
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMEMES: Machine learning framework for Enhanced MolEcular Screening†‡
Sarvesh
Mehta
a,
Siddhartha
Laghuvarapu§
a,
Yashaswi
Pathak§
a,
Aaftaab
Sethi
b,
Mallika
Alvala
 c and
U. Deva
Priyakumar
*a
c and
U. Deva
Priyakumar
*a
aCenter for Computational Natural Sciences and Bioinformatics, International Institute of Information Technology, Hyderabad 500 032, India. E-mail: deva@iiit.ac.in; Fax: +91 40 6653 1413; Tel: +91 40 6653 1161
bDepartment of Medicinal Chemistry, National Institute of Pharmaceutical Education and Research, Hyderabad 500 037, India
cSchool of Pharmacy and Technology Management, Narsee Monjee Institute of Management Sciences, Hyderabad, India
First published on 26th July 2021
Abstract
In drug discovery applications, high throughput virtual screening exercises are routinely performed to determine an initial set of candidate molecules referred to as “hits”. In such an experiment, each molecule from a large small-molecule drug library is evaluated in terms of physical properties such as the docking score against a target receptor. In real-life drug discovery experiments, drug libraries are extremely large but still there is only a minor representation of the essentially infinite chemical space, and evaluation of physical properties for each molecule in the library is not computationally feasible. In the current study, a novel Machine learning framework for Enhanced MolEcular Screening (MEMES) based on Bayesian optimization is proposed for efficient sampling of the chemical space. The proposed framework is demonstrated to identify 90% of the top-1000 molecules from a molecular library of size about 100 million, while calculating the docking score only for about 6% of the complete library. We believe that such a framework would tremendously help to reduce the computational effort in not only drug-discovery but also areas that require such high-throughput experiments.
Introduction
The drug discovery process is an extremely laborious process and the pipeline involves several steps each of which is both expensive and time consuming. The first step in the process after target identification and validation is to identify hit molecules, where potential strong binding drug-like molecules against a drug target are identified using computational methods. Once the hit molecules are identified, they are experimentally evaluated typically using biochemical assays towards lead identification. Further processes involve lead optimization, in vitro and in vivo evaluation, pre-clinical studies and clinical trials before the drug can be approved for use. The structure based drug design (SBDD) method, docking, is routinely used for identification of lead molecules.1–4 In the SBDD method, large libraries of ligands5–7 are virtually screened to determine their docking score against a drug target, which is a measure of the inter-molecular interaction between the target and the ligand.Recently new methods that use modern deep/reinforcement learning have been proposed to tackle problems in molecular sciences such as physical property prediction,8,9 drug design tasks,10 protein structure prediction,11–13 molecular simulations,14–16 and de novo molecule generation.17 Most of the deep learning models that tackle the problem of molecular generation are based on variational autoencoders,18–21 Generative Adversarial Networks22–24 and Reinforcement Learning.25–28 Although these models have been seen to perform really well in optimization of molecular properties such as the QED score (Quantitative Estimate of Drug likeliness) and log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P score (octanol–water partition coefficient), they have been shown to perform inadequately while optimizing objective functions involving docking calculations.29 Moreover, in a recent study by Gao and Coley,30 it was demonstrated that although the molecules generated by these methods are novel and diverse, they may be very difficult/infeasible to synthesize and hence cannot be of practical importance in a real-life drug discovery scenario.
P score (octanol–water partition coefficient), they have been shown to perform inadequately while optimizing objective functions involving docking calculations.29 Moreover, in a recent study by Gao and Coley,30 it was demonstrated that although the molecules generated by these methods are novel and diverse, they may be very difficult/infeasible to synthesize and hence cannot be of practical importance in a real-life drug discovery scenario.
In contrast to the molecules generated by deep generative models, molecule libraries enumerated via simple reactions can be novel, diverse and at the same time practically synthesized with a probability of ∼86%.30–32 In a recent study performed by Lyu et al.,31 96 million docking calculations were performed against the AmpC receptor. Among these the top ranked 1 million compounds (1% of the initial set) were systematically examined to identify hit molecules, which were further validated experimentally. In the same study, 138 million docking calculations were performed for the D4 dopamine receptor, which was used to show that the hit-rates fell almost monotonically with the docking-score. Although, Lyu et al. docked compounds in the order of 108, it is still a small fraction when compared to the 1.6 billion molecules enumerated in the ZINC Library. Moreover, their study also shows that hits for a target can be identified using only the top fraction of the ligands with respect to the docking score. Hence, a sampling method that can efficiently search the chemical space for high docking scores would speed up the process.
Recently, Gentile et al. proposed a deep learning based method “Deep Docking” to augment the process of SBDD.33 In this work, iterative docking is performed on a small portion of large libraries. The obtained values are used to train ligand-based QSAR models, which are used to predict the scores of the remaining ligands in the library. A cut-off is set to identify the hits among these predicted molecules. Molecules are then randomly sampled from these hits to further train the QSAR model for the next iteration. In this manner, the authors claim that with docking up to 50 times fewer molecules, 60% of the top scoring molecules can be retrieved.
