
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConformational interplay in hybrid peptide–helical aromatic foldamer macrocycles†
Sebastian
Dengler
 ,
Pradeep K.
Mandal
,
Lars
Allmendinger
,
Céline
Douat
and
Ivan
Huc
*
,
Pradeep K.
Mandal
,
Lars
Allmendinger
,
Céline
Douat
and
Ivan
Huc
*
Department of Pharmacy and Center for Integrated Protein Science, Ludwig-Maximilians-Universität, Butenandtstraße 5-13, D-81377 Munich, Germany. E-mail: ivan.huc@cup.lmu.de
First published on 27th July 2021
Abstract
Macrocyclic peptides are an important class of bioactive substances. When inserting an aromatic foldamer segment in a macrocyclic peptide, the strong folding propensity of the former may influence the conformation and alter the properties of the latter. Such an insertion is relevant because some foldamer–peptide hybrids have recently been shown to be tolerated by the ribosome, prior to forming macrocycles, and can thus be produced using an in vitro translation system. We have investigated the interplay of peptide and foldamer conformations in such hybrid macrocycles. We show that foldamer helical folding always prevails and stands as a viable means to stretch, i.e. unfold, peptides in a solvent dependent manner. Conversely, the peptide systematically has a reciprocal influence and gives rise to strong foldamer helix handedness bias as well as foldamer helix stabilisation. The hybrid macrocycles also show resistance towards proteolytic degradation.
Introduction
Ribosomal translation of mRNA into peptidic sequences has been shown to be tolerant to a great variety of peptide chain modifications and non-peptidic appendages.1 In turn, this tolerance has enabled the development of nucleic-acid-encoded display selection methods of non-natural peptides.2 Non-native peptide modifications may indeed bring advantages, e.g. with respect to bioavailability, resistance to proteolytic degradation, or achieving high binding affinity for a given biological target. For example, a beneficial modification is to introduce an electrophilic chloroacetamide at the peptide N-terminus in combination with the encoding of one mandatory cysteine in the peptide sequence.2 Spontaneous substitution of the chlorine by the thiol of the cysteine then gives access to libraries of ribosomally expressed thioether macrocycles, which have been shown to be better candidates than linear peptides as high affinity protein ligands. For the same purpose, libraries of peptide sequences derived from natural amino acids may also be macrocyclised through post-translational modifications.3Recently, it was shown that ribosomal translation tolerates peptide appendages consisting of aromatic amide foldamer sequences of quinoline-based monomer QXxx and pyridine-based monomer P (Fig. 1a).4–6 These appendages are far larger and more remote from peptides than what had been previously envisaged. They can be placed either at the N-terminus, i.e. on the peptide chain initiator,4,5 or within the sequence on an α-amino acid side chain.6 They can also be included within thioether peptide macrocycles such as previously described compound 3 (Fig. 1b). Interest for such peptide modifications stems from the high propensity of QXxx/P sequences to adopt stable helically folded conformations, particularly in water.7,8 The folding propensity of Q units is so high that they promote folding in otherwise flexible units, including α-amino acids,9,10 when these flexible units are inserted within Qn oligomers. The folding propensity of P units is weaker. For example, the rate of interconversion between right-handed (P) and left-handed (M) helical conformers increases when the proportion of P units within a Qn sequence increases,11 and P units promote less ordered conformations in organic solvents.12 P units were required for ribosomal expression, presumably because sequences must transiently unfold to transit through the ribosome exit tunnel.4 In macrocyclic aromatic foldamer–peptide hybrids, the first role of the foldamer is thus to bring prevalent folding information that may influence the peptide conformation. This approach can therefore be related to other synthetic appendages used to induce peptide conformations, e.g. molecular rotors,13 or α-helix staples.14 It also relates to the observation that natural macrocyclic peptides are the object of multiple biosynthetic modifications that expand their functions beyond what can be achieved with proteinogenic α-amino acids only.
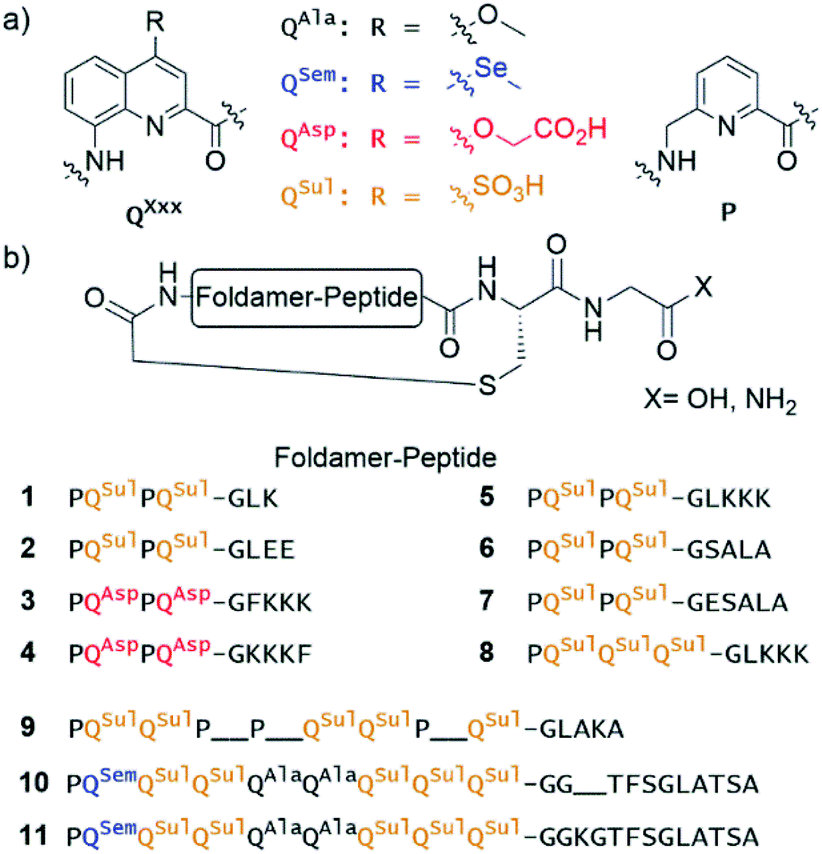 | ||
| Fig. 1 (a) Foldamer aromatic building blocks colour coded according to their side chain composition. (b) Sequence of macrocyclic foldamer–peptide hybrids synthesised on solid-phase. Most of the macrocycles possess a C-terminal carboxylic acid at the exception of 5 and 8 which possess a primary amide. | ||
Herein, we present an investigation of the interplay of foldamer and peptide conformations within such hybrid macrocycles upon varying parameters such as foldamer and peptide segment length and side-chain composition as well as the proportion of P and Q units within the foldamer. We find that, within macrocycles, foldamer helices may have a strong impact on peptide conformation. For example, we introduce the concept of peptide stretching via helical foldamer folding. We show that, concomitantly to conformational effects, the foldamers confer enhanced resistance of the peptides to proteolytic degradation. Furthermore, we observe a systematic reciprocal effect of the peptide whose chirality controls the foldamer helix handedness. The peptide loop also stabilises the foldamer helix conformation, thus acting as a sort of staple. Altogether, our results highlight the singular nature of these macrocyclic hybrids and the interest in involving them in future nucleic acid-encoded display selection experiments.
Results and discussion
Design and synthesis
Eleven hybrid macrocycles were considered that can be divided into two groups (Fig. 1). As a first group, compounds 1–7 and 8 consist of a PQPQ or PQQQ tetraamide foldamer segment, respectively, and peptide chains of variable length and composition. With three consecutive Q units, the foldamer helix of 8 was expected to be conformationally more stable, yet able to undergo handedness reversal in water.15 The QSul monomer was considered instead of the QAsp of previously described compound 3,4 because it confers very good water solubility as well as crystal growth ability.16 The intracyclic peptide segments ranged from three to six amino acids. Depending on the amino acid sequence and on the OH or NH2 C-terminal functionality, these eight compounds encompassed a variety of charge states at neutral pH, between −5 for 2 and +1 for 5.In a second group, we considered longer sequences with a foldamer segment encompassing nine P or Q units, and intracycle peptide segments ranging from five to thirteen amino acids. Compounds 9–11 thus comprise exactly five more aromatic units than 1–8, which amounts to exactly two helix turns.17 Thus, the aromatic helices differ in length across the two series but the anchor points of the peptides on the helices are positioned similarly so as to form a loop on one face of the helix. In 10 and 11, the eight consecutive Q units will confer the aromatic helix with such a high stability that handedness reversal is expected to remain kinetically inert in water.18 In contrast, hybrid macrocycle 9 has a mix of P and Q units, including two consecutive Ps, which was predicted to allow for helix handedness reversal in water at room temperature.11 Compound 9 also has a much shorter peptide segment than 10 and 11, whose peptides differ only by two additional amino acids in 11. The QSem unit was introduced to facilitate X-ray crystal structure elucidation using the anomalous scattering of Se, but this was not implemented as X-ray quality crystals of 10 and 11 were not obtained.
