
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA general approach to 2,2-disubstituted indoxyls: total synthesis of brevianamide A and trigonoliimine C†
Fan
Xu
 and
Myles W.
Smith
*
and
Myles W.
Smith
*
Department of Biochemistry, UT Southwestern Medical Center, 5323 Harry Hines Blvd, Dallas, Texas 75390, USA. E-mail: myles.smith@utsouthwestern.edu
First published on 23rd September 2021
Abstract
The indoxyl unit is a common structural motif in alkaloid natural products and bioactive compounds. Here, we report a general method that transforms readily available 2-substituted indoles into 2,2-disubstituted indoxyls via nucleophile coupling with a 2-alkoxyindoxyl intermediate and showcase its utility in short total syntheses of the alkaloids brevianamide A (7 steps) and trigonoliimine C (6 steps). The developed method is operationally simple and demonstrates broad scope in terms of nucleophile identity and indole substitution, tolerating 2-alkyl substituents and free indole N–H groups, elements beyond the scope of most prior approaches. Spirocyclic indoxyl products are also accessible via intramolecular nucleophilic trapping.
Introduction
Indoxyls, also known as indolin-3-ones, are important structural units in biologically active small molecules and natural alkaloids (1–5, indoxyl highlighted in red, Fig. 1A), and serve as versatile precursors to related N-heterocyclic compounds (e.g., 6).1 These include the highly cytotoxic duocarmycins (e.g., 3) and the potent μ-opoid receptor agonist mitrogynine pseudoindoxyl (4), which have received interest from the medical community as payloads in antibody–drug conjugates or potential analgesics, respectively.2 2,2-Disubstituted indoxyls bear a fully substituted center at the C-2 position and represent a core challenge in the synthesis of targets such as iboluteine (1) and brevianamide A (2). While many creative methods toward 2,2-disubstituted indoxyls have issued from the synthetic community, including cyclization/trapping of 2-alkynyl aryl azides3 or -nitroarenes,4 aryne heteroannulation,5 interrupted Ugi reactions,6 and many others,7 challenges still remain in accessing such motifs bearing two alkyl substituents at C-2, as would be required for 1–2.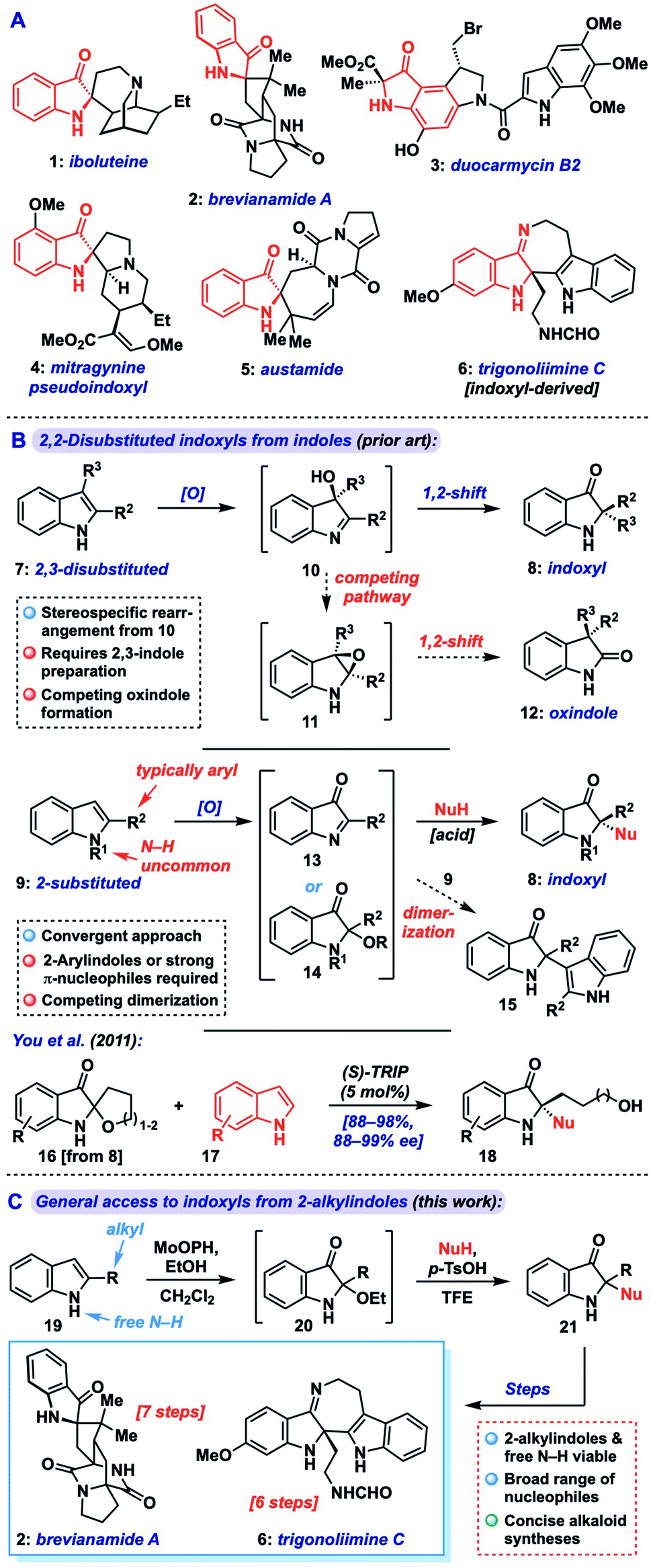 | ||
| Fig. 1 (A) Bioactive indoxyl natural products. (B) Approaches to 2,2-disubstituted indoxyls via indole dearomatization. (C) Our general method to 2,2-disubstituted indoxyls from 2-alkylindoles and its application in alkaloid synthesis. | ||
One of the most convenient ways to access 2,2-disubstituted indoxyls (8) is via dearomatization of readily available indole starting materials.8 Broadly speaking, such reactions fall into two classes: those that begin with (or proceed via) a 2,3-disubstituted indole (7), and those that begin with a 2-substituted indole (9) and introduce the second substituent through an additional nucleophilic component (Fig. 1B). The former approach proceeds via oxidation of the disubstituted indole 7 to a 3-hydroxyindolenine (10), followed by Wagner–Meerwein-type 1,2-shift of the C-3 substituent to C-2 with concomitant formation of the indoxyl ketone.9 Indeed, this approach has been widely explored, including in alkaloid total synthesis,10 but can suffer from competing rearrangement into the 3,3-disubstituted oxindole isomer 12 (via transient epoxide 11) in a manner that can be highly substrate- and condition-specific. Additionally, the preparation of the required 2,3-disubstituted indole can often be step-intensive.