In this work, a novel Machine learning framework for Enhanced MolEcular Screening (MEMES) based on Bayesian optimization is proposed for efficient sampling of molecules during the SBDD process. In the framework, the initial set of molecules are first featurized and represented as molecular vectors. These are then clustered using the K-means clustering algorithm. A small set of molecules are sampled from each cluster to build an initial diverse set of ligands, and their docking scores are calculated. A Gaussian process is trained as a surrogate function for the protein–ligand docking score. Two variants of the MEMES framework, ExactMEMES and DeepMEMES, are introduced depending upon the choice of the surrogate function used (see the Methods section). The initial training set is iteratively updated by sampling a small portion of molecules not previously sampled based on an acquisition function, and the process is repeated, until the maximum number of allowed docking calculations is reached. The proposed framework successfully samples a very high fraction of the top hits for a given protein and molecular library, while only calculating docking scores for 6% of the complete molecular library. Further, extensive analysis has been carried out to show the robustness of the framework on different proteins and molecular libraries with varying size.
Method
In this section, the various components in the proposed framework (Fig. 1) are explained. The docking methods, ligand libraries, and target receptors used in the MEMES framework are described in the section Docking methodology. In the section Molecular representation, the choice of different molecular embedding techniques used in this work is explained in detail. Further, Bayesian optimization, the techniques used to approximate the protein–ligand scoring function and point selection methods are explained.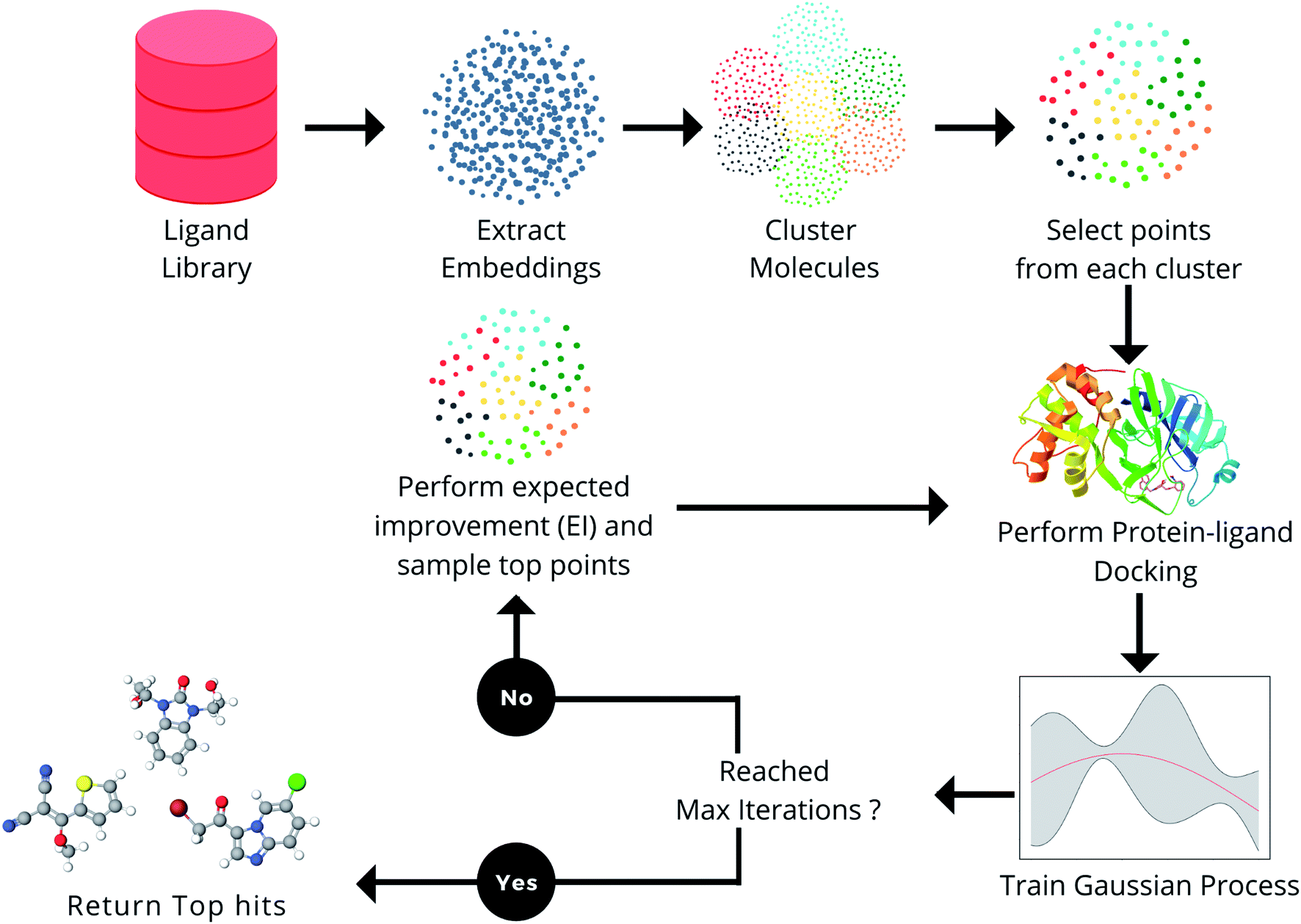 | ||
| Fig. 1 Overview of the proposed method, MEMES. | ||
Docking methodology
Molecular docking is useful in drug discovery projects to identify potential inhibitors against a protein receptor from small molecule libraries. The first step is ligand preparation, and protein preparation that was carried out using AutoDock 4.2 (AD 4)34 in this study. Three different small-molecule libraries of varying sizes were used in this study. First is the Zinc-250K dataset used earlier in molecular generation studies28,35,36 which contains 250000 drug-like molecules obtained from the ZINC15 database.7 Second is the Enamine dataset37 containing screening compounds that are grouped into different collections. Enamine HTS Collection containing 2106952 molecules is used in this study. The last one is the Ultra Large Docking Library introduced by Lyu et al.,31 which contains 96 million molecules docked against the AmpC β-lactamase (AmpC) receptor. Target proteins, Tau-Tubulin Kinase 1 (PDB ID: 4BTK) and SARS-CoV-2 Mpro complexed with an N3 inhibitor (PDB ID: 6LU7) used for evaluating the MEMES framework were obtained from the Research Collaboratory for Structural Bioinformatics-Protein Data Bank (RCSB-PDB).38 The next step in molecular docking is grid map generation carried using AutoGrid 4 utility in AutoDock. Finally, docking calculations were performed, keeping the protein active site rigid to get the docking score. Detailed information about the docking methodology is given in ESI Methods.‡
Molecular representation
The first step in the pipeline is to represent molecules as fixed-dimensional vectors. It is essential to choose vector representation techniques that effectively represent molecular structures and are sensitive to different atom types and bond connectivities. In this work, we performed trials with three molecular embedding techniques – ECFP,39 Mol2Vec40 and CDDD.41Bayesian optimization
Bayesian optimization is a technique used to optimize black-box functions that are expensive to evaluate.45,46 In recent years, Bayesian optimization has seen widespread applications in the field of chemistry, ranging from latent space optimization in molecular generation to reaction optimization for chemical synthesis.20,35,47,48 There are two main components in Bayesian optimization, a surrogate function which is a statistical model that can be used to approximate the black box, and an acquisition function to determine the next points to the sample. In this work, Gaussian Process Regression (ExactGP) and Deep Gaussian Process (DeepGP) are used as surrogate functions in ExactMEMES and DeepMEMES variants, respectively, and expected improvement49 is used as an acquisition function.| f(x1:k) ∼ Normal(μ0(x1:k),Σ0(x1:k,x1:k)) | (1) |
In eqn (1) the mean vector is obtained by evaluation of the mean function μ0 at each point xi and the covariance matrix is obtained by evaluation of the covariance function or kernel Σ at each pair of points xi and xj. The kernel function should have a property that the points closer should have strong correlation and the resulting covariance matrix is positive semi-definite. Suppose the prior distribution is constructed for n points. For a point x at k = n + 1, the distribution is obtained from Baye's rule -
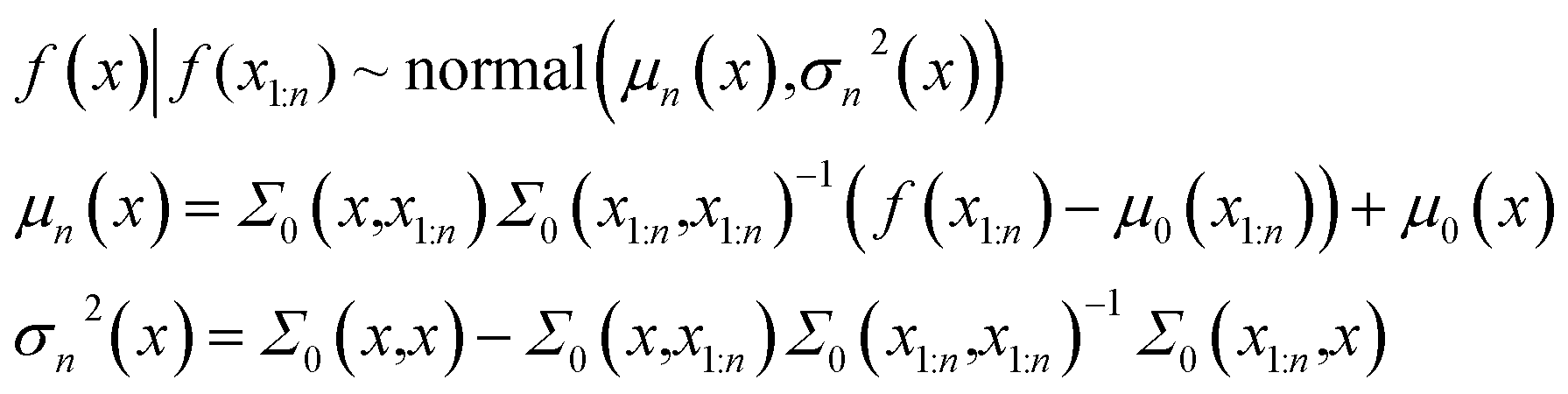 | (2) |
The conditional probability distribution is called the posterior probability distribution. For faster computations, the matrix inversions are obtained through Cholesky decompositions and solving a system of linear equations. In this work, the kernel function is chosen to be Radial Basis Function (RBF).50 The implementation of exact Gaussian processes in GPyTorch51 is used in this work.
The deep Gaussian process is a type of deep belief network where every hidden unit is a Gaussian process. The output of the l − 1th layer is used as the input to the lth layer. It can be defined as the composition of functions. Formally we can define DGP for a training data set of k points x1, …, xk whose function values are known represented in a vector y, as
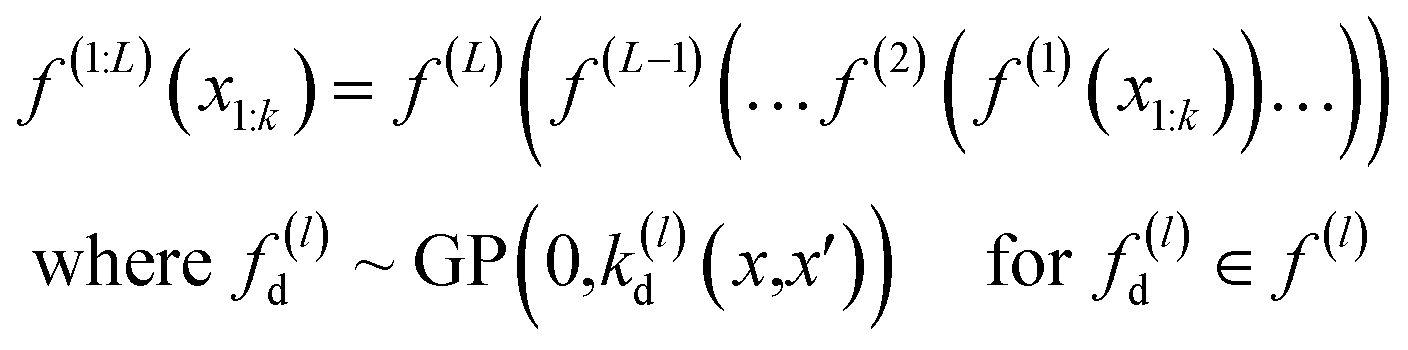 | (3) |
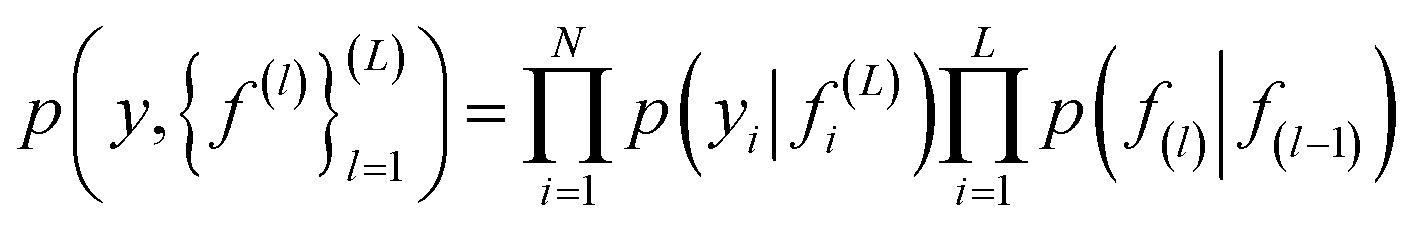 | (4) |
In eqn (4) the first term corresponds to likelihood, and the second corresponds to the GP prior. Non linear transformation is applied on the output of every hidden layer due to which the exact inference is not tractable.52 To overcome this problem various numbers of approximations have been developed such as expected propagation,53 variational auto-encoded deep Gaussian processes,54 and doubly stochastic variational inference for deep Gaussian processes.55 In this work, doubly stochastic variational inference is used here. The implementation of deep Gaussian processes in Gpytorch51 is used in this work.
