
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTime-resolved analysis of photoluminescence at a single wavelength for ratiometric and multiplex biosensing and bioimaging†
Qi
Wu‡
a,
Peiling
Dai‡
a,
Yun
Wang
a,
Jin
Zhang
a,
Meng
Li
a,
Kenneth Yin
Zhang
*a,
Shujuan
Liu
a,
Wei
Huang
 *abc and
Qiang
Zhao
*a
*abc and
Qiang
Zhao
*a
aState Key Laboratory of Organic Electronics and Information Displays, Jiangsu Key Laboratory for Biosensors, Institute of Advanced Materials (IAM), Institute of Flexible Electronics (Future Technology), Nanjing University of Posts & Telecommunications, Nanjing 210023, P. R. China. E-mail: iamyzhang@njupt.edu.cn; iamqzhao@njupt.edu.cn
bFrontiers Science Center for Flexible Electronics (FSCFE), Shaanxi Institute of Flexible Electronics (SIFE), Northwestern Polytechnical University (NPU), Xi'an 710072, China
cKey Laboratory of Flexible Electronics (KLOFE), Institute of Advanced Materials (IAM), Nanjing Tech University (NanjingTech), 30 South Puzhu Road, Nanjing 211816, China. E-mail: wei-huang@njtech.edu.cn
First published on 26th July 2021
Abstract
Simultaneous analysis of luminescence signals of multiple probes can improve the accuracy and efficiency of biosensing and bioimaging. Analysis of multiple signals at different wavelengths usually suffers from spectral overlap, possible energy transfer, and difference in detection efficiency. Herein, we reported a polymeric luminescent probe, which was composed of a phenothiazine-based fluorescent compound and a phosphorescent iridium(III) complex. Both luminophores emitted at around 600 nm but their luminescence lifetimes are 160 times different, allowing time-resolved independent analysis. As the fluorescence was enhanced in response to oxidation by hypochlorite and the phosphorescence was sensitive toward oxygen quenching, a four-dimensional relationship between luminescence intensity, fluorescence/phosphorescence ratio, hypochlorite concentration, and oxygen content was established. In cellular imaging, time-resolved photoluminescence imaging microscopy clearly showed the independent fluorescence response toward hypochlorite and phosphorescence response toward oxygen in separated time intervals. This work opens up a new idea for the development of multiplex biosensing and bioimaging.
Introduction
Photoluminescence bioimaging tracks emissive probes in biological environments to create visual images by analyzing and processing luminescence signals, giving structural and functional information of biological molecules,1–7 organelles,8–11 living cells,12–16 tissues,17–20 and organs.21,22 Laser scanning confocal microscopy has been widely used for analyzing luminescence intensity of a probe to construct images, which simply and directly reflect the spatial distribution and local concentration of the probe at the subcellular level.23,24 Once the luminescence intensity of the probe is correlated with its specific interaction with functional biomarkers, luminescence imaging can locate and quantify the marker while continuously monitoring the process of life activities involved in the cell.25–29 In order to improve the sensing and imaging accuracy and efficiency, it is usually necessary to employ multiple luminophores and simultaneously analyze two or more luminescence signals, thereby allowing one signal to be used as an internal standard,30–33 or multiple signals to respond toward different targets.34–36In practical applications, the luminophores simultaneously used are designed to have different emission wavelengths, so that they can be readily distinguishable by placing optical filters before the detector.37,38 However, there are some limitations seriously interfering with the sensing and imaging during wavelength-resolved analysis. Firstly, many organic fluorophores and phosphorescent transition metal complexes exhibited broad emission spectra. It is difficult to completely avoid spectral overlap, so it is usually necessary to sacrifice the brightness to avoid signal leakage and loss of sensitivity. Secondly, as the luminophores are separated in wavelength, energy transfer from the higher energy luminophore to the lower energy one is possible, resulting in undetectable high-energy signals.39,40 Thirdly, the refractive index, reflectance, and absorbance of photons of different wavelengths in biological samples are different. Hence, the loss of photons when passing through biological samples and the number of photons reaching the detector strongly depends on the wavelength.41 As shown in Fig. 1a, when dealing with wavelength-resolved two luminescence signals in cellular imaging, increasing intralipid in the culture medium caused reduced detection efficiency of photons at both 500 and 600 nm. The intensity ratio of the two wavelengths is also influenced by the amount of the intralipid due to their different transmittance, indicating that luminophores emitting at different wavelengths are not suitable for ratiometric imaging.