Alternatively, from a 2-substituted indole (9), oxidation can yield an electrophilic 3-oxoindolenine (13) or its 2-hydroxy/alkoxyindoxyl equivalent (14) prior to iminium trapping with a nucleophile, often mediated by Lewis or Brønsted acids. Aside from being complementary to the prior rearrangement approach, this strategy offers the advantage of being convergent, provided sufficient generality is available for the nucleophilic and electrophilic components. While this approach has been explored,11 a key issue in such reactions is avoiding simple dimerization of the indole fragment via attack of the nucleophilic indole starting material onto 13 during oxidation to yield an adduct like 15. In fact, many methods explicitly target such dimers (or trimers) because of this problem.12 Additionally, where cross-couplings are possible, the overwhelming majority of such methods are only suitable for 2-aryl substituted systems and/or require N-substitution, likely due to the instability of the corresponding 2-alkylimine/iminium intermediates (vide infra).11 Though a handful of exceptions exist,11a,h,j,m these approaches also typically employ highly nucleophilic coupling partners like indoles or pyrroles (Mayr nucleophilicity parameter, N = 5.5–7)13 presumably to ensure efficient iminium trapping. Among these methods, You and coworkers reported a notable asymmetric coupling of indoles with spiroindoxyls that relies on a hydroxyalkyl chain as reversible iminium trap (see 16, Scheme 1B, bottom), building off of racemic work by Kobayashi,11a which necessarily limits the scope to specific 2-(hydroxyalkyl) substrates (18).11d
Thus, a general approach which can tolerate a range of 2-alkyl substituents, a broader selection of nucleophiles, and not require N-protection would be highly desirable. This should expedite the synthesis of complex targets such as trigonoliimine C (6) and is especially evident when considering application to indoxyl alkaloids such as iboluteine (1) or brevianamide A (2), neither of which incorporate a 2-aryl unit or N-substitution. Herein, we describe the development of a general approach that transforms readily available 2-substituted indoles into 2,2-disubstituted indoxyls via nucleophile coupling with a 2-alkoxyindoxyl intermediate. The method demonstrates broad scope in terms of nucleophile identity and indole substitution, tolerating 2-alkyl substituents and free indole N–H groups. The utility of our approach is highlighted in concise syntheses of the alkaloids brevianamide A (2) and trigonoliimine C (6).
Results and discussion
To begin these efforts, we sought a robust, alkyl-group-tolerant entry to an activated intermediate such as 13 or 14 from 2-alkylindoles, which are either commercially available or accessible in one step using the C–H alkylation method developed by Bach and coworkers.14 Out of these options, we considered 2-alkoxyindoxyl intermediates to be the most feasible precursors. Although we screened several potential methods for their preparation,15 we found that the most user-friendly and scalable option was to treat the indole substrate with oxodiperoxymolybdenum(pyridine)(hexamethylphosphoric triamide)16 (MoOPH, 22, 2.0 equiv.) in a mixture of CH2Cl2 and EtOH at room temperature.Similar oxodiperoxymolybdenum(VI) complexes had been reported for N-acylindole oxidations by Sakamoto17a,b and later by Jimenez,17c along with a single example of the use of 22 with a free N–H 2-alkylindole substrate.17d Using these mild oxidation conditions, phthalimide-containing model substrate 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18 was converted to N–H 2-ethoxyindoxyl 24 in 74% after rapid chromatographic purification on triethylamine-deactivated silica gel (Table 1). In general, however, N–H 2-ethoxyindoxyl compounds displayed variable stability to chromatographic purification (often more severe than 24), which led us to develop a two-step, one-purification protocol from indole to final indoxyl product (vide infra); for optimizing the nucleophile coupling part of that sequence, however, we chose to utilize pure 24.
18 was converted to N–H 2-ethoxyindoxyl 24 in 74% after rapid chromatographic purification on triethylamine-deactivated silica gel (Table 1). In general, however, N–H 2-ethoxyindoxyl compounds displayed variable stability to chromatographic purification (often more severe than 24), which led us to develop a two-step, one-purification protocol from indole to final indoxyl product (vide infra); for optimizing the nucleophile coupling part of that sequence, however, we chose to utilize pure 24.
|
|
|||
|---|---|---|---|
| Entry | Deviation from above | 25 (%) | 26 (%) |
| a Reactions conducted on 0.1 mmol of 24; 1H NMR yields with 1,3,5-trimethoxybenzene as internal standard. b Isolated yield. | |||
| 1 | None | 98 (95)b | 0 |
| 2 | HFIP as solvent | 99 (98)b | 0 |
| 3 | CH2Cl2 as solvent | 19 | 27 |
| 4 | THF as solvent | 0 | 85 |
| 5 | CH3CN as solvent | 6 | 14 |
| 6 | TFA as catalyst | 81 | 0 |
| 7 | TMSOTf as catalyst | 56 | 0 |
| 8 | TMSCl as catalyst | 58 | 0 |
| 9 | TiCl4 as catalyst | 94 | 0 |
| 10 | BF3·OEt2 as catalyst | 86 | 0 |
| 11 | 5 mol% p-TsOH·H2O | 56 | 0 |
| 12 | 3.0 equiv. nucleophile | (80)b | 0 |
| 13 | 1.5 equiv. nucleophile | 72 (63)b | 0 |
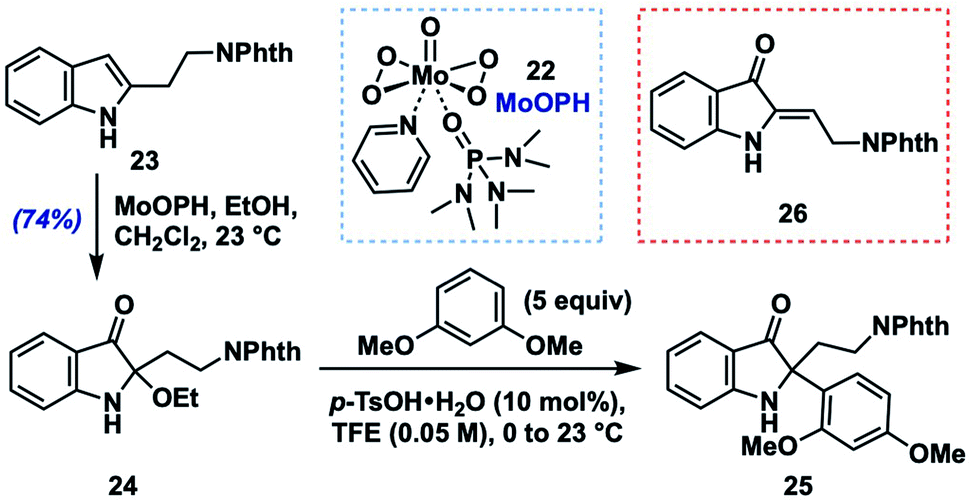
We began by screening conditions for coupling 24 with 1,3-dimethoxybenzene (5 equiv.) as a moderately nucleophilic partner (N = 2.48).13a Initial efforts to employ a catalytic amount of p-toluenesulfonic acid (p-TsOH, 10 mol%) in several common solvents (entries 3–5) at 0 °C to room temperature showed that a key issue was simple elimination of the ethoxy group (presumably via an iminium ion) to give (Z)-enamine 26.19 We found, however, that the fluoroalcohol solvents 2,2,2-trifluoroethanol (TFE) and 1,1,1,3,3,3-hexafluoroisopropanol (HFIP), which are known to stabilize cationic intermediates,20 gave none of the elimination byproduct and provided excellent yields of the desired aryl coupling product 25 (HFIP: 99%; TFE: 98%; entries 1 and 2). In fact, upon screening different acids in TFE, we found that acid identity was not especially important, with several Brønsted acids and Lewis acids (entries 6–10) all providing product to varying degrees (e.g., trifluoroacetic acid: 81%; TiCl4: 94%). In the case of Lewis acids in TFE, we consider these as likely precursors of the corresponding Brønsted acid (e.g., TfOH from TMSOTf) or perhaps able to engage in Lewis acid-assisted Brønsted acid21 catalysis with the protic solvent (e.g., BF3·OEt2). To assess whether p-TsOH in TFE was simply able to reprotonate any enamine 26 that formed to the iminium ion in order to funnel it to product 25, we subjected 26 to the standard reaction conditions (entry 1). Only decomposition of 26 was noted with no 25 formed, suggesting that the TFE solvent either stabilizes the intermediate iminium ion or increases the rate of its reaction with the aryl nucleophile (or both). Efforts to employ a lower loading of nucleophile and acid catalyst (entries 11–13) showed a moderate reduction in yield with 3 equivalents of arene (80%) but a more significant drop with 1.5 equivalents of nucleophile (72%) or 5 mol% p-TsOH (56%). Ultimately, we settled on the use of 10 mol% p-TsOH in TFE as a mild, user-friendly, and relatively inexpensive set of conditions with which to explore the generality of this method.22
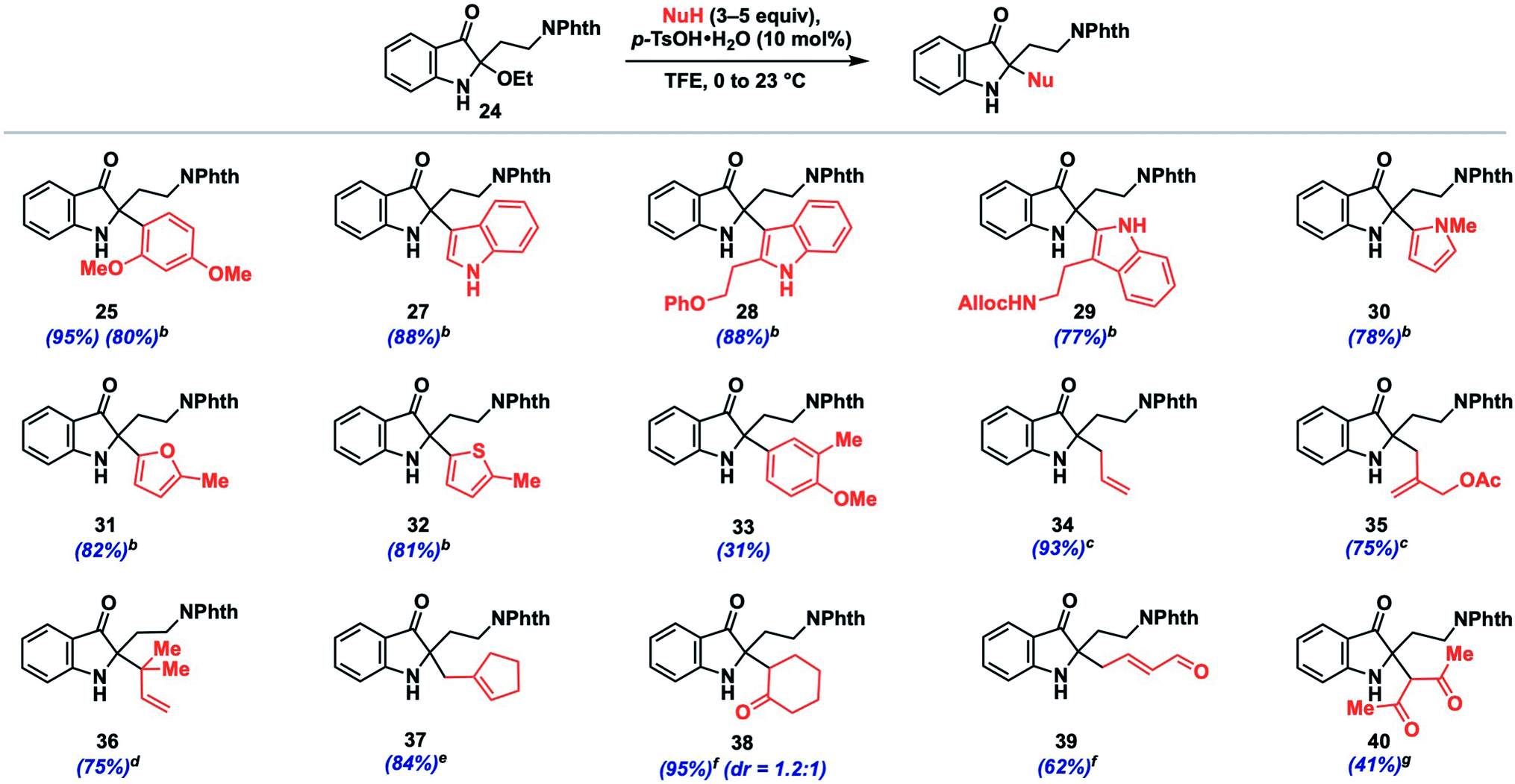
We initiated such studies by exploring the nucleophile scope using purified 24 as the iminium precursor (Table 2). Pleasingly, the reaction tolerated a range of arene and π-nucleophiles. For highly nucleophilic arene partners (N > 5) like indoles and pyrrole, we found that 3 equivalents of nucleophile could be employed, while maintaining high yields (77–88%, 27–30). Other heteroarene partners like 2-methylfuran (82%, 31) and 2-methylthiophene (81%, 32) were also competent under such conditions. A variety of allylations could be conducted with allylsilane, allystannane, or nucleophilic alkene partners to give olefin-containing products (34–37, 75–93%). The coupling efficiency with functionalized nucleophilic partners yielding 35 bearing an allylic acetoxy group (75%) or highly hindered ones which forge vicinal fully-substituted centers (75%, 36) is noteworthy.
| a Reactions conducted on 0.1 mmol of 24 using 5 equiv. of nucleophile unless otherwise noted. b 3 equiv. nucleophile. c From the corresponding allyltrimethylsilane. d n-Prenyltributylstannane as nucleophile. e Methylenecydopentane as nucleophile. f From the corresponding silylenol ether. g Acetylacetone as nucleophile. |
|---|
|
Additionally, we found that carbonyl nucleophiles in the form of silyl enol ethers could couple effectively, while a relatively acidic β-diketone was also a viable substrate; the products from these couplings provide versatile ketone (38, 40) or enal (39) functionalities for further elaboration. Finally, although the nucleophile scope is greater than that observed for related indoxyl syntheses,11,12 we did note that less nucleophilic coupling partners such as anisole (N = −1.18)13b and 2-methylanisole gave either no observed product or only a moderate yield (31%, 33), respectively. Based on our experience, nucleophiles with N > 1 tended to react effectively unless steric factors became dominant (see ESI† for unsuccessful partners).
Next, we explored the tolerance of the method toward different indole substituents using 1,3-dimethoxybenzene and allyltrimethylsilane as representative nucleophiles (Table 3). As mentioned above, the stability of the intermediate 2-ethoxyindoxyl to purification was variable, so the nucleophile coupling reaction was performed on this crude intermediate following work-up to provide a convenient two-step, one-purification transformation. Using an n-butyl group as a model 2-alkyl substituent, the method tolerated a range of substituents on the indole nucleus, including electron-withdrawing (e.g., CN, NO2, CF3, halogen, 41–52; 56–77%, 2 steps) and electron-donating (Me, OMe, 53, 62–63), though the efficiency for the latter systems was lower (37–50%, 2 steps). Substrates bearing N-substitution in the form of benzyl or methyl groups also performed well in the chemistry (62–67, 70–72; 37–78%, 2 steps), and were on average slightly higher yielding compared to their N–H congeners. Side-chains containing benzyloxy (54, 66–67), acetoxy (57), phenyl (56), cyclopropane (64–65), and fluoro (72) functionality were well tolerated, as was a simple 2-methyl substituent (58–59; 37–96%, 2 steps). A substrate containing an acid sensitive Boc-protected amino acid moiety also survived the coupling conditions, providing products 68 and 69 in good yields (65–67%, 2 steps) but with low diastereoselectivity (dr = 1.3–1.4:1).
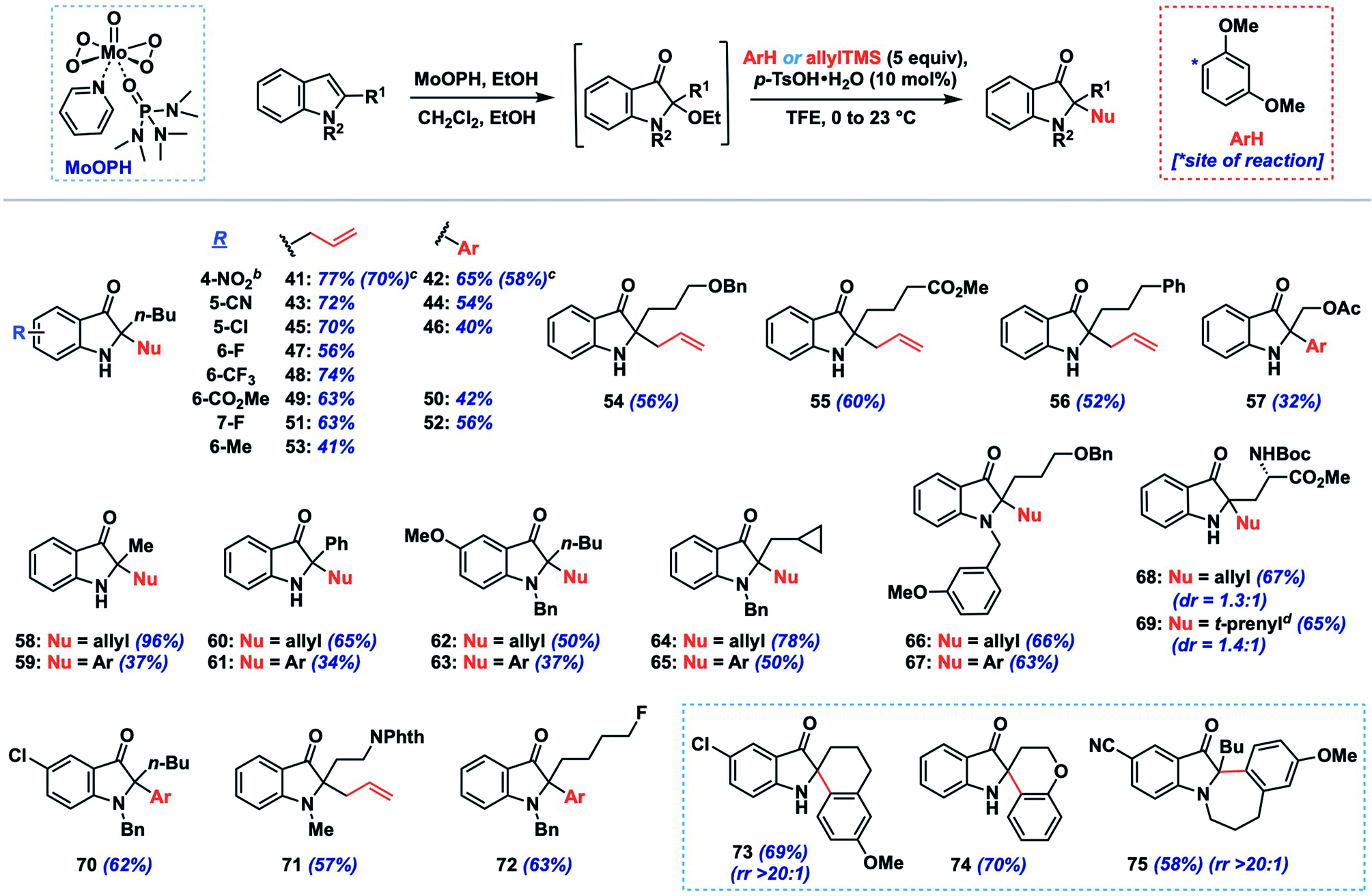
Although not extensively explored here, given our focus on 2-alkylindole systems, we did demonstrate that 2-phenylindole could be converted to indoxyls 60 and 61 in moderate yield (34–65%, 2 steps) using our protocol. Additionally, more complex spirocyclic products (73–74) could be generated in good yield (69–70%, 2 steps) by incorporating nucleophilic arenes into the side-chain, while a fused 7-membered indoxyl was formed via cyclization of an N-tethered methoxyarene (75, 58%). Finally, larger-scale preparations of products 41 and 42 (0.88 g, 4.0 mmol) proceeded in only slightly lower yields than our screening results (70 vs. 77%, 58 vs. 65%, respectively), suggesting that the reaction can provide useful quantities of indoxyl products if required.
With the scope of the method defined, we returned to our original goal of its application to complex molecule synthesis (Scheme 1). As an initial test, we targeted the preparation of the core structure of indoxyl-containing Iboga alkaloids like iboluteine (1)23 represented by spirocyclic Boc-amine 76. We were able to access this polycyclic indoxyl from cyclohexanone adduct 38via a deprotection/reductive amination/protection sequence (Scheme 1A). The two initial steps appeared to converge the mixture of diastereomers of 38 to one observable product, presumably via epimerization α-to the ketone or imine. The final structure and stereochemistry of 76 were confirmed by X-ray crystallographic analysis.
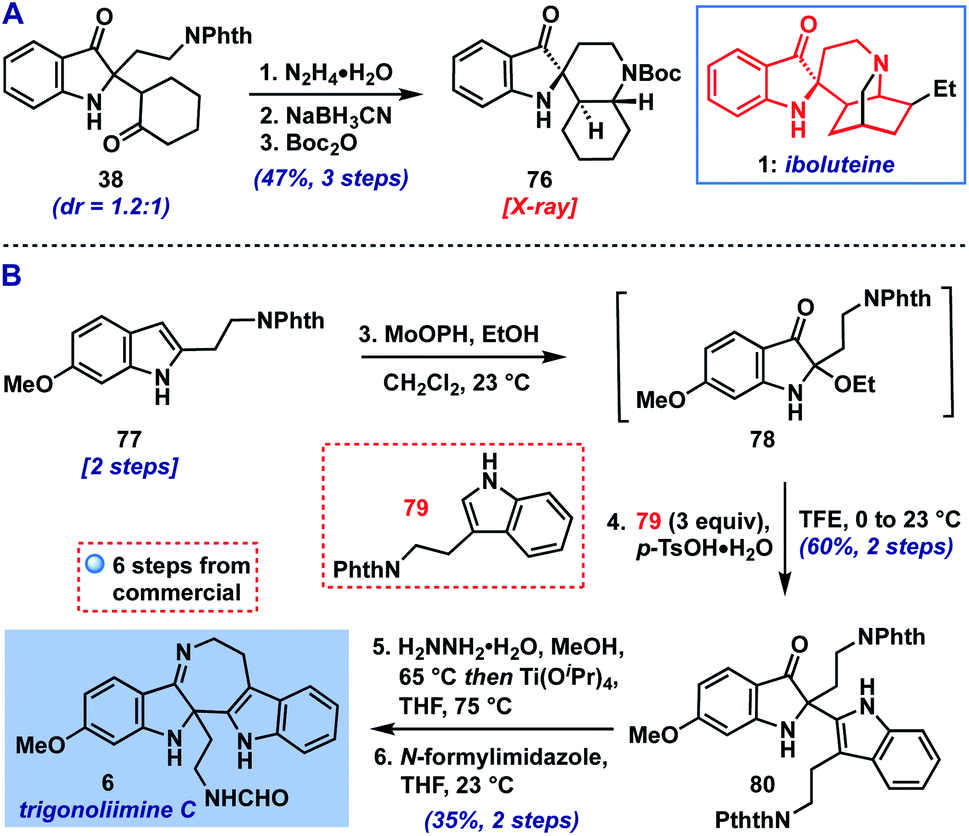 | ||
| Scheme 1 (A) Synthesis of iboluteine core structure (76). (B) Total synthesis of trigonoliimine C (6). | ||
Next, we set our sights on the synthesis of trigonoliimine C (6), an indole alkaloid isolated from the leaves of Trigonostemon lii24 that displays moderate anticancer activity (Scheme 1B).25d Trigonoliimine C (6) has been the subject of three prior total syntheses by the groups of Tambar, Movassaghi, and Ramana, wherein its imine motif was accessed via intramolecular indoxyl-amine condensation.25 Our synthesis of 6 began with the advancement of indole 77 (prepared in two steps from commercial materials, see ESI†) to 2-ethoxyindoxyl intermediate 78, followed by coupling with protected tryptamine 7926 (3 equiv.). This provided the indole addition product 80, a known precursor to 6,25b in 60% yield over the two steps (0.5 mmol scale). Thus, following the reported 2-step sequence,25b,c we completed our synthesis of trigonoliimine C (6) in a total of 6 steps (longest linear sequence).
Finally, we targeted the preparation of the more challenging brevianamide A (2) (Scheme 2), a bicyclo[2.2.2]diazaoctane alkaloid isolated from the fungus Penicillium brevicompactum in 1969 and shown to possess antifeedant activity against a number of crop pests.27 Though many illuminating biogenetic studies and syntheses of targets in this family have been reported over the last four decades,28 the total synthesis of brevianamide A has only very recently been achieved by Lawrence and coworkers (inset, Scheme 2).29 These authors described an elegant biomimetic approach to 2, involving a 1,2-shift to putative indoxyl 90 from transient 3-hydroxyindolenine intermediate 93 (cf.10 → 8, Fig. 1B), followed by a tautomerization/intramolecular [4 + 2] sequence. Overall, their approach provides a short 7-step synthesis of this natural product and its minor diastereomer, brevianamide B. For our part, we hoped to access 90 in a complementary fashion without the need for C-3/C-2-migration by utilizing a 2-substituted indole precursor. Thus, beginning with N-protected amino acid 8130 (one step from commercial L-propargylglycine), we could couple its carboxylic acid with imine 8329,31via activation with Ghosez's reagent32 (82) under modified literature conditions33 in 63% yield; here, heating the mixture of imine 83 and putative acid chloride proved crucial to achieving reasonable yields of enamide 84. Next, a Sonogashira coupling with 2-iodoaniline delivered 2-alkynyl aniline 85 (70%), which could be smoothly cyclized to indole 87 using JohnPhosAu(MeCN)SbF6 (86) in CH2Cl2 at room temperature in 80% yield.34 With 87 in hand, the stage was set for our key transformation to indoxyl 89. In the event, oxidation yielded the expected 2-ethoxyindoxyl intermediate which was stable enough for partial chromatographic purification in this case. Subsequent treatment with prenylstannane 88 under the usual conditions generated the desired product 89 as a mixture of diastereomers (dr = 1.2:1) in 45% yield over the two steps. It is noteworthy that our method is able to tolerate the presence of multiple functionalities in 87 and that reverse prenylation occurs efficiently despite forming two adjacent fully-substituted centers. Although separation of each diastereomer could in principle lead to a single enantiomer of brevianamide A, we elected to advance the diastereomeric mixture together since, in a racemic sense, the diastereomers would converge to the same intermediate at the stage of heterodiene 91. Thus, treatment with NH3 in MeOH29 was able to deprotect the phthalimide and cyclize to diketopiperazine 90, which was stable enough to be purified on silica gel if desired.35 We found, however, that simply concentrating the reaction mixture and subjection to Lawrence's conditions29 (LiOH in H2O) in the same pot was sufficient to deliver brevianamide A (2) in 54% yield, completing a 7-step total synthesis of this complex alkaloid. In our hands, we were unable to isolate any brevianamide B (formed via [4 + 2] on the opposite heterodiene face), though we did note very small amounts by 1H NMR or LC-MS analysis of the crude reaction mixture. As expected, our final sample of 2 displayed only minor enantioenrichment on the basis of its optical rotation (∼9% ee expected based on the 1.2:1 dr of 89).
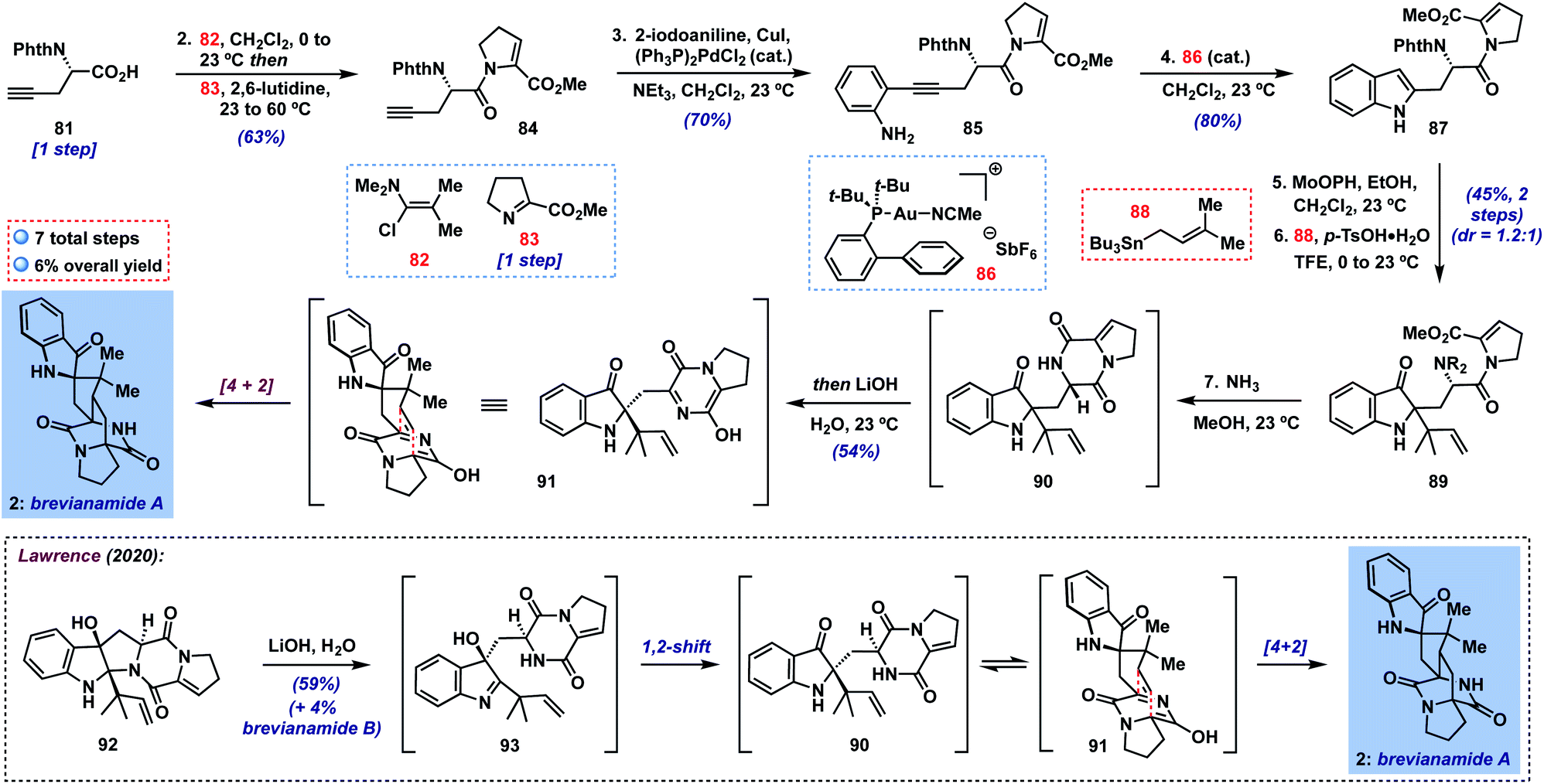 | ||
| Scheme 2 7-step total synthesis of brevianamide A (2). | ||
As a final test of the potential of the present method, we conducted preliminary investigations into an asymmetric variant.36 Initial screens using several chiral Brønsted and Lewis acid catalysts in the coupling of model 2-ethoxyindoxyl 24 with 1,3-dimethoxybenzene have yielded proof of concept (Scheme 3; see ESI† for screening details). Namely, using BINOL-derived N-triflyl phosphoramide 94 (10 mol%) in TFE at 0 °C, we have been able to isolate product 25 in excellent yield (95%) with modest enantioselectivity (24% ee). The low level of asymmetric induction is likely due to the requirement for a fluoroalcohol solvent for productive reaction with 2-alkylindole substrates, which presumably interferes with hydrogen bonding/ion-pairing between the chiral catalyst and the iminium intermediate. Future studies will seek to improve upon this result.
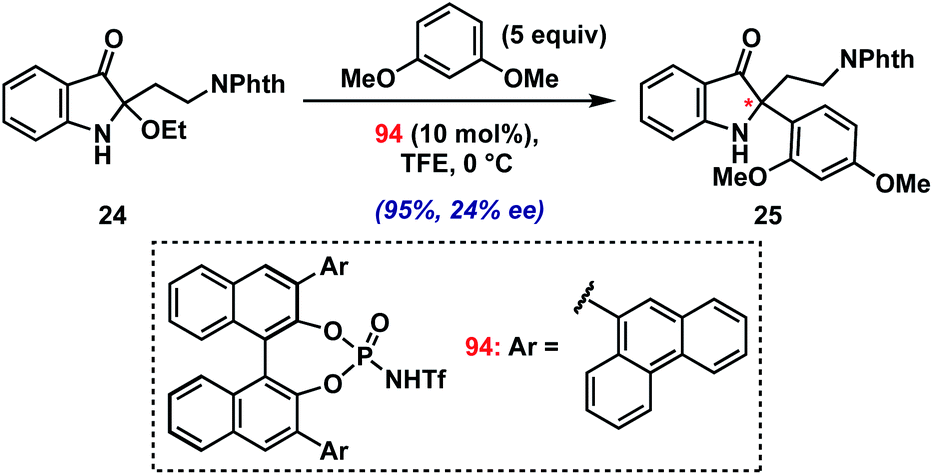 | ||
| Scheme 3 Preliminary asymmetric indoxyl synthesis. | ||
Conclusions
In summary, we have developed an operationally simple method for the conversion of 2-substituted indoles to 2,2-disubstituted indoxyls that is characterized by a broad substrate scope in terms of both indole and nucleophile partners. Our method provides convenient and relatively unique access to such heterocycles with 2,2-dialkyl substitution and free N–H groups, properties which should make it appealing for medicinal chemistry applications. We have showcased its utility in concise total syntheses of trigonoliimine C (6 steps) and brevianamide A (7 steps), being amongst the shortest approaches to these targets reported to date. Future studies will focus on the use of aerobic oxidation and asymmetric catalysis to provide an efficient, enantioselective route to such motifs. Our hope is that the tools communicated here will inspire further exploration of 2,2-disubstituted indoxyls as medicinal agents.Data availability
Data for all compounds in this manuscript are available in the ESI,† which includes experimental details, characterization data, and copies of 1H and 13C NMR spectra. Crystallographic data for compound 76 have been deposited under CCDC 2092889.Author contributions
F. X. and M. W. S. conceived the project. F. X. conducted the experimental studies and M. W. S. directed the research. M. W. S. composed the manuscript with input from F. X., and F. X. compiled the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by UT Southwestern through the W. W. Caruth Jr. Scholarship and the Welch Foundation (I-2045). We thank the Tambar, Ready, Qin, DeBrabander, Chen, and Falck groups (UT Southwestern) for generous access to equipment and chemicals, as well as helpful discussions. We are grateful to Dr Feng Lin, Dr Eric Weaver (UT Arlington), and Dr Vincent Lynch (UT Austin) for assistance with NMR studies, high-resolution mass spectrometry, and X-ray crystallographic analysis, respectively.Notes and references
- For reviews, see: (a) S. Y. Ryabova and V. G. Granik, Pharm. Chem. J., 1995, 29, 809 CrossRef; (b) R. Dalpozzo, G. Bartoli and G. Bencivenni, Chem. Soc. Rev., 2012, 41, 7247 RSC; (c) R. Dalpozzo, Adv. Synth. Catal., 2017, 359, 1772 CrossRef CAS; (d) P. S. Dhote, P. Patel, K. Vanka and C. Ramana, Org. Biomol. Chem., 2021 10.1039/d1ob01285a , accepted manuscript.
- (a) K. S. MacMillan and D. L. Boger, J. Med. Chem., 2009, 52, 5771 CrossRef CAS PubMed; (b) Z. Jukes, G. R. Morais, P. M. Loadman and K. Pors, Drug Discovery Today, 2021, 26, 577 CrossRef CAS PubMed; (c) A. Váradi, G. F. Marrone, T. C. Palmer, A. Narayan, M. R. Szabó, V. Le Rouzic, S. G. Grinnell, J. J. Subrath, E. Warner, S. Kalra, A. Hunkele, J. Pagirsky, S. O. Eans, J. M. Medina, J. Xu, Y.-Xi. Pan, A. Borics, G. W. Pasternak, J. P. McLaughlin and S. Majumdar, J. Med. Chem., 2016, 59, 8381 CrossRef PubMed.
- Indoxyls from 2-alkynyl arylazides: (a) A. Wetzel and F. Gagosz, Angew. Chem., Int. Ed., 2011, 50, 7354 CrossRef CAS PubMed; (b) W. Yong, P. Li, R. Sheng, W. Rao and X. Zhang, ChemistrySelect, 2018, 3, 11696 CrossRef CAS; (c) P. Li, R. Sheng, Z. Zhou, G. Hu and X. Zhang, Eur. J. Org. Chem., 2020, 2146 CrossRef CAS.
- Indoxyls from 2-alkynyl nitroarenes: (a) R.-R. Liu, S.-C. Ye, C.-J. Lu, G.-L. Zhuang, J.-R. Gao and Y.-X. Jia, Angew. Chem., Int. Ed., 2015, 54, 11205 CrossRef CAS PubMed; (b) W. Fu and Q. Song, Org. Lett., 2018, 20, 393 CrossRef CAS PubMed; (c) W. Fu, Y. Zhou and Q. Song, Chem.–Asian J., 2018, 13, 2511 CrossRef CAS PubMed; (d) H. Meng, Z. Xu, Z. Qu, H. Huang and G.-J. Deng, Org. Lett., 2020, 22, 6117 CrossRef CAS PubMed.
- (a) K. Okuma, N. Matsunaga, N. Nagahora, K. Shioji and Y. Yokomori, Chem. Commun., 2011, 47, 5822 RSC; (b) R. O. Torres-Ochoa, T. Buyck, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2018, 57, 5679 CrossRef CAS PubMed.
- J. S. Schneekloth, J. J. Kim and E. J. Sorensen, Tetrahedron, 2009, 65, 3096 CrossRef CAS PubMed.
- (a) K. Higuchi, K. Masuda, T. Koseki, M. Hatori, M. Sakamoto and T. Kawasaki, Heterocycles, 2007, 73, 641 CrossRef CAS; (b) Y. Goriya and C. V. Ramana, Chem. Commun., 2013, 49, 6376 RSC; (c) T.-G. Chen, P. Fang, X.-L. Hou and L.-X. Dai, Synthesis, 2015, 47, 134 CrossRef CAS; (d) C. V. Suneel Kumar and C. V. Ramana, Org. Lett., 2015, 17, 2870 CrossRef CAS PubMed; (e) Z. Xia, J. Hu, Y.-Q. Gao, Q. Yao and W. Xie, Chem. Commun., 2017, 53, 7485 RSC; (f) A. V. Aksenov, D. A. Aksenov, N. A. Aksenov, E. V. Aleksandrova and M. Rubin, J. Org. Chem., 2019, 84, 12420 CrossRef CAS PubMed; (g) X. Wang, J. Zhang, Y. He, D. Chen, C. Wang, F. Yang, W. Wang, Y. Ma and M. Szostak, Org. Lett., 2020, 22, 5187 CrossRef CAS PubMed.
- For reviews on indole dearomatization, see: (a) S. P. Roche, J.-J. Youte Tendoung and B. Tréguier, Tetrahedron, 2015, 71, 3549 CrossRef CAS; (b) C. Zheng and S.-L. You, Chem, 2016, 1, 830 CrossRef CAS; (c) F.-T. Sheng, J.-Y. Wang, W. Tan, Y.-C. Zhang and F. Shi, Org. Chem. Front., 2020, 7, 3967 RSC.
- (a) B. Witkop and J. B. Patrick, J. Am. Chem. Soc., 1951, 73, 2188 CrossRef CAS; (b) X. Zhang and C. S. Foote, J. Am. Chem. Soc., 1993, 115, 8867 CrossRef CAS; (c) Y. Liu and W. W. McWhorter, J. Org. Chem., 2003, 68, 2618 CrossRef CAS PubMed; (d) S. Lerch, L.-N. Unkel and M. Brasholz, Angew. Chem., Int. Ed., 2014, 53, 6558 CrossRef CAS PubMed; (e) W. Ding, Q.-Q. Zhou, J. Xuan, T.-R. Li, L.-Q. Lu and W.-J. Xiao, Tetrahedron Lett., 2014, 55, 4648 CrossRef CAS; (f) E. Schendera, S. Lerch, T. von Drathen, L.-N. Unkel and M. Brasholz, Eur. J. Org. Chem., 2017, 3134 CrossRef CAS; (g) H. Lauwick, Y. Sun, H. Akdas-Kilig, S. Dérien and M. Achard, Chem.–Eur. J., 2018, 24, 7964 CrossRef CAS PubMed.
- (a) A. J. Hutchison and Y. Kishi, J. Am. Chem. Soc., 1979, 101, 6786 CrossRef CAS; (b) R. M. Williams, T. Glinka, E. Kwast, H. Coffman and J. K. Stille, J. Am. Chem. Soc., 1990, 112, 808 CrossRef CAS; (c) P. S. Baran and E. J. Corey, J. Am. Chem. Soc., 2002, 124, 7904 CrossRef CAS PubMed; (d) J. Jeon, S. E. Lee and C.-H. Cheon, Org. Lett., 2021, 23, 2169 CrossRef CAS PubMedFor application of the hydroxyindolenine 1,2-shift, see: (e) Y. Liu and W. W. McWhorter, J. Am. Chem. Soc., 2003, 125, 4240 CrossRef CAS PubMed.
- (a) M. J. Buller, T. G. Cook and Y. Kobayashi, Heterocycles, 2007, 72, 163 CrossRef CAS; (b) K. Higuchi, Y. Sato, S. Kojima, M. Tsuchimochi, K. Sugiura, M. Hatori and T. Kawasaki, Tetrahedron, 2010, 66, 1236 CrossRef CAS; (c) L. Li, M. Han, M. Xiao and Z. Xie, Synlett, 2011, 1727 CAS; (d) Q. Yin and S.-L. You, Chem. Sci., 2011, 2, 1344 RSC; (e) A. Parra, R. Alfaro, L. Marzo, A. Moreno-Carrasco, J. L. García Ruano and J. Alemán, Chem. Commun., 2012, 48, 9759 RSC; (f) M. Rueping, R. Rasappan and S. Raja, Helv. Chim. Acta, 2012, 95, 2296 CrossRef CAS; (g) S. K. Guchhait, V. Chaudhary, V. A. Rana, G. Priyadarshani, S. Kandekar and M. Kashyap, Org. Lett., 2016, 18, 1534 CrossRef CAS PubMed; (h) X. Liu, X. Yan, Y. Tang, C.-S. Jiang, J.-H. Yu, K. Wang and H. Zhang, Chem. Commun., 2019, 55, 6535 RSC; (i) X. Ding, C. Dong, Z. Guan and Y. He, Angew. Chem., Int. Ed., 2019, 58, 118 CrossRef CAS PubMed; (j) X. Liu, X. Yan, J.-H. Yu, Y.-D. Tang, K. Wang and H. Zhang, Org. Lett., 2019, 21, 5626 CrossRef CAS PubMed; (k) A. Singh, S. Vanaparthi, S. Choudhary, R. Krishnan and I. Kumar, RSC Adv., 2019, 9, 24050 RSC; (l) X. Jiang, B. Zhu, K. Lin, G. Wang, W.-K. Su and C. Yu, Org. Biomol. Chem., 2019, 17, 2199 RSC; (m) X. Yan, Y.-D. Tang, C.-S. Jiang, X. Liu and H. Zhang, Molecules, 2020, 25, 419 CrossRef PubMed.
- For representative examples of the ease of oxidative indole dimerization/trimerization: (a) Y.-B. Kong, J.-Y. Zhu, Z.-W. Chen and L.-X. Liu, Can. J. Chem., 2013, 92, 269 CrossRef; (b) F. Lin, Y. Chen, B. Wang, W. Qin and L. Liu, RSC Adv., 2015, 5, 37018 RSC; (c) C. Zhang, S. Li, F. Bureš, R. Lee, X. Ye and Z. Jiang, ACS Catal., 2016, 6, 6853 CrossRef CAS; (d) X.-Y. Zhou, X. Chen, L.-G. Wang, D. Yang and J.-H. Li, Synlett, 2018, 29, 835 CrossRef CAS; (e) B. Deka, M. L. Deb, R. Thakuria and P. K. Baruah, Catal. Commun., 2018, 106, 68 CrossRef CAS; (f) J. Kothandapani, S. M. K. Reddy, S. Thamotharan, S. M. Kumar, K. Byrappa and S. S. Ganesan, Eur. J. Org. Chem., 2018, 2762 CrossRef CAS.
- (a) H. Mayr, T. Bug, M. F. Gotta, N. Hering, B. Irrgang, B. Janker, B. Kempf, R. Loos, A. R. Ofial, G. Remennikov and H. Schimmel, J. Am. Chem. Soc., 2001, 123, 9500 CrossRef CAS PubMed; (b) H. Mayr, B. Kempf and A. R. Ofial, Acc. Chem. Res., 2003, 36, 66 CrossRef CAS PubMed; (c) S. Lakhdar, M. Westermaier, F. Terrier, R. Goumont, T. Boubaker, A. R. Ofial and H. Mayr, J. Org. Chem., 2006, 71, 9088 CrossRef CAS PubMed.
- (a) L. Jiao and T. Bach, J. Am. Chem. Soc., 2011, 133, 12990 CrossRef CAS PubMed; (b) L. Jiao and T. Bach, Synthesis, 2014, 46, 35 Search PubMed.
- Other common oxidants were screened: under similar conditions mCPBA gave some desired product but more byproducts; H2O2 and t-BuOOH gave no reaction (even at 40 °C). A photocatalyzed aerobic oxidation based on that reported by Jiang and co-workers was successful but did not prove scalable above 0.2 mmol, see: ref. 12c.
- E. Vedejs and S. Larsen, Org. Synth., 1986, 64, 127 CrossRef CAS.
- (a) C.-S. Chien, T. Suzuki, T. Kawasaki and M. Sakamoto, Chem. Pharm. Bull., 1984, 32, 3945 CrossRef CAS; (b) C.-S. Chien, T. Takanami, T. Kawasaki and M. Sakamoto, Chem. Pharm. Bull., 1985, 33, 1843 CrossRef CAS; (c) C. I. Altinis Kiraz, T. J. Emge and L. S. Jimenez, J. Org. Chem., 2004, 69, 2200 CrossRef CAS PubMed; (d) A. Karadeolian and M. A. Kerr, J. Org. Chem., 2010, 75, 6830 CrossRef CAS PubMed.
- M. E. Kieffer, L. M. Repka and S. E. Reisman, J. Am. Chem. Soc., 2012, 134, 5131 CrossRef CAS PubMed.
- The stereochemistry of (Z)-26 was confirmed by 1D nOe experiments..
- For a review, see: (a) I. Colomer, A. Chamberlain, M. Haughey and T. J. Donohoe, Nat. Rev. Chem., 2017, 1, 0088 CrossRef CAS ; For the use of p-TsOH/TFE to avoid a competing elimination, see: ; (b) Y. Aota, Y. Doko, T. Kano and K. Maruoka, Eur. J. Org. Chem., 2020, 1907 CrossRef CAS ; For the use of HFIP as solvent in similar reactions with indole nucleophiles, see: ref. 11l..
- For a review, see: H. Yamamoto and K. Futatsugi, Angew. Chem., Int. Ed., 2005, 44, 1924 CrossRef CAS PubMed.
- TFE was selected as solvent given its lower cost compared to HFIP: TFE = $137 per L; HFIP = $207 per L (prices obtained from Oakwood chemical using 1 kg bottles; June 2021).
- Isolation: (a) R. Goutarel and M. M. Janot, Ann. Pharm. Fr., 1953, 11, 272 CAS ; For a recent total synthesis, see: ; (b) G. Zhao, X. Xie, H. Sun, Z. Yuan, Z. Zhong, S. Tang and X. She, Org. Lett., 2016, 18, 2447 CrossRef CAS PubMed.
- C.-J. Tan, Y.-T. Di, Y.-H. Wang, Y. Zhang, Y.-K. Si, Q. Zhang, S. Gao, X.-J. Hu, X. Fang, S.-F. Li and X.-J. Hao, Org. Lett., 2010, 12, 2370 CrossRef CAS PubMed.
- (a) X. Qi, H. Bao and U. K. Tambar, J. Am. Chem. Soc., 2011, 133, 10050 CrossRef CAS PubMed; (b) S. Han and M. Movassaghi, J. Am. Chem. Soc., 2011, 133, 10768 CrossRef CAS PubMed; (c) B. N. Reddy and C. V. Ramana, Chem. Commun., 2013, 49, 9767 RSC; (d) S. Han, K. C. Morrison, P. J. Hergenrother and M. Movassaghi, J. Org. Chem., 2014, 79, 473 CrossRef CAS PubMed.
- P. Caramenti, R. K. Nandi and J. Waser, Chem.–Eur. J., 2018, 24, 10049 CrossRef CAS PubMed.
- (a) A. J. Birch and J. J. Wright, J. Chem. Soc. D, 1969, 644 RSC; (b) R. R. M. Paterson, M. J. S. Simmonds, C. Kemmelmeier and W. M. Blaney, Mycol. Res., 1990, 94, 538 CrossRef CAS.
- For reviews, see: (a) K. A. Miller and R. M. Williams, Chem. Soc. Rev., 2009, 38, 3160 RSC; (b) J. M. Finefield, J. C. Frisvad, D. H. Sherman and R. M. Williams, J. Nat. Prod., 2012, 75, 812 CrossRef CAS PubMed; (c) K. R. Klas, H. Kato, J. C. Frisvad, F. Yu, S. A. Newmister, A. E. Fraley, D. H. Sherman, S. Tsukamoto and R. M. Williams, Nat. Prod. Rep., 2018, 35, 532 RSC . For representative diaza[2.2.2]octane alkaloid syntheses, see: ; (d) R. M. Williams and R. J. Cox, Acc. Chem. Res., 2003, 36, 127 CrossRef CAS PubMed; (e) B. M. Trost, N. Cramer and H. Bernsmann, J. Am. Chem. Soc., 2007, 129, 3086 CrossRef CAS PubMed; (f) S. B. Herzon and A. G. Myers, J. Am. Chem. Soc., 2005, 127, 5342 CrossRef CAS PubMed; (g) P. S. Baran, B. D. Hafensteiner, N. B. Ambhaikar, C. A. Guerrero and J. D. Gallagher, J. Am. Chem. Soc., 2006, 128, 8678 CrossRef CAS PubMed; (h) F. Frebault, N. S. Simpkins and A. Fenwick, J. Am. Chem. Soc., 2009, 131, 4214 CrossRef CAS PubMed; (i) E. V. Mercado-Marin and R. Sarpong, Chem. Sci., 2015, 6, 5048 RSC.
- R. C. Godfrey, N. J. Green, G. S. Nichol and A. L. Lawrence, Nat. Chem., 2020, 12, 615 CrossRef CAS PubMed.
- G. Deng, J. Chen, W. Sun, K. Bian, Y. Jiang and T.-P. Loh, Adv. Synth. Catal., 2018, 20, 3900 CrossRef.
- P. Huy, J.-M. Neudörfl and H.-G. Schmalz, Org. Lett., 2011, 13, 216 CrossRef CAS PubMed.
- B. Haveaux, A. Dekoker, M. Rens, A. R. Sidani, J. Toye and L. Ghosez, Org. Synth., 1979, 59, 26 CrossRef.
- Q. Li and I. B. Seiple, J. Am. Chem. Soc., 2017, 139, 13304 CrossRef CAS PubMed.
- (a) C. Wang and J. Sperry, Org. Lett., 2011, 13, 6444 CrossRef CAS PubMed; (b) A. Gimeno, A. Rodríguez-Gimeno, A. B. Cuenca, C. Ramírez de Arellano, M. Medio-Simón and G. Asensio, Chem. Commun., 2015, 51, 12384 RSC.
- The dihydro congener of 83 was reported to be unable to be isolated due to its instability, see: J. M. Finefield, J. C. Frisvad, D. H. Sherman and R. M. Williams, J. Nat. Prod., 2012, 75, 812 CrossRef CAS PubMed.
- For related asymmetric indoxyl syntheses proceeding via nucleophile addition to 2-aryl-3-oxoindolenines, see: ref. 11e and 11f (a) M. Rueping, S. Raja and A. Núñez, Adv. Synth. Catal., 2011, 353, 563 CrossRef CAS; (b) J.-S. Li, Y.-J. Liu, G.-W. Zhang and J.-A. Ma, Org. Lett., 2017, 19, 6364 CrossRef CAS PubMed; (c) C.-L. Dong, X. Ding, L.-Q. Huang, Y.-H. He and Z. Guan, Org. Lett., 2020, 22, 1076 CrossRef CAS PubMed . For examples involving 2-alkyl substitution, see: ref. 11d and 11j..
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2092889. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03533a |
| This journal is © The Royal Society of Chemistry 2021 |