Improvement at a point x is defined as
| I = max(0,f(x) − f*) | (5) |
| EI(x) = exp[max(0,f(x) − f*)] | (6) |
Using the reparameterization trick, x = μ + σε and integrating over the distribution, it can be shown that expected improvement can be obtained as
| EI(x) = (μ(x) − f* − ζ)Φ(Z) + σ(x)ϕ(Z) | (7) |
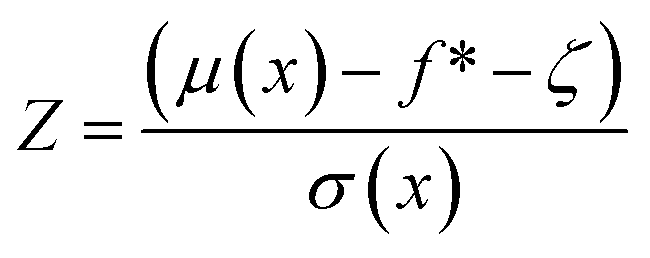 | (8) |
Results and discussion
In this section, the capability of the MEMES framework to sample a set of molecules having a highly negative docking score and high overlap with the actual top hit molecules while only performing docking calculations on only 6% of the molecules in the complete library is demonstrated. Further, the capability of the proposed method to sample a diverse set of molecules is shown. In this work, the performance of the MEMES framework is evaluated on two different surrogate functions ExactGP and DeepGP. Since ExactGP cannot be extended to be used on ultra large docking libraries due to computational constraints, in the subsequent subsection, the performances of ExactGP and DeepGP as the choice of surrogate function in the MEMES framework are compared to validate the performance of DeepMEMES against ExactMEMES. In the following subsection, the performance of the MEMES framework with DeepGP is demonstrated on large docking libraries. Finally, the robustness of the MEMES framework is demonstrated by applying it on molecular libraries with sizes ranging from 2 million to 96 million compounds.The framework proposed in this work “MEMES” is based on Bayesian optimization (Fig. 1). Firstly, in the MEMES method, all the ligands in the library are represented as fixed dimension feature vectors. Secondly, a small fraction of molecules are chosen to be the initial set. To ensure that this “initial set” is diverse and representative of the complete molecular library, a K-means clustering56 is performed on the pre computed feature vectors and molecules are uniformly sampled from each of the resulting clusters. Docking scores for each molecule in the initial set are computed against the given target receptor. A Gaussian process49,52,57 is then trained on this initial set. A new set of molecules is then picked from the rest of the dataset based on the“expected improvement” values calculated using the trained Gaussian process (see Methods). The docking score of these molecules are computed and these are added to the initial training set and the Gaussian process is retrained. The procedure is repeated iteratively, until the computational budget is reached or no improvement is observed.
MEMES identifies 95+% of top candidates by sampling only 6% of the dataset
The Zinc-250K dataset contains 250000 drug like molecules obtained from the ZINC 15 database.7 The ExactMEMES (MEMES framework with ExactGP) was applied on the Zinc-250K dataset against two protein receptors: Tau-Tubulin Kinase 1 (TTBK1) an attractive target protein to combat many neurodegenerative diseases such as Alzheimer's and the main protease (Mpro) of SARS-CoV-2, responsible for the outbreak of COVID-19. As the ExactGP used in this framework cannot be applied to a very large molecular library, the ZINC-250K dataset was selected to assess the performance of ExactMEMES.
Virtual screening docking calculations were performed to identify molecules that have high docking scores against a target receptor, i.e. to find top hits. It is also desired that the top hits identified in this process are diverse and span the complete molecular library. Here, we show that the ExactMEMES framework (with only 6% docking calculations) is able to sample molecules that have highly negative docking scores, and have high overlap with actual top hits of the given molecular library. This demonstrates that the MEMES framework not only identifies molecules exhibiting high negative docking scores but most of the top molecules in the complete library.
Fig. 2a and b show the mean docking score of actual top molecules in the molecular library, top molecules sampled by the ExactMEMES framework with Mol2Vec, CDDD and ECFP as molecular featurizer techniques and those by a random sampling method, against TTBK1 and SARS-CoV-2 Mpro respectively. The top 20 docking hits in the complete docking library for both the target receptors are given in ESI Fig. S1 and S2.‡ For ExactMEMES and random sampling, 15000 (∼6% of the complete molecular library) docking calculations were performed. From Fig. 2 it is quite evident that the ExactMEMES method significantly outperforms the random sampling baseline and matches the mean docking score of actual top compounds present in the molecular library. Fig. 2 also shows that ExactMEMES with CDDD featurization outperforms ExactMEMES with Mol2Vec and ECFP featurization techniques. The distribution of the docking scores of top sampled molecules is given in ESI Fig. S3‡ and the number of top docking hits identified by MEMES across different clusters is given in ESI Fig. S4.‡ Also the distribution of the top hits missed by the proposed method is given in ESI Fig. S5,‡ which shows that the current method is not biased in identifying top hits with respect to the values of the binding affinity.
