
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reversible redox controlled acids for cationic ring-opening polymerization†
Michael J.
Supej
 ,
Elizabeth A.
McLoughlin
,
Jesse H.
Hsu
and
Brett P.
Fors
*
,
Elizabeth A.
McLoughlin
,
Jesse H.
Hsu
and
Brett P.
Fors
*
Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, USA. E-mail: bpf46@cornell.edu
First published on 6th July 2021
Abstract
Advancements in externally controlled polymerization methodologies have enabled the synthesis of novel polymeric structures and architectures, and they have been pivotal to the development of new photocontrolled lithographic and 3D printing technologies. In particular, the development of externally controlled ring-opening polymerization (ROP) methodologies is of great interest, as these methods provide access to novel biocompatible and biodegradable block polymer structures. Although ROPs mediated by photoacid generators have made significant contributions to the fields of lithography and microelectronics development, these methodologies rely upon catalysts with poor stability and thus poor temporal control. Herein, we report a class of ferrocene-derived acid catalysts whose acidity can be altered through reversible oxidation and reduction of the ferrocenyl moiety to chemically and electrochemically control the ROP of cyclic esters.
Introduction
The physical properties and functionalities of polymeric materials are directly correlated to their structure and architecture.1–3 While living polymerization methodologies in conjunction with carefully chosen and timed monomer additions produce well-defined materials (e.g., block copolymers2), the ability to control chain growth with an external stimulus could lead to many advanced structures and architectures with potentially interesting physical properties. These externally controlled polymerization methodologies rely on changes in chemical reactivity upon application of an external stimulus (chemical,4–17 electrochemical,18–20 photochemical,21–30 thermal,31–33 mechanochemical34–37), which precisely regulates the incorporation of monomers at a growing polymer chain end. In addition to promoting the synthesis of advanced structures and architectures,30,45 the spatiotemporal control afforded by these externally controlled polymerizations has enabled the development of new lithographic38–42 and 3D printing technologies.43,44The development of methods for externally controlled ring-opening polymerization (ROP) of cyclic esters and carbonates is of particular interest as the polymers that are formed are biocompatible and degradable alternatives to petroleum-derived polyolefins. State-of-the-art methods for externally controlled ROP have involved the use of redox-switchable coordination-insertion catalysts, which have been pioneered by Byers7,10–12,20 and Diaconescu,8,9,13–17 or the use of photoacids, which induce polymerization through an activated monomer mechanism. Photoacid generators (PAGs)46 have been pivotal to the fields of lithography and microelectronics development; however, PAG-mediated polymerizations are not reversible and only provide temporal control over polymer initiation rather than chain growth. To overcome this challenge and develop a reversible photoacid, Boyer and Read de Alaniz have independently used merocyanine-based catalysts.47,48 However, slow thermal reversion of the spiropyran to the protonated merocyanine limits the degree of temporal control in these systems. Similarly, Hecht and Liao have each reported catalysts for photoswitchable ROP,49,50 but limitations related to catalytic efficiency and reversibility were encountered in these systems as well. On this basis, the discovery of an acid catalyst that can be reversibly activated by an external stimulus for ROP remains a challenge.
We postulated that temporal control over acid-catalysed, cationic ROP could be achieved by designing a reversible, redox-controlled acid whose pKa could be altered through changes in oxidation state.51,52 Specifically, by tethering ferrocene to an acidic functional group53,54 we envisaged a system where the pKa would decrease upon oxidation from Fe(II) to Fe(III) and subsequently initiate ROP by an activated monomer mechanism (Fig. 1). Importantly, reduction of the ferrocenium species back to ferrocene would restore the original acidity of the molecule and deactivate the catalyst, affording reversible termination and thus temporal control over the polymerization.
