
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic supramolecular self-assembly of platinum(II) complexes perturbs an autophagy–lysosomal system and triggers cancer cell death†
Ka-Chung
Tong
ab,
Pui-Ki
Wan
ab,
Chun-Nam
Lok
 ab and
Chi-Ming
Che
*ab
ab and
Chi-Ming
Che
*ab
aState Key Laboratory of Synthetic Chemistry, Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, China. E-mail: cmche@hku.hk
bLaboratory for Synthetic Chemistry and Chemical Biology Limited, Units 1503-1511, 15/F., Building 17W, Hong Kong Science Park, New Territories, Hong Kong, China
First published on 29th September 2021
Abstract
Self-assembly of platinum(II) complexes to form supramolecular structures/nanostructures due to intermolecular ligand π–π stacking and metal–ligand dispersive interactions is widely used to develop functional molecular materials, but the application of such non-covalent molecular interactions has scarcely been explored in medical science. Herein is described the unprecedented biological properties of platinum(II) complexes relevant to induction of cancer cell death via manifesting such intermolecular interactions. With conjugation of a glucose moiety to the planar platinum(II) terpyridyl scaffold, the water-soluble complex [Pt(tpy)(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CArOGlu)](CF3SO3) (1a, tpy = 2,2′:6′,2′′-terpyridine, Glu = glucose) is able to self-assemble into about 100 nm nanoparticles in physiological medium, be taken up by lung cancer cells via energy-dependent endocytosis, and eventually transform into other superstructures distributed in endosomal/lysosomal and mitochondrial compartments apparently following cleavage of the glycosidic linkage. Accompanying the formation of platinum-containing superstructures are increased autophagic vacuole formation, lysosomal membrane permeabilization, and mitochondrial membrane depolarization, as well as anti-tumor activity of 1a in a mouse xenograft model. These findings highlight the dynamic, multi-stage extracellular and intracellular supramolecular self-assembly of planar platinum(II) complexes driven by modular intermolecular interactions with potential anti-cancer application.
CArOGlu)](CF3SO3) (1a, tpy = 2,2′:6′,2′′-terpyridine, Glu = glucose) is able to self-assemble into about 100 nm nanoparticles in physiological medium, be taken up by lung cancer cells via energy-dependent endocytosis, and eventually transform into other superstructures distributed in endosomal/lysosomal and mitochondrial compartments apparently following cleavage of the glycosidic linkage. Accompanying the formation of platinum-containing superstructures are increased autophagic vacuole formation, lysosomal membrane permeabilization, and mitochondrial membrane depolarization, as well as anti-tumor activity of 1a in a mouse xenograft model. These findings highlight the dynamic, multi-stage extracellular and intracellular supramolecular self-assembly of planar platinum(II) complexes driven by modular intermolecular interactions with potential anti-cancer application.
Introduction
The [Pt(terpy)Cl]+ complex has a unique position in coordination chemistry. It is the first metallointercalator manifesting an intercalative binding interaction between the base pairs of DNA.1,2 This complex has a low aqueous solubility, undergoes hydrolysis of the chloride ligand and reacts with thiolate to give [Pt(tpy)(SR)]+ which was reported to exhibit inhibitory activity on thioredoxin reductase3 and anti-leukemia activity.4 The underlying anti-cancer mechanisms of action of PtII terpyridine complexes, however, remain to be explored.For decades, the platinum(II) terpyridine scaffold has been extensively used in the design of functional materials through supramolecular self-assembly processes.5–9 The ordered molecular architectures formed from PtII terpyridine complexes are orchestrated by intermolecular dispersive PtII⋯ligand and ligand⋯ligand π–π stacking interactions.10–12 By manipulating the dynamic assembly/disassembly processes under cellular conditions,13–16 pincer platinum(II) complexes have been reported as bio-imaging probes for organelle-specific labeling. Nonetheless, the therapeutic potential of such intermolecular self-assembly reactions and the as-assembled superstructures in cellulo are scarcely studied. On the other hand, there are examples of organic-based self-assembled peptides17–22 and aromatic carbohydrate amphiphiles23 found to induce cancer cell death upon enzyme-triggered supramolecular self-assembly.
Coordination of the σ-donating acetylide ligand confers a good stability to the platinum(II) terpyridine complex against ligand substitution reactions.24,25 Taking advantage of the biocompatibility and hydrophilicity of the glucose unit, functionalization of the platinum(II) terpyridine complex with a glycosylated arylacetylide ligand could lead to a stable amphiphilic PtII complex for molecular assembly and at the same time enable the biological activity of the PtII complex to be activated through enzymatic deglycosylation.26 Herein is described the supramolecular assembly of platinum(II) terpyridyl acetylide complexes [Pt(tpy)(CCAr)]+ (tpy = 2,2′:6′,2′′-terpyridine or 4,4′,4′′-tri-tert-butyl-2,2′:6′,2′′-terpyridine, Ar = C6H4-R; Fig. 1) with one of these complexes, 1a, found to induce autophagic vacuole formation and lysosomal cell death. Complex 1a was found to undergo self-assembly to form nanoparticles, display transformable superstructures upon enzymatic action in cellulo and exhibit selective cytotoxicity towards cancer cells relative to normal fibroblasts. The accumulation of platinum-containing superstructures in endosomes/lysosomes and mitochondria consequently leads to disruption of lysosomal integrity, perturbation of autophagy and cancer cell death.
 | ||
| Fig. 1 Chemical structures of the platinum(II) terpyridyl acetylide complexes. | ||
Results
Synthesis, characterization and binding studies
The platinum(II) complexes (1a–1d and 2; Fig. 1) were synthesized by reacting [Pt(tpy)Cl](CF3SO3) (where tpy = terpyridine or tert-butyl substituted terpyridine (tBu3tpy)) with the corresponding HCCAr arylacetylide ligands through a ligand exchange reaction.27 Experimental procedures and characterization data (NMR spectroscopic data, high-resolution ESI-MS and elemental analyses) of the complexes are detailed in the ESI (Fig. S1–S10†). All of the PtII complexes are air-stable in the solid state under ambient conditions over a month. Complex 1a bearing a glucose moiety is water-soluble with solubility of 8.6 g L−1. It is stable in aqueous buffer solution containing glutathione, against hydrolysis in physiological buffer and in RPMI 1640 culture medium for 24 h, as revealed by UPLC/MS analysis (Fig. S11–S13†).
The UV-vis absorption data and spectra of the [Pt(tpy)(CCAr)](CF3SO3) complexes are summarized in Fig. S14 and Table S1.† In DMSO, complexes 1a–1d and 2 show intense absorption bands at 260–360 nm (ε = 34.8–46.6 × 103 mol−1 dm3 cm−1) and a broad absorption band at λ > 370 nm (ε = 2.9–4.9 × 103 mol−1 dm3 cm−1). The lowest-energy absorption band for 1d (λmax = 488 nm) with a hydroxyl substituent, and those of the glycosylated complexes 1a (λmax = 457 nm; glucose) and 1b (λmax = 458 nm; peracetylated glucose) all display a marked red-shift of 20–50 nm relative to 1c (λmax = 438 nm).28
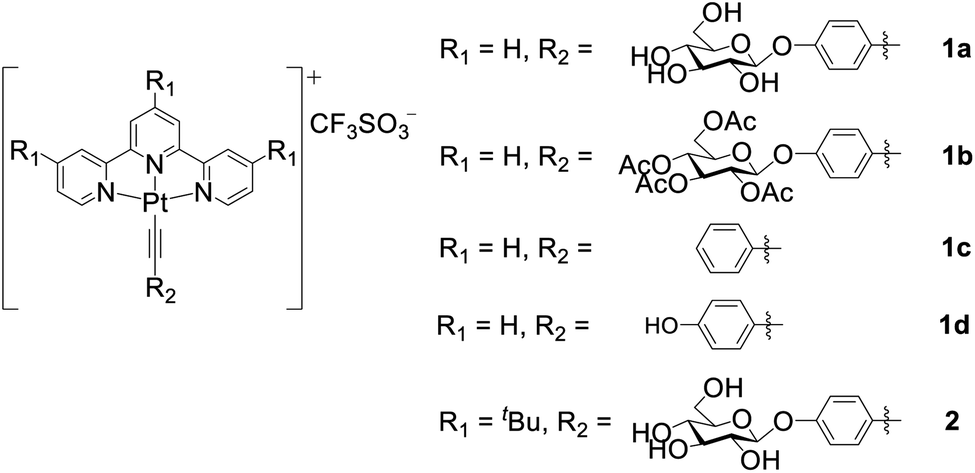
The binding of the PtII complexes with DNA was studied by UV-vis absorption spectroscopy. The absorption spectrum of 1a displays significant hypochromism (16.8%) and a bathochromic shift of the lowest-energy absorption band (λmax = 445 nm) by 42 nm upon the addition of calf thymus DNA (ctDNA) (Fig. 2a). The tBu3tpy PtII analogue 2 exhibits minor hypochromism (6.4%) with a red shift of only 8 nm at λmax of 426 nm (Fig. 2b). In the case of the telomeric G-quadruplex (HTelo) DNA (Fig. S15†), 1a exhibits similar hypochromism (19.6%) at λmax of 345 nm with red-shifted absorption bands (400–600 nm). These spectral changes together with the isosbestic points for 1a can be attributed to the intercalation of the unsubstituted PtII terpyridyl motif between the DNA base pairs or guanine tetrad.