Of note, compounds 1–7 may in principle be expressed by the ribosome using a PQPQ-GF initiator, whereas 8–11 are more rigid (8) or longer (9–11) than what the ribosome has been shown to tolerate until now.4,5 One may nevertheless consider introducing large and rigid foldamer helices, such as those of 8–11 into hybrid macrocycles through a post-translational modification, i.e. after ribosomal translation of the peptide.3 For the purpose of the present study, all compounds were produced by chemical synthesis. The microwave-assisted solid phase synthesis (SPS) of the non-cyclic N-chloroacetamido-terminated foldamer peptide hybrid precursors was performed using established protocols.4,8 The precursors of 5 and 8 were synthesised by iterative coupling on low-loading (LL) Rink amide resin with the use of HBTU for peptide couplings and couplings on the aliphatic amine of P units, and acid chloride activation for couplings on the aromatic amines of Q units (see ESI and Fig. S1† for details).8 The C-terminal primary amide of both 5 and 8 was found to hydrolyse under the conditions used for structural investigations. Consequently, most other oligomers were prepared on LL Fmoc-Gly-Wang resin to yield acid-terminated sequences. We also investigated the benefits of the ProTide™ resin, which contains a PEG and polystyrene core and possesses excellent swelling properties. We found that using the commercially available LL Cl-MPA ProTide™ resin resulted in significant improvements in SPS recovered crude yield and product purity (see ESI†). The precursors of the longest hybrid molecules 9–11 were thus synthesised on this resin.
The non-cyclic precursors of 1, 3, 4 and 9 were prepared using identical SPS protocols to those used for the precursors of 5 and 8. For the precursors of 2, 6, and 7, which share the same foldamer segment, as well as for the precursors of 10 and 11, a fragment condensation approach was developed and implemented. This consisted in the SPS and purification of an Fmoc-foldamer-G-OH segment and then of the coupling of this entire segment to the N-terminal amine of the resin-bound peptide using a phosphonium-based coupling reagent (Fig. S1†). This strategy allows for faster diversification of either the peptide or the foldamer parts, which could be independently constructed and assembled in a second stage. After chloroacetylation and final resin cleavage, the non-cyclic precursors produced by fragment condensation were recovered in high purity and good yield (see ESI†).
Macrocylisations were performed via substitution of the chlorine by the Cys thiol side chain at low concentration (100 μM) with triethylamine as base (see ESI†). The PQPQ-containing sequences 1–5 macrocylised within 30 min in aqueous medium. Reverse phase HPLC (RP-HPLC) monitoring showed that, under the same conditions, it took 3 h to observe full conversion to sequences 6–8, and one day for 10 and 11. RP-HPLC analysis of crude 11 showed two peaks with the same integration. The corresponding products were separated by semi-preparative RP-HPLC. ESI-MS, NMR and circular dichroism (CD) spectroscopies allowed us to identify these compounds as the right-handed P-11 and the left-handed M-11 diastereomers. As planned in the initial design, interconversion between these compounds is kinetically hampered in water at room temperature. The separation of P-10 and M-10 could not be achieved.
Sequence 9, which possesses a short peptide chain with respect to its foldamer segment, did not cyclise in water, but slowly cyclised in DMF. RP-HPLC analysis showed reaction completion after 36 hours. This result unveils that compound 9 may cyclise only when its foldamer helix is partly unfolded, a situation that required both multiple P units and a solvent like DMF that is less favourable to aromatic helix folding. This peculiar behaviour has been further investigated as reported below.
Helix folding and handedness bias in solution
Circular dichroism (CD) spectra of hybrid macrocycles 1–9 all showed bands in the 260–460 nm region (Fig. 2), where the peptides do not absorb light, providing qualitative evidence of helical folding of the foldamer segments, and of helix handedness bias. Thus, we can infer that in macrocycles 1–8 that contain a tetraamide foldamer, and in macrocycle 9 that contains a nonaamide foldamer with multiple P units, helix handedness is dynamic in solution and is biased by the L-chirality of the peptide despite the fact that the helices themselves bear no stereogenic centre. This type of remote communication of stereogenic information requires a transmission mechanism along the macrocycle, a phenomenon that has been observed in other systems.19–23 The positive sign of the CD bands at 331 or 408 nm indicate that P-helicity is favoured regardless of peptide length and sequence.24 In contrast, the diastereomeric conformers P-11 and M-11 obtained by chromatographic separation showed opposite CD spectra that did not evolve with time. In this case, helix handedness is kinetically inert. The same kinetic inertness may be expected for the related macrocycle 10. Thus, the flat CD spectrum of 10 reflects the fact that P-10 and M-10 were recovered as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture after SPS and that their CD spectra cancel each other. The chiral centres on the peptide may in principle stabilise one or the other helical sense, but this influence does not operate because helix handedness inversion is kinetically locked on the laboratory time scale.
:1 mixture after SPS and that their CD spectra cancel each other. The chiral centres on the peptide may in principle stabilise one or the other helical sense, but this influence does not operate because helix handedness inversion is kinetically locked on the laboratory time scale.
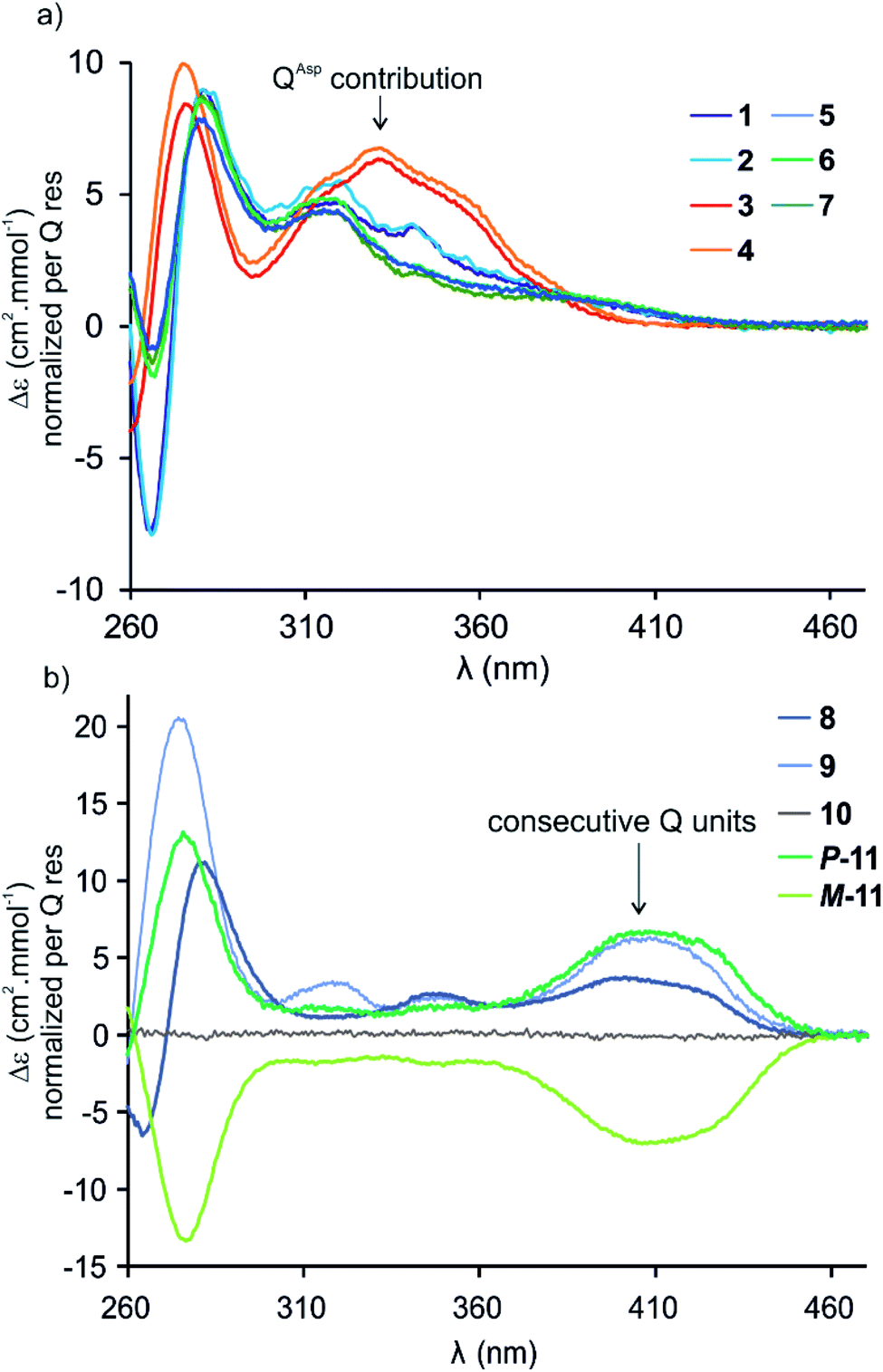 | ||
| Fig. 2 CD spectra of 1–11 in water/acetonitrile (75:25, v/v) at 20 °C. (a) Sequences 1–7 composed of a PQPQ segment. (b) Sequences 8–11 containing consecutive Q units. The P and M helical conformers of 11 are stable and were separated by RP-HPLC. The flat CD spectrum of 10 indicates a 50:50 mixture of P- and M-helical conformers. | ||
By comparison with other chiral Qn oligomers18,24 the intensity of CD spectra of 1–9 (Δε values normalised by Q residue in Fig. 2) suggest that helix handedness bias is strong. Nevertheless, it is difficult to precisely quantify diastereomeric excess on the sole basis of CD intensity because the CD bands vary according to the foldamer sequence. For instance, the band at 408 nm is observed only when consecutive Q units are present (Fig. 2b). Bands at 331 or 320 nm are observed depending on whether Q units are QAsp (3–4), or QSul (1–2) and (5–7), respectively (Fig. 2a).
NMR spectroscopy provided additional evidences of foldamer helical folding and helix handedness bias (Fig. 3). Two typical indicators are the spreading of resonances over a large range of chemical shift values and significant upfield shifts of the signals of protons shielded by ring current effects associated with aromatic stacking within the helix. Typically, the signal of one quinoline H3 proton of PQPQ foldamers 1–2 and 5–7 is found near 8.7 ppm, and the other near 7.9–7.6 ppm (Fig. S3†). In foldamers 3 and 4 which bear different side chains on their Q units, H3 signals are shifted to 7.6–7.4 ppm for one and 6.7–6.5 ppm for the other, but the chemical shift difference nevertheless remains. This difference shows that only one quinoline H3 proton is exposed to ring current effects mediated by aromatic stacking, in agreement with solid state structures of the helices (see below).
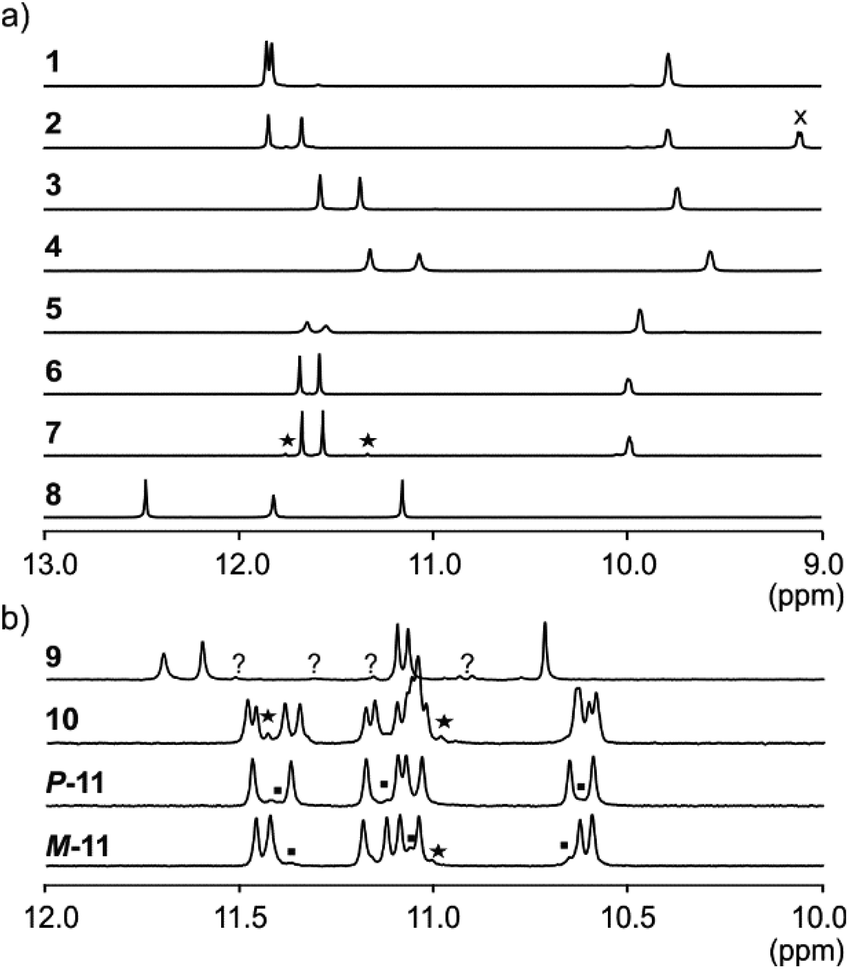 | ||
| Fig. 3 Excerpts from the 500 MHz 1H NMR spectra of (a) compounds 1–8 recorded at 25 °C in H2O/CD3CN 75:25 (v/v) and (b) compounds 9 and 10, 11 recorded at 25 °C in H2O/D2O 9:1 (v/v), and H2O/CD3CN 1:1 (v/v), respectively. The spectra show amide resonances i.e. typically all NHs belonging to Q and some NHs belong to P units. Other NHs belonging to P units are found at lower field and overlap with aromatic resonances. X indicates the signal of an aromatic proton. Resonances marked with stars have been interpreted as impurities. Squares in the spectra of P-11 and M-11 correspond to the other diastereoisomer resulting from incomplete separation. Resonances marked with (?) in 9 could be an impurity or the other diastereomeric conformer. | ||
The 1H NMR aromatic amide signals of P-11 and M-11 were found to differ (Fig. 3b). Their purity confirmed the good separation of these two compounds by RP-HPLC. The chemical shift differences between aromatic NH resonances of these two compounds is remarkable considering that the corresponding protons lie within the helix and not in close proximity of the peptide stereogenic centre. This highlights the fact that diastereomeric conformers should be distinguishable by NMR when they exchange slowly on the NMR timescale. The foldamer helices of 8 and 9 are both expected to undergo slow handedness inversion on the NMR time scale, as do previously described sequences as short as Ac-Q3-G-OH and Ac-QPQPQ-G-OH.4 The presence of a single set of signals in the 1H NMR spectra of 8 and 9 (Fig. 3) thus reflects that handedness bias is quantitative, as far as NMR can detect, in these two cases.
The NMR spectra of 1–7 also show a single set of signals. This may reflect either quantitative helix handedness bias, i.e. exchange is slow but the other diastereomer is not abundant enough to be detected, or that exchange remains fast even at low temperatures. Indeed, fast P/M-helix equilibrium on the NMR timescale might be expected for these compounds on the basis that Ac-PQAspPQAsp-G-OH undergoes fast exchange at room temperature.4 We probed the anisochronicity of the main chain NH–CH2-aryl methylene protons in the centre of the PQSulPQSul foldamer segment of 1, 2, 5 and 7 (Fig. S5†). Total correlation spectroscopy (TOCSY) spectra recorded at different temperatures showed distinct signals for the two protons and variable Δδ values. However, this information alone remains inconclusive: Clayden et al. have thoroughly illustrated how a chemical shift difference Δδ between diastereotopic protons is compatible with fast exchange combined with helix handedness bias.25 Thus the observed anisochronous CH2 signals do not directly inform about helix handedness dynamics and the extent of handedness bias. We therefore decided to synthesise achiral compounds to assess whether exchange is slow or fast on the NMR timescale and whether the peptide loop may stabilise the helix.