 | ||
| Fig. 1 Ferrocenyl acids afford chemical and electrochemical control over the ring-opening polymerization (ROP) of cyclic esters. | ||
Results and discussion
To test our hypothesis, we examined the ROP of caprolactone (CL) catalysed by substituted ferrocenes in both the absence and presence of oxidant (Table 1). We selected benzyl alcohol (BnOH) as the initiator for its relatively high oxidation potential55 and its ability to provide an NMR label at the polymer chain end. Combining CL, BnOH, and either ferrocenyl phosphonic (1a), ferrocenyl (phenyl)phosphinic (1b), or ferrocenyl (phenyl)phosphonic (1c) acid showed no polymerization (see ESI Table S1,† entries 1–3). These results demonstrate that these acids in their neutral state are not competent catalysts for polymerization of CL. Significantly, addition of 1.2 equivalents of AgBF4 relative to the ferrocenyl acid in all three of these reactions efficiently initiated ROP and gave polymers with good agreement between theoretical and experimental number average molar masses (Mns) and low dispersity (Đ) values (Table 1, entries 1–3).56 It is worth noting that analogous reactivity was also observed in dichloromethane (DCM) (Table 1, entry 6) and the polymer obtained had a lower dispersity value. Furthermore, no polymerization was observed in the absence of ferrocenyl acid (Table 1, entry 7). In the absence of BnOH initiator, we observed 2% monomer conversion, which we attribute to the presence of trace water (Table 1, entry 9). The ferrocenyl carboxylic acid catalysts 2a and 2b showed little to no catalytic activity in either the presence or absence of oxidant (Table 1, entries 4–5; ESI Table S1,† entries 4–5), demonstrating that the identity of the acid is pivotal for efficient polymerization.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Acid catalyst | [CL]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[BnOH]:[FcXH]:[Ox] :[BnOH]:[FcXH]:[Ox] |
Conversion | M n,Theo (kg mol−1) | M n,Exp (kg mol−1) | Đ |
| a [CL] = 9.1 M (neat), 1.0 mol% [FcXH], 1.2 mol% AgBF4, 23 °C. b [CL] = 4.8 M (in DCM). c CL was metered in at a rate of 0.1 mL per hour. d [CL] = 4.98 M (in DCM), 0.2 M Bu4NBF4, 2.0 mA constant current, RVC anode, RVC cathode. e [CL] = 4.98 M (in DCM), 0.2 M Bu4NBF4, no applied current, RVC anode, RVC cathode. f M n,Theo = [CL]/[BnOH] × MWCL × conversion + MWBnOH. g M n,Exp determined by gel permeation chromatography with a multiangle light scattering (MALS) detector. | ||||||
| 1a | 1a | 100:1:1:1.2 |
52% | 6.0 | 5.6 | 1.15 |
| 2a | 1b | 100:1:1:1.2 |
36% | 4.1 | 3.2 | 1.14 |
| 3a | 1c | 100:1:1:1.2 |
45% | 5.9 | 4.9 | 1.15 |
| 4a | 2a | 100:1:1:1.2 |
<5% | — | — | — |
| 5a | 2b | 100:1:1:1.2 |
<5% | — | — | — |
| 6b | 1b | 100:1:1:1.2 |
35% | 4.1 | 4.0 | 1.07 |
| 7b | 1b | 100:1:1:0 |
0% | — | — | — |
| 8b | 1b | 100:1:0:1.2 |
0% | — | — | — |
| 9b | 1b | 100:0:1:1.2 |
2% | — | 10.6 | 1.09 |
| 10b,c | 1b | 200:1:1:1.2 |
49% | 11.3 | 10.9 | 1.26 |
| 11b | 1b | 100:1:2:2.4 |
65% | 7.5 | 6.3 | 1.13 |
| 12b | 1b | 53:1:1:1.2 |
87% | 5.3 | 4.2 | 1.09 |
| 13b | 1b | 26:1:1:1.2 |
>99% | 3.0 | 2.3 | 1.09 |
| 14b | 1b | 11:1:1:1.2 |
>99% | 1.3 | 1.3 | 1.05 |
| 15d | 1c | 51:1:0.5:— |
65% | 2.8 | 3.8 | 1.08 |
| 16d | 1c | 51:1:0:— |
>99% | 8.8 | 5.9 | 1.86 |
| 17e | 1c | 51:1:0.5:— |
0% | — | — | — |

Based on these experiments, we identified the redox responsive nature of our ferrocenyl acid catalysts as the key component to achieving temporal control over ROP. Thus, we first sought to understand the impact that oxidation state has on the acidity of the FcXH species. To this end, the pKa of 1a, 1b, 1c, and 2a were determined in DMSO-d6 by titration with bases whose conjugate acids have known pKa values in DMSO (Table S8†). The pKas of the oxidized species 1a+, 1b+, 1c+, and 2a+ were then determined through square scheme thermochemical analysis using the pKa values of the 1a, 1b, 1c, and 2a and relevant electrochemical values [i.e., E1/2(FcXH/FcXH+) and E1/2(FcX−/FcX)] (Fig. 2).57–59 As shown in Table S8,† the ferrocenyl carboxylic acid 2a is the least acidic, having a pKa of 10.8 in DMSO. The ferrocenyl phosphonic acid 1a and the ferrocenyl (phenyl)phosphinic acid 1b have similar pKa values of ca. 8.5 in DMSO. The ferrocenyl (phenyl)phosphonic acid 1c is the most acidic FcXH species, having a pKa of 7.2 in DMSO. Upon oxidation, these pKa values decrease by ca. 3–5 units. Although the pKa of the ferrocenyl carboxylic acid 2a decreases the most upon oxidation (ΔpKa = 5.1), the oxidized species (2a+) is the least acidic FcXH+ species, having a pKa of ca. 6 in DMSO. The inability of 2a+ to polymerize CL suggests that the oxidized FcXH+ species must have a pKa < 6 to efficiently catalyse ROP. In support of this hypothesis is the observation that phenylphosphonic acid (pKaca. 8) is unable to catalyse the polymerization of CL (Table S2,† entry 1). The oxidized ferrocenyl phosphonic (1a+), ferrocenyl (phenyl)phosphinic (1b+), and ferrocenyl (phenyl)phosphonic (1c+) acid catalysts have similar pKa values of ∼4 in DMSO, which is in good agreement with the pKa value of diphenyl phosphoric acid, which is known to efficiently catalyse ROP (Fig. 2).60,61 These detailed measurements suggest that redox acid controlled ROP can be realized by designing acid catalysts whose pKa values switch from greater than to less than ca. 5 upon application of an external stimulus.