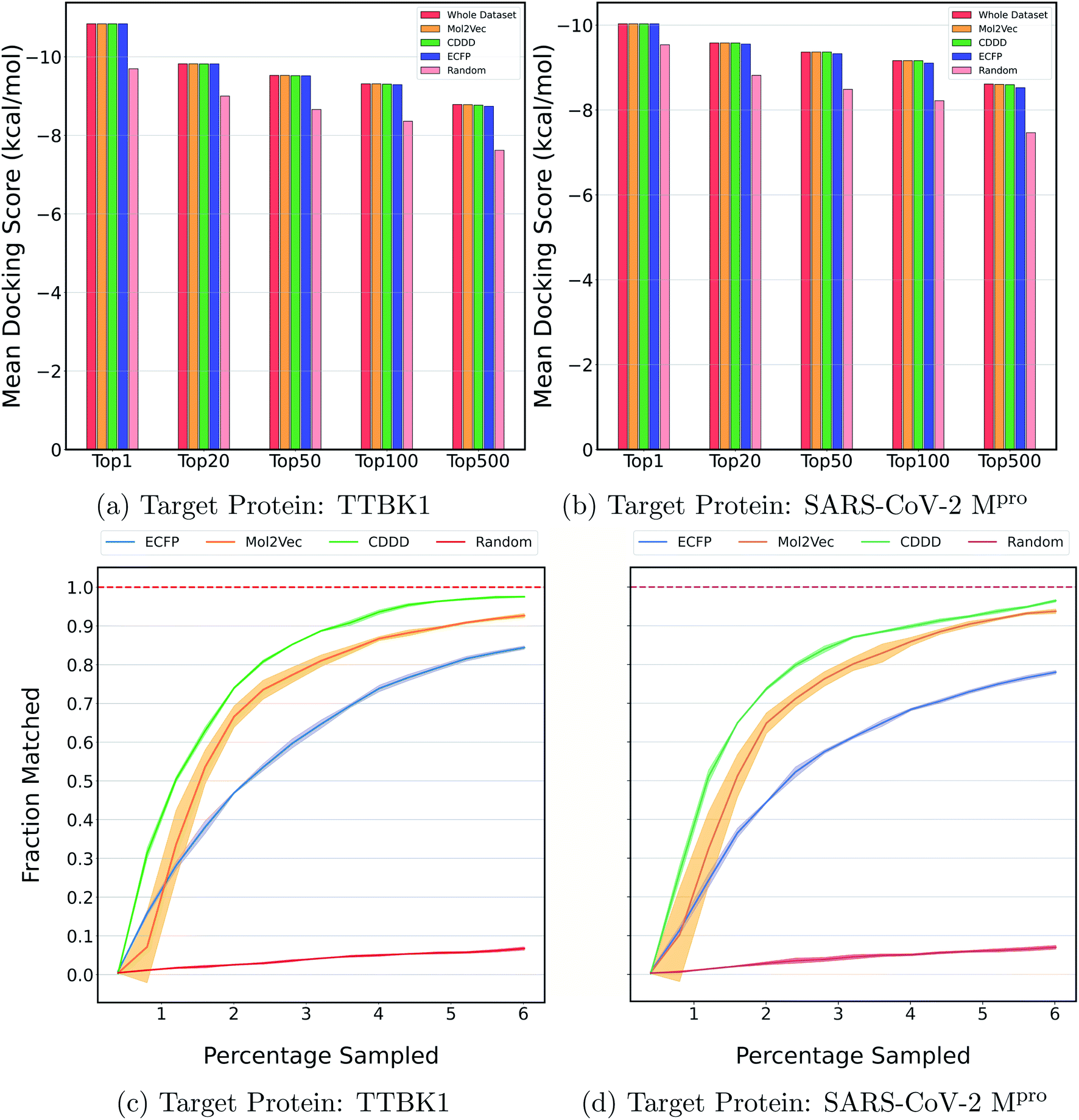 | ||
| Fig. 2 Performance on Zinc-250K using ExactMEMES against both target receptors. (a) and (b) compare the mean docking score of top hits sampled by MEMES and random sampling against the mean docking score of actual top hits in the library. (c) and (d) show the fraction of the top 500 sampled molecules that are actual top hits against the percentage of the dataset sampled. The reported results are an average of 3 runs and the shaded region represents standard deviation across these runs. | ||
Fig. 2c and d show the fraction of top 500 sampled molecules that are actual top hits for receptors TTBK1 and SARS-CoV-2 Mpro against the percentage of molecules sampled from the docking library using ExactMEMES and random sampling (see ESI Fig. S6‡ for similar analysis of top 100 sampled molecules). Fig. 2c and d show that ExactMEMES significantly outperforms random sampling and almost shows a complete overlap with the actual top hits when the percentage sampled is around 6%. Further intersection of the top 500 molecules sampled by the ExactMEMES framework, random sampling, and actual top hits for receptors TTBK1 and SARS-CoV-2 Mpro from the molecular library is shown in Fig. 3. ESI Table S1‡ demonstrates the overlap results for top 100, and top 500 molecules for all molecular embeddings (Mol2Vec, CDDD, and ECFP).
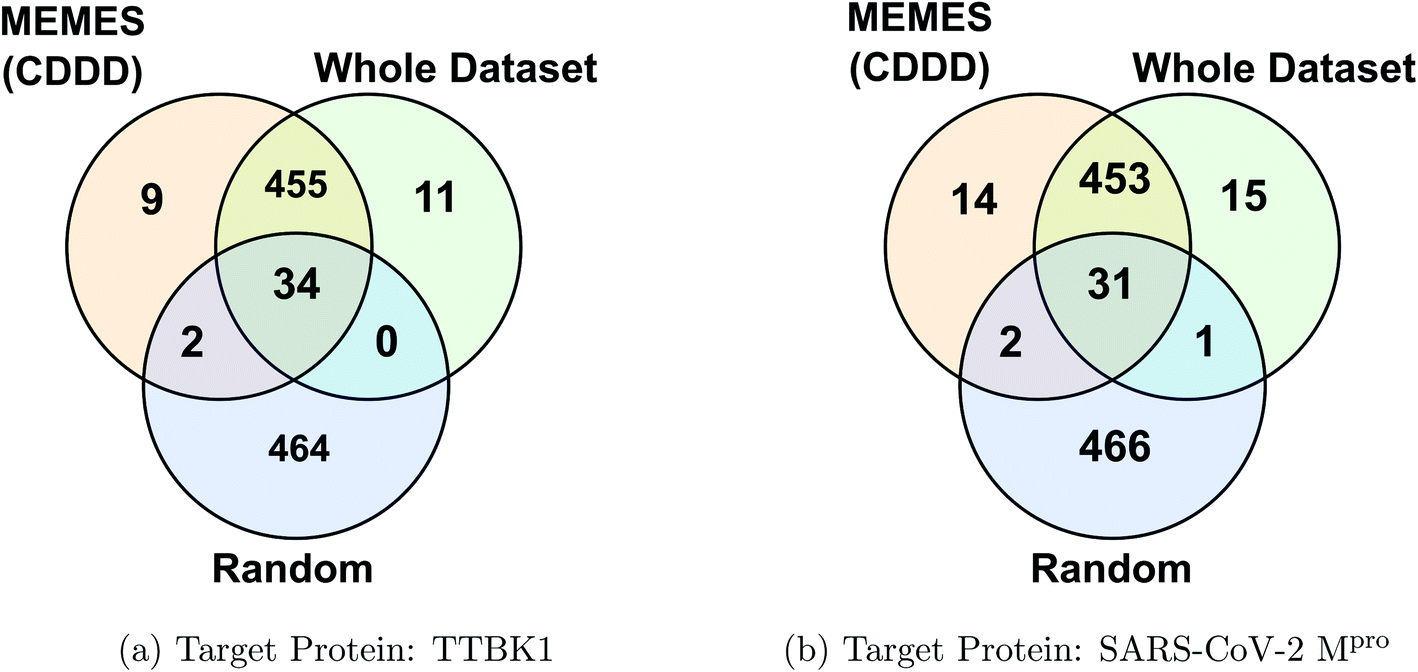 | ||
| Fig. 3 Venn diagram showing the intersection of the top 500 molecules identified by the MEMES framework and actual top 500 hits from the Zinc-250K docking library (the statistics shown are for one of the three runs). | ||
ExactMEMES vs. DeepMEMES
The above results show the ability of the ExactMEMES framework to identify top hits only by performing docking of less than 6% of the complete docking library but it cannot be applied on large docking libraries due to computation constraints. Therefore to overcome this issue, the DeepMEMES variant of the proposed framework is introduced. In this section, the performance of DeepMEMES is compared against that of ExactMEMES on the Zinc-250K docking library.Fig. 4 shows the comparison of the fraction of the molecules matched with actual top hits of the docking library between DeepMEMES and ExactMEMES using Mol2Vec as molecular embedding (see ESI Fig. S7‡ for comparison results with CDDD as the featurization technique). From Fig. 4, we can infer that DeepMEMES has comparable performance with ExactMEMES. See ESI Discussion 1‡ for the performance of DeepMEMES on Zinc-250K. Performance comparison between DeepMEMES and Deep Docking (study by Gentile et al.) in terms of ability to identify top hits and time is provided in ESI Discussion 2.‡ Further sections show the application of DeepMEMES on different molecular libraries to assess its performance on large datasets.