 | ||
| Fig. 2 Plots of the UV-vis absorption titrations of complex (a) 1a or (b) 2 with increasing concentration of ctDNA. ITC trace and binding curve of the binding of (c) 1a and (d) 2 to ctDNA. Fits were obtained using a one-site binding model. | ||
The DNA-binding affinity of the PtII terpyridyl complexes was determined by isothermal titration calorimetry (ITC). From the binding studies with ctDNA, glycosylated 1a with an unsubstituted tpy ligand (KD: 0.48 μM) displays a 27.5-fold lower equilibrium dissociation constant (KD) than 2 (KD: 13.2 μM) coordinating with bulky tBu3tpy (Fig. 2c and d). Intriguingly, 1a demonstrates 4.3-fold higher binding affinity (KD: 19.6 μM) toward the DNA duplex bearing cytosine–cytosine (CC) mismatched base pairs compared with 2 (KD: 83.3 μM) (Fig. S16†). Complex 1d containing hydroxyl-substituted arylacetylide shows 3.7- and 2.5-fold lower dissociation constants for calf thymus (KD: 0.13 μM) and guanine–cytosine (GC)-rich (KD: 29.3 μM) DNAs compared with 1a (GC-rich DNA, KD: 74.1 μM; Fig. S17†). Collectively, these findings reveal that the planar scaffold of platinum(II) terpyridyl arylacetylide with less steric hindrance contributes to the stronger DNA binding affinity. Fluorescence quenching titration of human serum albumin (HSA) with increasing concentrations of the PtII complexes 1a or 1d revealed the decrease in fluorescence of tryptophan residues with the binding constants determined to be 23.4 and 18.3 μM (Fig. S18†), respectively.
Self-assembly properties and conversion of superstructures induced by β-glucosidase in aqueous buffer
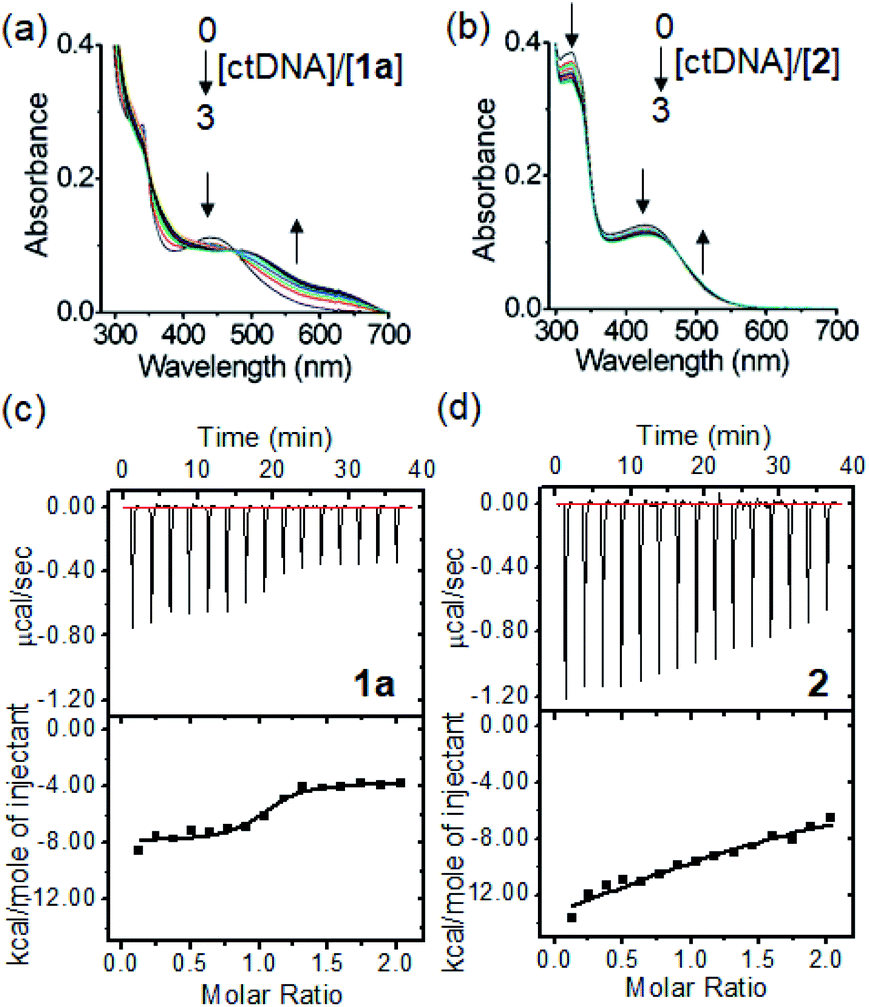
The morphologies of the PtII complexes in aqueous solutions containing 1% DMSO were examined by transmission electron microscopy (TEM) (Fig. 3a). Complex 1a displayed spherical-shaped nanoparticles with an average hydrodynamic diameter of 139.6 ± 2.5 nm (Fig. 3c(i)). In stark contrast, irregular networks of fibrous structures with diameters of 10–60 nm and length spanning over 10 μm were formed for 1b. Complex 1c, without a para-substituent on the arylacetylide ligand, displayed regular-shaped microwires with diameters of approximately 140–170 nm. For 1d, wire-like structures with branched and curved morphologies (with a length of approximately 8–30 μm and a width of 150–200 nm) were observed. A selected area electron diffraction pattern of the wire-like structure showed a d spacing of 3.32 Å (Fig. S19†), suggesting the presence of intermolecular Pt⋯Pt interaction between the platinum(II) complex molecules.10 Compared to 1a, 2 with a bulky tBu3tpy ligand showed non-uniform microparticle-like aggregates. 1H NMR studies of 1a revealed that the proton peaks of 1a in deuterated DMSO are sharp and well-resolved (Fig. 3b). Upon increasing the D2O content, the aromatic proton peaks are broadened accompanied by a decrease in intensity and upfield shift, all of which are suggestive of the aggregation of the PtII complex driven by intermolecular interactions. The peak shape and intensity of protons on the glucose moiety (δ: 3.17–5.41 ppm), however, remained unaffected and the proton signals were slightly downfield shifted (Δδ 0.17–0.2 ppm). Nanoparticle tracking analysis showed that 1a formed nanoparticles with average hydrodynamic diameters of 105.6 ± 7.1 nm and 87.3 ± 4.6 nm in phosphate-buffered saline (Fig. S20†) and RPMI 1640 cell culture medium containing 10% fetal bovine serum (Fig. 3c(ii)), respectively.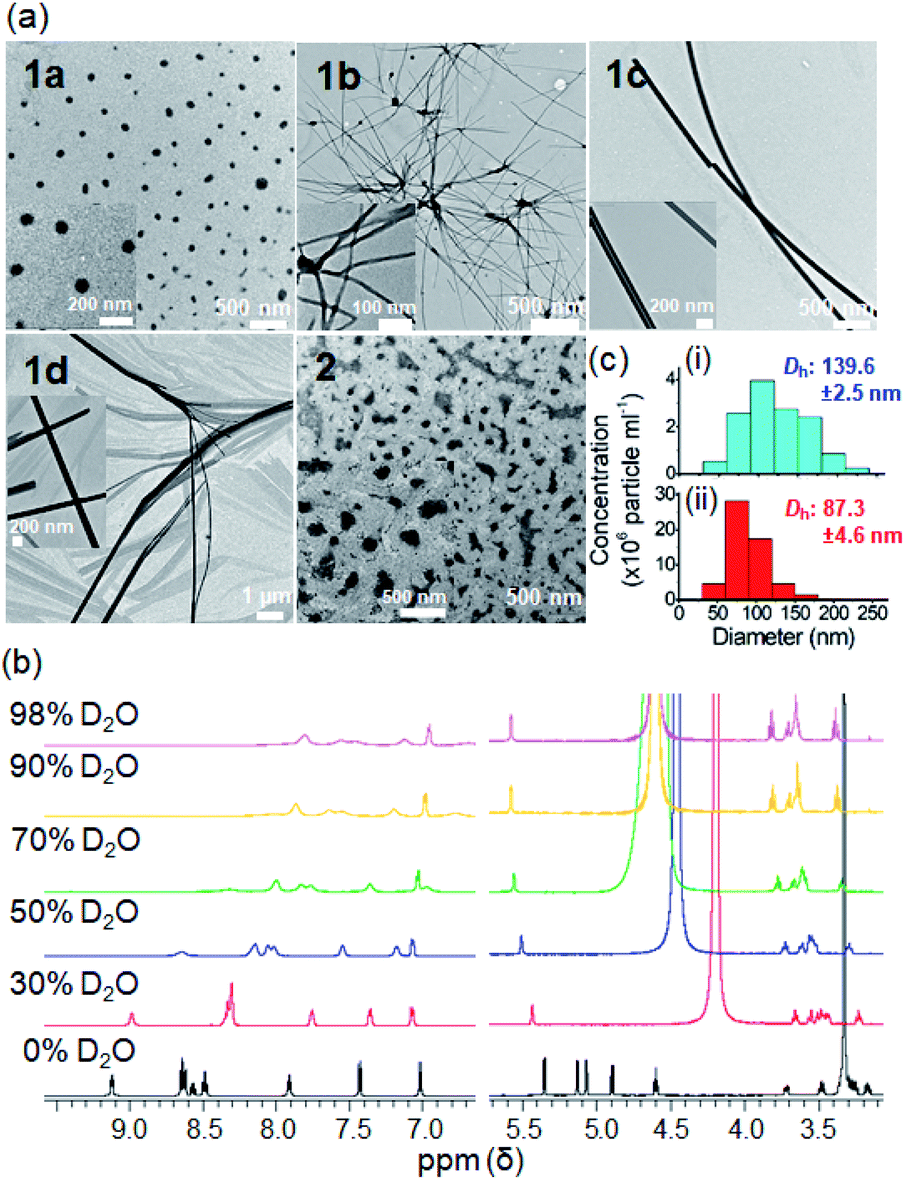 | ||
Fig. 3 (a) TEM images of complexes 1a–1d and 2 at 1 × 10−4 M in DMSO/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :99 v/v). (b) 1H NMR spectra of 1a (2 × 10−4 M) in DMSO-d6 with increasing D2O content. (c) Particle size distribution of nanoparticles formed by 1a in (i) aqueous solution containing 1% DMSO or (ii) RPMI 1640 culture medium containing 10% fetal bovine serum as determined by nanoparticle tracking analysis. :99 v/v). (b) 1H NMR spectra of 1a (2 × 10−4 M) in DMSO-d6 with increasing D2O content. (c) Particle size distribution of nanoparticles formed by 1a in (i) aqueous solution containing 1% DMSO or (ii) RPMI 1640 culture medium containing 10% fetal bovine serum as determined by nanoparticle tracking analysis. | ||