Stabilisation of the foldamer helix by the peptide loop
Macrocycle 13 (Fig. 4a) is achiral: it contains five glycines and a cysteamine instead of the cysteine of other macrocycles. It was prepared by SPS using commercially available cysteamine 2-chlorotrityl resin. Compound 12 was prepared as a non-cyclic control.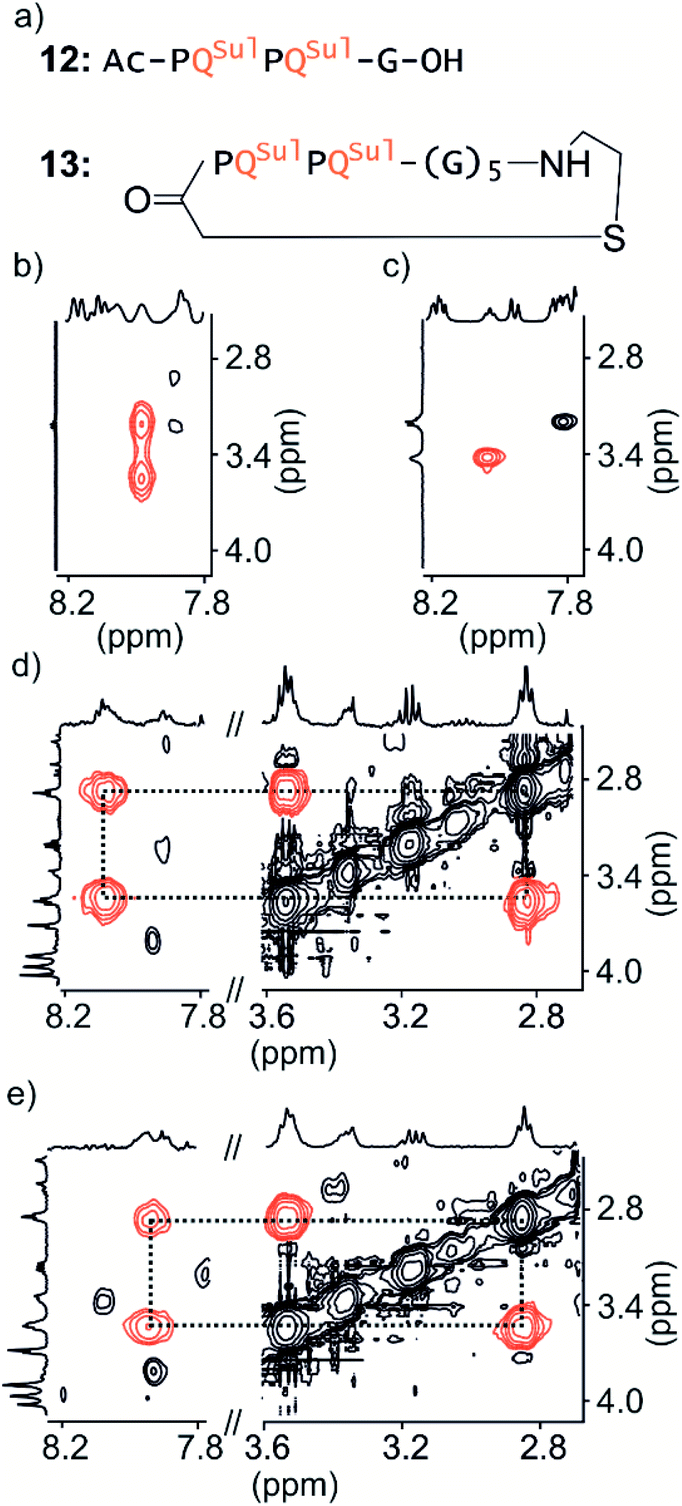 | ||
| Fig. 4 Parts of TOCSY spectra showing NH–CH2J-couplings of 12 (a) in 50 mM NaHCO3 in H2O/D2O (9:1 v/v) at −5 °C (b) and 25 °C (c) respectively and of 13 (a) in 12.5 mM NH4OAc in H2O/CD3CN (75:25 v/v) at 0 °C (d) and 25 °C (e) respectively. | ||
We have shown before that the benzylic diastereotopic CH2 protons of P units have different chemical shift values in the context of aromatic helices: they are anisochronous.8 However, if the P and M conformers equilibrate rapidly on the NMR timescale, only average signals are observed. In basic medium (50 mM NaHCO3), the TOCSY spectrum of 12 show no anisochronicity of these CH2 protons at room temperature indicating fast helix handedness inversion (Fig. 4c and S6†), as was previously observed for Ac-PQAspPQAsp-G-OH.4 Upon cooling to −5 °C, diastereotopic CH2 signals coupled to amide NH groups split (Fig. 4b), indicating that slow exchange is reached. Unfortunately, cyclic compound 13 is not well soluble in this medium and showed only broad peaks. Measurements were instead performed in H2O/CD3CN 75:25 (v/v). In the presence of acetonitrile, the signals of diastereotopic protons of 12 no longer split, even at −10 °C, indicating that helix handedness inversion is faster in the presence of acetonitrile. In contrast, the spectrum of 13 shows anisochronous signals for its diastereotopic protons even at 25 °C (Fig. 4d, e and S7†) with Δδ values up to 0.68 ppm. We thus conclude that the peptide loop slows down helix handedness inversion, i.e. that it stabilises the foldamer helix. This result also indicates that the single set of NMR signals of 1–7 reflect slow exchange combined with quantitative handedness control of the foldamer helix by the chiral peptide.
Solid state structure elucidation
The solid state structures of foldamer peptide macrocycles 1, 7 and 9 were elucidated by single crystal X-ray crystallography, in addition to the previously described structure of 3.4 Racemic crystallography26 had proven essential in the case of 3 as well as for other chiral foldamers and peptides.9,27 In anticipation, D-1 and D-9 were synthesised and mixed with the L-enantiomer to produce racemic mixtures. Eventually, this proved unnecessary in the case of 1 since single crystals of D-1 diffracting at atomic resolution (0.8 Å) were obtained (Fig. S9†). The structure was solved in space group P65. The image shown in Fig. 5a is actually the mirror-image conformation of D-1 to allow for comparison with the L-enantiomers of the other structures. We also obtained crystals of 7 that diffracted at 1.06 Å, and the structure could be solved in space group P1 with eight independent molecules in the asymmetric unit (Fig. 5c, d and S10†). In the structures of 1 and 3, and in the eight independent molecules of 7, the aromatic foldamer PQPQ segment was found to invariably adopt the same compact helical conformation whose ends are connected by a peptide loop. Changing loop size and sequence has no visible effect on foldamer conformation. The helices are right-handed in agreement with the sign of the CD bands in solution (Fig. 2a).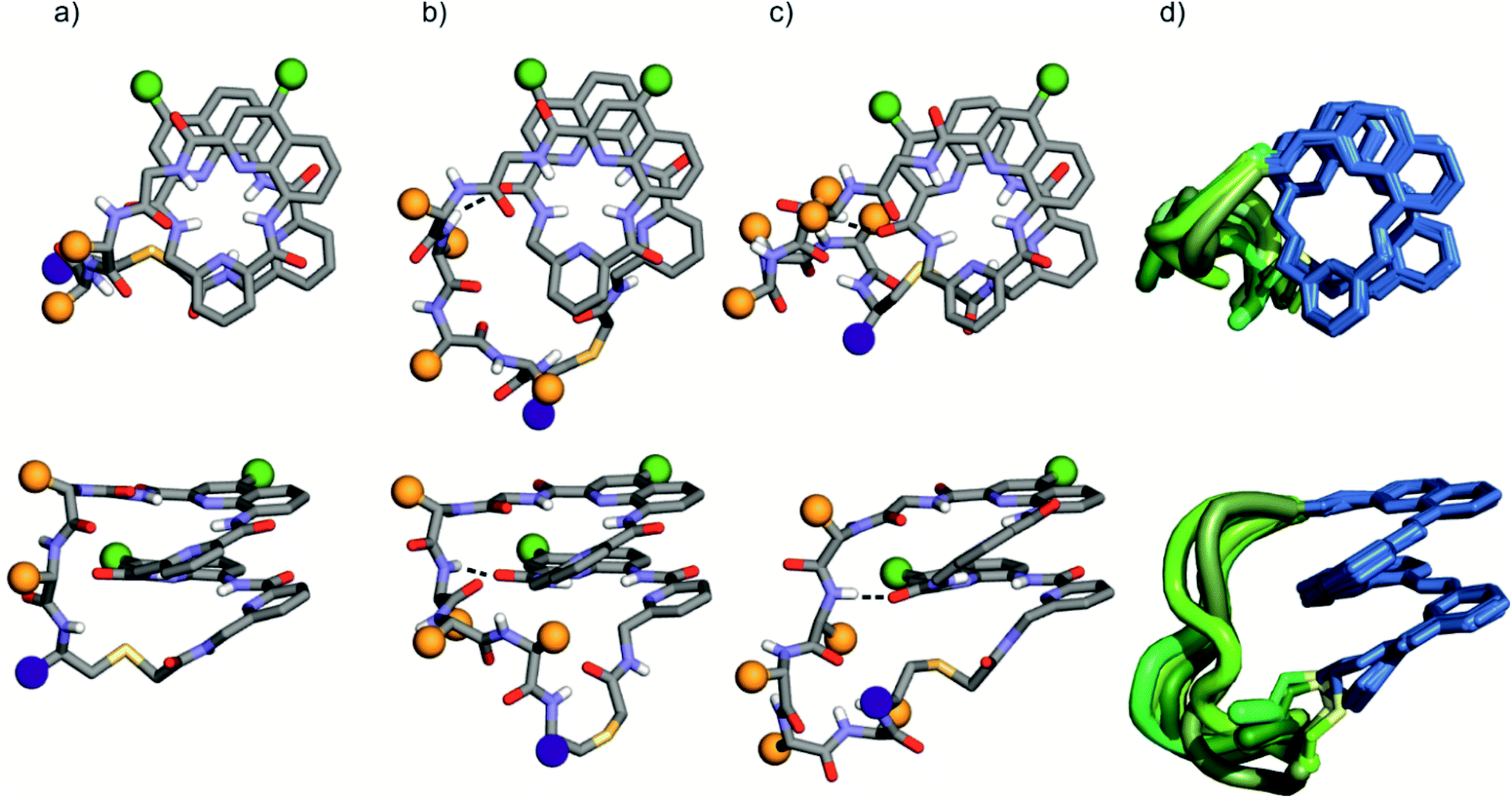 | ||
| Fig. 5 Solid-state structures of 1 (a), 3 (b), and 7 (c) in ball and stick representation shown in top views (above) and side views (below). Amino acid side chains in the peptide loop are represented as orange spheres. Foldamer side chains are shown as green spheres, and peptide C-termini are shown as purple spheres. Peptide-foldamer hydrogen bonding is shown as dashed lines. (d) Overlay of eight different conformers obtained from the crystal structure of 7. The foldamer is represented in blue, the thioether linker in yellow, and the peptide loops in different green tones. | ||
An inter-segment hydrogen bond between the NH of the third amino acid of the peptide segment and an amide CO of the foldamer segment had been previously seen in the structure of 3.4 This interaction was invoked as a possible mechanism for the transmission of stereochemical information from the peptide to the foldamer handedness. The same hydrogen bond can be observed in the structure of 3 and in six out of the eight structures of 7, and also in the structure of 9 (see below). However, no such interactions are observed within the shorter peptide loop of 1. How stereochemical information is conveyed from the peptide to the foldamer is thus not always apparent. The mechanism may not necessarily entail the stabilisation of the observed favoured diastereomeric conformer but may instead rest on the destabilisation of the less favoured conformer. Furthermore, stereochemical information may also be conveyed directly through the main chain.20,21 The crystal structures also reveal that all peptide side chains point away from the foldamer. This hints at the possibility that handedness bias is independent from the peptide sequence, as seems to be the case for all macrocycles in this study, despite their sequence and charge state variability.
