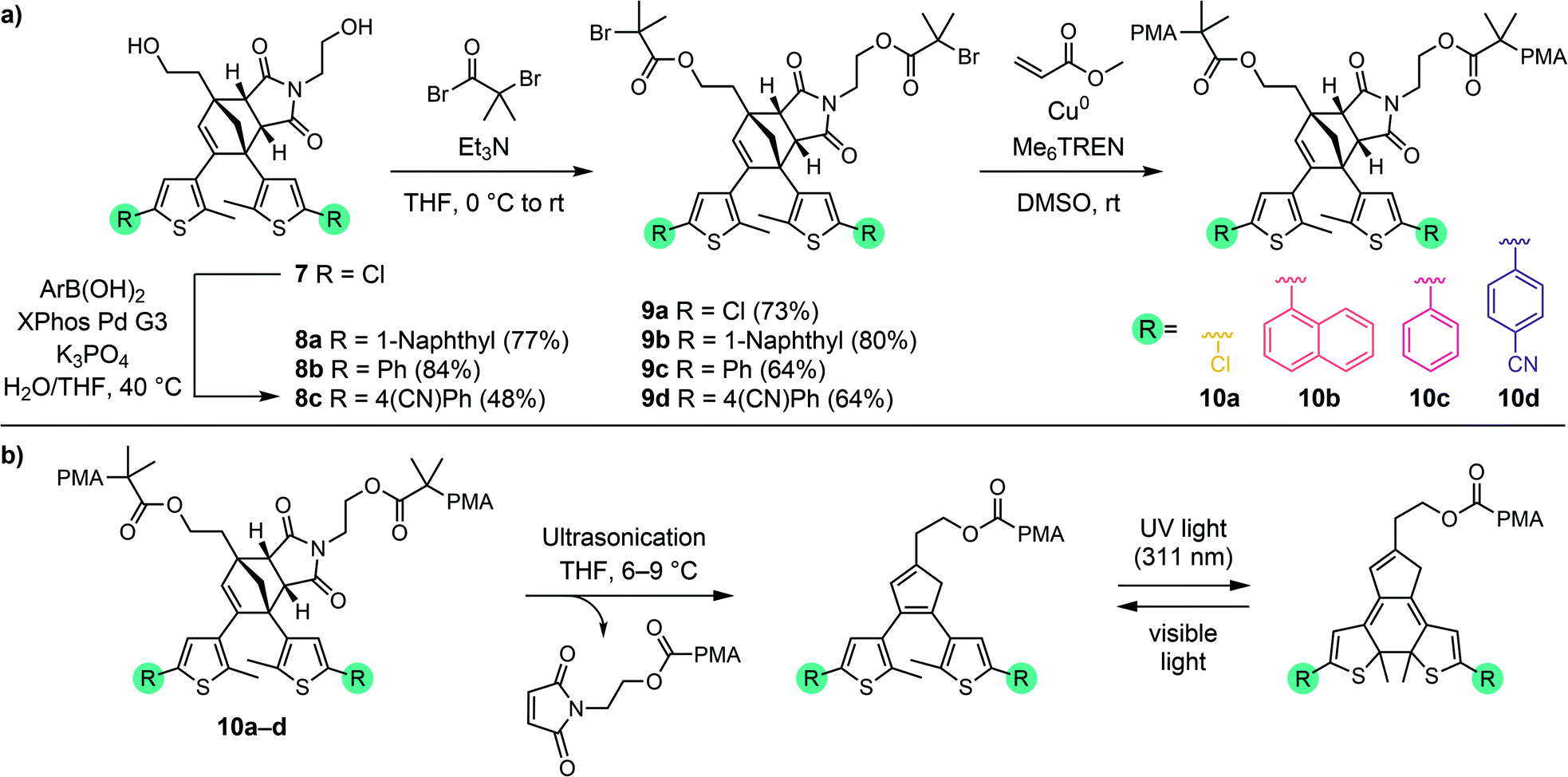 | ||
| Scheme 3 (a) Synthesis of polymers 10a–d containing a chain-centered mechanophore with varied substitution, and (b) unveiling of DAE photoswitches by ultrasound-induced mechanical force and subsequent photoswitching. | ||
The reactivity of the masked DAE mechanophores was initially investigated experimentally by subjecting solutions of each polymer (2 mg mL−1 in THF) to combinations of pulsed ultrasonication (6–9 °C, 1 s on/2 s off, 11.6 W cm−2) and UV irradiation as illustrated in Scheme 3b. Ultrasonication generates mechanical forces on polymers maximized near the chain midpoint where the mechanophores are installed, enabling convenient evaluation of mechanochemical reactivity.41 The untreated polymer solutions are colorless, and moreover, completely unresponsive to UV irradiation (311 nm for 60 s) as evidenced by the absence of any perceptible changes in UV-vis absorption after exposure to UV light (Fig. 1). The polymer solutions also remain completely colorless after ultrasonication for 120 min. However, when the polymers previously subjected to ultrasound-induced mechanical activation are irradiated with UV light under the same conditions as above, the solutions become intensely colored with absorption maxima ranging from 444 nm to 585 nm depending on the substitution of the thienyl groups on the cyclopentadiene–maleimide mechanophores. Monitoring the mechanochemical transformations over the course of each sonication experiment by subjecting the polymer solutions to UV irradiation at various time points reveals a progressive increase in optical density in the visible region with increasing duration of ultrasonication (Fig. S3†). The sequence of mechanical