The self-assembly behavior of the platinum(II) terpyridyl acetylide complexes at different pH and upon enzymatic cleavage of the glycosidic linkage by β-glucosidase (β-glu) was studied. TEM images revealed that 1a formed nanoparticles with diameters of 150–180 nm in phosphate-buffered saline (PBS; pH 7.4) (Fig. 4a), the morphology of which was disrupted to give irregular-shaped aggregates under acidic conditions (pH 5.8). Following the incubation with β-glu (0.5 U mL−1) at 37 °C for 24 h, nanorod-like structures with a diameter of 0.2–0.5 μm were observed. Under the same experimental conditions, UPLC/MS analysis indicated that more than 70% of 1a (t = 2.48 min; m/z 707.143) was converted into 1d (t = 2.90 min; m/z 545.091) after incubation with β-glu for 24 h (Fig. 4b(iv) and c) compared with only ca. 2% conversion in the buffer control (Fig. 4b(iii)). These findings are indicative of the cleavage of the glycoside unit of 1a to give 1d by β-glu and which may result in different assembled molecular structures. A schematic illustration of the β-glu-mediated cleavage of the glucose from 1a is shown in Fig. 4d.
 | ||
| Fig. 4 Study of the glycosidic bond cleavage from the glycosylated platinum(II) complex. (a) TEM images of the as-assembled structures of 1a at different pH and/or with β-glu (0.5 U mL−1) after 24 h. (b) UPLC/MS analysis of 1a in different media for 24 h. (i) 1a in PBS (pH 7.4), (ii) 1d in PBS (pH 7.4), (iii) 1a in PBS (pH 5.8) and (iv) 1a with β-glu (0.5 U mL−1) in PBS (pH 5.8). (c) Time-dependent release of 1d in the absence or presence of β-glu. (d) Schematic illustration of the β-glucosidase-mediated cleavage of glucose in 1a. | ||
In vitro anti-cancer properties and cellular uptake
The anti-proliferation activity (half-maximal inhibitory concentration, IC50 values) of the platinum(II) complexes against various human cancerous and normal cell lines was examined by naphthol blue black (NBB) assay, with cisplatin as a reference. After 72 h treatment (Table 1), complexes 1a–1d displayed higher anti-proliferative activities (IC50: 1.5–48.4 μM) towards different cancer cell lines compared to normal lung fibroblast (CCD-19Lu; IC50: 36.8 to >100 μM). The anti-proliferative activity of complex 1a bearing the glucose moiety was slightly higher than that of 1b having peracetylated glucose by 1.4- to 2.4-fold in most of the cancer cell lines. Complex 1d, the aglycone of 1a, showed comparable inhibition of cancer cell growth to that of 1a and a moderate anti-proliferation effect on CCD-19Lu cells. The time-dependent cell growth inhibition of complexes 1a and 1b was evaluated (Fig. 5a and Table S2†). Both complexes were relatively inactive (IC50: >100 μM) toward different cell lines (NCI-H460 lung, PLC liver and HCT116 colorectal carcinomas) 24 h post-treatment. 1a (IC50: 54.6–74.5 μM) exhibited a slightly more inhibitory effect than 1b (IC50: 90.7–97.4 μM) after 48 h.| Complex | IC50 (μM) | |||||
|---|---|---|---|---|---|---|
| NCI-H460 | A2780 | PLC | HCT116 | MDA-MB-231 | CCD-19Lu | |
| a The anti-proliferative activity of the complexes was determined by NBB assay. NCI-H460 = non-small cell lung carcinoma; A2780 = ovarian carcinoma; PLC = primary liver carcinoma; HCT116 = colorectal carcinoma; MDA-MB-231 = triple-negative breast cancer carcinoma; CCD-19Lu = normal lung fibroblast. | ||||||
| 1a | 7.1 ± 0.3 | 8.7 ± 0.2 | 17.2 ± 1.2 | 7.5 ± 0.6 | 23.8 ± 5.8 | >100 |
| 1b | 17.0 ± 1.2 | 3.66 ± 0.3 | 24.2 ± 2.1 | 16.2 ± 1.3 | 48.4 ± 3.6 | >100 |
| 1c | 1.5 ± 0.1 | 4.6 ± 0.1 | 2.6 ± 0.2 | 3.3 ± 0.2 | 1.7 ± 0.3 | 36.8 ± 4.6 |
| 1d | 7.8 ± 0.1 | 8.9 ± 0.6 | 28.1 ± 3.3 | 10.1 ± 1.5 | 44.9 ± 6.2 | 65.9 ± 7.0 |
| Cisplatin | 6.9 ± 0.5 | 8.9 ± 0.7 | 9.1 ± 1.2 | 5.1 ± 0.4 | 19.7 ± 1.6 | 30.8 ± 4.9 |
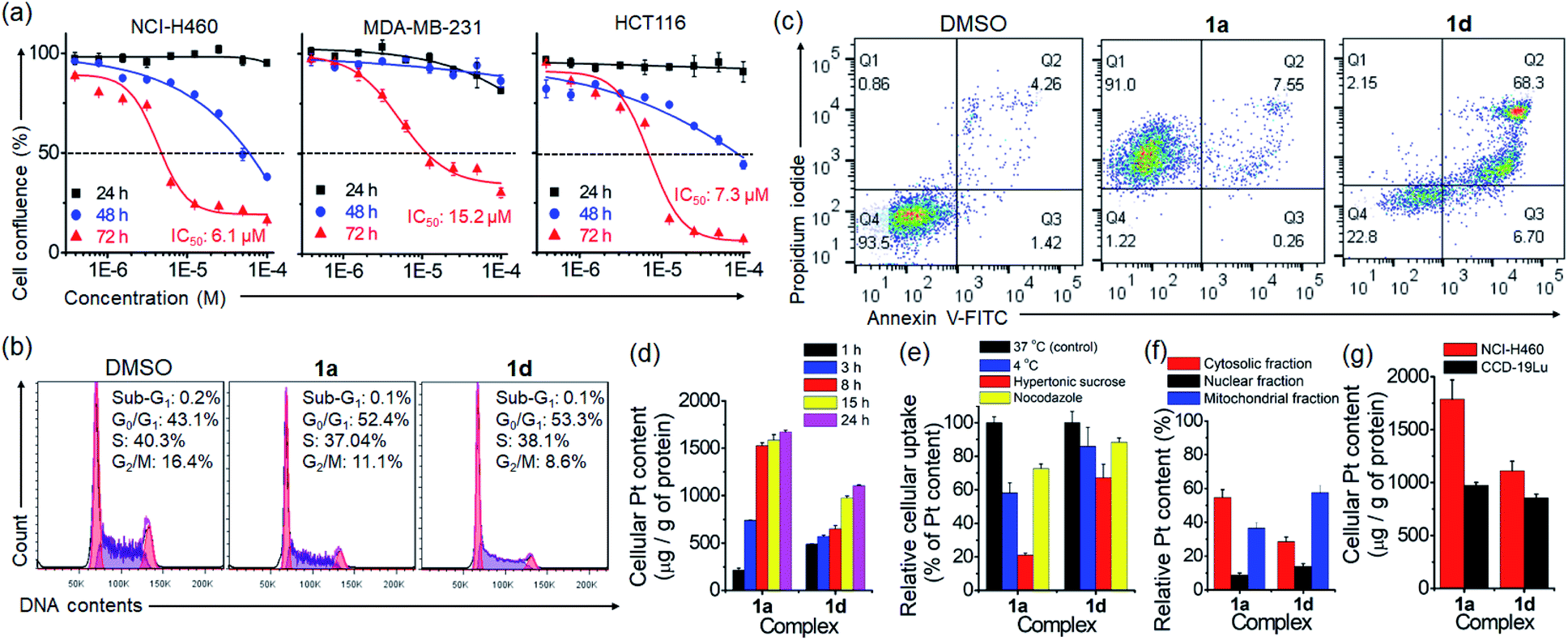 | ||
| Fig. 5 (a) Time-dependent anti-proliferative activity of 1a on human lung (NCI-H460), breast (MDA-MB-231) and colorectal (HCT116) cancer cells for 24, 48 and 72 h. Flow cytometric analyses of the (b) cell cycle progression (24 h) and (c) cell death (72 h) of NCI-H460 cells treated with 1a or 1d. (d) Time-dependent uptake of different platinum(II) complexes in NCI-H460 cells. (e) Energy- or endocytosis-dependence of the uptake and (f) subcellular distribution of platinum content of 1a and 1d in NCI-H460 cells. (g) Cellular platinum content in 1a- and 1d-treated NCI-H460 lung cancer and CCD-19Lu normal lung fibroblast cells. | ||