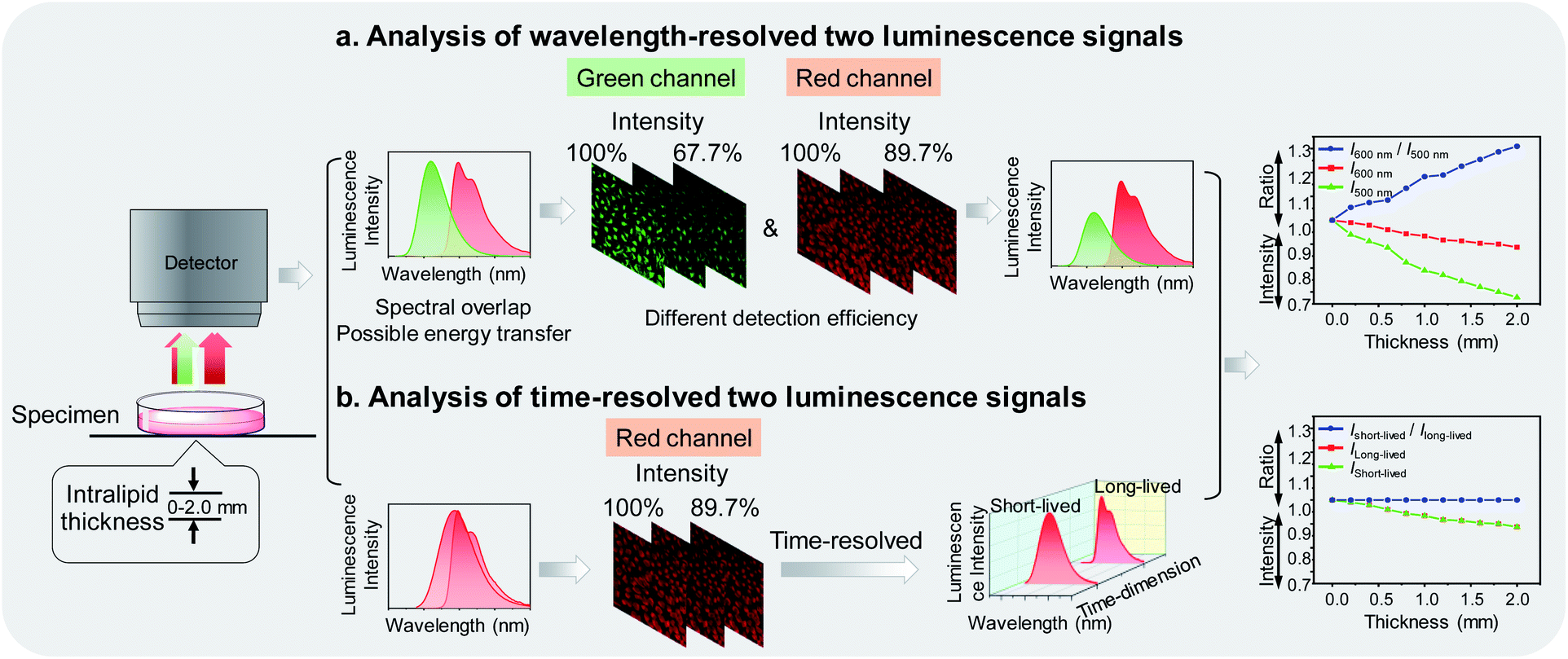 | ||
| Fig. 1 (a) Analysis of wavelength-resolved two luminescence signals. The luminescence intensity at 500 and 600 nm showed different decrease trends as the increase of intralipid in the medium. (b) Analysis of time-resolved two luminescence signals. Time-resolved technique allows independent analysis two luminescence signals at the same wavelength. | ||
To address these concerns, in this work, we proposed to analyze multiple photoluminescence signals at the same wavelength via a time-resolved technique. A new dual-emissive polymeric probe com-posed of two luminophores was designed and synthesized. The two luminophores emitted at almost the same wavelength to ensure limited energy transfer and consistent photon-detection efficiency, but exhibited different luminescence lifetimes, enabling independent analysis of each spectrum via a time-resolved technique42 (Fig. 1b). Although the amount of the intralipid in the culture medium caused photon loss during imaging, the ratio of short-lived luminescence signals over long-lived ones remained unchanged. The two luminophores were designed to respond toward hypochlorite and oxygen, respectively. Hypochlorite plays an important role in the destruction of pathogens in the immune system43 and unregulated cellular production of hypochlorite may be associated with various diseases.44 Cellular oxygen contents are kept in a certain range and hypoxia is an important feature of many diseases including tumors.45 Therefore, sensing and imaging cellular hypochlorite and oxygen are of great importance and have attracted much attention.31,38,46,47 Under constant oxygen conditions, the polymeric probe was used for time-resolved ratiometric sensing and imaging of hypochlorite by using the oxygen-sensitive luminescence as an internal standard. When the oxygen content changed, the probe was used for simultaneously sensing and imaging of hypochlorite and oxygen via time-resolved luminescence analysis.