 | ||
| Fig. 2 (a) Square scheme thermochemical analysis for ferrocenyl acid pKa determination, (b) relative acidities of the neutral (solid marker) and oxidized (dashed marker) forms of the ferrocenyl acids used in this study. | ||
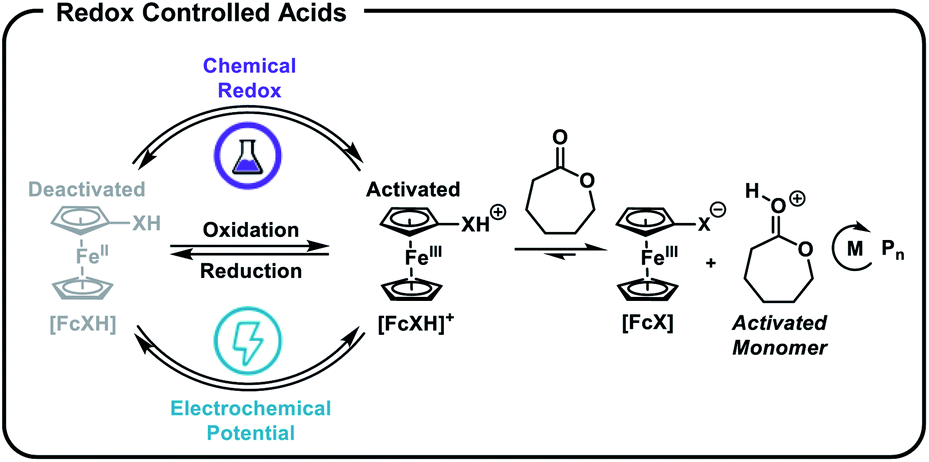
We next investigated the living characteristics of redox acid controlled ROP. By varying the monomer-to-alcohol ratio, we targeted a range of Mns and synthesized polymers that maintained good agreement between experimental and theoretical Mns and low Đ values (Table 1, entries 10–14). Furthermore, while monitoring the polymerization of CL under optimized reaction conditions,62 we observed that Mn grows linearly with conversion (Fig. 3a). The chain-end fidelity of the resulting polyesters was assessed through the synthesis of block copolymers. Using our standard conditions, we generated a 2.3 kg mol−1 poly(CL) macro-initiator which was isolated and then re-subjected to identical reaction conditions to polymerize δ-valerolactone (VL). The resulting 5.5 kg mol−1 poly(CL-b-VL) diblock copolymer shows a monomodal distribution with a low Đ value of 1.09 (Fig. 3b). This data demonstrates the ability of our method to generate degradable polyester block polymers and shows that it is competent for monomers other than CL.