The effect of PtII terpyridyl acetylide complexes on the cell cycle progression of NCI-H460 lung cancer cells was examined by using flow cytometry. Treatment of cells with 1a or 1d for 24 h induced a slight increase in G0/G1 populations by 9.3% and 10.2%, respectively (Fig. 5b). In contrast, cisplatin-treated cells exhibited a 53.7% increase in G2/M populations (Fig. S21†). Prolonging the treatment time with 1a or 1d to 48 h caused no significant increase in cell populations compared with those at 24 h (Fig. S22†). The DNA damage response as the double-strand breaks was examined by immunofluorescence staining of phosphorylated histone protein (γ-H2AX) (Fig. S23†). In the DMSO vehicle group, γ-H2AX immunofluorescence was undetectable. Notably, the nuclei of 1a- or 1d-treated cells displayed some significant fluorescent γ-H2AX foci at 24 h post-treatment. The number and intensity of γ-H2AX foci were even more markedly increased in cells exposed to cisplatin, which was reported to activate the DNA damage response.29 Cell death examination by flow cytometry using annexin V/propidium iodide (PI) double staining indicated that a majority of 1a-treated NCI-H460 lung cancer cells displayed necrotic cell death (91%; annexin V-negative and PI-positive) after a 72 h treatment (Fig. 5c). However, cell death of 1d-treated NCI-H460 cells mainly present as late apoptosis (68.3%; annexin V-positive and PI-positive). A similar stage in late apoptosis (87.2%; annexin V-positive and PI-positive) was also found in cisplatin-treated cells under the same experimental conditions (Fig. S24†).
Cellular uptake and distribution of the platinum complexes in NCI-H460 cells were examined through determining the platinum content by using ICP-MS. As shown in Fig. 5d and S25,† the time-dependent uptake profile of different complexes revealed that the cellular Pt content increased progressively and became steady after 15 h. Notably, 1a treatment resulted in the highest intracellular Pt level after 24 h, exhibiting ca. 3.2-, 8- and 1.6-fold higher Pt levels than 1b, 1c and 1d (Fig. S25†). Among the compounds tested, 1a showed the lowest lipophilicity (logP) of −0.88 (Fig. S26†). A significant decrease in Pt levels in cells was observed upon incubation at 4 °C (77.1%) or upon inhibition of endocytosis by hypertonic sucrose (27.4%) or microtubule disrupting agent nocodazole (41.9%) (Fig. 5e), suggesting the involvement of energy-dependent endocytosis in the cellular uptake of glycosylated 1a. In the subcellular distribution study, most of the Pt content in 1a-treated cells was found in the cytosolic fraction (55%), and less in the mitochondrial (37%) and nuclear (9%) fractions (Fig. 5f). Meanwhile, for 1d treatment, the Pt content was found mostly in the mitochondrial fraction (58%), followed by the cytosolic (29%) and nuclear (14%) fractions. By comparing the uptake difference between cancer and normal cells, Pt accumulation in 1a- and 1d-treated NCI-H460 cancer cells was, respectively, 1.9- and 1.3-fold higher than that in CCD-19Lu normal fibroblast cells (Fig. 5g).
Aggregation of the platinum(II) complexes in cellulo
Given the self-assembling properties of platinum(II) terpyridyl complexes, we investigated whether these complexes could self-assemble into superstructures in live cells. With bio-TEM imaging, 1a-treated cancer cells (NCI-H460 lung and MDA-MB-231 breast cancer; Fig. 6a and S27†) showed electron-dense structures with the majority present as irregular-shaped nanofibrils in cytosolic vesicle-like structures, presumably the endosomes and/or lysosomes.30 A small portion of aggregates with size of 70–500 nm was also observed in the mitochondria-like structures (Fig. S28†). The presence of elemental platinum in these superstructures was confirmed by energy-dispersive X-ray (EDX) analysis (Fig. 6b, S27b and S28c†). Under the same incubation conditions, 1d-treated cells showed only platinum-containing aggregates in the mitochondria (Fig. 6c and d). It is worth noting that these superstructures, however, were hardly observed in 1a- or 1d-treated CCD-19Lu normal lung fibroblast cells (Fig. 6e). Based on UPLC/MS analysis of 1a-treated NCI-H460 lung cancer cells, 1d was the predominant species over 1a (20:1 in peak area; Fig. S29†), apparently derived from deglycosylation of 1a.
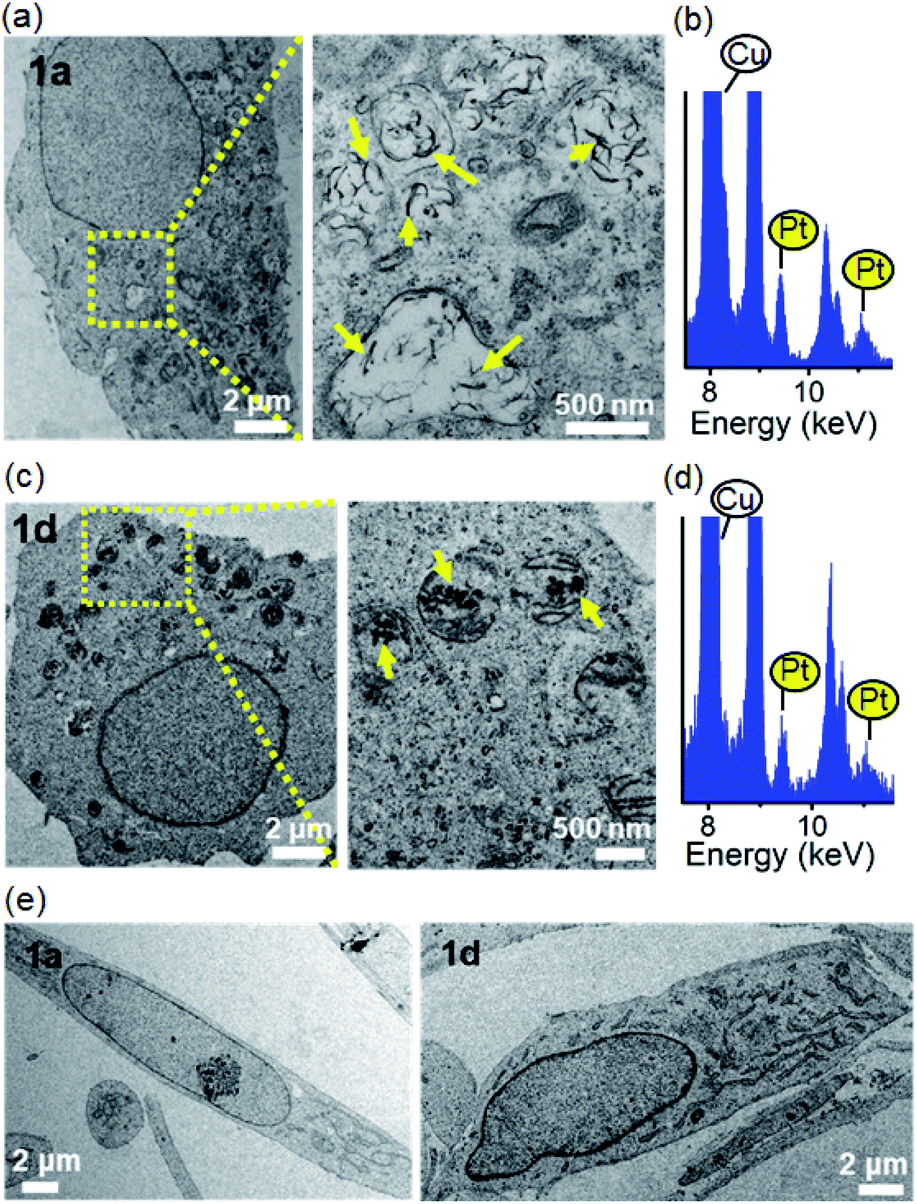 | ||
| Fig. 6 Bio-TEM images of (a and c) NCI-H460 lung cancer cells or (e) CCD-19Lu normal lung fibroblasts after incubation with 1a or 1d (10 μM) for 24 h. The yellow arrows in the magnified yellow box region highlight the electron-dense nanostructures inside the cell. (b and d) EDX spectra of the electron-dense superstructures in 1a- or 1d-treated cells (platinum signals are highlighted in yellow). | ||
Lysosomal membrane permeabilization, autophagy perturbation and mitochondrial dysfunction
We hypothesized that accumulation of nanostructures in the late endosomes or lysosomes may perturb the lysosomal structure and function. The integrity of the lysosomes in 1a-treated cells was examined by acridine orange (AO), LysoTracker and pepstatin A–BODIPY FL staining using confocal microscopy. AO is a lysosomotropic dye which emits red fluorescence inside the normal acidic compartment of lysosomes but shows green fluorescence when bound to DNA or in the cytosol. As depicted in Fig. 7a, distinct red fluorescent dots representing lysosomes were observed in vehicle- and 1d-treated cells. However, the red fluorescence of AO was markedly reduced in 1a-treated cells, indicative of the loss of acidic lysosomes. Staining of vehicle-treated cells with LysoTracker Deep Red, another pH-sensitive fluorescent probe, revealed a punctate red fluorescence pattern in the perinuclear region (Fig. 7b). However, 1a-treated cells showed significant diminishment of the red fluorescent puncta. In contrast, the mitochondrial mass and pattern revealed by MitoTracker Green staining were negligibly affected (Fig. 7b). These findings indicated that 1a treatment specifically resulted in disruption of lysosomes. Different from 1a, 1d-treated cells still showed lysosomal and mitochondrial fluorescence (Fig. 7a and b). When cells were co-stained with LysoTracker Deep Red and pepstatin A–BODIPY FL, a green fluorescent probe specific to the lysosomal proteases (cathepsins D and E), 1a treatment resulted in diffused green fluorescence in the cytosol with diminished red fluorescence in contrast with the clear vesicular (lysosomal), colocalized red and green fluorescent staining in vehicle- or 1d-treated cells (Fig. 7c). This result further indicates the disruption of lysosomes upon 1a treatment with leakage of lysosomal contents into the cytoplasm, a typical scenario linked to “lysosomal cell death”.31 However, NCI-H460 cells treated with cisplatin showed negligible damage to the cellular lysosomes as revealed by the comparable staining pattern of acridine orange, LysoTracker deep red and pepstatin A–BODIPY FL to the saline vehicle (Fig. S30†).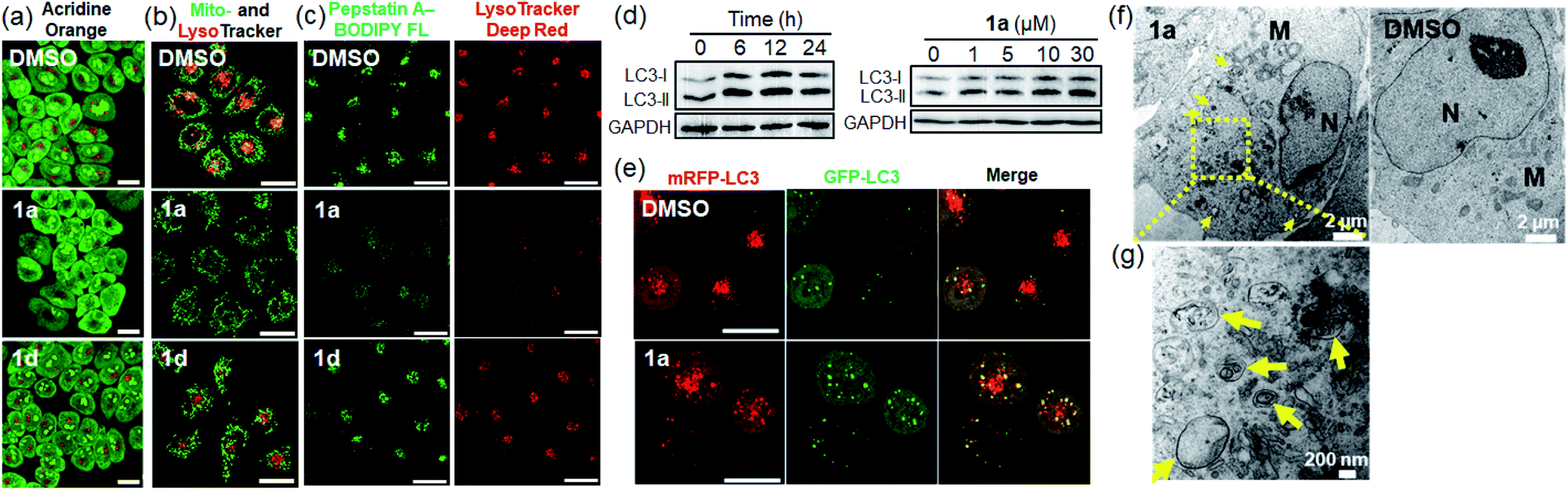 | ||
| Fig. 7 Lysosomal disruption and disruption of autophagic flux in NCI-H460 cells after 24 h-treatment with 1a or 1d (10 μM) as determined by (a) acridine orange (10 μM), (b) LysoTracker Deep Red and MitoTracker Green (50 nM), and (c) pepstatin A–BODIPY FL conjugate (1 μM) and LysoTracker Deep Red staining using confocal imaging microscopy. Scale bar = 20 μm. (d) Immunoblotting analysis of autophagy protein markers (LC3) in cells after 1a treatment in a time- (left) and dose- (right) dependent manner. (e) Confocal images of NCI-H460 cells stably expressing mRFP-GFP-LC3 following treatment with 1a for 24 h. Scale bar = 20 μm. (f) Bio-TEM images of the accumulation of autophagosomes in 1a-treated NCI-H460 cells at 24 h. (g) Magnified view of the yellow box region showing the formation of double-membraned autophagosomes (M: mitochondria; N: nucleus). | ||
The lysosome is central to cellular degradative activities via autophagy,32 and its impairment perturbs the autophagic process which is particularly active in cancer cells having a high rate of protein turnover for cell survival.33 Western blotting analysis of 1a-treated cells displayed increased levels of the lipidated form of autophagic vacuole protein marker LC3 (LC3-II) in a time- and dose-dependent manner (Fig. 7d). The 1a-treated cells also showed an increase in immunofluorescence staining of LC3 puncta (Fig. S31†), both of which are indicative of increased formation of autophagic vacuoles. Similarly, NCI-H460 cells treated with cisplatin also showed a dose-dependent increase in the level of LC3-II (Fig. S32†). The effect of 1a treatment on autophagy was monitored using NCI-H460 lung cancer cells stably transfected with mRFP-GFP tandem fluorescent protein tagged LC3 (tfLC3) as the autophagic vacuole localization marker.34 Cells exposed to 1a for 24 h resulted in increased green and red puncta representing LC3-labelled autophagic vacuoles (Fig. 7e). The increased number of colocalized green and red puncta (yellow puncta; Pearson's colocalization coefficient of 0.64) suggests the induced formation of the intermediate autophagic vacuoles (autophagosomes) without maturation into acidified autolysosomes. TEM examination of the ultrastructure and morphology of 1a-treated cells revealed a marked increase in the number of double-membraned cytosolic vesicles (Fig. 7f, g and S33†), consistent with the typical morphology of autophagosomes.35 An unbiased thermal proteome profiling experiment36,37 showed that in NCI-H460 lung cancer cells, a number of proteins directly involved in endosomal/lysosomal functions (e.g. Ras-related protein Rap-1b (RAP1B), palmitoyl-protein thioesterase 1 (PPT1), transferrin receptor protein 1 (TFRC) and protein numb homolog (NUMB)) were stabilized (ΔTm ≥ +1 °C) upon treatment with 1a compared with the vehicle control (Table S3†), the results of which are in line with the findings of cellular uptake of 1avia endocytosis (Fig. 5e), and perturbation of the endosomal–lysosomal and autophagy system by 1a treatment (Fig. 7).