activation followed by UV irradiation is critical for color formation, consistent with the mechanophore design in which the retro-[4 + 2] cycloaddition reaction of the cyclopentadiene–maleimide Diels–Alder adduct gates the photoinduced 6π electrocyclic ring-closing reaction of the unmasked DAE photoswitch. The results of the solution-phase experiments expose two important points about the mechanophore design. First, the specificity observed in the reaction sequence as illustrated by the UV-vis absorption spectra acquired after various mechanochemical and photochemical treatments of the polymers demonstrates a clear advance over our prior mechanophore design.30 Second, the modularity of the masked DAE photoswitch is evident in the range of absorption achieved that spans the visible spectrum, enabled by late-stage functionalization of the mechanophore and leveraging the extensive structure–property relationships developed for DAE molecules.31
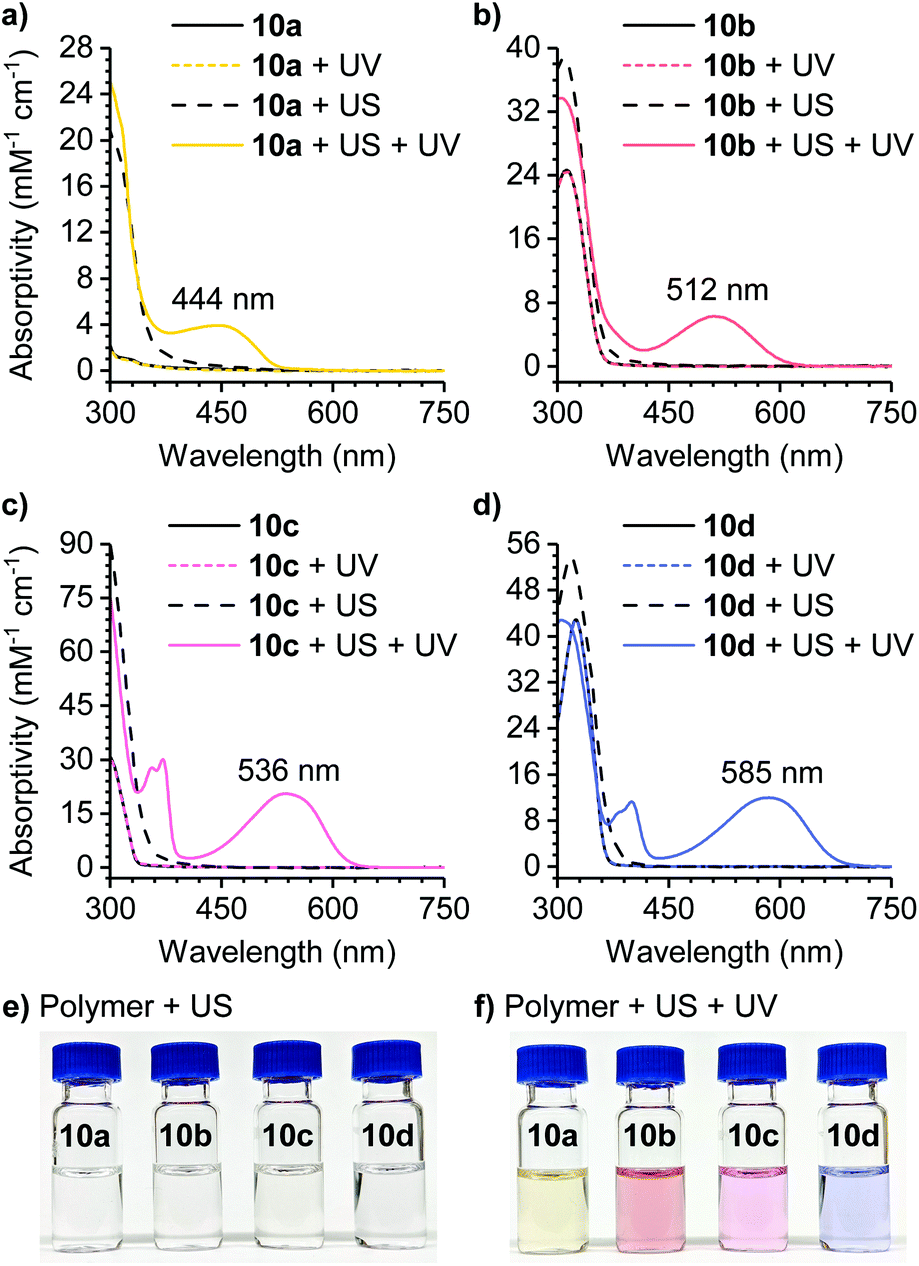 | ||
| Fig. 1 Characterization of mechanically gated photoswitching. (a–d) UV-vis absorption spectra of polymers 10a–d (2 mg mL−1 in THF) before and after 120 min of ultrasonication (US) and/or irradiation with UV light (311 nm, 60 s). Photographs of the polymer solutions (e) after ultrasonication, and (f) the same solutions after irradiation with UV light (311 nm, 120 s). The solutions of polymers 10a and 10b were concentrated 5× after ultrasonication for the photographs. | ||