The eight molecules of 7 highlight some differences in the long peptide loop conformation (Fig. 5d and S10†) which may contain intrapeptide hydrogen bonds. When the peptide loop is short with respect to helix length as in 1, its conformational freedom is reduced because the helical folding of the foldamer amounts to stretching the peptide segment. Peptide stretching was also obvious in the crystal structure of 9 which was obtained concurrently (Fig. 6). In this case, crystals of the pure enantiomer only diffracted at a resolution of 2.4 Å whereas the racemate produced crystals that diffracted at 1.2 Å. The X-ray structure of the racemate could be solved and refined in the P1 space group. The asymmetric unit contains three molecules of each enantiomer where L-9 adopted a canonical right-handed helix conformation and its D-counterpart the opposite handedness (Fig. 6a and S11†). Thus, the foldamer helix folds despite the relatively short peptide which is stretched as a result. The six conformers were overlaid after inverting the three D-enantiomers (Fig. 6b), showing high conservation of the helix shape and minimal variations of the peptide loop. A certain analogy may be drawn between the effect of the foldamer and the effect of the multiple staples that have been shown to stabilise α-helical peptide conformations.14 Inserting a rigid component in a peptide macrocycle may favour an extended or a helical peptide conformation, depending on the rigid component's length.
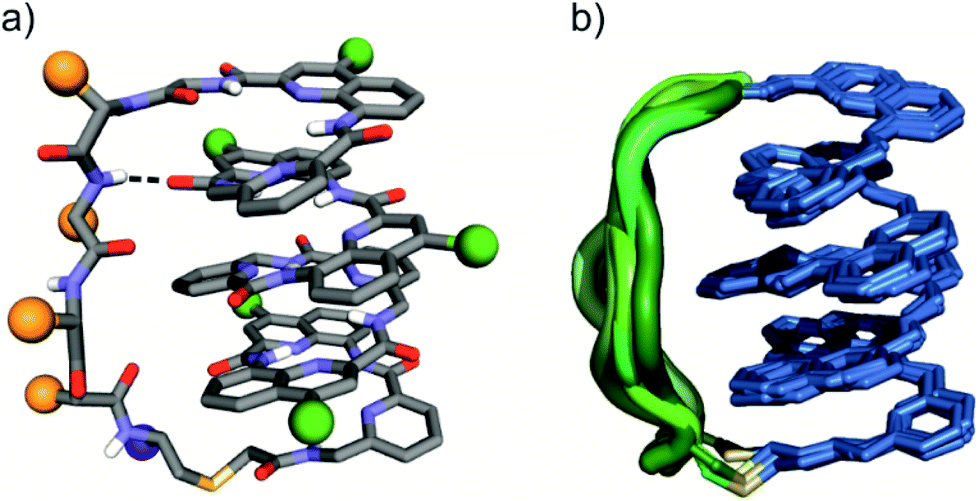 | ||
| Fig. 6 (a) Solid state structure of 9 in ball and stick representation; amino acid side chains in the peptide loop are represented as orange spheres; foldamer side chains are shown as green spheres; the C-terminus is shown as a purple sphere; and a peptide–foldamer hydrogen bond is shown as a dashed line. (b) Overlay of six different conformers obtained from the crystal structure of 9; the foldamer represented in blue, the thioether linker is represented in yellow and the peptide loops are represented in different green tones. | ||
The peptide ϕ and ψ angles of the solid state structure of 1, 3, 7 and 9 were measured and reported in a Ramachandran plot28 (Table S1,†Fig. 7). The plot showed that a wide range of values, typical of α-helices and β-sheets, are covered. All of these values are within the classical favoured regions. No values were found within the left-handed α-helix area, unlike when individual amino acids are inserted within Qn sequences.10 Thus, the influence of the foldamer helices on the peptides do not extend to the induction of disfavoured conformations.