In addition, the cellular mitochondria of 1a- and 1d-treated cells were found with partial swelling, distorted membrane and disordered cristae compared to those having an intact morphology in the vehicle control from TEM imaging (Fig. S28 and S34†). The changes in mitochondrial membrane potential (MMP) were determined using JC-1 staining.38 As revealed by flow cytometric analysis (Fig. S35†), cells treated with the vehicle control displayed red fluorescence with a majority of cells (90.4%) in the JC-1 aggregate form, indicating a normal mitochondrial polarization state. In contrast, both 1a- and 1d-treated cells displayed a moderate decrease in red fluorescence by ca. 21% indicating mitochondrial depolarization after 24 h. For cisplatin treatment, an only 8.3% decrease in red fluorescence of JC-1 relative to the saline vehicle was observed (Fig. S35†). Measurement of intracellular oxidant levels using a fluorogenic probe, DCFH-DA, revealed that compared with the vehicle control, 1a- and 1d-treated cells showed only minor increments (almost 1.2-fold) in DCF fluorescence intensity (Fig. S36†).
In vivo inhibition of tumor growth and increased autophagic vacuole formation in tumor xenografts
The in vivo anti-tumor efficacy of 1a was examined on a nude mouse model bearing NCI-H460 lung cancer xenografts. Owing to the limited aqueous solubility of 1d, only 1a was studied. The tumor-bearing mice were randomly divided into two groups (n = 6) and administered with 1a (25 mg kg−1) or PBS vehicle via intravenous (i.v.) injection for 14 days. As shown in Fig. 8a, c and d, the tumor volume and tumor weight of 1a-treated mice were reduced by 66% and 53% compared with those of the vehicle control group, respectively, after 14 days. Tumor-bearing mice receiving cisplatin (1.5 mg kg−1, i.v.) under a similar administration regimen were used as a positive control for comparison, and significant suppression of tumor growth with 90% and 88% reduction in the tumor volume and tumor weight, respectively, was found. None of the mice died or displayed any sign of weight loss throughout the treatment period (Fig. 8b). No obvious histopathological lesion was observed in the major organs (heart, liver, spleen, lungs and kidneys) of 1a- and PBS vehicle-treated mice after 14 days (Fig. 8e). Additionally, blood biochemistry analysis showed that the plasma levels of alanine aminotransferase (ALT) and creatinine (CREA) in 1a-treated mice were comparable to those in the vehicle control (Fig. 8f), in which ALT and CREA are liver and kidney damage biomarkers, respectively. All these results reveal the low systemic toxicity of 1a. Hematoxylin and eosin (H&E) stained tumor tissue sections from 1a-treated mice revealed a marked decrease in the nucleus-to-cytoplasm ratios and noticeable nuclear fragmentation (Fig. 8g). A significant reduction of immunohistochemical staining for the cell proliferation marker protein Ki-67 was also observed in the same group of tumor specimens. In addition, compared with the vehicle control (Fig. S37†), TEM analysis of 1a-treated tumors revealed a number of double- and multi-membrane bound vesicles containing nanofibril-like materials (Fig. 8h and S38†). These are attributable to the increased autophagic vacuole formation in tumor xenografts. | ||
| Fig. 8 In vivo anti-tumor effect of complex 1a (25 mg kg−1) and cisplatin (1.5 mg kg−1) in a nude mouse model bearing NCI-H460 lung cancer xenografts. (a) Tumor volume and (b) body weight of mice (n = 6). (c) Weight and (d) photograph of the dissected tumors harvested from mice after 14 days of treatment. Data are expressed as the mean ± standard error; **p < 0.01. (e) H&E stained images of major organs harvested from mice after 14 days of treatment; scale bar = 50 μm. (f) Blood biochemistry analysis of alanine aminotransferase (ALT; upper) and creatinine (lower) in different groups of mice after 14 days of treatment. (g) H&E (left) and Ki-67 antigen (right) stained images of tumor tissues from mice after 14 days of treatment; scale bar = 50 μm. The yellow arrows indicate the nuclear fragmentation as shown in the enlarged area. (h) Representative TEM images of the ultrastructure of NCI-H460 tumors harvested from 1a-treated mice. | ||
Discussion
Supramolecular self-assembly of amphiphiles into superstructures has generated considerable interest in biomedical science. Such molecular amphiphiles, as exemplified by the amphiphilic peptides or aromatic carbohydrates that assemble into fibrous superstructures upon biological triggers (e.g., pH variation or enzymatic action), impair the organelle-specific functions and hence impede the cancer cell proliferation.23,39,40 In the literature, nanostructures/particles made of iron oxide, gold, quantum dots or titanium dioxide have been exploited as potential therapeutics in modulating the autophagic process by impairing the functional pH gradient and membrane permeability of lysosomes through an endocytosis-mediated uptake mechanism.41 In this work, we demonstrated the use of supramolecular self-assembly of a platinum(II) complex, as exemplified by a glucose-appended platinum(II) terpyridyl arylacetylide, as an anti-cancer strategy to perturb the autophagy–lysosomal system and trigger cancer cell death (Fig. 9).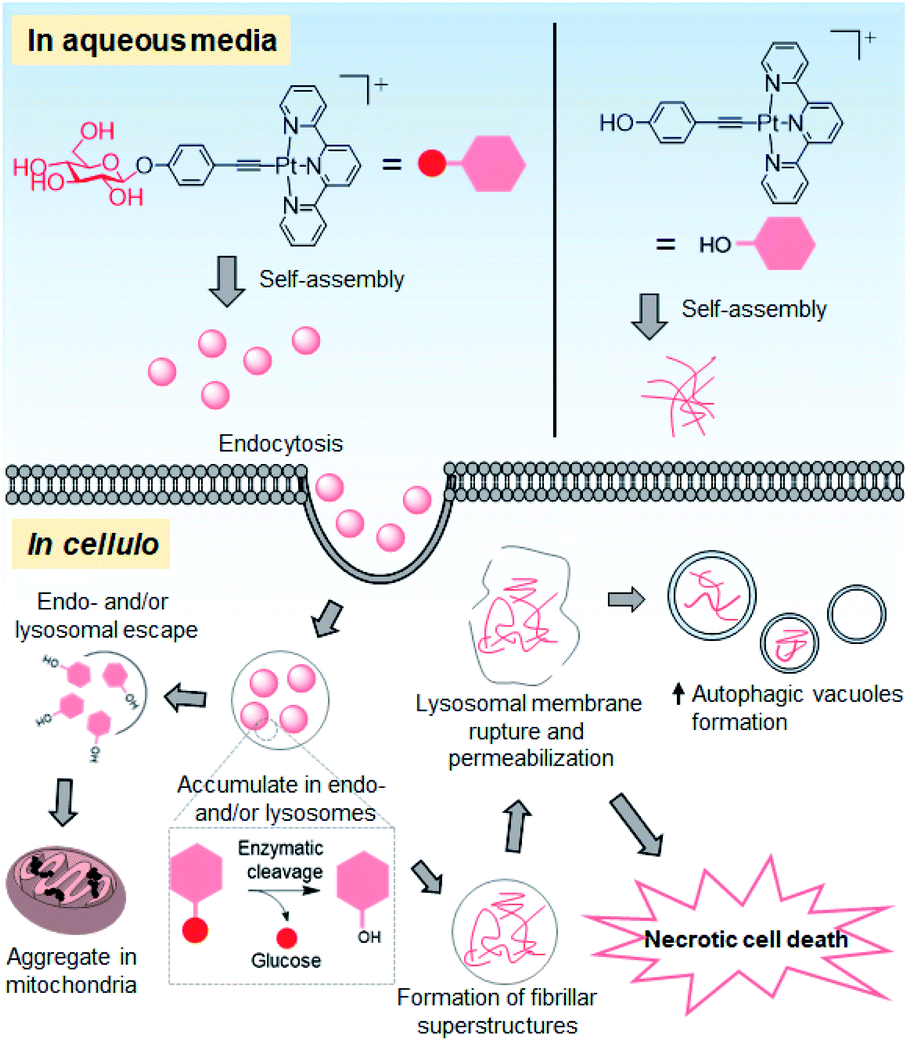 | ||
| Fig. 9 Schematic drawing of the proposed anti-cancer mechanisms of action of 1a. | ||
The planar PtII terpyridyl motif suggests an intercalative DNA binding interaction, in line with the pronounced hypo- and bathochromic spectral changes of 1a with respect to 2 that contains a tBu3tpy ligand in the UV-vis absorption titration with ctDNA (Fig. 2a and b). The stable structural scaffold of the platinum(II) terpyridyl complex governed by the strong σ-coordination of the alkynyl ligand could endow the complex to have a non-covalent intercalative interaction with DNA. Nonetheless, the majority of cellular Pt contents in the 1a (55%)- and 1d (58%)-treated NCI-H460 lung cancer cells was found to distribute in the cytosol and mitochondria, respectively, rather than in the nucleus (Fig. 5f). At the same molar concentrations, 1a and 1d elicited a much lower degree of DNA damage response (i.e. lower number of fluorescent γH2AX foci) compared with those treated with cisplatin (Fig. S23†) that can form a covalent platinum–DNA adduct.29 The fragmented nuclei observed in the tumor tissue of 1a-treated mice (Fig. 8g) could be attributed to apoptosis-associated DNA fragmentation42 that might contribute to the in vivo anti-tumor action of 1a, although the possibility of ligand dissociation of 1a and the canonical Pt(II) DNA-directed anti-cancer effect occurring in vivo for the mice treated with this platinum complex for 14 days cannot be excluded.
The different superstructures for 1a and 1d formed under cellular conditions can be attributed to the different uptake mechanism(s) and cellular localization of these two platinum(II) complexes as well as the cleavage of the glycosidic linkage of 1a under acidic and hydrolase-containing conditions. Glucose conjugation of the platinum(II) terpyridyl arylacetylide complex not only improves the inherent aqueous solubility but also renders the conjugate amphiphilic for supramolecular self-assembly. Presumably driven by the intermolecular interactions of the platinum(II) terpyridyl arylacetylide moiety and hydrogen bonding of the glucose unit in aqueous medium, complex 1a assembles into nanoparticles with a size of about 100 nm in serum-containing PBS and culture medium (Fig. S20† and 3c(ii)). These as-assembled nanostructures facilitate the uptake of 1a into NCI-H460 cells through energy-dependent endocytosis, as evidenced by the significant reduction in the cellular Pt level (up to 80%) upon incubation at low temperature (4 °C) or inhibition of endocytosis (Fig. 5e). The lower extent of reduction by endocytic blockade of up to 31% for 1d treatment suggests that the uptake is primarily through passive diffusion. Endocytosis governs the cellular uptake of nanoparticles and involves vesicular trafficking along the endosomal and lysosomal compartments.43 Compared to normal cells, such multi-step processes are enhanced and dysregulated in cancer cells.44 As supported by the bio-TEM images, accumulated platinum-containing nanofibrils were observed in the deformed endosomal and/or lysosomal vesicles of 1a-treated NCI-H460 lung and MDA-MB-231 breast cancer cells (Fig. 6a and S27†). The associated impairment of lysosomal structures was supported by a significant reduction in lysosomal staining with the fluorescent probes acridine orange and LysoTracker, and leakage of lysosomal probe pepstatin A–BODIPY FL into the cytosol (Fig. 7a–c). These findings are distinct from 1d treatment showing a comparable lysosomal staining pattern to that of the vehicle control (Fig. 7a–c) and formation of only platinum-containing aggregates in the mitochondria (Fig. 6c). Lysosomes are acidic (pH 4.5–6.5) and rich in hydrolytic enzymes such as glucosidase.32,45In vitro, 1a undergoes enzymatic cleavage of the glycosidic linkage by β-glucosidase to afford 1d in acidic medium as revealed by UPLC/MS analysis. Under the same conditions, 1a forms nanoparticles at pH 7.4 while 1d itself (i.e., deglycosylated 1a) forms nanorod structures at pH 5.8 (Fig. 4). These findings are in line with the in cellulo observation that 1a forms extracellular nanoparticles in the cell culture medium at physiological pH (Fig. 3c(ii)), and 1a-treated cells show nanofibrils in the endosomal and/or lysosomal vesicles of cancer cells (Fig. 6a and S27†), presumably via disassembly of particle structures followed by deglycosylation of 1a and reassembly in the acidic vesicles (Fig. 9). These superstructures were hardly observed in the 1a-treated normal fibroblasts (Fig. 6e), possibly attributed to the lower endocytic efficiency44 and/or β-glucosidase activity46 found in normal cells compared with lung cancer cells.
Emerging evidence suggests the modulation of autophagy by self-assembled nanomaterials.47 Indeed, aside from the lysosomal impairment, 1a treatment induces autophagic vacuole formation in NCI-H460 lung cancer cells in vitro and tumor tissues in vivo, as evidenced by the increased levels of the lipidated form of the LC3 (LC3-II) protein and formation of characteristic autophagosomes (Fig. 7d–g, 8h, S31, S33 and S38†), which presumably functions to encapsulate the foreign nanomaterials formed in situ. The majority of 1a-treated cells underwent necrotic cell death (Fig. 5c), which may be linked to the significant lysosomal rupture and leakage of degradative enzymes into the cytosol.31 In contrast, treatment of cells with 1d, the aglycone of 1a, resulted in depolarization of mitochondrial membrane potential (Fig. S35†), negligible lysosomal impairment and autophagic vacuolization (Fig. 7a–c and S34†), which in turn resulted in apoptotic cancer cell death (Fig. 5c). These biological observations delineate the role of the glucose unit in directing the assembling of the platinum(II) terpyridyl complex into superstructures in physiological medium (Fig. 3c and 4a), endosomal/lysosomal targeting in cellulo (Fig. 6a and S27†), lysosomal membrane permeabilization (Fig. 7a–c) and subsequent cancer cell death (Fig. 5c). Our results exemplify the supramolecular assembly of the planar platinum(II) complex in relation to cancer cell killing.
Conclusions
We present the supramolecular self-assembly properties of a planar alkynylplatinum(II) terpyridine complex with anti-cancer action via perturbing the autophagy–lysosomal system. Conjugation of a hydrophilic glucose moiety to the platinum(II) terpyridyl complex affords a water-soluble compound which self-assembles into nanoparticles in the cell culture medium and enters the cancer cells through endocytosis. Further intracellular transformation of the superstructures results in lysosomal disruption and is accompanied by cancer cell death. Importantly, the amphiphilic conjugate exhibits effective in vivo anti-tumor activity. Our study highlights the exploitation of supramolecular assemblies of platinum(II) complexes in an interdisciplinary area of platinum chemistry yet to be developed and sheds new light on developing a new class of metal-based anti-cancer agents by harnessing the intrinsic self-assembly properties.Ethical statement
All animal experiments were conducted under the guidelines approved by the Committee on the Use of Live Animals in Teaching and Research of the University of Hong Kong.Data availability
All experimental supporting data and procedures are available in the ESI.†Author contributions
C.-M. C. designed and supervised the project; K.-C. T. and P.-K. W. designed and performed the experiments; K.-C. T., P.-K. W. and C.-N. L. carried out the data analysis and prepared the manuscript, and all authors contributed to revising the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge the funding support from “Laboratory for Synthetic Chemistry and Chemical Biology” under the Health@InnoHK Program launched by Innovation and Technology Commission, The Government of Hong Kong Special Administrative Region of the People's Republic of China as well as the Innovation and Technology Fund (ITS/488/18). We acknowledge The University of Hong Kong Li Ka Shing Faculty of Medicine Centre for PanorOmic Sciences Imaging and Flow Cytometry Core for the confocal microscopy imaging and flow cytometry facilities. We thank Dr Yi Man Eva Fung for her assistance with the thermal proteome profiling experiment. We thank Mr Frankie Yu-Fee Chan of the Electron Microscope Unit of HKU for his technical assistance with the EDX and SAED measurements.References
- K. W. Jennette, S. J. Lippard, G. A. Vassiliades and W. R. Bauer, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 3839–3843 CrossRef CAS PubMed.
- A. H. Wang, J. Nathans, G. van der Marel, J. H. van Boom and A. Rich, Nature, 1978, 276, 471–474 CrossRef CAS PubMed.