A series of control experiments was performed to further characterize the reactivity of the cyclopentadiene–maleimide mechanophore. In order to further support the structural identity of the colored species generated after sequential ultrasonication and UV photoirradiation of the polymers above, we synthesized small molecule model compound 11o to serve as an analog of the photoswitch implicated in the mechanical activation of polymer 10c containing a phenyl-substituted masked DAE mechanophore. Irradiation of model compound 11o in acetone-d6 with 311 nm light for 60 min produces an intensely purple-colored solution that was analyzed by 1H NMR spectroscopy (Fig. 2a). The 1H NMR spectrum obtained after photoirradiation is consistent with nearly quantitative conversion of 11o to ring-closed isomer 11c (Fig. S4†). Critically, the UV-vis absorption spectrum of 11c in THF closely matches the spectrum of polymer 10c obtained after ultrasonication and UV irradiation (Fig. 2b). Furthermore, additional GPC measurements using an in-line UV-vis detector were performed on polymer 10c after being subjected to ultrasonication and UV photoirradiation. A peak monitored at 491–581 nm, encompassing the λmax of the anticipated ring-closed DAE isomer, is observed with a retention time that coincides with the low molecular weight polymer peak in the RI traces, indicating that polymer chain scission is accompanied by the generation of a polymer-bound photoswitch, consistent with the expected reactivity (Fig. S5†). Finally, the same sequential ultrasonication and UV irradiation treatment was applied to a control polymer analogous to 10c but containing the Diels–Alder adduct at the end of the polymer chain, which is not subjected to mechanical force during ultrasonication.3 No change in visible absorption is detected under these conditions (Fig. S6†), confirming the mechanical origin of the reactivity observed for polymers 10a–d containing a chain-centered mechanophore.
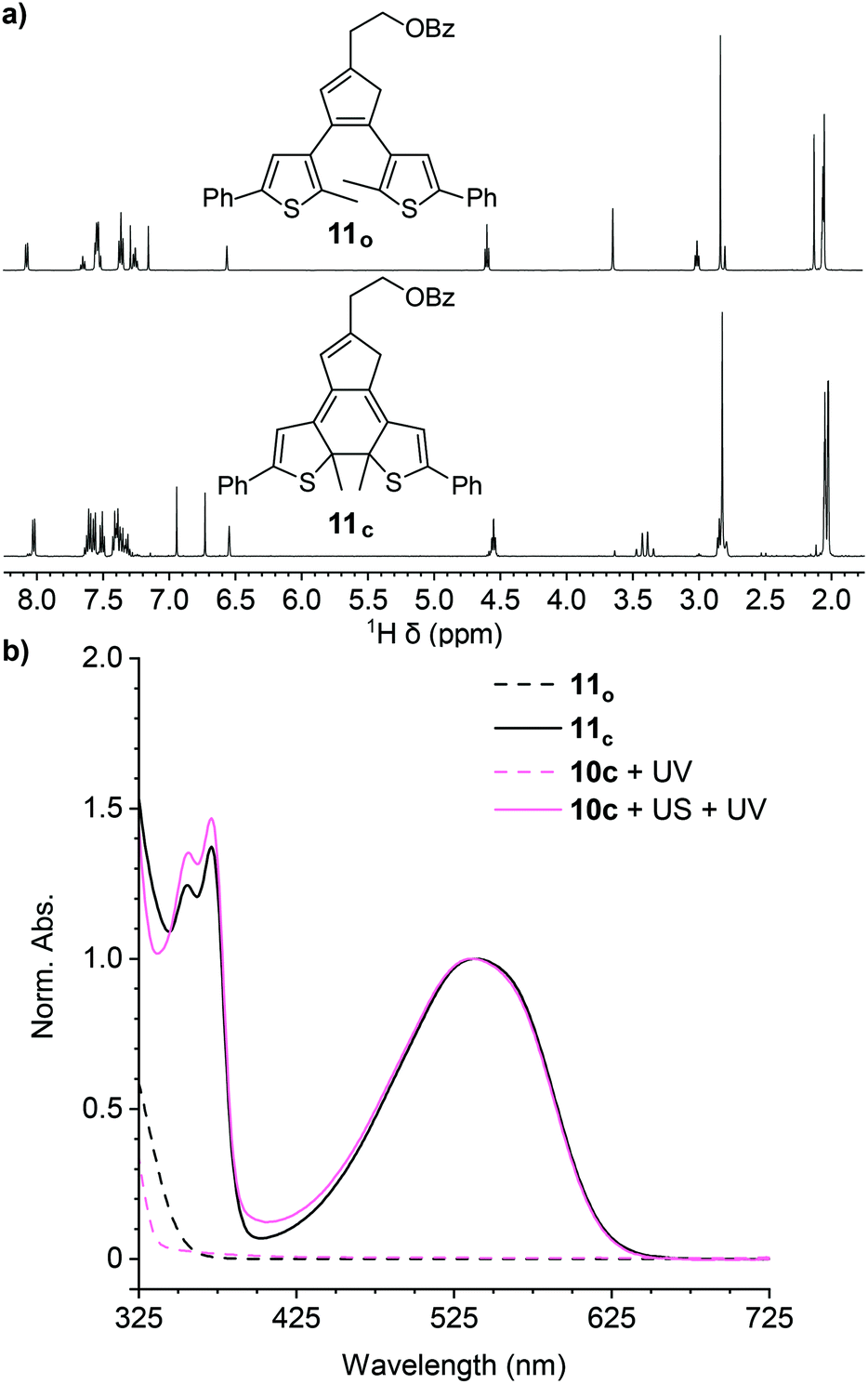 | ||
| Fig. 2 (a) Partial 1H NMR spectra (500 MHz, acetone-d6) of model small molecule 11o (top) and the ring-closed isomer 11c (bottom) generated nearly quantitatively upon irradiation with UV light (311 nm, 60 min). (b) UV-vis absorption spectra of 11o and 11c compared to polymer 10c in THF before and after ultrasonication (US) and irradiation with UV light. The matching absorption spectra indicate that the same ring-closed DAE structure exemplified by small molecule 11c is generated from mechanochemical activation and subsequent UV photoirradiation of polymer 10c. | ||
Encouraged by the solution-phase reactivity of the mechanophores, we sought to investigate mechanically gated photoswitching in solid polymeric materials. A PMA network incorporating 1.5 mol% of a cyclopentadiene–maleimide mechanophore crosslinker analogous to the structure in 10c was synthesized via free radical polymerization. Likewise, a control PMA network was synthesized similarly using 1.5 mol% of a mechanochemically inactive ethylene glycol dimethacrylate crosslinker and 1.5 mol% of a monoacrylate-functionalized mechanophore comonomer (Fig. 3a, see the ESI for details). Compression of both polymeric materials for 5 min with a hydraulic press followed by irradiation with 311 nm light for 5 min produces a purple color in the active, mechanophore-crosslinked material indicating successful generation of the ring-closed DAE photoswitch (Fig. 3b). The photochromic behavior of the mechanically activated material was still evident one week after the original compression experiment (Fig. S7†). Consistent with the solution-phase experiments, no color change was observed in the active network upon irradiation with UV light without first applying mechanical force (Fig. S8†). On the other hand, no color was produced by the control network after subjecting it to the same sequence of compression and UV photoirradiation, indicating that transfer of mechanical force across the cyclopentadiene–maleimide junction of the mechanophore is necessary to induce the retro-[4 + 2] cycloaddition reaction and reveal the DAE photoswitch.
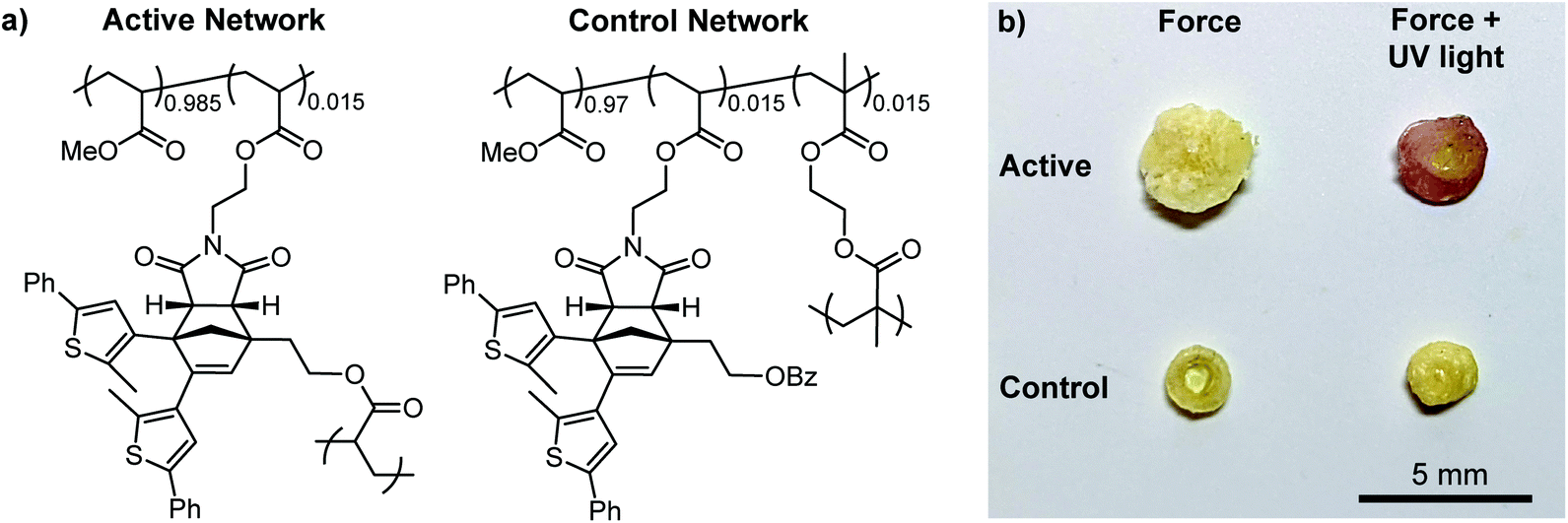 | ||
| Fig. 3 (a) Structures of mechanochemically active and control PMA networks incorporating the masked photoswitch Diels–Alder adduct as either a crosslinker or a pendant group, respectively. (b) Photographs of the active and control networks after compression for 5 min (left) before and (right) after irradiation with 311 nm UV light. The purple color of the active sample after UV irradiation demonstrates the successful realization of mechanically gated photoswitching in the solid state. No color change is observed in the control sample after irradiation with UV light. | ||
Conclusions
We have designed a masked diarylethene (DAE) mechanophore for mechanically gated photoswitching that overcomes prior limitations and presents a highly modular platform for accessing color-tunable molecular force probes. Mechanical activation of the cyclopentadiene–maleimide Diels–Alder adduct facilitates a formal retro-[4 + 2] cycloaddition reaction to reveal a latent DAE photoswitch, which is transformed via a 6π electrocyclic ring-closing reaction under UV light to generate an intensely colored isomer. Unlike many conventional mechanochromic mechanophores, the mechanically gated photoswitching strategy enables the mechanical history of a material to be recorded and read on-demand using light. Moreover, by decoupling the mechanochemical reaction from the chromogenic response, the design overcomes limitations of mechanochemical specificity encountered by other mechanophores, which in many cases produce the same visual signal upon exposure to alternative stimuli. We present a robust synthetic strategy that enables straightforward access to the cyclopentadiene–maleimide mechanophore, which is diversified via late stage Pd-catalyzed cross-coupling reactions to modulate the photophysical properties of the masked DAE photoswitch. Solution-phase ultrasonication experiments performed on a small library of functionally diverse mechanophores showcase the excellent selectivity of the compounds upon exposure to combinations of mechanical force and UV light. A chromogenic response is only observed upon sequential mechanochemical activation followed by UV photoirradiation, revealing colored DAE isomers with absorption spectra that span the visible region of the electromagnetic spectrum. We also demonstrate mechanically gated photoswitching in solid polymeric materials for the first time, illustrating the potential of this mechanophore platform for a variety of stress reporting/recording, encryption, and pattering applications.Author contributions