 | ||
| Fig. 3 (a) Plot of Mnvs. monomer conversion for the cationic ring-opening polymerization of ε-caprolactone [CL] = 4.8 M (in DCM), 1.0 mol% BnOH, 1.0 mol% [FcXH], 1.2 mol% AgBF4, 23 °C, (b) synthesis and GPC traces of poly(caprolactone) and poly(caprolactone-block-valerolactone). | ||
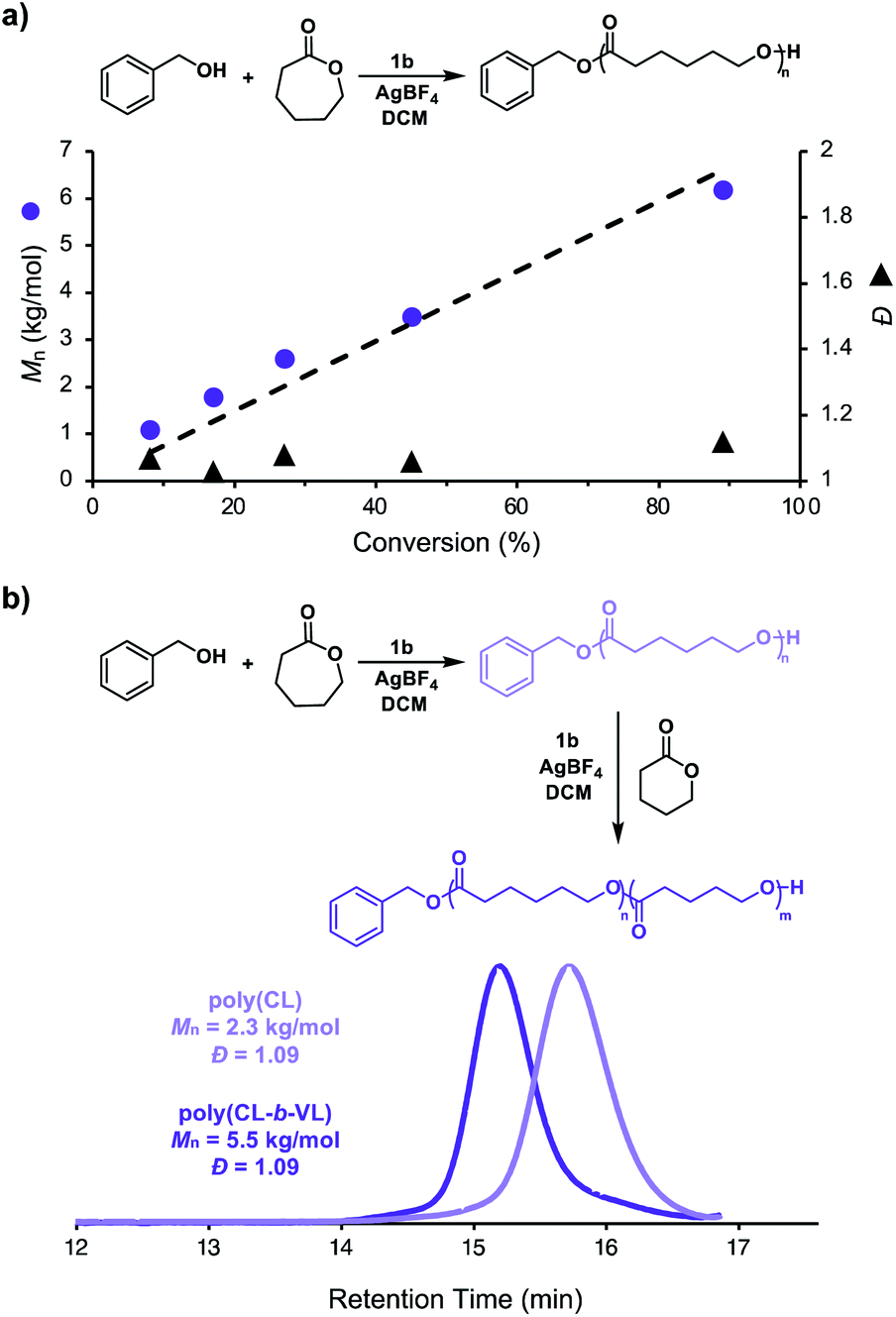
Having shown that this system exhibits living characteristics, we next aimed to demonstrate temporal control over ROP. While addition of a chemical oxidant will oxidize the ferrocenyl acid to facilitate polymerization, we posited that subsequent addition of a chemical reductant would prompt reduction of the ferrocenyl acid to reversibly halt polymerization. Our thermochemical analysis on the acidic characteristics of the ferrocenyl acids illustrated that catalyst 1c would be the most ideal candidate for temporally controlled polymerization as it proved to be both the most acidic and most soluble ferrocenyl acid catalyst. To test our hypothesis, we first added AgBF4 to a solution of CL, BnOH, and ferrocenyl acid (1c) in DCM. After approximately 3.5 h, cobaltocene (CoCp2) was added to the reaction to reversibly terminate the polymerization. This overall process was repeated two times, demonstrating excellent temporal control over the polymerization using chemical redox reagents (Fig. 4a).