- K. Becker, C. Herold-Mende, J. J. Park, G. Lowe and R. H. Schirmer, J. Med. Chem., 2001, 44, 2784–2792 CrossRef CAS PubMed.
- W. D. McFadyen, L. P. Wakelin, I. A. Roos and V. A. Leopold, J. Med. Chem., 1985, 28, 1113–1116 CrossRef CAS PubMed.
- I. Eryazici, C. N. Moorefield and G. R. Newkome, Chem. Rev., 2008, 108, 1834–1895 CrossRef CAS PubMed.
- C. Y.-S. Chung and V. W.-W. Yam, J. Am. Chem. Soc., 2011, 133, 18775–18784 CrossRef CAS PubMed.
- M. C.-L. Yeung and V. W.-W. Yam, Chem. Sci., 2013, 4, 2928–2935 RSC.
- S. Y.-L. Leung, K. M.-C. Wong and V. W.-W. Yam, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2845–2850 CrossRef CAS PubMed.
- K. Y. Kim, J. Kim, C. J. Moon, J. Liu, S. S. Lee, M. Y. Choi, C. Feng and J. H. Jung, Angew. Chem., Int. Ed., 2019, 58, 11709–11714 CrossRef CAS PubMed.
- F. Camerel, R. Ziessel, B. Donnio, C. Bourgogne, D. Guillon, M. Schmutz, C. Iacovita and J. P. Bucher, Angew. Chem., Int. Ed., 2007, 46, 2659–2662 CrossRef CAS PubMed.
- Y.-S. Wong, F. C.-M. Leung, M. Ng, H.-K. Cheng and V. W.-W. Yam, Angew. Chem., Int. Ed., 2018, 57, 15797–15801 CrossRef CAS PubMed.
- H.-L. Au-Yeung, S. Y.-L. Leung, A. Y.-Y. Tam and V. W.-W. Yam, J. Am. Chem. Soc., 2014, 136, 17910–17913 CrossRef CAS PubMed.
- M. Mauro, A. Aliprandi, D. Septiadi, N. S. Kehr and L. De Cola, Chem. Soc. Rev., 2014, 43, 4144–4166 RSC.
- A. Aliprandi, M. Mauro and L. De Cola, Nat. Chem., 2016, 8, 10–15 CrossRef CAS PubMed.
- C. Y.-S. Chung, S. P.-Y. Li, M.-W. Louie, K. K.-W. Lo and V. W.-W. Yam, Chem. Sci., 2013, 4, 2453–2462 RSC.
- J. L.-L. Tsai, T. Zou, J. Liu, T. Chen, A. O.-Y. Chan, C. Yang, C.-N. Lok and C.-M. Che, Chem. Sci., 2015, 6, 3823–3830 RSC.
- J. Li, K. Shi, Z. F. Sabet, W. Fu, H. Zhou, S. Xu, T. Liu, M. You, M. Cao, M. Xu, X. Cui, B. Hu, Y. Liu and C. Chen, Sci. Adv., 2019, 5, eaax0937 CrossRef CAS PubMed.
- J. Zhou, X. Du, N. Yamagata and B. Xu, J. Am. Chem. Soc., 2016, 138, 3813–3823 CrossRef CAS PubMed.
- Z. Feng, H. Wang, X. Chen and B. Xu, J. Am. Chem. Soc., 2017, 139, 15377–15384 CrossRef CAS PubMed.
- Z. Feng, H. Wang, S. Wang, Q. Zhang, X. Zhang, A. A. Rodal and B. Xu, J. Am. Chem. Soc., 2018, 140, 9566–9573 CrossRef CAS PubMed.
- Z. Feng, X. Han, H. Wang, T. Tang and B. Xu, Chem, 2019, 5, 2442–2449 CAS.
- M. Pieszka, S. Han, C. Volkmann, R. Graf, I. Lieberwirth, K. Landfester, D. Y. W. Ng and T. Weil, J. Am. Chem. Soc., 2020, 142, 15780–15789 CrossRef CAS PubMed.
- R. A. Pires, Y. M. Abul-Haija, D. S. Costa, R. Novoa-Carballal, R. L. Reis, R. V. Ulijn and I. Pashkuleva, J. Am. Chem. Soc., 2015, 137, 576–579 CrossRef CAS PubMed.
- W. Lu, B. X. Mi, M. C. Chan, Z. Hui, C.-M. Che, N. Zhu and S.-T. Lee, J. Am. Chem. Soc., 2004, 126, 4958–4971 CrossRef CAS PubMed.
- V. Ramu, S. Gautam, A. Garai, P. Kondaiah and A. R. Chakravarty, Inorg. Chem., 2018, 57, 1717–1726 CrossRef CAS PubMed.
- E. C. Calvaresi and P. J. Hergenrother, Chem. Sci., 2013, 4, 2319–2333 RSC.
- D.-L. Ma, T.-Y. Shum, F. Zhang, C.-M. Che and M. Yang, Chem. Commun., 2005, 4675–4677 RSC.
- G. S.-M. Tong, Y.-C. Law, S. C.-F. Kui, N. Zhu, K. H. Leung, D. L. Phillips and C.-M. Che, Chem.–Eur. J., 2010, 16, 6540–6554 CrossRef CAS PubMed.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- P. Wang, M. A. Rahman, Z. Zhao, K. Weiss, C. Zhang, Z. Chen, S. J. Hurwitz, Z. G. Chen, D. M. Shin and Y. Ke, J. Am. Chem. Soc., 2018, 140, 2478–2484 CrossRef CAS PubMed.
- P. Boya and G. Kroemer, Oncogene, 2008, 27, 6434–6451 CrossRef CAS PubMed.
- P. Saftig and J. Klumperman, Nat. Rev. Mol. Cell Biol., 2009, 10, 623–635 CrossRef CAS PubMed.
- E. White, J. Clin. Invest., 2015, 125, 42–46 CrossRef PubMed.
- S. Kimura, T. Noda and T. Yoshimori, Autophagy, 2007, 3, 452–460 CrossRef CAS PubMed.
- N. Mizushima, T. Yoshimori and B. Levine, Cell, 2010, 140, 313–326 CrossRef CAS PubMed.
- M. M. Savitski, F. B. Reinhard, H. Franken, T. Werner, M. F. Savitski, D. Eberhard, D. Martinez Molina, R. Jafari, R. B. Dovega, S. Klaeger, B. Kuster, P. Nordlund, M. Bantscheff and G. Drewes, Science, 2014, 346, 1255784 CrossRef PubMed.
- H. Franken, T. Mathieson, D. Childs, G. M. Sweetman, T. Werner, I. Togel, C. Doce, S. Gade, M. Bantscheff, G. Drewes, F. B. Reinhard, W. Huber and M. M. Savitski, Nat. Protoc., 2015, 10, 1567–1593 CrossRef CAS PubMed.
- S. T. Smiley, M. Reers, C. Mottola-Hartshorn, M. Lin, A. Chen, T. W. Smith, G. D. Steele Jr and L. B. Chen, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 3671–3675 CrossRef CAS PubMed.
- A. Brito, P. M. R. Pereira, D. Soares da Costa, R. L. Reis, R. V. Ulijn, J. S. Lewis, R. A. Pires and I. Pashkuleva, Chem. Sci., 2020, 11, 3737–3744 RSC.
- A. N. Shy, B. J. Kim and B. Xu, Matter, 2019, 1, 1127–1147 CrossRef PubMed.
- L. Guo, N. He, Y. Zhao, T. Liu and Y. Deng, Theranostics, 2020, 10, 3206–3222 CrossRef CAS PubMed.
- C. Stadelmann and H. Lassmann, Cell Tissue Res., 2000, 301, 19–31 CrossRef CAS PubMed.
- I. Canton and G. Battaglia, Chem. Soc. Rev., 2012, 41, 2718–2739 RSC.
- S. L. Schmid, J. Cell Biol., 2017, 216, 2623–2632 CrossRef CAS PubMed.
- T. Dinur, K. M. Osiecki, G. Legler, S. Gatt, R. J. Desnick and G. A. Grabowski, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 1660–1664 CrossRef CAS PubMed.
- J.-Y. Moon, S.-W. Kim, G.-M. Yun, H.-S. Lee, Y.-D. Kim, G.-J. Jeong, I. Ullah, G.-J. Rho and B.-G. Jeon, Anim. Cells Syst., 2015, 19, 295–304 CrossRef CAS.
- K. Peynshaert, B. B. Manshian, F. Joris, K. Braeckmans, S. C. De Smedt, J. Demeester and S. J. Soenen, Chem. Rev., 2014, 114, 7581–7609 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Instrumentation, experimental procedures, synthesis and characterization data, details of biological studies, and supplementary figures and tables. See DOI: 10.1039/d1sc02841c |
| This journal is © The Royal Society of Chemistry 2021 |