M. J. R. conceptualized the project and provided guidance during all stages. R. W. B. and M. J. R. designed the research. R. W. B. performed the experiments. R. W. B. and M. J. R. analyzed the data and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Caltech is gratefully acknowledged. We thank the Center for Catalysis and Chemical Synthesis of the Beckman Institute at Caltech for access to equipment. We also thank Dr Mona Shahgholi and Dr David VanderVelde for technical assistance, and Dr Xiaoran Hu for helpful discussions.References
- M. K. Beyer and H. Clausen-Schaumann, Chem. Rev., 2005, 105, 2921–2948 CrossRef CAS.
- M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Chem. Rev., 2009, 109, 5755–5798 CrossRef CAS.
- J. Li, C. Nagamani and J. S. Moore, Acc. Chem. Res., 2015, 48, 2181–2190 CrossRef CAS.
- J. M. J. Paulusse and R. P. Sijbesma, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5445–5453 CrossRef CAS.
- P. A. May and J. S. Moore, Chem. Soc. Rev., 2013, 42, 7497–7506 RSC.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martínez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72 CrossRef CAS PubMed.
- J. Sung, M. J. Robb, S. R. White, J. S. Moore and N. R. Sottos, J. Am. Chem. Soc., 2018, 140, 5000–5003 CrossRef CAS PubMed.
- A. R. Sulkanen, J. Sung, M. J. Robb, J. S. Moore, N. R. Sottos and G. Liu, J. Am. Chem. Soc., 2019, 141, 4080–4085 CrossRef CAS PubMed.
- G. Kim, V. M. Lau, A. J. Halmes, M. L. Oelze, J. S. Moore and K. C. Li, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10214–10222 CrossRef CAS.
- A. Piermattei, S. Karthikeyan and R. P. Sijbesma, Nat. Chem., 2009, 1, 133–137 CrossRef CAS.
- P. Michael and W. H. Binder, Angew. Chem., Int. Ed., 2015, 54, 13918–13922 CrossRef PubMed.
- Z. Chen, J. A. M. Mercer, X. Zhu, J. A. H. Romaniuk, R. Pfattner, L. Cegelski, T. J. Martinez, N. Z. Burns and Y. Xia, Science, 2017, 357, 475–479 CrossRef CAS.
- J. Yang, M. Horst, J. A. H. Romaniuk, Z. Jin, L. Cegelski and Y. Xia, J. Am. Chem. Soc., 2019, 141, 6479–6483 CrossRef CAS PubMed.
- B. R. Boswell, C. M. F. Mansson, J. M. Cox, Z. Jin, J. A. H. Romaniuk, K. P. Lindquist, L. Cegelski, Y. Xia, S. A. Lopez and N. Z. Burns, Nat. Chem., 2021, 13, 41–46 CrossRef CAS.
- M. B. Larsen and A. J. Boydston, J. Am. Chem. Soc., 2013, 135, 8189–8192 CrossRef CAS PubMed.
- X. Hu, T. Zeng, C. C. Husic and M. J. Robb, J. Am. Chem. Soc., 2019, 141, 15018–15023 CrossRef CAS PubMed.
- Z. Shi, Q. Song, R. Göstl and A. Herrmann, Chem. Sci., 2021, 12, 1668–1674 RSC.
- R. Göstl and R. P. Sijbesma, Chem. Sci., 2016, 7, 370–375 RSC.
- C. Baumann, M. Stratigaki, S. P. Centeno and R. Göstl, Angew. Chem., Int. Ed., 2021, 60, 13287–13293 CrossRef CAS PubMed.
- M. Karman, E. Verde-Sesto and C. Weder, ACS Macro Lett., 2018, 7, 1028–1033 CrossRef CAS.
- C. P. Kabb, C. S. O'Bryan, C. D. Morley, T. E. Angelini and B. S. Sumerlin, Chem. Sci., 2019, 10, 7702–7708 RSC.
- Y. Chen, A. J. H. Spiering, S. Karthikeyan, G. W. M. Peters, E. W. Meijer and R. P. Sijbesma, Nat. Chem., 2012, 4, 559–562 CrossRef CAS PubMed.
- Y. Chen, G. Mellot, D. van Luijk, C. Creton and R. P. Sijbesma, Chem. Soc. Rev., 2021, 50, 4100–4140 RSC.
- M. J. Robb, T. A. Kim, A. J. Halmes, S. R. White, N. R. Sottos and J. S. Moore, J. Am. Chem. Soc., 2016, 138, 12328–12331 CrossRef CAS.
- Z. Wang, Z. Ma, Y. Wang, Z. Xu, Y. Luo, Y. Wei and X. Jia, Adv. Mater., 2015, 27, 6469–6474 CrossRef CAS.
- K. Imato, T. Kanehara, T. Ohishi, M. Nishihara, H. Yajima, M. Ito, A. Takahara and H. Otsuka, ACS Macro Lett., 2015, 1307–1311 CrossRef CAS.
- R. W. Barber, M. E. McFadden, X. Hu and M. J. Robb, Synlett, 2019, 30, 1725–1732 CrossRef CAS.
- K. Imato, M. Nishihara, T. Kanehara, Y. Amamoto, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2012, 51, 1138–1142 CrossRef CAS PubMed.
- J. Wang, T. B. Kouznetsova, R. Boulatov and S. L. Craig, Nat. Commun., 2016, 7, 13433 CrossRef PubMed.
- X. Hu, M. E. McFadden, R. W. Barber and M. J. Robb, J. Am. Chem. Soc., 2018, 140, 14073–14077 CrossRef CAS PubMed.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- R. Stevenson and G. De Bo, J. Am. Chem. Soc., 2017, 139, 16768–16771 CrossRef CAS.
- S. J. Alward and A. G. Fallis, Can. J. Chem., 1984, 62, 121–127 CrossRef CAS.
- M. K. Beyer, J. Chem. Phys., 2000, 112, 7307–7312 CrossRef CAS.
- I. M. Klein, C. C. Husic, D. P. Kovács, N. J. Choquette and M. J. Robb, J. Am. Chem. Soc., 2020, 142, 16364–16381 CrossRef CAS PubMed.
- L. N. Lucas, J. van Esch, R. M. Kellogg and B. L. Feringa, Chem. Commun., 1998, 2313–2314 RSC.
- V. Lemieux, S. Gauthier and N. R. Branda, Angew. Chem., Int. Ed., 2006, 45, 6820–6824 CrossRef CAS PubMed.
- N. C. Bruno, M. T. Tudge and S. L. Buchwald, Chem. Sci., 2013, 4, 916–920 RSC.
- W. R. Browne, J. J. D. de Jong, T. Kudernac, M. Walko, L. N. Lucas, K. Uchida, J. H. van Esch and B. L. Feringa, Chem.–Eur. J., 2005, 11, 6430–6441 CrossRef CAS PubMed.
- S. Pu, T. Yang, J. Xu, L. Shen, G. Li, Q. Xiao and B. Chen, Tetrahedron, 2005, 61, 6623–6629 CrossRef CAS.
- K. L. Berkowski, S. L. Potisek, C. R. Hickenboth and J. S. Moore, Macromolecules, 2005, 38, 8975–8978 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthetic procedures, GPC chromatograms, DFT calculations, photographs, UV-vis and NMR spectra. See DOI: 10.1039/d1sc02890a |
| This journal is © The Royal Society of Chemistry 2021 |