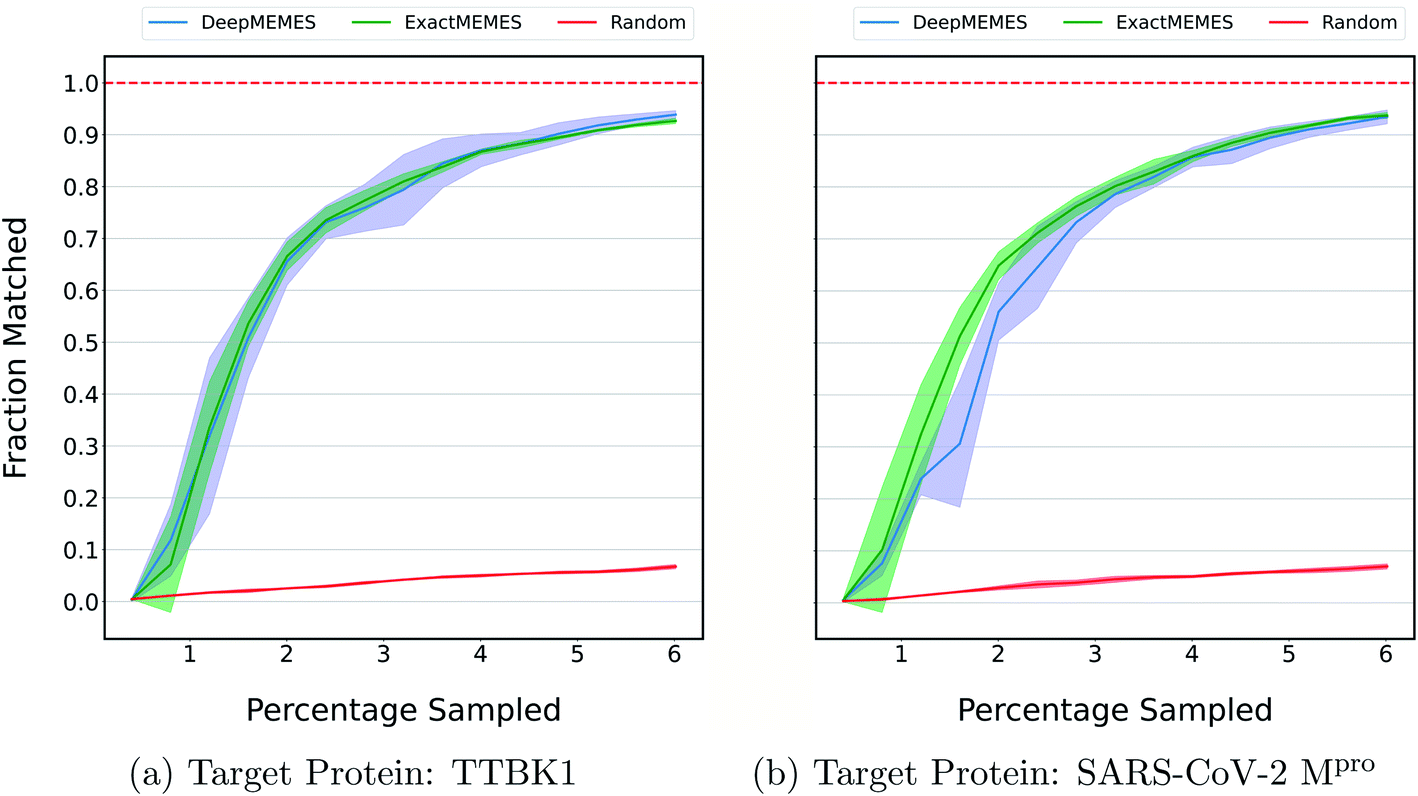 | ||
| Fig. 4 To compare the performance of ExactMEMES and DeepMEMES, a fraction of the top 500 molecules sampled that are actual top hits from the Zinc-250K dataset is plotted against the percentage of the dataset sampled (see ESI Fig. S7‡ for similar analysis for top 100 molecules). Mol2Vec as a featurization technique was used for this comparison. The reported trial results are an average of 3 runs and the shaded region represents standard deviation across these runs. | ||
MEMES framework on large libraries
In real life drug discovery experiments, to find a hit against a target receptor, usually ultra large docking libraries are screened. Hence, it is essential to validate the performance of the MEMES method on docking libraries that mimic real-life use cases. As ExactMEMES cannot be applied on large docking libraries due to computational constraints and since both are comparable in performance, DeepMEMES framework performance was demonstrated on two large docking libraries Enamine37 HTS Collection (2 million molecules) and an Ultra Large Docking Library31 (96 million molecules).Enamine dataset
The Enamine dataset37 consists of collections of compounds that are used in virtual screening. Enamine HTS Collection containing 2106952 screening compounds was chosen to illustrate the performance of DeepMEMES. The DeepMEMES framework is applied on Enamine HTS Collection to demonstrate that the top docking hits can be identified only by docking a small fraction of the complete library against the target receptor TTBK1.
Fig. 5a shows the fraction of top 500 sampled molecules that are actual top hits sampled from the docking library using the DeepMEMES framework (using Mol2Vec, CDDD and ECFP embedding). ESI Fig. S9‡ provides similar analysis of the top 100 sampled molecules and ESI Fig. S10‡ shows the distribution of the docking scores of top hits sampled. Fig. 5b shows the overlap of the top 500 molecules sampled using the DeepMEMES framework (Mol2Vec), random sampling and actual top hits for target protein TTBK1 (ESI Table S2‡ demonstrates detailed overlap results for Mol2Vec, CDDD and ECFP featurization techniques). From Fig. 5a and b, we can infer that a high percentage of molecules sampled by DeepMEMES matches with the actual top hits by performing only 125000 docking calculations, which is ∼6% of the chosen docking library. Fig. 5a shows that the DeepMEMES framework with Mol2Vec embedding outperformed the CDDD and ECFP embeddings and hence was chosen for further trials with ultra large docking libraries.
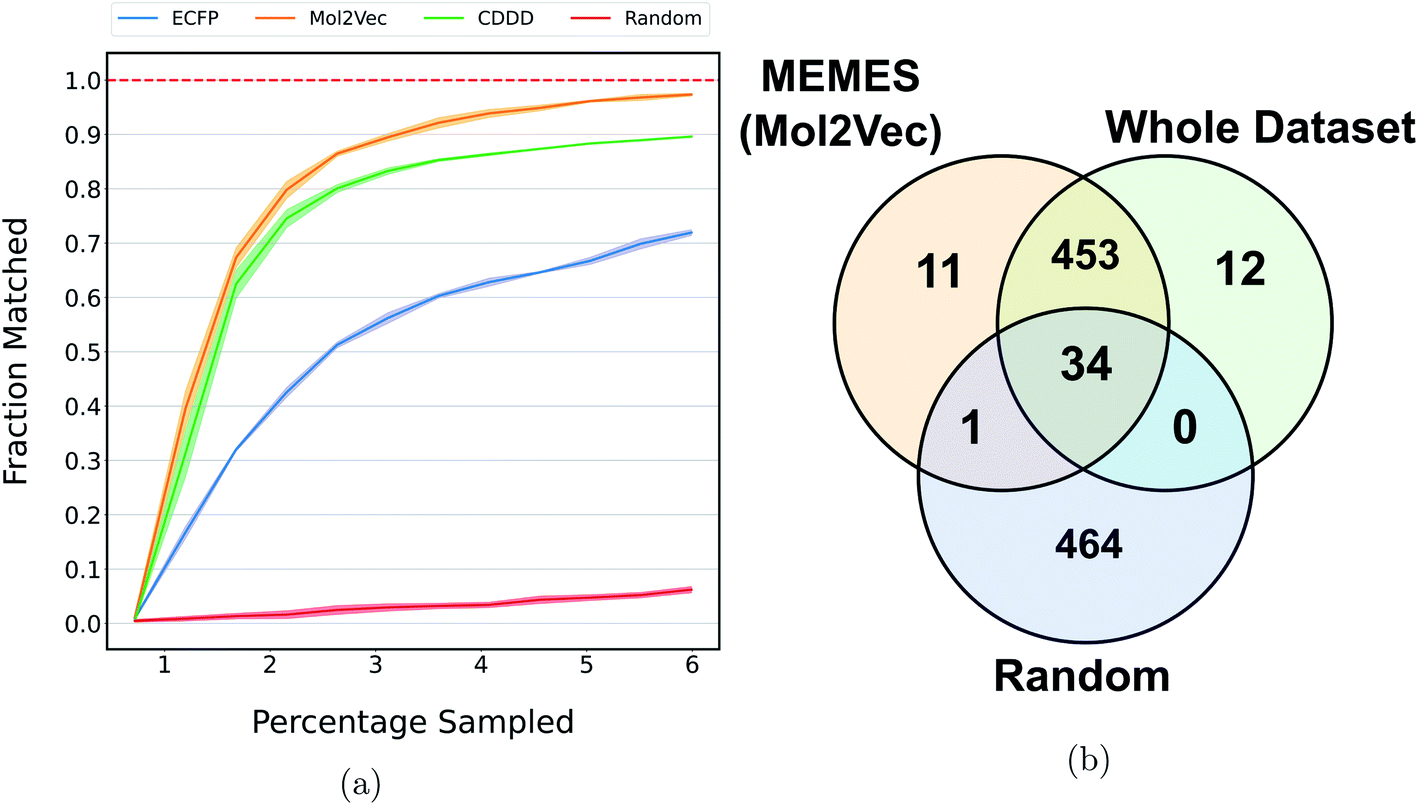 | ||
| Fig. 5 (a) and (b) show the performance of DeepMEMES on the Enamine dataset against target protein TTBK1. (a) shows the fraction of the top 500 sampled molecules that are actual top hits in the library. The reported results are an average of 3 runs and the shaded region represents standard deviation across these runs. The Venn diagram (b) demonstrates the overlap of top 500 hits DeepMEMES(Mol2Vec), random sampling and the whole dataset (the statistics shown are for one of the three runs). | ||
The libraries of compounds used for virtual ligand screening campaigns are fairly large. To substantially reduce the computational cost and deal with molecules with desired physico-chemical properties which are more in line with known drug profiles, rule-based filtering is often employed.58,59 Depending on the objectives of a given project, different filtering criteria are applied based on parameters such as the molecular weight, hydrogen bond acceptor and donors, rotatable bonds, logP, Pan-assay interference compounds (PAINS) and the topological polar surface area (TPSA) among many others.60 TPSA is a popular descriptor in medicinal chemistry that is used for filtering molecules with blood–brain barrier crossing tendency.61 Trials were performed on the Enamine dataset using the MEMES framework combined with rule based filtering to demonstrate that such filtering techniques improve the efficiency of the proposed framework (see ESI Discussion 3‡ for more details).