Results and discussion
The dual-emissive polymeric probe P1 was designed by incorporation of a phenothiazine-based fluorescent compound 1, a phosphorescent iridium(III) complex 2, and polyethylene glycol (PEG) into the side chains on the polyethylene backbone (Fig. 2a and S1†). Compound 1 was weakly emissive owing to the photoinduced electron transfer (PET) from the electron-rich sulfur atom to the electron-deficient pyridinium.48 In response to hypochlorite, which oxidizes the thioether to sulfoxide, compound 1 exhibited significant fluorescence enhancement at around 600 nm with a lifetime of 2.5 ns (Fig. S2†). Complex 2 emitted at the same wavelength with a much longer lifetime of about 0.4 μs owing to its phosphorescence nature. Meanwhile, the phosphorescence of complex 2 was readily quenched by molecular oxygen via triplet–triplet energy transfer (Fig. S2†). Both compound 1 and complex 2 were excitable at 405 nm, which is the most widely used excitation laser source in confocal microscopy. PEG was used to improve the water solubility and biocompatibility. Polymeric luminescent probes P2 and P3 (Fig. 2a), where complex 2 and compound 1 was absent, respectively, were also synthesized for comparison studies. All the small molecular intermediates and monomers were characterized via1H and 13C nuclear magnetic resonance (NMR), matrix assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry (MS) and the polymers were synthesized via radical polymerization and characterized via gel permeation chromatography (GPC). The contents of compound 1 and complex 2 in P1 were about 6% and 1% (molar ratio) calculated according to the absorption spectra. The averaged molecular weight (Mw) of P1 was about 133![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 with a polydispersity index (PDI) of 1.13. P1 was completely soluble in aqueous solution and suitable for bioimaging.
000 g mol−1 with a polydispersity index (PDI) of 1.13. P1 was completely soluble in aqueous solution and suitable for bioimaging.
 | ||
| Fig. 2 (a) Chemical structures. (b–d) Luminescence spectra (b), excited-state decay curves (c) and relative occurrence of fluorescence and phosphorescence (d) of P1 in PBS in the presence of 0–10 μM NaClO. (e) Isolated fluorescence and phosphorescence spectra of P1 in PBS in the presence of 0–10 μM NaClO. (f) Luminescence spectra of P1 in PBS under an atmosphere containing 0–100% O2 co-balanced with 100%–0 N2. (g) Isolated fluorescence and phosphorescence spectra of P1 in PBS under N2, air, and O2 atmospheres. | ||
The luminescence response of P1 toward hypochlorite was investigated in phosphate buffer saline (PBS) under ambient conditions. Upon photoexcitation at 405 nm, P1 showed a mixed emission spectrum at 500–700 nm which was composed of the fluorescence of compound 1 and the phosphorescence of complex 2. Addition of hypochlorite led to luminescence enhancement of P1 (Fig. 2b) because of oxidation-induced fluorescence turn-on of compound 1. Similar luminescence enhancement was also observed in the polymeric probe P2 but not in P3 in which compound 1 was absent (Fig. S3†). The remarkable luminescence response was specific toward hypochlorite in preference to other reactive oxygen species and biothiols (Fig. S4†). Luminescence lifetime analysis revealed biexponential decay of P1 with lifetimes of about 2.7 and 500.0 ns corresponding to compound 1 and complex 2, respectively (Fig. 2c). The proportion of short-lived fluorescence (If) at 600 nm in the spectrum increased from 11.7% to 78.0% upon addition of hypochlorite (Fig. 2d). The proportion of long-lived phosphorescence (Ip) decreased from 88.3% to 22.0% when the fluorescence was turned on by hypochlorite. According to these ratios, the fluorescence and phosphorescence were isolated from the total luminescence via computational calculation. The isolated spectra showed that the fluorescence was significantly enhanced while the phosphorescence was insensitive to hypochlorite (Fig. 2e). Using the phosphorescence signal as an internal standard, the intensity ratio (If/Ip) of fluorescence over phosphorescence increased from 0.13 to 3.5 (about 27 fold) when the concentration of hypochlorite was increased from 0 to 10 μM. Further addition of hypochlorite did not change the total luminescence spectrum as well as the intensity ratio.
The luminescence response of P1 toward oxygen was recorded in PBS in the absence of hypochlorite under an O2/N2 mixed atmosphere with different O2 contents. When increasing and reducing the O2 content, the emission at about 600 nm was quenched and enhanced, respectively (Fig. 2f), owing to the O2-induced phosphorescence quenching of complex 2. Luminescence lifetime analysis showed that while the fluorescence lifetime remained unchanged, the phosphorescence lifetime was shortened in pure O2 and elongated in pure N2. Meanwhile, the phosphorescence proportion (88.3% in air) in the total emission spectra was reduced to 84.5% and increased to 93.0%, respectively (Fig. 2g). Similar response toward oxygen was also observed for P3 but not for hypochlorite-oxidized P2 in which the phosphorescent iridium(III) complex was absent (Fig. S5†).