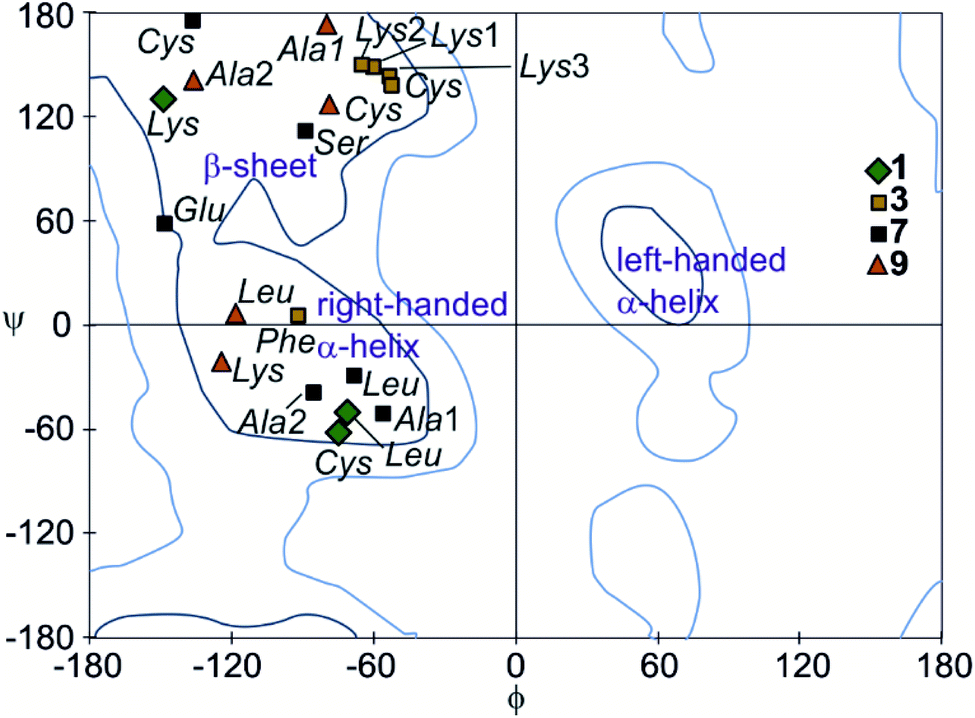 | ||
| Fig. 7 Peptide chain dihedral angles (phi and psi) obtained from X-ray crystal structures of compounds 1, 3, 7, and 9 shown in a Ramachandran plot. Dark blue lines represent favoured regions; light blue lines represent allowed regions. | ||
Foldamer folding induces peptide extension
The stretched peptide in the solid state conformation of 9 likely explains why the macrocyclisation step that produces 9 did not work in water, unlike the other macrocyclisation reactions reported here, and why macrocyclisation of 9 proceeded smoothly in DMF. The foldamer segment of 9 contains four flexible P units out of nine monomers. The P units were introduced to accelerate helix handedness inversion and allow for helix sense bias to take place in water. The helix destabilising effect of P units is enhanced in organic solvents to the point that (PQ)n oligomers do not adopt a canonical helix conformation in chloroform but instead include 90° kinks between PQ dimers.12 Conversely, water stabilises canonical helix conformations7 and (PQ)n oligomers are normally folded in water.8 Compound 9 may not have cyclised in water precisely because of its well-folded structure, so the peptide would have had to stretch for cyclisation to take place. In contrast, the foldamer may not be fully folded or not always folded in DMF, thus allowing for cyclisation without stretching the peptide (Fig. 8a). To test this hypothesis, we measured the CD spectrum of D-9 in DMF and monitored its change as water is progressively added (Fig. 8b). Note that D-9 has opposite chirality to 9 and thus an opposite CD spectrum (compound D-9 was available, and a resynthesis of L-9 for this experiment was thus unneeded). We found that a weaker CD signal is observed in pure DMF than in water, and that its sign suggests P-handedness, thus confirming a different folding behaviour in this solvent. Upon adding water, the CD signal progressively evolves to that of the one handed M-helix characterised by NMR and X-ray crystallography. In contrast, no such solvent-induced variation is observed when performing the same experiment with shorter macrocycle 8 (Fig. S4†), whose PQ3 segment folds regardless of solvent. These results thus allow to formulate the original concept of peptide extension directed by solvent-induced aromatic foldamer folding.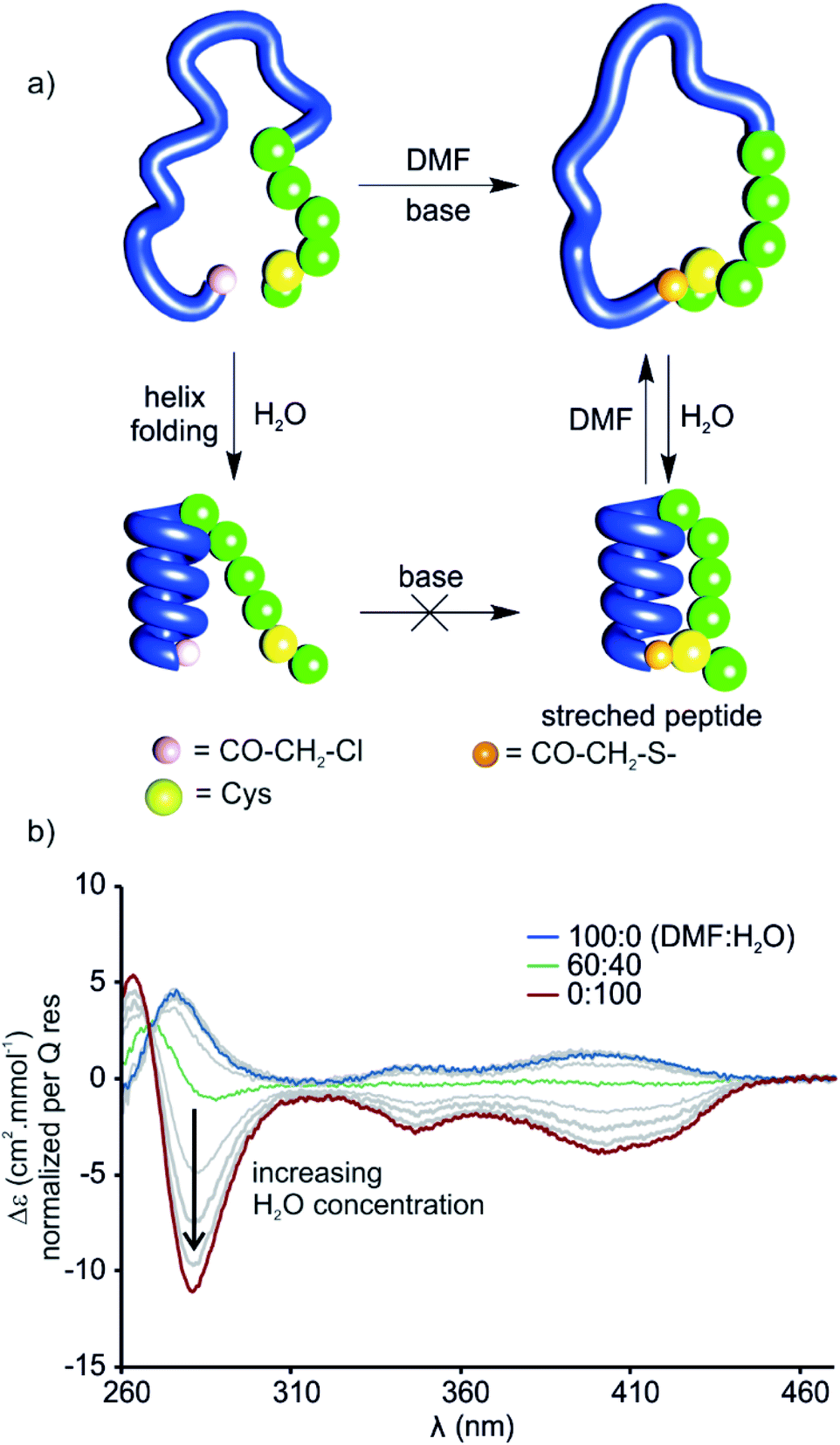 | ||
| Fig. 8 (a) Cartoon representation of the folding of a macrocyclic foldamer–peptide hybrid (compound D-9, foldamer segment depicted as a blue strand, α-amino acid residues represented as green spheres) in DMF and water before and after cyclisation. Thioether formation (the cysteine thiol reacts with chloroacetamide under basic conditions) progressed rapidly in DMF whereas in water no cyclisation was observed. Folding of the helix could be induced in an aqueous environment which resulted in stretching of the peptide segment in the macrocycle. (b) CD-spectra of D-9 in various DMF/H2O mixtures at 25 °C. M-helical handedness of the foldamer is promoted by increasing water concentration (grey lines represent increasing water concentration in 10% steps). | ||
The foldamer confers resistance to proteolytic degradation
We further evaluated the proteolytic stability of the macrocyclic foldamer-peptide hybrids. Improved stability in biological media would be of interest for their potential future translatability as therapeutic molecules as demonstrated for other peptidic macrocycles, for example in the field of protein–protein interaction modulation. We focused on sequences 1, 3 and 4, the latter two being sequence isomers, and assessed their degradation by three proteases, namely α-chymotrypsin, trypsin and pronase E (Fig. 9 and S12†). Chymotrypsin cleaves amide bonds on the C-terminal side of hydrophobic and/or aromatic residues (Leu or Phe). Trypsin cleaves on the C-terminal side of positively charged residues (Lys). Pronase E acts at many different sites of α-peptides. In order to assess the importance of macrocyclisation in resistance towards proteolytic degradation, non-cyclic analogues 14, 15 (Fig. 9) and 16 (see ESI†) were synthesised as reference compounds for 1, 4 and 3, respectively. To avoid problems with oxidation, Cys was replaced by Ala in the acyclic molecules.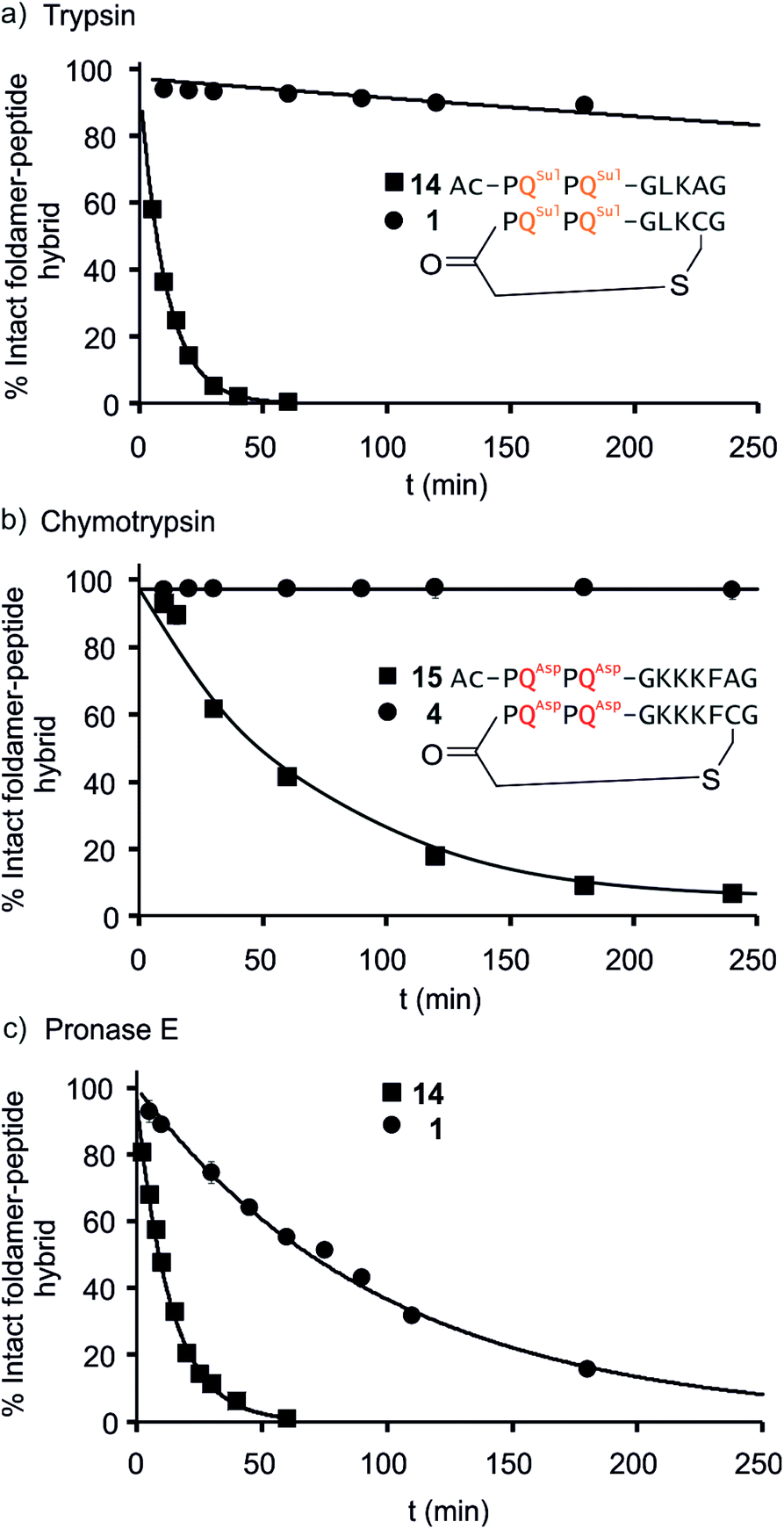 | ||
| Fig. 9 Representative enzymatic degradation of foldamer–peptide hybrids in the presence of (a) trypsin E; (b) α-chymotrypsin; and (c) pronase E. The decreasing abundance of intact macrocyclic compound 1 [(a) and (c)] and compound 4 [(b)] (circles) are shown along with their respective non-cyclic analogues 14 and 15 (squares). The percentage of intact foldamer–peptide hybrid was calculated as the area under the HPLC chromatogram corresponding to the foldamer–peptide hybrid at each time point relative to t = 0. Lines are for guiding the eye only, they do not represent a fit. | ||
The kinetics of proteolysis was monitored by RP-LC-MS.29,30 Representative degradation curves are depicted in Fig. 8 and S13.† In all cases, acyclic analogues containing cognate cleavage sites were degraded rapidly and had disappeared within one to four hours under the conditions used. In contrast, macrocycle 4 was completely resistant to degradation by chymotrypsin (Fig. 9b) and above 90% of 1 was still present in trypsin-containing medium after 250 min (Fig. 9a). Compound 1 was more prone to degradation by pronase E, with a half-life of t1/2 = 69 min (Fig. 9c). But this degradation was still considerably slower than that of its acyclic analogue 14 (t1/2 = 9 min). Compound 3 was more sensitive than 1 to the action of trypsin – due to the fact that it possesses three Lys residues instead of one – and pronase E, but its degradation was nevertheless slower (t1/2 = 41 min) than that of its acyclic analogue 16 (t1/2 = 3.6 min, Fig. S12†).
LC-MS analyses also allowed us to characterise the degradation products (Fig. S13–S17†). The action of pronase E on macrocycle 1 induced the loss of central Leu–Lys dipeptide, which entails hydrolysis after Gly and before Cys, but it is unclear which of these two cleavages occurs first since the second cleavage is faster and intermediates do not accumulate (Fig. S13†). The non-cyclic analogue 14 evolved into the Ac-PQSulPQSul-G fragment and intermediates show that cleavage occur at multiple sites. As anticipated, the action of trypsin on 14 induced the loss of the Ala–Gly dipeptide (Fig. S14†). The same dipeptide was released during the incubation of 15 with chymotrypsin (Fig. S15†). As mentioned above, macrocycles 1 and 4 are much less prone to these cleavages. For sequences 3 and 16 LC-MS analysis confirmed that trypsin cleaves at different lysine residues whereas pronase has a different degradation pattern (Fig. S16 and S17†). Overall, these experiments highlight that proteolytic degradation is dependent on which protease and which peptide sequence are involved, but that in all cases, significant resistance is conferred by insertion of the peptide in a hybrid macrocycle containing a foldamer helix. These results bode well for the possible use of such macrocycles in vivo.
Conclusions
Our results demonstrate extensive conformational interplay between peptides and helical aromatic foldamers within hybrid macrocycles. The peptide loop stabilises the foldamer helix conformations and quantitatively biases its handedness. Conversely, aromatic helix folding restricts the peptide conformation to an extended structure when the length of the two segments (peptide vs. folded helix) match. We also showed that proteolytic degradation of the peptide is slowed down considerably within these hybrid macrocycles. Altogether these results suggest that hybrid foldamer–peptide macrocycles extend the range of properties beyond what can be achieved with peptide macrocycles alone. Further investigation may concern the contribution of the foldamer segment to enhance the cell penetration of the peptide,31 or to enhance the peptide's ability to bind to a protein target. Progress along these lines is being made and will be reported in due course.Data availability
Crystallographic data for 1, 7, and 9 has been deposited at the CCDC under accession numbers 2002474, 2010173 and 201013, respectively, and can be obtained from https://www.ccdc.cam.ac.uk.Author contributions
S. D. carried out most of the experimental work. P. K. M. assisted with crystal growth, X-ray data collection, crystal structure elucidation and refinement. L. A. carried out NMR data collection. C. D. contributed to synthesis and CD studies, and co-supervised the work. I. H. conceptualised and supervised the study. S. D., C. D. and I. H. wrote the manuscript. All authors commented the data and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr J. Basquin and Dr G. Pompidor for assistance in synchrotron measurement at the PXII beamline at Swiss Light Source (SLS, Villigen, Switzerland) and the P13 beamline operated by EMBL Hamburg at PETRA III storage ring (DESY, Hamburg, Germany) respectively.Notes and references
- C. J. Noren, S. J. Anthony-Cahill, M. C. Griffith and P. G. Schultz, Science, 1989, 244, 182 CrossRef CAS PubMed; K. Josephson, M. C. T. Hartman and J. W. Szostak, J. Am. Chem. Soc., 2005, 127, 11727 CrossRef PubMed; Y. V. Schlippe, M. C. Hartman, K. Josephson and J. W. Szostak, J. Am. Chem. Soc., 2012, 134, 10469 CrossRef PubMed; Y. Sako, J. Morimoto, H. Murakami and H. Suga, J. Am. Chem. Soc., 2008, 130, 7232 CrossRef PubMed; H. Neumann, K. Wang, L. Davis, M. Garcia-Alai and J. W. Chin, Nature, 2010, 464, 441 CrossRef PubMed; T. Kawakami, K. Ogawa, T. Hatta, N. Goshima and T. Natsume, ACS Chem. Biol., 2016, 11, 1569 CrossRef PubMed.
- Y. Goto, A. Ohta, Y. Sako, Y. Yamagishi, H. Murakami and H. Suga, ACS Chem. Biol., 2008, 3, 120 CrossRef CAS PubMed; Y. Yamagishi, I. Shoji, S. Miyagawa, T. Kawakami, T. Katoh, Y. Goto and H. Suga, Chem. Biol., 2011, 18, 1562 CrossRef PubMed; S. A. K. Jongkees, S. Caner, C. Tysoe, G. D. Brayer, S. G. Withers and H. Suga, Cell. Chem. Biol., 2017, 24, 381 CrossRef PubMed; T. Passioura, W. Liu, D. Dunkelmann, T. Higuchi and H. Suga, J. Am. Chem. Soc., 2018, 140, 11551 CrossRef PubMed.
- P. Timmerman, J. Beld, W. C. Puijk and R. H. Meloen, ChemBioChem, 2005, 6, 821 CrossRef CAS PubMed; C. Heinis, T. Rutherford, S. Freund and G. Winter, Nat. Chem. Biol., 2009, 5, 502 CrossRef PubMed; S. S. Kale, C. Villequey, X. D. Kong, A. Zorzi, K. Deyle and C. Heinis, Nat. Chem., 2018, 10, 715 CrossRef PubMed; D. E. Streefkerk, M. Schmidt, J. H. Ippel, T. M. Hackeng, T. Nuijens, P. Timmerman and J. H. van Maarseveen, Org. Lett., 2019, 21, 2095 CrossRef PubMed; G. K. Mothukuri, S. S. Kale, C. L. Stenbratt, A. Zorzi, J. Vesin, J. Bortoli Chapalay, K. Deyle, G. Turcatti, L. Cendron, A. Angelini and C. Heinis, Chem. Sci., 2020, 11, 7858 RSC.
- J. M. Rogers, S. Kwon, S. J. Dawson, P. K. Mandal, H. Suga and I. Huc, Nat. Chem., 2018, 10, 405 CrossRef CAS PubMed.
- C. Tsiamantas, S. Kwon, C. Douat, I. Huc and H. Suga, Chem. Commun., 2019, 55, 7366 RSC.
- C. Tsiamantas, S. Kwon, J. M. Rogers, C. Douat, I. Huc and H. Suga, Angew. Chem., Int. Ed., 2020, 59, 4860 CrossRef CAS PubMed.
- T. Qi, V. Maurizot, H. Noguchi, T. Charoenraks, B. Kauffmann, M. Takafuji, H. Ihara and I. Huc, Chem. Commun., 2012, 48, 6337 RSC.
- B. Baptiste, C. Douat-Casassus, K. Laxmi-Reddy, F. Godde and I. Huc, J. Org. Chem., 2010, 75, 7175 CrossRef CAS PubMed.
- M. Kudo, V. Maurizot, B. Kauffmann, A. Tanatani and I. Huc, J. Am. Chem. Soc., 2013, 135, 9628 CrossRef CAS PubMed.
- X. Hu, P. K. Mandal, B. Kauffmann and I. Huc, ChemPlusChem, 2020, 85, 1580–1586 CrossRef CAS PubMed.
- M. Vallade, P. Sai Reddy, L. Fischer and I. Huc, Eur. J. Org. Chem., 2018, 5489 CrossRef CAS.
- N. Delsuc, F. Godde, B. Kauffmann, J.-M. Léger and I. Huc, J. Am. Chem. Soc., 2007, 129, 11348 CrossRef CAS PubMed.
- D. B. Diaz, S. D. Appavoo, A. F. Bogdanchikova, Y. Lebedev, T. J. McTiernan, G. dos Passos Gomes and A. K. Yudin, Nat. Chem., 2021, 13, 218 CrossRef CAS PubMed.
- Y. H. Lau, P. De Andrade, Y. Wu and D. R. Spring, Chem. Soc. Rev., 2014, 44, 91 RSC; G. L. Verdine and G. J. Hilinski, Methods Enzymol., 2012, 503, 3 Search PubMed; C. E Schafmeister, J. Po and G. L. Verdine, J. Am. Chem. Soc., 2000, 122, 5891 CrossRef CAS.
- J. Buratto, C. Colombo, M. Stupfel, S. J. Dawson, C. Dolain, B. Langlois d'Estaintot, L. Fischer, T. Granier, M. Laguerre, B. Gallois and I. Huc, Angew. Chem., Int. Ed., 2014, 53, 883 CrossRef CAS PubMed.
- X. Hu, S. J. Dawson, P. K. Mandal, X. de Hatten, B. Baptiste and I. Huc, Chem. Sci., 2017, 8, 3741 RSC.
- H. Jiang, J.-M. Léger and I. Huc, J. Am. Chem. Soc., 2003, 125, 3448 CrossRef CAS PubMed.
- S. J. Dawson, Á. Mészáros, L. Pethő, C. Colombo, M. Csékei, A. Kotschy and I. Huc, Eur. J. Org. Chem., 2014, 4265 CrossRef CAS.
- J. Clayden, A. Lund, L. Vallverdú and M. Helliwel, Nature, 2004, 431, 966 CrossRef CAS PubMed; R. A. Brown, V. Diemer, S. J. Webb and J. Clayden, Nat. Chem., 2013, 5, 853 CrossRef PubMed; F. Lister, B. Le Bailly, S. J. Webb and J. Clayden, Nat. Chem., 2017, 9, 420 CrossRef.
- C. Tsiamantas, X. de Hatten, C. Douat, B. Kauffmann, V. Maurizot, H. Ihara, M. Takafuji, N. Metzler-Nolte and I. Huc, Angew. Chem., Int. Ed., 2016, 55, 6848 CrossRef CAS PubMed.
- Z. J. Kinney, V. C. Kirind and C. Scott Hartley, Chem. Sci., 2019, 10, 9057 RSC.
- R. Ishidate, A. J. Markvoort, K. Maeda and E. Yashima, J. Am. Chem. Soc., 2019, 141, 7605 CrossRef CAS PubMed; T. Ikai, S. Kawabata, F. Mamiya, D. Taura, N. Ousaka and E. Yashima, J. Am. Chem. Soc., 2020, 142, 21913 CrossRef.
- E. Suarez-Picado, E. Quinoa, R. Riguera and F. Freire, Angew. Chem., Int. Ed., 2020, 59, 4537 CrossRef CAS PubMed; F. Freire, J. M. Seco, E. Quinoa and R. Riguera, Angew. Chem., Int. Ed., 2011, 50, 11692 CrossRef PubMed.
- A. M. Kendhale, L. Poniman, Z. Dong, K. Laxmi-Reddy, B. Kauffmann, Y. Ferrand and I. Huc, J. Org. Chem., 2011, 76, 195 CrossRef CAS PubMed.
- J. Clayden, A. Castellanos, J. Solà and G. A. Morris, Angew. Chem., Int. Ed., 2009, 48, 5962 CrossRef CAS PubMed.
- For a review, see: J. Jacques, A. Collet and S. H. Wilen in Enantiomers, racemates and resolutions, Krieger, Malabar, 3rd edn, 1994 Search PubMed.
- D. F. Kreitler, Z. Yao, J. D. Steinkruger, D. E. Mortenson, L. Huang, R. Mittal, B. R. Travis, K. T. Forest and S. H. Gellman, J. Am. Chem. Soc., 2019, 141, 1583 CrossRef CAS; D. E. Mortenson, J. D. Steinkruger, D. F. Kreitler, D. V. Perroni, G. P. Sorenson, L. Huang, R. Mittal, H. G. Yun, B. R. Travis, M. K. Mahanthappa, K. T. Forest and S. H. Gellman, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13144 CrossRef PubMed; C. Toniolo, C. Peggion, M. Crisma, F. Formaggio, X. Shui and D. S. Eggleston, Nat. Struct. Biol., 1994, 1, 908 CrossRef PubMed; K. Mandal, B. L. Pentelute, D. Bang, Z. P. Gates, V. Y. Torbeev and S. B. H. Kent, Angew. Chem., Int. Ed., 2012, 51, 1481 CrossRef PubMed; P. Mateus, N. Chandramouli, C. D. Mackereth, B. Kauffmann, Y. Ferrand and I. Huc, Angew. Chem., Int. Ed., 2020, 59, 5797 CrossRef PubMed; G. Lautrette, B. Wicher, B. Kauffmann, Y. Ferrand and I. Huc, J. Am. Chem. Soc., 2016, 138, 10314 CrossRef PubMed; M. Lee, J. Shim, P. Kang, M.-G. Choia and S. H. Choi, Chem. Commun., 2016, 52, 5950 RSC.
- G. N. Ramachandran, C. Ramakrishnan and V. Sasisekharan, J. Mol. Biol., 1963, 7, 95 CrossRef CAS.
- J. D. Sadowsky, J. K. Murray, Y. Tomita and S. H. Gellman, ChemBioChem, 2007, 8, 903 CrossRef CAS PubMed.
- E. Teyssières, J.-P. Corre, S. Antunes, C. Rougeot, C. Dugave, G. Jouvion, P. Claudon, G. Mikaty, C. Douat, P. L. Goossens and G. Guichard, J. Med. Chem., 2016, 59, 8221 CrossRef PubMed.
- J. Iriondo-Alberdi, K. Laxmi-Reddy, B. Bouguerne, C. Staedel and I. Huc, ChemBioChem, 2010, 11, 1679 CrossRef CAS PubMed; M. Oba, ChemBioChem, 2019, 20, 2041 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic protocols, details of crystallographic studies, characterisation of new compounds. CCDC 2002474, 2010131 and 2010173. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03640h |
| This journal is © The Royal Society of Chemistry 2021 |