Ultra large docking library
In a recent study, Lyu et al.31 introduced a large compound library containing 96 million molecules. The whole library was docked to find potential molecules against the AmpC β-lactamase (AmpC) receptor. The DeepMEMES framework with Mol2Vec as molecular embedding was applied to this molecular library to show that top docking hits can be identified by performing docking calculations on a fraction of the complete library. Fig. 6a shows the fraction of top 1000 sampled molecules that are actual top hits sampled from the docking library using DeepMEMES (Mol2Vec), and Fig. 6c shows the comparison of the distribution of the docking scores. Similar analysis for the top 500 and top 5000 molecules is given in ESI Fig. S11 and S12.‡Fig. 6b shows the overlap of the top 1000 molecules sampled using the DeepMEMES framework (using Mol2Vec embedding), random sampling, and actual top hits for target protein AmpC. ESI Table S3‡ demonstrates the overlap results for the top 500, top 1000 and top 5000 molecules for three runs. From Fig. 6a–c we can infer that 90% of molecules sampled by the DeepMEMES framework matches the actual top hits only by performing 5800000 docking calculations, ∼6% of the complete library. It is a significant improvement over random sampling where only 5.5% of sampled molecules matches actual top hits.
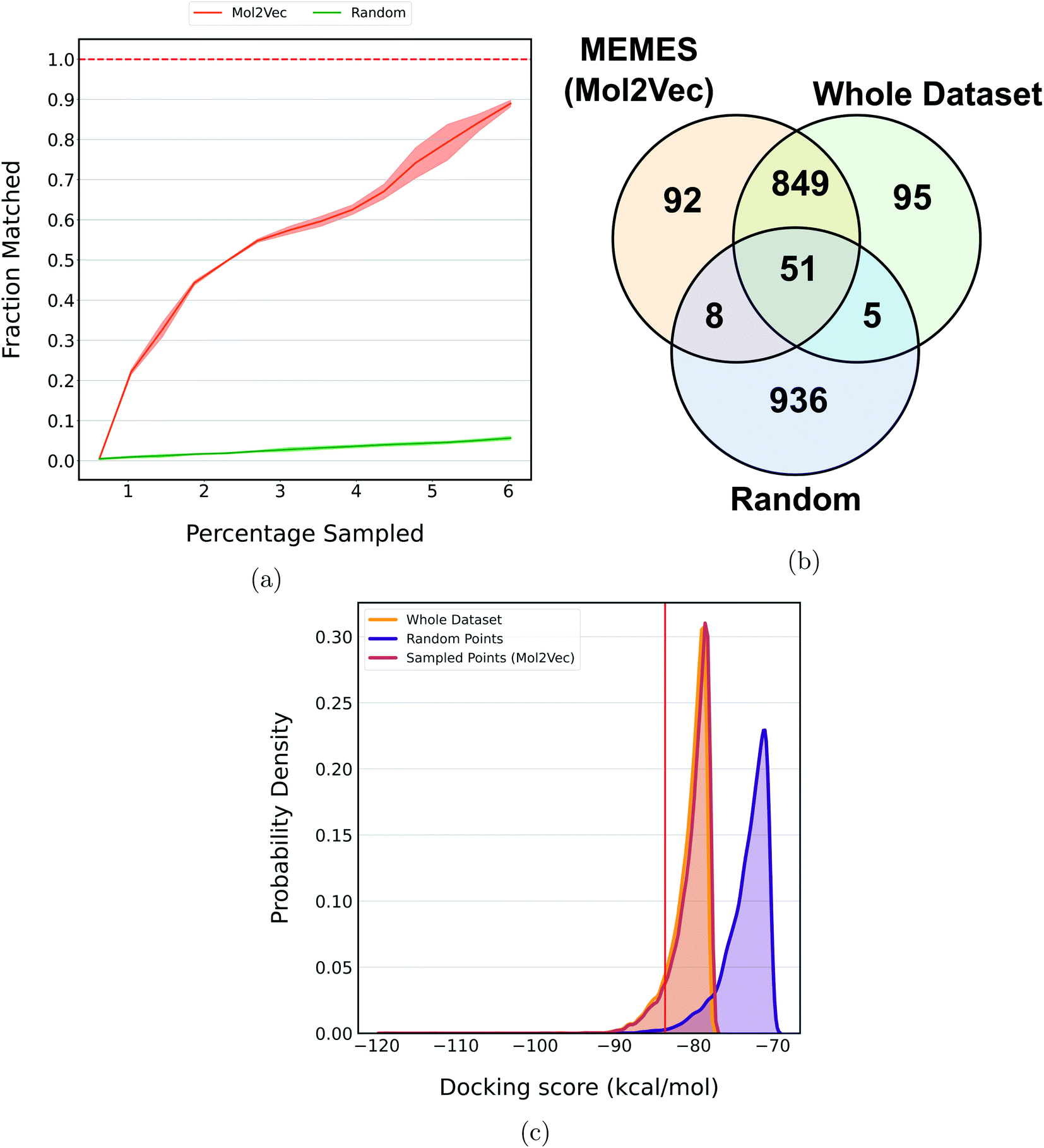 | ||
| Fig. 6 (a)–(c) show the performance of DeepMEMES on an Ultra Large Docking Library against target protein AmpC. (a) shows the fraction of the top 1000 sampled molecules that are actual top hits in the library. The result shown is an average over three runs. The Venn diagram (b) demonstrates the overlap of the top 1000 hits identified by DeepMEMES (Mol2Vec), random sampling and the whole dataset. (c) shows the distribution of the docking scores for top 10000 molecules sampled by DeepMEMES, random sampling and the whole dataset. The vertical red line denotes the cutoff docking score for the top 1000 hits (the distribution plot and Venn diagram are made from one of the three runs). | ||
Effect of the docking library size on the performance of DeepMEMES
The previous section shows the application of DeepMEMES on Enamine HTS collection37 and an ultra large docking library31 for target protein TTBK1 and AmpC, respectively. The purpose of this exercise is to demonstrate the robustness of the proposed framework on docking libraries of varying sizes. K-means clustering was performed on an ultra large docking library,31 creating 1000 clusters, and subsets of different sizes ranging from 2 million to 96 million were created by uniformly sampling from each of the resulting clusters. Finally, DeepMEMES performance was assessed on each of the resulting subsets.Fig. 7 shows the fraction match of the sampled molecules that matches actual top hits for different docking library sizes. 85–95% of the molecules sampled by the DeepMEMES framework with Mol2Vec featurization matches the actual top hits irrespective of the docking library's size, demonstrating the consistent performance of the proposed framework.
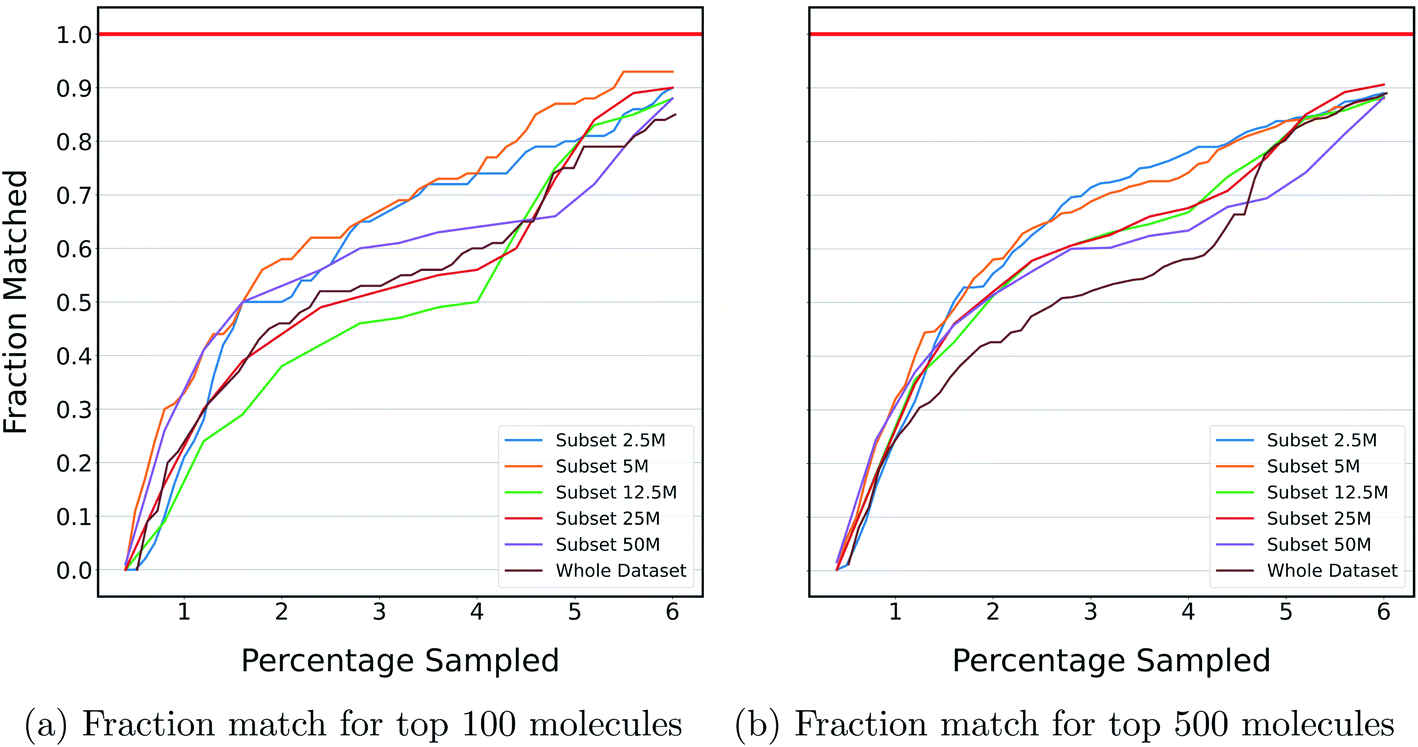 | ||
| Fig. 7 Fraction of top molecules sampled by DeepMEMES (with Mol2Vec as the featurization technique) that matches with actual top hits from the corresponding subsets against the percentage of the dataset sampled. | ||
In summary, high throughput virtual screening requires exhaustive evaluation of each molecule in a complete docking library to find potential candidate molecules. In this study, the MEMES framework based on Bayesian optimization for efficient sampling of the chemical space for high throughput exercises is proposed. We showcase the MEMES framework application in hit identification, i.e., to sample molecules with high docking scores against target receptors. Two variants of the MEMES framework are introduced, ExactMEMES and DeepMEMES, depending on the choice of surrogate function. Various MEMES runs were performed with Mol2Vec, CDDD and ECFP as molecular featurization techniques, and with different sized molecular libraries ranging from 2 million to 96 million to find hit molecules against different target receptors to showcase the efficiency of the proposed framework. The MEMES framework was able to identify more than 90% of the actual top hits while only calculating the docking score for about 6% of the complete molecular library showing the robustness of the proposed framework. In this work, MEMES framework application was demonstrated on virtual screening of molecular libraries, but it can also be applied on other screening applications where exhaustive evaluation is infeasible.
Data availability
The data that support the findings of this study are available from the corresponding author upon request.Author contributions
UDP conceptualized the problem; SM, SL, YP and UDP designed the ML methodology; AS and MA designed the docked methodology. SM performed the investigations and data analysis. SM, AS, SL, YP and UDP wrote the manuscript. UDP supervised the project. All authors reviewed the manuscript.Conflicts of interest
International Institute of Information Technology, Hyderabad has filed provisional patent application for the use of the MEMES framework in high-throughput screening exercises, with U. D. P., S. M., S. L., and Y. P. listed as inventors. Provisional patent application No.: 202041050608. Application status: awaiting complete specification (provisional patent filed). The funders did not have any role in the design, idea, data collection, analysis, interpretation, writing of the manuscript or decision to submit it for publication.Acknowledgements
We thank Professor Brian Shoichet for making the data on docking calculations on AmpC protein available to us. We acknowledge the financial support through the DST-SERB grant (no. CVD/2020/000343) and IHub-Data, IIIT Hyderabad. This work was partially funded by Intel Corp. as part of its Pandemic Response Technology Initiative (PRTI).References
- H. R. Schmidt, R. M. Betz, R. O. Dror and A. C. Kruse, Structural basis for σ 1 receptor ligand recognition, Nat. Struct. Mol. Biol., 2018, 25, 981–987 CrossRef CAS PubMed.
- P. D. Lyne, Structure-based virtual screening: an overview, Drug discovery today, 2002, 7, 1047–1055 CrossRef CAS.
- T. Cheng, Q. Li, Z. Zhou, Y. Wang and S. H. Bryant, Structure-based virtual screening for drug discovery: a problem-centric review, AAPS J., 2012, 14, 133–141 CrossRef CAS PubMed.
- J. D. McCorvy, K. V. Butler, B. Kelly, K. Rechsteiner, J. Karpiak, R. M. Betz, B. L. Kormos, B. K. Shoichet, R. O. Dror and J. Jin, et al., Structure-inspired design of β-arrestin-biased ligands for aminergic GPCRs, Nat. Chem. Biol., 2018, 14, 126 CrossRef CAS PubMed.
- A. Gaulton, L. J. Bellis, A. P. Bento, J. Chambers, M. Davies, A. Hersey, Y. Light, S. McGlinchey, D. Michalovich and B. Al-Lazikani, et al., ChEMBL: a large-scale bioactivity database for drug discovery, Nucleic Acids Res., 2012, 40, D1100–D1107 CrossRef CAS.
- L. C. Blum and J.-L. Reymond, 970 million drug-like small molecules for virtual screening in the chemical universe database GDB-13, J. Am. Chem. Soc., 2009, 131, 8732–8733 CrossRef CAS.
- T. Sterling and J. J. Irwin, ZINC 15–ligand discovery for everyone, J. Chem. Inf. Model., 2015, 55, 2324–2337 CrossRef CAS.
- Y. Pathak, S. Laghuvarapu, S. Mehta and U. D. Priyakumar, Chemically interpretable graph interaction network for prediction of pharmacokinetic properties of drug-like molecules, Proceedings of the AAAI Conference on Artificial Intelligence, 2020, pp 873–880 Search PubMed.
- S. Laghuvarapu, Y. Pathak and U. D. Priyakumar, Band nn: A deep learning framework for energy prediction and geometry optimization of organic small molecules, J. Comput. Chem., 2020, 41, 790–799 CrossRef CAS PubMed.
- J. Vamathevan, D. Clark, P. Czodrowski, I. Dunham, E. Ferran, G. Lee, B. Li, A. Madabhushi, P. Shah and M. Spitzer, et al., Applications of machine learning in drug discovery and development, Nat. Rev. Drug Discovery, 2019, 18, 463–477 CrossRef CAS.
- R. Aggarwal, A. Gupta, V. Chelur, C. Jawahar and U. D. Priyakumar, DeepPocket: Ligand Binding Site Detection and Segmentation using 3D Convolutional Neural Networks, 2021, DOI:10.26434/chemrxiv.14611146.v1.
- Y. B. L. Samaga, S. Raghunathan and U. D. Priyakumar, SCONES: Self-Consistent Neural Network for Protein Stability Prediction Upon Mutation, 2021, DOI:10.26434/chemrxiv.14729445.v1.
- A. W. Senior, R. Evans, J. Jumper, J. Kirkpatrick, L. Sifre, T. Green, C. Qin, A. Žídek, A. W. Nelson and A. Bridgland, et al., Improved protein structure prediction using potentials from deep learning, Nature, 2020, 577, 706–710 CrossRef CAS.
- P. Pattnaik, S. Raghunathan, T. Kalluri, P. Bhimalapuram, C. V. Jawahar and U. D. Priyakumar, Machine learning for accurate force calculations in molecular dynamics simulations, J. Phys. Chem. A, 2020, 124, 6954–6967 CrossRef CAS PubMed.
- F. Noé, A. Tkatchenko, K.-R. Müller and C. Clementi, Machine learning for molecular simulation, Annu. Rev. Phys. Chem., 2020, 71, 361–390 CrossRef.
- S. Manzhos and T. Carrington Jr, Neural network potential energy surfaces for small molecules and reactions, Chem. Rev., 2020 DOI:10.1021/acs.chemrev.0c00665.
- V. Bagal, R. Aggarwal, P. Vinod and U. D. Priyakumar, LigGPT: Molecular Generation using a Transformer-Decoder Model, 2021, DOI:10.26434/chemrxiv.14561901.v1.
- D. P. Kingma and M. Welling, Auto-encoding variational bayes, 2013, arXiv preprint arXiv:1312.6114.
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, Automatic chemical design using a data-driven continuous representation of molecules, ACS Cent. Sci., 2018, 4, 268–276 CrossRef.