 | ||
| Fig. 4 (a) Monomer conversion vs. time plot illustrating temporal control over ROP using AgBF4 to initiate polymerization and CoCp2 to reversibly terminate polymerization, (b) monomer conversion vs. time plot illustrating temporal control over ROP using anodic potential to initiate polymerization and cathodic potential to reversibly terminate polymerization. | ||
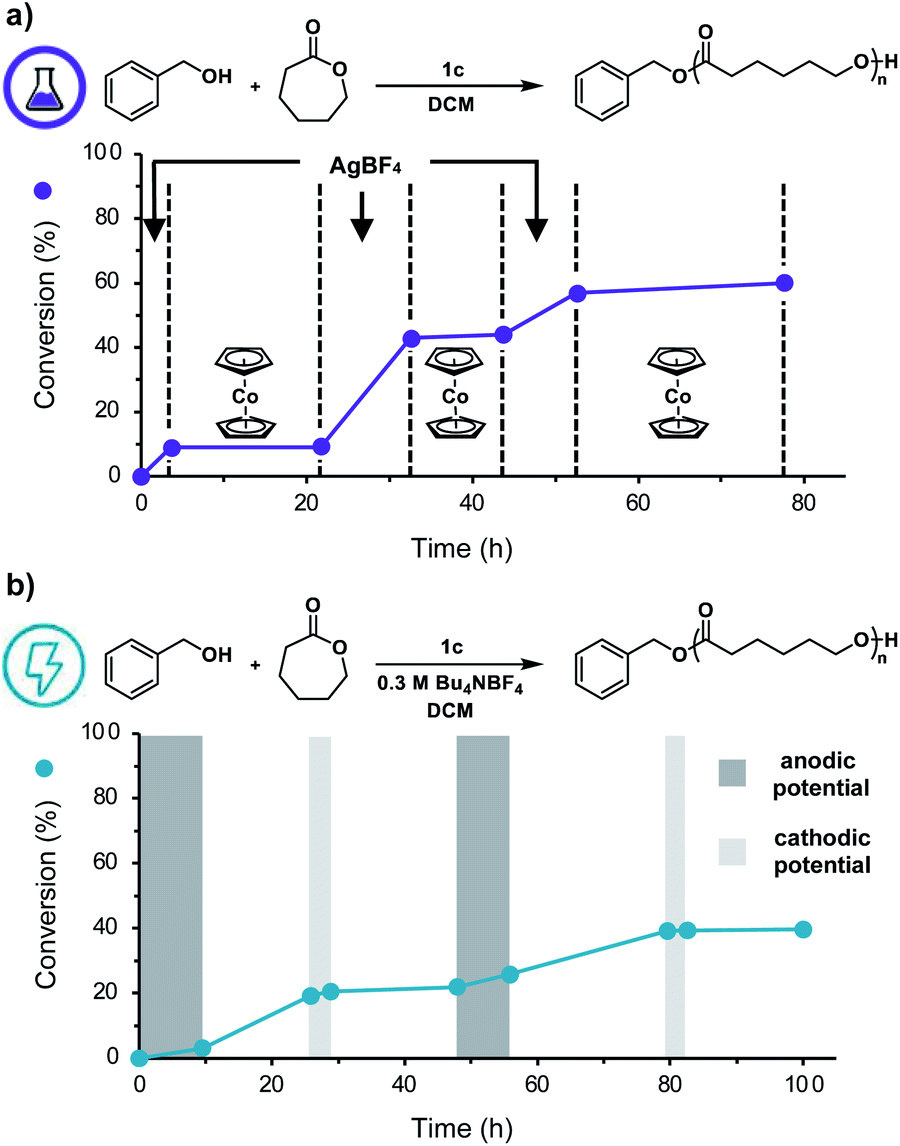
We next sought to replace these chemical redox reagents with an electrochemical stimulus. Application of 2.0 mA of constant current to a solution of DCM, CL, BnOH, 1c, and tetrabutylammonium tetrafluoroborate (Bu4NBF4) in a divided electrochemical cell fitted with reticulated vitreous carbon (RVC) electrodes, for 2.5 hours resulted in efficient polymerization (Table 1, entry 15), yielding a polymer with good agreement between experimental and theoretical Mns and a low Đ value. Importantly, no polymerization was observed in the absence of an anodic current and only uncontrolled polymerization of CL occurred in the absence of catalyst 1c (Table 1, entries 16, 17).
To further adapt this electrochemical system for temporal control, cyclic voltammetry was performed to identify the E1/2 value of 1c in DCM (0.78 V vs. Ag/AgCl, Fig. S14†). Noting this value, we applied a constant anodic potential of 1.4 V vs. Ag/AgCl for 9.5 h to a DCM solution of CL, BnOH, 1c, and Bu4NBF4 in a divided electrochemical cell equipped with a platinum anode and an RVC cathode. After 16 h, a constant cathodic potential of 0.0 V vs. Ag/AgCl was applied for 3.0 h to reversibly terminate the polymerization. This cycling between anodic and cathodic potential was repeated once more, demonstrating excellent temporal control using an electrochemical stimulus (Fig. 4b). It should be noted that while both chemical and electrochemical methods afforded temporal control, we noticed increased tailing to lower molecular weights during electrochemical on/off experiments (Fig. S11b†). We hypothesize this observation is due to competitive oxidation of alcohol polymer chain-ends. These results illustrate the power of our redox acid platform for both efficiently controlling polymerization and enabling temporal control over polymerization—thus overcoming a challenge associated with traditional acid-mediated processes.
Conclusions
In conclusion, we have developed a class of redox-active ferrocenyl acids that can be used to temporally control the cationic ROP of cyclic esters. The ability of these acids to reversibly promote polymerization depends upon their pKa values in the neutral and oxidized state, suggesting that other switchable acid catalysts may be realized using similar principles. These redox acid controlled systems show excellent temporal control over the polymerization, mitigating shortcomings of traditional acid catalysed polymerizations. Furthermore, the initial chemically controlled methodology was extended to electrochemical control. When compared to other electrochemically controlled ROPs20,63 which utilize sensitive metal catalysts, these bench stable acid catalysts prove advantageous; however, the present system remains comparatively limited in molecular weight. We anticipate that this class of redox acid controlled polymerizations will serve as the foundation for further advancements and discovery in the field of redox acid mediated processes.Author contributions
M. J. S. and B. P. F. conceived this project. M. J. S. and J. H. H. conducted all polymer synthesis and optimization experiments. E. A. M. conducted all thermochemical analysis experiments. The final version of the manuscript was written with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSF under award number CHE-1752140. Additionally, this work made use of the NMR Facility at Cornell University that was supported in part by the NSF under Award CHE-1531632. E. A. M. thanks the Center for Sustainable Polymers for a Postdoctoral Fellowship. B. P. F. thanks the Alfred P. Sloan foundation for a Sloan Research Fellowship.Notes and references
- D. M. Rosenbaum and D. R. Liu, J. Am. Chem. Soc., 2003, 125, 13924–13925 CrossRef CAS PubMed.
- F. S. Bates, M. A. Hillmyer, T. P. Lodge, C. M. Bates, K. T. Delaney and G. H. Fredrickson, Science, 2012, 336, 434–440 CrossRef CAS PubMed.
- N. Corrigan, M. Ciftci, K. Jung and C. A. J. M. Boyer, Angew. Chem., Int. Ed., 2021, 60, 1748–1781 CrossRef CAS PubMed.
- C. K. A. Gregson, I. J. Blackmore, V. C. Gibson, N. J. Long, E. L. Marshall and A. J. P. White, Dalton Trans., 2006, 25, 3134–3140 RSC.
- C. K. A. Gregson, V. C. Gibson, N. J. Long, E. L. Marshall, P. J. Oxford and A. J. P. White, J. Am. Chem. Soc., 2006, 128, 7410–7411 CrossRef CAS PubMed.
- E. M. Broderick, N. Guo, C. S. Vogel, C. Xu, T. Miller, K. Meyer, P. Mehrkhodavandi and P. L. Diaconescu, J. Am. Chem. Soc., 2011, 133, 9278–9281 CrossRef CAS PubMed.
- A. B. Biernesser, K. R. D. Chiaie, J. B. Curley and J. A. Byers, Angew. Chem., Int. Ed., 2016, 55, 5251–5254 CrossRef CAS PubMed.
- X. Wang, A. Thevenon, J. L. Brosmer, I. Yu, S. I. Khan, P. Mehrkhodavandi and P. L. Diaconescu, J. Am. Chem. Soc., 2014, 136, 11264–11267 CrossRef CAS PubMed.
- E. M. Broderick, N. Guo, T. Wu, C. S. Vogel, C. Xu, J. T. Miller, K. Meyer, T. Cantat and P. L. Diaconescu, Chem. Commun., 2011, 47, 9897–9899 RSC.
- K. R. Delle Chiaie, L. M. Yablon, A. B. Biernesser, G. R. Michalowski, A. W. Sudyn and J. A. Byers, Polym. Chem., 2016, 7, 4675–4681 RSC.
- K. R. Delle Chiaie, A. B. Biernesser, M. A. Ortuño, B. Dereli, D. A. Iovan, M. J. T. Wilding, B. Li, C. J. Cramer and J. A. Byers, Dalton Trans., 2017, 46, 12971–12980 RSC.
- M. A. Ortuño, B. Dereli, K. R. D. Chiaie, A. B. Biernesser, M. Qi, J. A. Byers and C. J. Cramer, Inorg. Chem., 2018, 57, 2064–2071 CrossRef PubMed.
- J. Wei and P. L. Diaconescu, Acc. Chem. Res., 2019, 52, 415–424 CrossRef CAS PubMed.
- A. Lai, Z. C. Hern and P. L. Diaconescu, ChemCatChem, 2019, 11, 4210–4218 CrossRef CAS.
- R. Dai and P. L. Diaconescu, Dalton Trans., 2019, 48, 2996–3002 RSC.
- S. M. Quan and P. L. Diaconescu, Chem. Commun., 2015, 51, 9643–9646 RSC.
- S. M. Quan, X. Wang, R. Zhang and P. L. Diaconescu, Macromolecules, 2016, 49, 6768–6778 CrossRef CAS.
- B. M. Peterson, S. Lin and B. P. Fors, J. Am. Chem. Soc., 2018, 140, 2076–2079 CrossRef CAS PubMed.
- Y. Wang, M. Fantin and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 376–381 CrossRef CAS.
- M. Qi, Q. Dong, D. Wang and J. A. Byers, J. Am. Chem. Soc., 2018, 140, 5686–5690 CrossRef CAS PubMed.
- N. Corrigan, S. Shanmugam, J. Xu and C. Boyer, Chem. Soc. Rev., 2016, 45, 6165–6212 RSC.
- T. G. McKenzie, Q. Fu, M. Uchiyama, K. Satoh, J. Xu, C. Boyer, M. Kamigaito and G. G. Qiao, Adv. Sci., 2016, 3, 1500394 CrossRef PubMed.
- S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2015, 137, 9988–9999 CrossRef CAS PubMed.
- S. Dadashi-silab, S. Doran and Y. Yagci, Chem. Rev., 2016, 116, 10212–10275 CrossRef CAS PubMed.
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS PubMed.
- B. P. Fors, J. T. Trotta and B. P. Fors, Synlett, 2016, 27, 702–713 CrossRef.
- K. Murata, K. Saito, S. Kikuchi, M. Akita and A. Inagaki, Chem. Commun., 2015, 51, 5717–5720 RSC.
- J. C. Theriot, C.-H. Lim, H. Yang, M. D. Ryan, C. B. Musgrave and G. M. Miyake, Science, 2016, 352, 1082–1086 CrossRef CAS PubMed.
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS PubMed.
- B. M. Peterson, V. Kottisch, M. J. Supej and B. P. Fors, ACS Cent. Sci., 2018, 4, 1228–1234 CrossRef CAS PubMed.
- I. V. O. Reetz and Y. Yagci, Prog. Polym. Sci., 1998, 23, 1485–1538 CrossRef.
- S. Naumann and M. R. Buchmeiser, Catal. Sci. Technol., 2014, 4, 2466–2479 RSC.
- S. Naumann and M. R. Buchmeiser, Macromol. Rapid Commun., 2014, 35, 682–701 CrossRef CAS PubMed.
- Z. Wang, X. Pan, L. Li, M. Fantin, J. Yan, Z. Wang, Z. Wang, H. Xia and K. Matyjaszewski, Macromolecules, 2017, 50, 7940–7948 CrossRef CAS.
- Z. Wang, X. Pan, J. Yan, S. Dadashi-Silab, G. Xie, J. Zhang, Z. Wang, H. Xia and K. Matyjaszewski, ACS Macro Lett., 2017, 6, 546–549 CrossRef CAS.
- T. G. McKenzie, E. Colombo, Q. Fu, M. Ashokkumar and G. G. Qiao, Angew. Chem., Int. Ed., 2017, 56, 12302–12306 CrossRef CAS PubMed.
- H. Mohapatra, M. Kleiman and A. P. Esser-Kahn, Nat. Chem., 2017, 9, 135–139 CrossRef CAS.
- J. E. Poelma, B. P. Fors, G. F. Meyers, J. W. Kramer and C. J. Hawker, Angew. Chem., Int. Ed., 2013, 52, 6844–6848 CrossRef CAS PubMed.
- B. P. Fors, J. E. Poelma, M. S. Menyo, M. J. Robb, D. M. Spokoyny, J. W. Kramer, J. H. Waite and C. J. Hawker, J. Am. Chem. Soc., 2013, 135, 14106–14109 CrossRef CAS PubMed.
- J. Clausmeyer, W. Schuhmann and N. Plumeré, TrAC, Trends Anal. Chem., 2014, 58, 23–30 CrossRef CAS.
- H. Liu, S. Hoeppener and U. S. Schubert, Adv. Eng. Mater., 2016, 18, 890–902 CrossRef CAS.
- C. Xia, R. C. Advincula, A. Baba and W. Knoll, Chem. Mater., 2004, 16, 2852–2856 CrossRef CAS.
- N. D. Dolinski, Z. A. Page, E. B. Callaway, F. Eisenreich, R. V. Garcia, R. Chavez, D. P. Bothman, S. Hecht, F. W. Zok and C. J. Hawker, Adv. Mater., 2018, 30, 1800364 CrossRef PubMed.
- J. J. Schwartz and A. J. Boydston, Nat. Commun., 2019, 10, 791 CrossRef CAS PubMed.
- Z. Zhang, T. Zeng, L. Xia, C. Hong, D. Wu and Y. You, Nat. Commun., 2018, 9, 2577 CrossRef PubMed.
- C. J. Martin, G. Rapenne, T. Nakashima and T. Kawai, J. Photochem. Photobiol., C, 2018, 34, 41–51 CrossRef CAS.
- C. Fu, J. Xu and C. Boyer, Chem. Commun., 2016, 52, 7126–7129 RSC.
- M. S. Zayas, N. D. Dolinski, J. L. Self, A. Abdilla, C. J. Hawker, C. M. Bates and J. Read de Alaniz, ChemPhotoChem, 2019, 3, 467–472 CrossRef CAS.
- F. Eisenreich, M. Kathan, A. Dallmann, S. P. Ihrig, T. Schwaar, B. M. Schmidt and S. Hecht, Nat. Catal., 2018, 1, 516–522 CrossRef CAS.
- X. Zhang, Q. Ma, Y. Jiang, S. Hu, J. Li and S. Liao, Polym. Chem., 2021, 12, 885–892 RSC.
- Y. Hu, A. P. Shaw, D. P. Estes and J. R. Norton, Chem. Rev., 2016, 116, 8427–8462 CrossRef CAS PubMed.
- E. S. Wiedner, M. B. Chambers, C. L. Pitman, R. M. Bullock, A. J. M. Miller and A. M. Appel, Chem. Rev., 2016, 116, 8655–8692 CrossRef CAS.
- F. Carre, J. Le Bideau, O. Oms and D. Leclercq, J. Organomet. Chem., 2004, 689, 2654–2661 CrossRef.
- R. P. Shekurov, V. A. Miluykov, D. R. Islamov, D. B. Krivolapov, O. N. Kataeva, T. P. Gerasimova, S. A. Katsyuba, G. R. Nasybullina, V. V. Yanilkin and O. G. Sinyashin, J. Organomet. Chem., 2014, 766, 40–48 CrossRef CAS.
- N. L. Weinberg and H. R. Weinberg, Chem. Rev., 1968, 68, 449–523 CrossRef CAS.
- A slight excess of AgBF4 is used to ensure complete oxidation of the ferrocenyl acid catalyst.
- E. A. McLoughlin, K. M. Waldie, S. Ramakrishnan and R. M. Waymouth, J. Am. Chem. Soc., 2018, 140, 13233–13241 CrossRef CAS.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed.
- A. Wu, J. Masland, R. D. Swartz, W. Kaminsky and J. M. Mayer, Inorg. Chem., 2007, 46, 11190–11201 CrossRef CAS PubMed.
- P. Christ, A. G. Lindsay, S. S. Vormittag, J. M. Neudörfl, A. Berkessel and A. C. O'Donoghue, Chem.–Eur. J., 2011, 17, 8524–8528 CrossRef CAS PubMed.
- K. Makiguchi, T. Satoh and T. Kakuchi, Macromolecules, 2011, 44, 1999–2005 CrossRef CAS.
- We observed a dependence of polymerization rate on the concentration of activated acid catalyst, where catalyst concentration was ultimately limited by solubility.
- Y. Zhong, Q. Feng, X. Wang, J. Chen, W. Cai and R. Tong, ACS Macro Lett., 2020, 9, 1114–1118 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03011f |
| This journal is © The Royal Society of Chemistry 2021 |