To use P1 for simultaneous detection of hypochlorite and oxygen content, a functional relationship between the luminescence of P1 and the two variables was established. According to the luminescence titration curves of P1 upon addition of hypochlorite, the fluorescence intensity showed a linear relationship with the concentration of hypochlorite in the range of 0–10 μM (Fig. 3a). As the phosphorescence was insensitive to hypochlorite, the If/Ip ratio also exhibited a linear relationship with the concentration of hypochlorite (Fig. 3b). In the absence of hypochlorite, the phosphorescence response toward oxygen followed Stern–Volmer equation49 (Ip0/Ip = 1 + KSV[O2]), giving a hyperbola relationship between the phosphorescence intensity and the oxygen content Ip = Ip0/(1 + KSV[O2]), where Ip0 and Ip are phosphorescence intensities in the absence and presence of O2, respectively, and KSV is the Stern–Volmer constant (Fig. 3c). During simultaneous detection of hypochlorite and oxygen, the total luminescence spectrum gave the sum of fluorescence and phosphorescence, and time resolved analysis distinguished between fluorescence and phosphorescence based on their different decay rates in time domain and indicated their proportions in the total spectrum. The data obtained from the luminescence titrations were fitted and followed the following two equations:
| If + Ip = 0.065 × [ClO−] + 0.26/(1 + 0.08 × [O2]) + 0.12 |
| If/Ip = (0.065 × [ClO−] + 0.012)/(0.26/(1 + 0.08 × [O2]) + 0.096) |
 | ||
| Fig. 3 (a) Luminescence titration curve of P1 upon addition of 0–10 μM NaClO. (b) Plot of the If/Ip ratio versus hypochlorite concentration for P1. (c) Plot of luminescence intensity versus oxygen content for P1. (d) Four-dimensional relationship between luminescence intensity, fluorescence/phosphorescence ratio, hypochlorite concentration, and oxygen content for P1. (e–g) Projections of the curved surface on each coordinate plane. (h) Experimental and calculated luminescence intensity of P1 in the presence of 0–10 μM NaClO under N2 atmosphere. (i) Experimental and calculated luminescence intensity of P1 in the presence 10 μM NaClO under 0–100% O2 atmosphere. (j) Excited-state decay curves of P1 in the presence of 0 (solid line) and 10 μM (dashed line) NaClO under N2 (red line) and O2 (blue line) atmospheres. (k) Relative occurrence of fluorescence (green) and phosphorescence (red) of P1 in the absence (−) and presence (+) of 10 μM NaClO under N2 and O2 atmospheres. | ||
A four-dimensional relationship between luminescence intensity, fluorescence/phosphorescence ratio, hypochlorite concentration, and oxygen content was established (Fig. 3d). The projections of the three-dimensional surface onto the three perpendicular two-dimensional coordinate systems were shown in Fig. 3e–g. Fig. 3e showed the linear relationship between the luminescence intensity and the hypochlorite concentration in pure N2 and O2. Fig. 3f showed the luminescence response toward oxygen in the presence and absence of hypochlorite (10 μM). Fig. 3g illustrated the relationship between the fluorescence/phosphorescence ratio and hypochlorite and oxygen. To evaluate the accuracy of the 4D relationship, luminescence titration against hypochlorite was performed in pure N2, and the luminescence response toward oxygen was investigated in the presence of 10 μM of hypochlorite. As shown in Fig. 3h and i, the experimental data were highly consistent with the calculated data based on the 4D relationship graph. Additionally, luminescence decay curves of P1 in the presence and absence of hypochlorite (10 μM) were measured under N2 and O2 atmospheres, respectively (Fig. 3j). As shown in Fig. 3k, the fluorescence and phosphorescence proportions were well in line with the calculated ones. The difference between the experimental and calculated values was slightly larger in the absence of hypochlorite compared with that in the presence of hypochlorite, because the experimental error increased when the fluorescence was too weak in the absence of hypochlorite.
The cellular imaging and sensing properties of P1 was investigated using human cervix epithelioid carcinoma (HeLa) cells as a mode cell line. The cytotoxicity of P1 toward HeLa cells was evaluated via the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) assay. HeLa cells maintained a viability >90% after incubation with P1 at a concentration of 300 μg mL−1 for 24 h (Fig. S6†), confirming negligible cytotoxic effect of P1 under the following imaging conditions. The microscopy imaging was performed under excitation at 405 nm, and the luminescence signals at 600 ± 25 nm were collected and analyzed. The microscopy luminesce images of HeLa cells treated with P1 are illustrated in Fig. 4a. HeLa cells incubated with P1 (200 μg mL−1, 37 °C, 1 h) showed cytoplasmic staining. Further incubation of the cells with hypochlorite for 30 min led to significant enhancement of intracellular luminescence, which has been attributed to the oxidation-induced fluorescence turn-on. However, it is difficult to quantitatively analyze the luminescence intensity against the hypochlorite concentration in the culture medium, because when the excitation power and slit width were fixed, the intracellular luminescence signals were either too weak to create clear images in the absence of hypochlorite or too strong that exceeded the detection limit of the detector when the fluorescence was turned on. Both the luminescence lifetime and the fluorescence/phosphorescence ratio were independent of the excitation power and thus high quality images were taken under suitable but different excitation powers.50 Time-resolved analysis of the images was performed via photoluminescence lifetime imaging microscopy (PLIM). The PLIM images showed that the luminescence of intracellular P1 exhibited a unique lifetime and distributed evenly in the cytoplasm (Fig. 4a). Statistical analysis revealed that the lifetimes of the intracytoplasmic P1 were normally distributed in the range of 300–600 ns with a mean of about 450 ns, which is fully in line with the phosphorescence lifetime of P1 (Fig. 4b). Further incubation with hypochlorite (10 μM, 30 min) gave rise to some short-lived pixels in the image and an additional distribution at around 150 ns appeared. The lifetime value was much longer than the fluorescence lifetime and has tentatively been assigned to the averaged fluorescence and phosphorescence lifetime of the hypochlorite-oxidized P1. The signals at about 150 ns became dominant when the hypochlorite concentration in the medium increased to 30 μM (Fig. 4a and b). The relative occurrence of the short-lived (<300 ns) and long-lived (>300 ns) signals linearly increased from 0.22 to 3.32 as the hypochlorite concentration in the medium increased from 0 to 30 μM (Fig. 4c). In another experiment, HeLa cells were pretreated with elesclomol (125 nM, 37 °C, 2 h) to induce production of endogenous hypochlorite.51 The cells were then incubated with P1 (200 μg mL−1, 37 °C, 1 h). The PLIM image showed substantial short-lived signals <300 ns (Fig. 4d and e), suggestive of oxidation of intracellular P1 by endogenous hypochlorite. The occurrence ratio of the short-lived signals over the long-lived signals (>300 ns) was about 1.29, which indicated that the amount of endogenously produced hypochlorite was almost the same as internalized amount during incubation of the cells with about 12 μM of hypochlorite for 30 min (Fig. 4c).
 | ||
| Fig. 4 (a) Luminescence and PLIM images of living HeLa cells incubated with P1 (200 μg mL−1, 37 °C, 1 h) followed by incubation with NaClO at different concentrations for 30 min. (b) Lifetime distributions in the PLIM images. (c) The occurrence ratio of the short-lived signals over the long-lived signals in the PLIM images. The “star” indicates the position for the imaging of endogenously produced hypochlorite in the fitted line. (d) Luminescence and PLIM images of living HeLa cells stimulated with elesclomol (125 nM, 37 °C, 2 h) followed by incubation with P1 (200 μg mL−1, 37 °C, 1 h). (e) Lifetime distributions in the PLIM image of endogenously produced hypochlorite. Scale bar: 20 μm. | ||
The utilization of P1 for analysis of intracellular oxygen content was then demonstrated. HeLa cells were preloaded with P1 (200 μg mL−1, 37 °C, 1 h), and then cultured under 2% O2 atmosphere for 30 min. Although luminescence images of the cells did not show remarkable changes, the luminescence lifetimes were extended from 450 ns to 480 ns (Fig. 5a and b). The oxygen content in the culture atmosphere was then increased to 95%. The intracellular luminescence became very dark owing to the quenching of phosphorescence. Clear image was obtained by turning up the laser power and increasing the slit width (Fig. 5a). PLIM analysis showed that the luminescence lifetimes were shortened to about 390 ns (Fig. 5a and b). The less occurrence indicated that the phosphorescence was quenched by 66%. After that, hypochlorite (5 μM) was added to the medium and the cells were further cultured under ambient condition (37 °C, air) for 30 min. As expected, the phosphorescence lifetime was restored to about 450 ns and additional lifetime distribution at about 150 ns appeared (Fig. 5c and d). The occurrence ratio of the short-lived signals over the long-lived signals was about 0.53, which was in line with the linear relationship in Fig. 4c. Further changing the oxygen content in the culture atmosphere to 2% or 95% O2 caused the corresponding phosphorescence response without interfering with the short-lived signals <300 ns (Fig. 5c and d). These results showed that the single-wavelength dual-emissive polymeric probe P1 can be used for simultaneously and independently imaging and analyzing multiple analytes in living cells via time-resolved photoluminescence analysis.
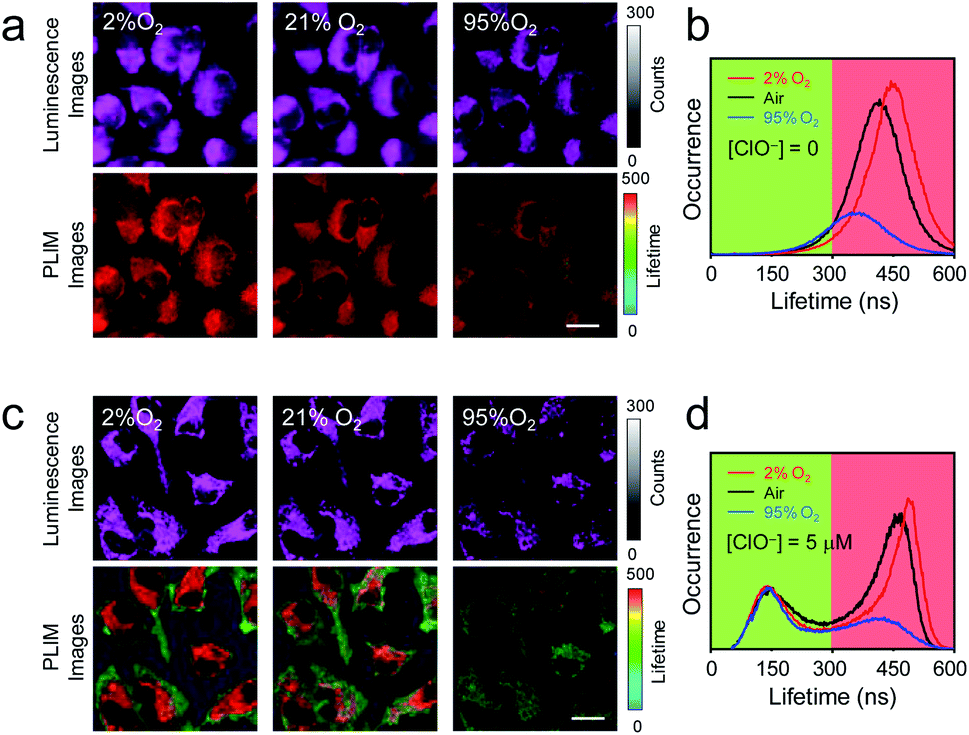 | ||
| Fig. 5 (a) Luminescence and PLIM images of living HeLa cells incubated with P1 (200 μg mL−1, 37 °C, 1 h) and then cultured under 2%, air, 95% oxygen conditions for 30 min. (b) Lifetime distributions in the PLIM images in (a). (c) Luminescence and PLIM images of living HeLa cells incubated with P1 (200 μg mL−1, 37 °C, 1 h), NaClO (5 μM, 37 °C, 30 min), and then cultured under 2%, air, 95% oxygen conditions for 30 min. (d) Lifetime distributions in the PLIM images in (c). Scale bar: 20 μm. | ||
Conclusions
Simultaneously analyzing signals of multiple luminescent probes effectively improves the accuracy and efficiency of luminescence biological detection and disease diagnosis. During signal processing, it is particularly important to avoid mutual interference of the signals from different probes. In the traditional method, identification of luminescence signals is usually based on their different wavelengths. In this work, we reported a novel method where identification of luminescence signals is based on their different decay rates. In the wavelength-resolved luminescence processing, luminescence intensity at different wavelengths is analyzed independently. The wavelength range covers the visible region and extends to the near-infrared region, usually being 400–900 nm with λmax/λmin to be 2.25. However, it is difficult to avoid spectral overlap and possible energy transfer between probes. Additionally, the difference in the penetration depth of light at different wavelengths in biological samples causes their detection efficiency to be different. In the time-resolved luminescence analysis, probes are allowed to emit at the same wavelength but different excited-state decay rates are required. The luminescence with different lifetimes is analyzed independently. We selected a phenothiazine-based fluorescent compound and a phosphorescent iridium(III) complex to construct a single-wavelength dual-emissive polymeric probe. The two luminophores emitted at almost the same wavelength but their luminescence lifetimes are 160 times different. The fluorescence was significantly enhanced upon selectively oxidation by hypochlorite and the phosphorescence was efficiently quenched by molecular oxygen. The luminescence spectrum gave the sum of fluorescence and phosphorescence, and analysis of the biexponential decay rates showed their proportions in the total spectrum. A four-dimensional relationship between luminescence intensity, fluorescence/phosphorescence ratio, hypochlorite concentration, and oxygen content was established, which allowed simultaneous quantitative determination of hypochlorite concentration and oxygen content by measuring the luminescence intensity and the fluorescence/phosphorescence ratio at a single wavelength. Owing to the excellent water solubility and biocompatibility, the single-wavelength dual-emissive polymeric probe was used for living cell imaging. Under ambient conditions, the phosphorescence remained unchanged. Time-resolved ratiometric sensing and imaging of intracellular hypochlorite was demonstrated using the phosphorescence signal as an internal standard. When the intracellular oxygen content changed, time-resolved photoluminescence analysis clearly showed the independent fluorescence response toward hypochlorite and phosphorescence response toward oxygen in separated time intervals. Using time-resolved photoluminescence imaging to analyze three or more analytes is in progress in our laboratory. The luminescence lifetimes of available probes varied from nanoseconds of fluorescent dyes to microseconds of transition metal complexes, to milliseconds of lanthanide chelates, to seconds of afterglow probes, giving τmax/τmin to be 109. The probes are designed to have the same emission wavelength in the red or near-infrared region to ensure the deep tissue penetration and the same photon detection efficiency.Author contributions
K. Y. Z., Q. Z. and W. H. devised the method. Y. W., J. Z., M. L. and S. L. contributed the initial implementation. K. Y. Z., Q. W., and P. D. designed, supervised, performed and analyzed the experiments. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank National Funds for Distinguished Young Scientists (61825503), National Natural Science Foundation of China (61975085) and Natural Science Foundation of Jiangsu Province of China (BK20190088) for financial support. Q. W. acknowledges the receipt of Postgraduate Research & Practice Innovation Program of Jiangsu Province (46030CX18036). K. Y. Z. acknowledges the receipt of Qing Lan Project of Jiangsu Province and 1311 Project of Nanjing University of Posts and Telecommunications.Notes and references
- N. Zhao, K. Kamijo, P. D. Fox, H. Oda, T. Morisaki, Y. Sato, H. Kimura and T. J. Stasevich, Nat. Commun., 2019, 10, 2947 CrossRef PubMed.
- Y. Li, H. Song, C. Xue, Z. Fang, L. Xiong and H. Xie, Chem. Sci., 2020, 11, 5889–5894 RSC.
- X. Wu, H. Li, E. Lee and J. Yoon, Chem, 2020, 6, 2893–2901 CAS.
- J. Wu, S. Zaccara, D. Khuperkar, H. Kim, M. E. Tanenbaum and S. R. Jaffrey, Nat. Methods, 2019, 16, 862–865 CrossRef CAS PubMed.
- E. K. Grant, D. J. Fallon, H. C. Eberl, K. G. M. Fantom, F. Z. C. Messenger, N. C. O. Tomkinson and J. T. Bush, Angew. Chem., Int. Ed., 2019, 58, 17322–17327 CrossRef CAS PubMed.
- Y. Ma, Q. Chen, X. Pan and J. Zhang, Top. Curr. Chem., 2021, 379, 10 CrossRef CAS PubMed.
- Q. Wu, K. Y. Zhang, P. Dai, H. Zhu, Y. Wang, L. Song, L. Wang, S. Liu, Q. Zhao and W. Huang, J. Am. Chem. Soc., 2020, 142, 1057–1064 CrossRef CAS PubMed.
- S. Li, X. Ling, Y. Lin, A. Qin, M. Gao and B. Z. Tang, Chem. Sci., 2018, 9, 5730–5735 RSC.
- T. Tamura, A. Fujisawa, M. Tsuchiya, Y. Shen, K. Nagao, S. Kawano, Y. Tamura, T. Endo, M. Umeda and I. Hamachi, Nat. Chem. Biol., 2020, 16, 1361–1367 CrossRef CAS PubMed.
- H. Zhu, J. Fan, J. Du and X. Peng, Acc. Chem. Res., 2016, 49, 2115–2126 CrossRef CAS PubMed.
- M. Y. Chin, J. A. Espinosa, G. Pohan, S. Markossian and M. R. Arkin, Cell Chem. Biol., 2021, 28, 320–337 CrossRef CAS PubMed.
- S. A. Jones, S.-H. Shim, J. He and X. Zhuang, Nat. Methods, 2011, 8, 499–505 CrossRef CAS PubMed.
- Y.-A. Lee, J.-J. Kim, J. Lee, J. H. J. Lee, S. Sahu, H.-Y. Kwon, S.-J. Park, S.-Y. Jang, J.-S. Lee, Z. Wang, W. L. Tam, B. Lim, N.-Y. Kang and Y.-T. Chang, Angew. Chem., Int. Ed., 2018, 57, 2851–2854 CrossRef CAS PubMed.
- A. Morozumi, M. Kamiya, S.-N. Uno, K. Umezawa, R. Kojima, T. Yoshihara, S. Tobita and Y. Urano, J. Am. Chem. Soc., 2020, 142, 9625–9633 CAS.
- Y. Zhang, K.-H. Song, S. Tang, L. Ravelo, J. Cusido, C. Sun, H. F. Zhang and F. M. Raymo, J. Am. Chem. Soc., 2018, 140, 12741–12745 CrossRef CAS PubMed.
- H. Xiao, P. Li and B. Tang, Chem.–Eur. J., 2021, 27, 1–20 CrossRef.
- Y. Yang, Y. Hu, W. Shi and H. Ma, Chem. Sci., 2020, 11, 12802–12806 RSC.
- H. Wan, J. Yue, S. Zhu, T. Uno, X. Zhang, Q. Yang, K. Yu, G. Hong, J. Wang and L. Li, Nat. Commun., 2018, 9, 1171 CrossRef PubMed.
- E. A. Owens, M. Henary, G. E. Fakhri and H. S. Choi, Acc. Chem. Res., 2016, 49, 1731–1740 CrossRef CAS PubMed.
- C. Li, G. Chen, Y. Zhang, F. Wu and Q. Wang, J. Am. Chem. Soc., 2020, 142, 14789–14804 CrossRef CAS PubMed.
- S. Zhu, S. Herraiz, J. Yue, M. Zhang, H. Wan, Q. Yang, Z. Ma, Y. Wang, J. He and A. L. Antaris, Adv. Mater., 2018, 30, 1705799 CrossRef.
- J. Huang and K. Pu, Chem. Sci., 2021, 12, 3379–3392 RSC.
- W. Xu, Z. Zeng, J.-H. Jiang, Y.-T. Chang and L. Yuan, Angew. Chem., Int. Ed., 2016, 55, 13658–13699 CrossRef CAS PubMed.
- G. Jiang, Y. Jin, M. Li, H. Wang, M. Xiong, W. Zeng, H. Yuan, C. Liu, Z. Ren and C. Liu, Anal. Chem., 2020, 92, 10342–10349 CrossRef CAS PubMed.
- K. Ren, R. Wu, A. P. K. K. K. Mudiyanselage, Q. Yu, B. Zhao, Y. Xie, Y. Bagheri, Q. Tian and M. You, J. Am. Chem. Soc., 2020, 142, 2968–2974 CrossRef CAS PubMed.
- S. Xu, H.-W. Liu, X. Yin, L. Yuan, S.-Y. Huan and X.-B. Zhang, Chem. Sci., 2019, 10, 320–325 RSC.
- W. An, L. S. Ryan, A. G. Reeves, K. J. Bruemmer, L. Mouhaffel, J. L. Gerberich, A. Winters, R. P. Mason and A. R. Lippert, Angew. Chem., Int. Ed., 2019, 58, 1361–1365 CrossRef CAS.
- S. Ye, H. Zhang, J. Fei, C. H. Wolstenholme and X. Zhang, Angew. Chem., Int. Ed., 2021, 60, 1339–1346 CrossRef CAS.
- J. Zhang, X. Chai, X.-P. He, H.-J. Kim, J. Yoon and H. Tian, Chem. Soc. Rev., 2019, 48, 683–722 RSC.
- Z. Chen, K. Y. Zhang, X. Tong, Y. Liu, C. Hu, Q. Yu and Q. Zhao, Adv. Funct. Mater., 2016, 29, 4386–4396 CrossRef.
- K. Y. Zhang, J. Zhang, Y. Liu, S. Liu, P. Zhang, Q. Zhao, Y. Tang and W. Huang, Chem. Sci., 2015, 6, 301–307 RSC.
- S. J. Park, Y. J. Kim, J. S. Kang, I. Y. Kim, K. S. Choi and H. M. Kim, Anal. Chem., 2018, 90, 9465–9471 CrossRef CAS PubMed.
- D. Y. Zhang, M. Azrad, W. Demark-Wahnefried, C. J. Frederickson, S. J. Lippard and R. J. Radford, ACS Chem. Biol., 2015, 10, 385–389 CrossRef CAS PubMed.
- J. A. Robson, M. Kubánková, T. Bond, R. A. Hendley, A. J. P. White, M. K. Kuimova and J. D. E. T. Wilton-Ely, Angew. Chem., Int. Ed., 2020, 59, 21431–21435 CrossRef CAS.
- M. Ren, Z. Li, B. Deng, L. Wang and W. Lin, Anal. Chem., 2019, 91, 2932–2938 CrossRef CAS PubMed.
- H. Li, W. Shi, X. Li, Y. Hu, Y. Fang and H. Ma, J. Am. Chem. Soc., 2019, 141, 18301–18307 CrossRef CAS PubMed.
- N. Li, C. Chang, W. Pan and B. Tang, Angew. Chem., Int. Ed., 2012, 23, 7426–7430 CrossRef.
- K. Y. Zhang, P. Gao, G. Sun, T. Zhang, X. Li, S. Liu, Q. Zhao, K. K.-W. Lo and W. Huang, J. Am. Chem. Soc., 2018, 140, 7827–7834 CrossRef CAS.
- J. Vuojola, U. Lamminmäki and T. Soukka, Anal. Chem., 2019, 81, 5033–5038 CrossRef PubMed.
- F. D. Maiolo and A. Painelli, J. Chem. Theory Comput., 2018, 14, 5339–5349 CrossRef PubMed.
- R. A. Weissleder, Nat. Biotechnol., 2001, 19, 316–317 CrossRef CAS.
- K. Y. Zhang, Q. Yu, H. Wei, S. Liu, Q. Zhao and W. Huang, Chem. Rev., 2018, 118, 1770–1839 CrossRef CAS.
- Y. Nosaka and A. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS.
- N. Hananya, O. Green, R. Blau, R. Satchi-Fainaro and D. Shabat, Angew. Chem., Int. Ed., 2017, 56, 11793–11796 CrossRef CAS PubMed.
- W. R. Wilson and M. P. Hay, Nat. Rev. Cancer, 2011, 11, 393–410 CrossRef CAS PubMed.
- B. Zhu, P. Li, W. Shu, X. Wang, C. Liu, Y. Wang, Z. Wang, Y. Wang and B. Tang, Anal. Chem., 2016, 88, 12532–12538 CrossRef CAS PubMed.
- W. Zhang, W. Liu, P. LI, J. Kang, J. Wang, H. Wang and B. Tang, Chem. Commun., 2015, 51, 10150–10153 RSC.
- H. Xiao, K. Xin, H. Dou, G. Yin, Y. Quan and R. Wang, Chem. Commun., 2015, 51, 1442–1445 RSC.
- X.-D. Wang and O. S. Wolfbeis, Chem. Soc. Rev., 2014, 43, 3666–3761 RSC.
- X. Pei, Y. Pan, L. Zhang and Y. Lv, Appl. Spectrosc. Rev., 2021, 56, 324–345 CrossRef CAS.
- K. Y. Zhang, T. Zhang, H. Wei, Q. Wu, S. Liu, Q. Zhao and W. Huang, Chem. Sci., 2018, 9, 7236–7240 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02811a |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2021 |