- Y. Pathak, K. S. Juneja, G. Varma, M. Ehara and U. D. Priyakumar, Deep learning enabled inorganic material generator, Phys. Chem. Chem. Phys., 2020, 22, 26935–26943 RSC.
- H. Dai, Y. Tian, B. Dai, S. Skiena and L. Song, Syntax-directed variational autoencoder for molecule generation, Proceedings of the International Conference on Learning Representations, 2018 Search PubMed.
- I. Goodfellow, J. Pouget-Abadie, M. Mirza, B. Xu, D. Warde-Farley, S. Ozair, A. Courville and Y. Bengio, Generative adversarial nets, Adv. Neural Inf. Process. Syst., 2014, 2672–2680 Search PubMed.
- N. De Cao and T. Kipf, MolGAN: An implicit generative model for small molecular graphs, 2018, arXiv preprint arXiv:1805.11973.
- T. Blaschke, M. Olivecrona, O. Engkvist, J. Bajorath and H. Chen, Application of generative autoencoder in de novo molecular design, Mol. Inf., 2018, 37, 1700123 CrossRef.
- X. Yang, J. Zhang, K. Yoshizoe, K. Terayama and K. Tsuda, ChemTS: an efficient python library for de novo molecular generation, Sci. Technol. Adv. Mater., 2017, 18, 972–976 CrossRef CAS.
- M. H. Segler, T. Kogej, C. Tyrchan and M. P. Waller, Generating focused molecule libraries for drug discovery with recurrent neural networks, ACS Cent. Sci., 2018, 4, 120–131 CrossRef CAS PubMed.
- G. L. Guimaraes, B. Sanchez-Lengeling, C. Outeiral, P. L. C. Farias and A. Aspuru-Guzik, Objective-reinforced generative adversarial networks (organ) for sequence generation models, 2017, arXiv preprint arXiv:1705.10843.
- J. You, B. Liu, Z. Ying, V. Pande and J. Leskovec, Graph convolutional policy network for goal-directed molecular graph generation, Adv. Neural Inf. Process. Syst., 2018, 6410–6421 Search PubMed.
- T. Cieplinski, T. Danel, S. Podlewska and S. Jastrzebski, We should at least be able to Design Molecules that Dock Well, 2020, arXiv preprint arXiv:2006.16955.
- W. Gao and C. W. Coley, The synthesizability of molecules proposed by generative models, J. Chem. Inf. Model., 2020, 60(12), 5714–5723 CrossRef CAS.
- J. Lyu, S. Wang, T. E. Balius, I. Singh, A. Levit, Y. S. Moroz, M. J. O'Meara, T. Che, E. Algaa and K. Tolmachova, et al., Ultra-large library docking for discovering new chemotypes, Nature, 2019, 566, 224–229 CrossRef CAS PubMed.
- A. Tomberg and J. Boström, Can “easy” chemistry produce complex, diverse and novel molecules?, Drug Discovery Today, 2020, 25, 2174–2181 CrossRef CAS PubMed.
- F. Gentile, V. Agrawal, M. Hsing, A.-T. Ton, F. Ban, U. Norinder, M. E. Gleave and A. Cherkasov, Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery, ACS Cent. Sci., 2020, 6(6), 939–949 CrossRef CAS.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- W. Jin, R. Barzilay and T. Jaakkola, Junction tree variational autoencoder for molecular graph generation, 2018, arXiv preprint arXiv:1802.04364.
- Z. Zhou, S. Kearnes, L. Li, R. N. Zare and P. Riley, Optimization of molecules via deep reinforcement learning, Sci. Rep., 2019, 9, 1–10 Search PubMed.
- Enamine, http://www.enamine.net/.
- RCSB, https://www.rcsb.org.
- D. Rogers and M. Hahn, Extended-connectivity fingerprints, J. Chem. Inf. Model., 2010, 50, 742–754 CrossRef CAS.
- S. Jaeger, S. Fulle and S. Turk, Mol2vec: unsupervised machine learning approach with chemical intuition, J. Chem. Inf. Model., 2018, 58, 27–35 CrossRef CAS PubMed.
- R. Winter, F. Montanari, F. Noé and D.-A. Clevert, Learning continuous and data-driven molecular descriptors by translating equivalent chemical representations, Chem. Sci., 2019, 10, 1692–1701 RSC.
- T. Mikolov, I. Sutskever, K. Chen, G. S. Corrado and J. Dean, Distributed representations of words and phrases and their compositionality, Adv. Neural Inf. Process. Syst., 2013, 3111–3119 Search PubMed.
- J. S. Delaney, ESOL: estimating aqueous solubility directly from molecular structure, J. Chem. Inf. Comput. Sci., 2004, 44, 1000–1005 CrossRef CAS PubMed.
- T. D. Challenge. Tox21 data challenge 2014, 2014 Search PubMed.
- M. Pelikan, D. E. Goldberg, E. Cantú-Paz, et al., BOA: The Bayesian optimization algorithm, Proceedings of the genetic and evolutionary computation conference GECCO-99, 1999, pp 525–532 Search PubMed.
- P. I. Frazier, A tutorial on bayesian optimization, 2018, arXiv preprint arXiv:1807.02811.
- R.-R. Griffiths and J. M. Hernández-Lobato, Constrained Bayesian optimization for automatic chemical design using variational autoencoders, Chem. Sci., 2020, 11, 577–586 RSC.
- B. J. Shields, J. Stevens, J. Li, M. Parasram, F. Damani, J. I. M. Alvarado, J. M. Janey, R. P. Adams and A. G. Doyle, Bayesian reaction optimization as a tool for chemical synthesis, Nature, 2021, 590, 89–96 CrossRef CAS.
- J. Snoek, H. Larochelle and R. P. Adams, Practical bayesian optimization of machine learning algorithms, Adv. Neural Inf. Process. Syst., 2012, 2951–2959 Search PubMed.
- A. Wilson and R. Adams, Gaussian process kernels for pattern discovery and extrapolation, Int. Conf. Mach. Learn., 2013, 1067–1075 Search PubMed.
- J. R. Gardner, G. Pleiss, D. Bindel, K. Q. Weinberger and A. G. Wilson, GPyTorch: Blackbox Matrix-Matrix Gaussian Process Inference with GPU Acceleration, Proceedings of the 32nd International Conference on Neural Information Processing Systems, Red Hook, NY, USA, 2018, pp. 7587–7597 Search PubMed.
- A. Damianou and N. Lawrence, Deep Gaussian processes, Artif. Intell. Stat., 2013, 207–215 Search PubMed.
- T. Bui, D. Hernández-Lobato, J. Hernandez-Lobato, Y. Li and R. Turner, Deep Gaussian processes for regression using approximate expectation propagation, Int. Conf. Mach. Learn., 2016, 1472–1481 Search PubMed.
- Z. Dai, A. Damianou, J. González and N. Lawrence, Variational auto-encoded deep Gaussian processes, 2015, arXiv preprint arXiv:1511.06455.
- H. Salimbeni and M. Deisenroth, Doubly stochastic variational inference for deep Gaussian processes, Adv. Neural Inf. Process. Syst., 2017, 4588–4599 Search PubMed.
- J. A. Hartigan and M. A. Wong, Algorithm AS 136: A k-means clustering algorithm, J. R. Stat. Soc. Ser. C Appl. Stat., 1979, 28, 100–108 Search PubMed.
- C. E. Rasmussen, Gaussian processes in machine learning, Summer School on Machine Learning, 2003, pp. 63–71 Search PubMed.
- C. A. Lipinski, Chapter 11 Filtering in Drug Discovery, Annu. Rep. Comput. Chem., 2005, 1, 155–168 CAS.
- M. A. Miteva, S. Violas, M. Montes, D. Gomez, P. Tuffery and B. O. Villoutreix, FAF-Drugs: free ADME/tox filtering of compound collections, Nucleic Acids Res., 2006, 34, W738–W744 CrossRef CAS PubMed.
- E. Lionta, G. Spyrou, D. Vassilatis and Z. Cournia, Structure-based virtual screening for drug discovery: principles, applications and recent advances, Curr. Top. Med. Chem., 2014, 14, 1923–1938 CrossRef CAS.
- S. Prasanna and R. Doerksen, Topological polar surface area: a useful descriptor in 2D-QSAR, Curr. Med. Chem., 2009, 16, 21–41 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor N. Sathyamurthy on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available: Tables of performance of ExactMEMES and DeepMEMES, performance comparison of MEMES with deep docking, figures of structure of top hits, distribution plots of binding affinities, distributions of molecular clusters, distributions of binding affinities of missed hits, fractions matched against the sampled percentage, protein–ligand complexes and protein–ligand interactions, and supplementary discussions and methods. See DOI: 10.1039/d1sc02783b |
| § Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |