
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLipid-mimicking phosphorus-based glycosidase inactivators as pharmacological chaperones for the treatment of Gaucher's disease†
Manuel
Scherer‡
a,
Andrés G.
Santana‡
 a,
Kyle
Robinson
a,
Steven
Zhou
b,
Hermen S.
Overkleeft
c,
Lorne
Clarke
b and
Stephen G.
Withers
*a
a,
Kyle
Robinson
a,
Steven
Zhou
b,
Hermen S.
Overkleeft
c,
Lorne
Clarke
b and
Stephen G.
Withers
*a
aDept. of Chemistry. University of British Columbia, Vancouver, British Columbia V6T 1Z1, Canada. E-mail: withers@chem.ubc.ca
bDept. of Medical Genetics. University of British Columbia, Vancouver, British Columbia V6H 3N1, Canada
cInstitute of Chemistry. Leiden University, Leiden, Netherlands
First published on 20th September 2021
Abstract
Gaucher's disease, the most prevalent lysosomal storage disorder, is caused by missense mutation of the GBA gene, ultimately resulting in deficient GCase activity, hence the excessive build-up of cellular glucosylceramide. Among different therapeutic strategies, pharmacological chaperoning of mutant GCase represents an attractive approach that relies on small organic molecules acting as protein stabilizers. Herein, we expand upon a new class of transient GCase inactivators based on a reactive 2-deoxy-2-fluoro-β-D-glucoside tethered to an array of lipid-mimicking phosphorus-based aglycones, which not only improve the selectivity and inactivation efficiency, but also the stability of these compounds in aqueous media. This hypothesis was further validated with kinetic and cellular studies confirming restoration of catalytic activity in Gaucher cells after treatment with these pharmacological chaperones.
Introduction
Gaucher's disease1 is a genetic condition resulting from deficiency of the lysosomal enzyme β-glucocerebrosidase (GCase EC: 4.2.1.25) with the concomitant accumulation of glucosylceramide. The deficiency of GCase is a consequence of a large number of individual variants within the GBA gene and the resultant clinical features of Gaucher's disease are heterogeneous even when patients share the same pathogenic variant.2 The recent population-based studies showing that GBA variants are over-represented in Parkinson disease patients has expanded the role and medical impact of GCase deficiency.3 A frequent mechanism underlying GCase deficiency is protein misfolding resulting in intracellular instability and altered subcellular trafficking. This has been aptly demonstrated for the two common GBA missense variants N370S (p.N409S) and L444P (p.L483P).4 Currently approved therapies for Gaucher's disease include enzyme replacement, effective only for the non-neuronal forms of the disease, as well as substrate-reduction therapy. Pharmacological chaperones (PCs) represent an additional potential therapeutic approach allowing the targeting of both systemic and neurological symptoms. This form of treatment utilizes small molecules, most commonly GCase competitive inhibitors, to promote and stabilize the folded conformation of mutant GCase,5 thereby assisting successful transit to the lysosome.6 While a number of potential chaperones have been evaluated, none have yet received clinical approval for the treatment of Gaucher's disease. The vast majority of GCase PCs developed to date have been reversible competitive inhibitors. In vitro studies have shown that these PCs increase the quantity of GCase within cells but suffer from prolonged inhibition of the enzyme within the lysosome. This is further exacerbated by the basic, lysosomotropic properties of these compounds. Potential means to avert this issue include development of reversible, non-competitive GCase ligands,7 as well as ligands that can be actively cleaved by GCase once it is correctly targeted to the lysosome. An alternative approach proposed by our group and others involves the generation of molecules that utilize the catalytic mechanism of GCase to form a transient covalent intermediate with the enzyme and subsequently undergo cleavage within the lysosome to release the active enzyme.8 To be successful as a therapeutic, the kinetics of this process require fine tuning. A major advantage of such an approach is that although the parent molecule is a potent GCase inhibitor, the cleaved and released end product will have no significant affinity for the active site. 2-Fluoroglucosides (2FGlc) with activated leaving groups are excellent candidates since they form a long-lived, but catalytically competent glycosyl-enzyme intermediate with retaining β-glycosidases such as GCase (Fig. 1).9 Early studies utilizing purified GCase showed that 2-deoxy-2-fluoro-β-D-glucosyl fluoride (2FGlcF) forms a 2-fluoroglucosyl-enzyme intermediate with a hydrolytic half-life of 1300 min and, when administered to rats, this molecule inactivates GCase in all organs, including the brain.10 These biodistribution and release kinetics are ideal, but the rate constant for GCase inactivation was found to be too low to be clinically useful. This has led to two separate approaches to potentially increase this rate constant. One involves replacement of the fluoride leaving group with an imidate that can be activated by protonation in the active site.11 The other involves replacement with a dialkyl phosphate leaving group in which the hydrophobic alkyl residues mimic the ceramide of the natural substrate glucosylceramide (Fig. 1), while the low pKa of the aglycone ensures rapid inactivation. Indeed, inactivation rates were >4000-fold higher than those of 2FGlcF, and with much greater selectivity over other glycosidases. Unfortunately, the dialkyl phosphates were unstable, with a half-life of ca. 15 min under physiological conditions. In order to minimize instability, a single phosphonate analogue bearing O-benzyl and C-benzyl substituents was synthesized and shown to indeed be more stable (t1/2 = 630 min) but was a less efficient GCase inactivator.8b Based on these proof of principle observations, we set out to find more stable and more active chaperones by exploring other phosphonate substituents that better mimic the natural substrate, along with the use of phosphoramidates and phosphordiamidates as leaving groups that may recruit additional stabilizing interactions through their nitrogen atom.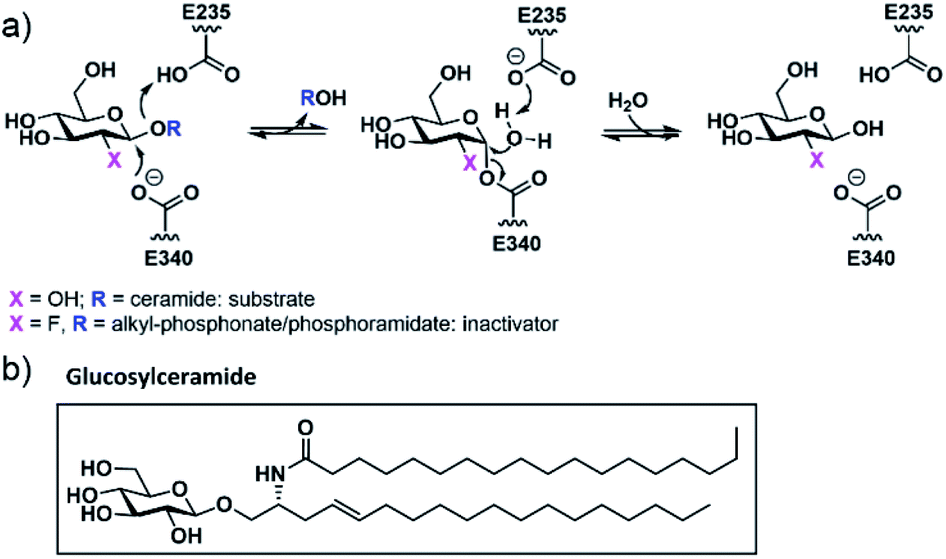 | ||
| Fig. 1 (a) GCase double-displacement mechanism acting on its natural substrate or a 2FGlc-derived inactivator; (b) chemical structure of glucosylceramide, GCase natural substrate. | ||
Results and discussion
Syntheses of the phosphorus-based aglycones and their 2FGlc-phosphonates, phosphordiamidates and phosphoramidates are shown in Scheme 1. 2FGlc phosphonates 1–4 were prepared via Koenigs-Knorr glycosylation of 3,4,6-tri-O-acetyl-2-deoxy-2-fluoro-α-D-glucopyranosyl bromide with the respective phosphonic acid mono esters 13a–13d. The glycosylation products 16a–16d were subsequently de-O-acetylated under methoxide-catalyzed conditions to furnish the desired inactivators 1–4. The acceptor phosphonic acid mono esters themselves (13a–13d) were readily synthesized from dialkyl phosphites 11a and 11b which, after alkylation, gave dialkyl alkylphosphonates 12a–12d. Finally, the use of either TMSI or NaI allowed the transformation of dialkyl alkylphosphonates 12a–12d into the corresponding phosphonic acid mono esters 13a–13d. 2FGlc phosphonate 5 was prepared from commercially available POCl2Ph onto which an octyl residue had been attached using Grignard conditions to give octyl phenyl-phosphoryl chloride 15a. Coupling of chloride 15a with 3,4,6-tri-O-acetyl-2-deoxy-2-fluoro-D-glucopyranose using Et3N provided per-O-acetylated glycosyl phosphonate 16e from which compound 5 was readily obtained by deprotection. Compounds 1–5, thus prepared, were evaluated as diastereomeric mixtures. Next, to check whether the phosphorus chirality in these diasteroisomeric compounds has any significant effect on the binding affinity towards the enzyme, a sample of 4 was subjected to stringent chromatography (see ESI†) yielding a 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of the two phosphorus diastereomers. With a ki/Ki value of 40.9 min−1 mM−1, the inactivation rate of this highly enriched fraction was only 1.1-fold greater than that of the 50:50 mixture of 4, indicating that the two stereoisomers bind the enzyme active site with very similar affinities.
:1 ratio of the two phosphorus diastereomers. With a ki/Ki value of 40.9 min−1 mM−1, the inactivation rate of this highly enriched fraction was only 1.1-fold greater than that of the 50:50 mixture of 4, indicating that the two stereoisomers bind the enzyme active site with very similar affinities.
 | ||
| Scheme 1 Synthesis of 2-fluorosugars bearing phosphorus-based aglycones: (a) 11b: diethyl phosphite, n-octanol, NaH, 160 °C; (b) 11a, NaH, THF, alkyl halide, 12a: with n-butyl iodide, 12b: with BnBr; (c) 12c: 11b, Na, toluene, 1-bromooctane, reflux; (d) 12d: 11b, Cs2CO3, DMF, BnBr, TBAI; (e) 13a, 13b and 13d: TMSI, CH2Cl2; (f) 13c: NaI, pyridine, reflux; (g) 3,4,6-tri-O-acetyl-2-deoxy-2-fluoro-D-glucopyranosyl bromide, THF/MeCN/CH2Cl2 or MeCN, Ag2CO316a: from 13a, 16b: from 13b, 16c: from 13c, 16d: from 13d; (h) 14b: n-octylamine, Et3N, toluene; (i) 14c: n-octanol, quant.; (j) 15a: from 14a with n-octylmagnesium bromide, Et2O/toluene (4:1); (k) Et3N, toluene, corresponding amine 15b: POCl3, n-octylamine, 15c: from 14b with aniline, 15d: from 14c with n-octylamine, 15e: from 14a with n-octylamine; 15f: from 14a with n-dodecylamine; (l) 3,4,6-tri-O-acetyl-2-deoxy-2-fluoro-D-glucopyranose, CH2Cl2, Et3N, 3 Å molecular sieves, 16e: from 15a, 16f: from 15b, 16g: from 15c, 16h: from 15d, 16i: from 15e, 16j: from 15f; (m) Na, MeOH, 1: from 16a, 2: from 16b, 3: from 16c, 4: from 16d, 5: from 16e, 6: from 16f, 7: from 16g, 8: from 16h, 9: from 16i, 10: from 16j; compounds 11a and 14a are commercially available. | ||
Evaluation of the 2FGlc phosphonates 1–5 (Fig. 2) as inactivators of GCase revealed a trend wherein increasing the hydrophobicity of the aglycone resulted in faster inactivation of GCase. This is presumably a consequence of the substrates becoming closer mimics of ceramide with its long hydrophobic chains (Fig. 1). Interestingly, and as seen previously, the presence of an aromatic residue is beneficial for the inactivation since octyl-benzyl derivative 4 was three times more active than the di-octyl analogue 3 (36.4 min−1 mM−1 for 4vs. 10.9 min−1 mM−1 for 3) (Table 1). As a consequence of this trend, there is an almost 20-fold increase of the second-order rate constant ki/Ki in going from the di-butyl derivative 1 to the octyl-benzyl analogue 4, even though their substituent inductive effects must be similar. Importantly, however, by altering the position of the oxygen atom (4 to 5), the rate constant ki can be further increased 5-fold (3.67 min−1 for 4vs. 18.6 min−1 for 5) while the Ki stays largely unaffected (101 μM for 4vs. 117 μM for 5), pushing the ki/Ki value up to 160 min−1 mM−1 (Table 1). The rise in this case is presumably a consequence of the greater electronegativity of the phenol (pKa ∼ 10) than the alkyl alcohol it replaced (pKa ∼ 15).
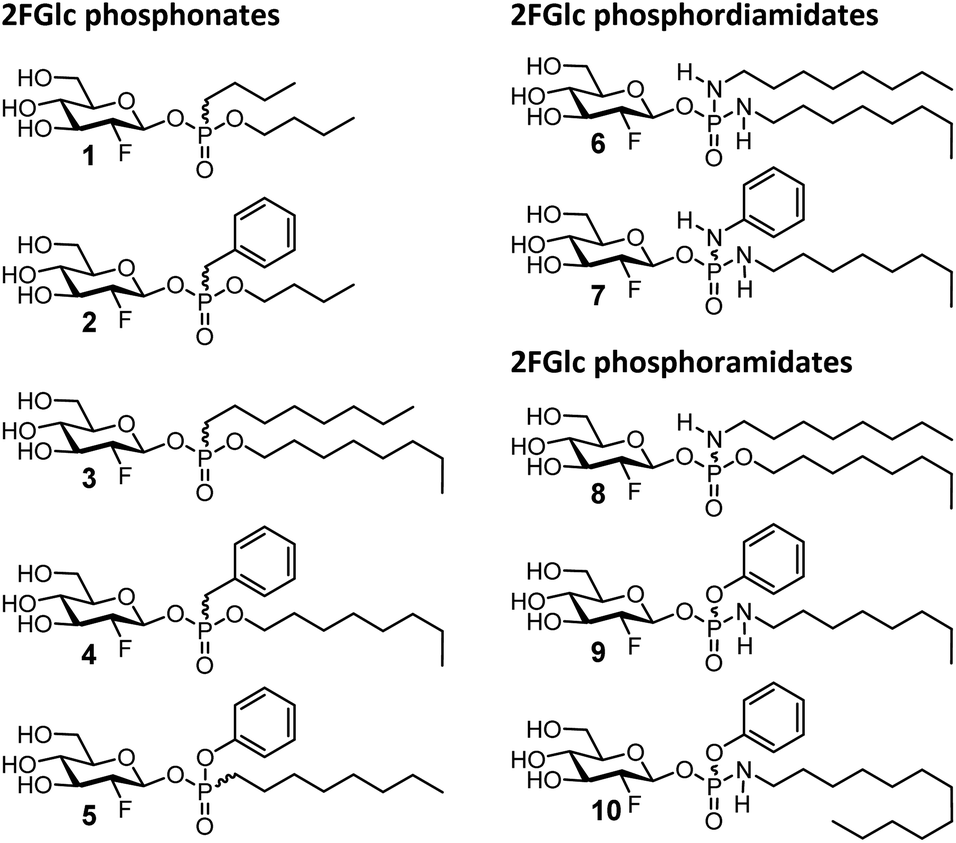 | ||
| Fig. 2 Glucosylceramide-mimicking 2-deoxy-2-fluoroglucopyranosyl phosphonates, phosphoramidates and phosphordiamidates 1–10 prepared in this study. | ||
|
|
k i [min−1] | K i [μM] | k i/Ki [min−1 mM−1] |
|---|---|---|---|
| a 37 °C, pH = 5.5 (for detailed procedure, see ESI). | |||
| 1 R = OBut R′ = But | 0.49 | 264 | 1.87 |
| 2 R = OBut R′ = Bn | 5.44 | 742 | 7.34 |
| 3 R = OOct R′ = Oct | 4.09 | 377 | 10.9 |
| 4 R = OOct R′ = Bn | 3.67 | 101 | 36.4 |
| 5 R = OPh R′ = Oct | 18.6 | 117 | 160 |
| 6 R = R′ = NHOct | 0.13 | 8.2 | 16.1 |
| 7 R = NHOct R′ = NHPh | 0.1 | 4.8 | 21.5 |
| 8 R = OOct R′ = NHOct | 7.45 | 188 | 39.5 |
| 9 R = OPh R′ = NHOct | 15 | 36.2 | 416 |
| 10 R = OPh R′ = NHDodec | 4.22 | 22.2 | 190 |

While the improvements achieved here were useful and in the right direction, we were intrigued by the possibilities of incorporating nitrogen atoms into the anomeric leaving group, not only because it would render closer mimics of the natural aglycone, glucosylceramide (Fig. 1), but also because of the potential of harnessing strong hydrogen bonding interactions with active site carboxylic acids, a strategy behind the impressive history of azasugar inhibitors of glycosidases. An added bonus is the built-in acid lability of phosphoramidates, minimising persistence in the lysosome through enhanced spontaneous hydrolysis.12 Fittingly, 2FGlc phosphordiamidates 6–7 were synthesized by a similar approach to that used for the phosphonates (Scheme 1). Coupling of the fluorinated glycone and aglycone moieties was achieved by reaction of phosphordiamidochloridates 15b and 15c with the anomeric hemiacetal of the otherwise protected 2FGlc to give intermediates 16f and 16g. The excellent β-selectivity observed presumably arose from the greater reactivity of the less sterically hindered beta anomer, followed by rapid interconversion of the alpha and beta anomers under the employed reaction conditions. These intermediates were subsequently deacetylated to provide 2FGlc phosphordiamidates 6–7 (Fig. 2). In parallel, 2FGlc phosphoramidates 8–10 were obtained in excellent overall yields of 49% up to 76% from commercially available POCl3 and POCl2OPh in three and four synthetic steps, respectively (Scheme 1). The approach involved the preparation of phosphoryldichlorides 14a–14c which were transformed into phosphoramidochloridates 15d–15f. Subsequent highly β-selective and high yielding anomeric phosphorylations provided compounds 16h–16j. These afforded 2FGlc phosphoramidates 8–10 after Zemplén deprotection (Fig. 2). Except for compounds 4 and 6, all final compounds were obtained as a 1:1 mixture of diastereomers at the phosphorus stereocenter and were enzymatically evaluated as such.
Consistent with expectations about improved binding, the 2FGlc phosphordiamidates 6 and 7 both proved to bind with high affinity, exhibiting Ki values of 8.2 μM and 4.8 μM, respectively. However, it appears that this high affinity derives from ground state binding since inactivation rates were also low, as reflected in the ki values of 0.13 min−1 and 0.10 min−1 and modest second-order rate constants of ki/Ki = 16.1 min−1 mM−1 for 6 and 21.5 min−1 mM−1 for 7 (Table 1). In order to speed up the inactivation step, we elected to replace one of the amines on the phosphorus atom with a more electron-withdrawing O-alkyl or O-phenyl substituent in the form of the 2FGlc phosphoramidates 8–10. The N-octyl-O-octyl reagent 8 did indeed bind better and react faster than the di-O-octyl derivative 3, even though the effect was a modest 4-fold increase in ki/Ki. However, by replacing the O-octyl with an O-Ph residue (compound 9), the ki value was increased and the Ki decreased resulting in a substantially higher second-order rate constant ki/Ki of 416 min−1 mM−1. Compound 9 combines the positive effects of the enhanced binding by the N–H (lowering the Ki), the interaction of the aromatic residue and the electron-withdrawing nature of the OPh (enhancing ki). In an attempt to further increase the ki/Ki value, the alkyl chain length of 9 was elongated from an n-octyl to an n-dodecyl residue (compound 10), but surprisingly, this actually decreased the second-order rate constant two-fold (Table 1). Quite possibly the longer alkyl chains promote the formation of aggregates making compound 10 less accessible to the enzyme. Unsurprisingly, the isomer of compound 8 in which the nitrogen atom replaces the glycosidic oxygen (octyl 2FGlc-phosphoramidate) exhibited a very low affinity for the enzyme and behaved as a poor competitive inhibitor, rather than as an inactivator (synthesis in ESI†).
In order to test stability under physiological conditions, 2FGlc phosphoramidate 9 was incubated at 37 °C in a phosphate buffer at pH 6.8 and the decrease of the rate constant of inactivation due to degradation of the inactivator was followed over time and used as an indirect measure of the remaining inactivator concentration. Under these conditions, 2FGlc phosphoramidate 9 showed a similar rate of hydrolysis and half-life as the previously synthesized 2FGlc phosphonate (0.0014 min−1vs. 0.0011 min−1 and t1/2 of 510 min vs. 630 min, respectively) and around 40 times enhanced stability compared to the original 2FGlc phosphates (t1/2 510 min vs. 15 min) (see ESI, Fig. S3†).8b
The pharmacological chaperone potential of this class of compounds was tested with phosphonate 4 as a representative molecule, since we know that phosphorus chirality is not a significant factor in determining affinity in this case. Mouse embryonic fibroblast (MEF) cells from transgenic mice expressing solely human L444P-containing GCase (a common variant underlying neuropathic Gaucher's disease) on a murine GCase knockout background13 were continuously treated for three days with daily media changes containing 100 μM 2FGlc phosphonate 4. After the three days, cells were washed, fresh media without inactivator was added and cells subsequently harvested at various time intervals. To assess the amounts of rescued active enzyme present, we used the fluorescent mechanism-based inactivator MDW933 that reacts only with active enzyme (Fig. 3).14 By running PAGE gels we can assess the amount of reactivated enzyme present from the intensity of the fluorescence, while subsequent Western blots of the same gel give insights into total mass of enzyme present. Total enzyme gradually increased to a maximum at 48 hours (Fig. 3, bottom), then appears to decrease. By contrast, relatively little active GCase is seen after 24 hours due to inactivation by the chaperone, but then it gradually increases to an apparent maximum at 48–72 hours due to steady reactivation through turnover of the 2-fluoroglucosyl enzyme (Fig. 3, top). In parallel both the GCase and hexosaminidase activities of the cells were measured during the four days after completion of treatment. Consistent with the pattern seen in the gels, activity increased over time, reaching 1.31 times the initial activity after 96 hours (see ESI, Table S1†). To account for variations in cell sampling all values were normalized relative to the hexosaminidase activity measured.7
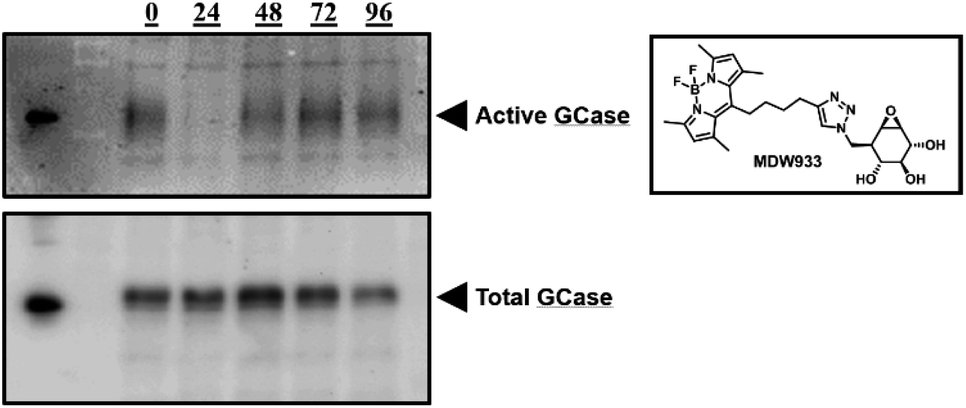 | ||
| Fig. 3 PAGE gels imaged with the fluorescent activity-based probe MDW933 (top) and Western blot (bottom), showing changes in active and total [GCase] over time (h) after treatment with phosphonate 4, respectively. Lane 0 represents untreated cells. | ||
Similar results were obtained on the same system with a dosing regimen involving treatment for 7 days, then monitoring for activity and protein concentration via MDW933 labelling and Western blots of PAGE gels, respectively (see ESI, Fig. S4†). This chaperoning is also seen in patient fibroblast cells expressing the L444P mutation, as seen in Table S2.† Essentially equivalent levels of chaperoning are seen as with in MEF cells. Finally, this behaviour is not restricted to the L444P mutation since studies of the N370S mutant in both MEF cells and in human fibroblasts revealed similar profiles of activity gain as seen in the MDW933 stained gels of Fig. S5.†
These results therefore confirm that the 2FGlc phosphonate 4 is indeed able to: (1) penetrate the fibroblast cells, and (2) inactivate GCase effectively. Fittingly, the recovery of the enzymatic activity seen over the course of seven days, due to gradual turnover of the covalently bound 2FGlc, is compatible with clinical practice.
Conclusions
In conclusion, ten novel 2FGlc glycosides with different phosphorus-based aglycones were prepared and their inactivation potential towards GCase analyzed. The 2FGlc phosphoramidate 9 proved to be a potent inactivator with a ki/Ki value of 416 min−1 mM−1 and was much more stable than the 2FGlc phosphates under physiological conditions. Cell assays confirmed that 2FGlc glycosides such as 4 are able to inactivate GCase in fibroblast cells and that the inactivated enzyme slowly recovers activity on a timescale compatible with treatment, making them promising candidates as pharmacological chaperones.Data availability
All data are included.Author contributions
Manuel Scherer and Andrés G. Santana performed all the synthesis and most of the kinetic analyses. Kyle Robinson performed some of the enzyme kinetic studies. Steven Zhou performed cell-based analyses, supervised by Lorne Clarke. Hermen Overkleeft generated the fluorescent activity based probe. Funding was acquired by Lorne Clarke and Stephen G. Withers. Manuscript writing was performed primarily by MS, AGS and SGW with input from LC.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the Canadian Institutes for Health Research (CIHR) and the Canadian Glycoscience Network GlycoNet. AGS would like to thank EMBO for a long-term postdoctoral fellowship (ALTF 1029-2013). MS thanks the Swiss National Science Foundation (SNSF) for a postdoctoral fellowship.Notes and references
- A. H. Futerman and G. van Meer, Nat. Rev. Mol. Cell Biol., 2004, 5, 554 CrossRef CAS PubMed.
- P. K. Mistry, G. López, R. Schiffmann, N. W. Barton, N. J. Weinreb and E. Sidransky, Mol. Genet. Metab., 2017, 120, 8 CrossRef CAS PubMed.
- (a) F. Atashrazm, D. Hammond, G. Perera, C. Dobson-Stone, N. Mueller, R. Pickford, W. Scott Kim, J. B. Kwok, S. J. G. Lewis, G. M. Halliday and N. Dzamko, Sci. Rep., 2018, 8, 15446 CrossRef PubMed; (b) M. Horowitz, D. Elstein, A. Zimran and O. Goker-Alpan, Hum. Mutat., 2016, 37, 1121 CrossRef CAS PubMed.
- G. A. Grabowski, Lancet, 2008, 372, 1263 CrossRef CAS.
- (a) R. Thomas and A. R. Kermode, Mol. Genet. Metab., 2019, 126, 83 CrossRef CAS PubMed; (b) J. M. Benito, J. M. García Fernández and C. O. Mellet, Expert Opin. Ther. Pat., 2011, 21, 885 CrossRef CAS PubMed.
- D. Reczek, M. Schwake, J. Schroder, H. Hughes, J. Blanz, X. Y. Jin, W. Brondyk, S. Van Patten, T. Edmunds and P. Saftig, Cell, 2007, 131, 770 CrossRef CAS PubMed.
- (a) E. D. Goddard-Borger, M. B. Tropak, S. Yonekawa, C. Tysoe, D. J. Mahuran and S. G. Withers, J. Med. Chem., 2012, 55, 2737 CrossRef CAS PubMed; (b) M. Aguilar-Moncayo, M. I. García-Moreno, A. Trapero, M. Egido-Gabas, A. Llebaria, J. M. García Fernandez and C. Ortiz Mellet, Org. Biomol. Chem., 2011, 9, 3698 RSC; (c) T. S. Rasmussen, S. Allman, G. Twigg, T. D. Butters and H. H. Jensen, Bioorg. Med. Chem. Lett., 2011, 21, 1519 CrossRef CAS PubMed.
- (a) C.-L. Kuo, W. W. Kallemeijn, L. T. Lelieveld, M. Mirzaian, I. Zoutendijk, A. Vardi, A. H. Futerman, A. H. Meijer, H. P. Spaink, H. S. Overkleeft, J. M. F. G. Aerts and M. Artola, FEBS J., 2019, 286, 584 CrossRef CAS PubMed; (b) B. P. Rempel, M. P. Tropak, D. J. Mahuran and S. G. Withers, Angew. Chem., Int. Ed., 2011, 50, 10381 CrossRef CAS PubMed.
- S. C. Miao, J. D. McCarter, M. E. Grace, G. A. Grabowski, R. Aebersold and S. G. Withers, J. Biol. Chem., 1994, 269, 10975 CrossRef CAS.
- (a) J. D. McCarter, M. J. Adam, N. G. Hartman and S. G. Withers, Biochem. J., 1994, 301, 343 CrossRef CAS PubMed; (b) C. P. Phenix, B. P. Rempel, K. Colobong, D. J. Doudet, M. J. Adam, L. A. Clarke and S. G. Withers, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10842 CrossRef CAS PubMed.
- M. T. C. Walvoort, W. W. Kallemeijn, L. I. Willems, M. D. Witte, J. M. F. G. Aerts, G. A. van der Marel, J. D. C. Codée and H. S. Overkleeft, Chem. Commun., 2012, 48, 10386 RSC.
- (a) I. Oney and M. Caplow, J. Am. Chem. Soc., 1967, 89, 6972 CrossRef CAS; (b) T. Mena-Barragán, A. Narita, D. Matias, G. Tiscornia, E. Nanba, K. Ohno, Y. Suzuki, K. Higaki, J. M. García Fernández and C. Ortiz Mellet, Angew. Chem., Int. Ed., 2015, 54, 11696 CrossRef PubMed.
- A. Sanders, H. Hemmelgarn, H. L. Melrose, L. Hein, M. Fuller and L. A. Clarke, Blood Cells, Mol., Dis., 2013, 51, 109 CrossRef CAS PubMed.
- M. D. Witte, W. W. Kallemeijn, J. Aten, K.-Y. Li, A. Strijland, W. E. Donker-Koopman, A. M. C. H. van den Nieuwendijk, B. Bleijlevens, G. Kramer, B. I. Florea, B. Hooibrink, C. E. M. Hollak, R. Ottenhoff, R. G. Boot, G. A. van der Marel, H. S. Overkleeft and J. M. F. G. Aerts, Nat. Chem. Biol., 2010, 6, 907 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic methods, NMR characterization, GCase inactivation kinetics. See DOI: 10.1039/d1sc03831a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |