
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilicon as a powerful control element in HDDA chemistry: redirection of innate cyclization preferences, functionalizable tethers, and formal bimolecular HDDA reactions†
Mandy
Lynn
,
Merrick
Pierson Smela
 and
Thomas R.
Hoye
*
and
Thomas R.
Hoye
*
Department of Chemistry, University of Minnesota, 207 Pleasant St. SE, Minneapolis, MN 55455, USA. E-mail: hoye@umn.edu
First published on 6th October 2021
Abstract
The 1,3-diyne and diynophile in hexadehydro-Diels–Alder (HDDA) reaction substrates are typically tethered by linker units that consist of C, O, N, and/or S atoms. We describe here a new class of polyynes based on silicon-containing tethers that can be disposed of and/or functionalized subsequent to the HDDA reaction. The cyclizations are efficient, and the resulting benzoxasiloles are amenable to protodesilylation, halogenation, oxygenation, and arylation reactions. The presence of the silicon atom can also override the innate mode of cyclization in some cases, an outcome attributable to a β-silyl effect on the structure of intermediate diradicals. Overall, this strategy equates formally to an otherwise unknown, bimolecular HDDA reaction and expands the versatility of this body of aryne chemistry.
Introduction
The generation of highly versatile, reactive o-benzyne intermediates from triyne or tetrayne substrates and their subsequent capture by various trapping agents – i.e., the hexadehydro-Diels–Alder (HDDA) cascade1 – is a growing area of research.2 In essentially all cases, the starting diyne and diynophile have been tethered by a linker unit that consists of C, N, and/or O atoms.2 In 2018, we reported a formal bimolecular HDDA reaction using erasable sulfur-based linkers (Fig. 1a, 1→3).3 This inspired us to explore other heteroatom-based tethers that could be readily accessed, stable to the (thermal) HDDA reaction conditions, and usefully manipulated following cyclization. | ||
| Fig. 1 (a) Previous studies of HDDA cascade reactions using erasable sulfur-based linkers. (b) In this report we disclose formal bimolecular HDDA reactions using disposable and/or functionalizable silicon linkers. | ||
Silicon tethers have long been recognized as versatile linkers to support various intramolecular transformations such as radical cyclization, cycloaddition, and ring-closing metathesis.4 Notably, Suzuki and co-workers recently demonstrated the use of silicon tethering to enhance the efficiency of trapping reactions of classically generated benzynes (e.g., by the Kobayashi protocol5) by rendering the benzyne capture event intramolecular.6 However, silicon-tethered HDDA reactions are an under-explored area of research.7 In the lone previous report, an intriguing preference for initial HDDA rather than tetradehydro-Diels–Alder (TDDA) cyclization in substrates capable of either was observed. Here we describe the chemistry of a novel class of HDDA substrates that contain disposable and/or functionalizable silicon tethers (Fig. 1b, 4→6). In addition to hypothesized outcomes, several unexpected reaction selectivities were revealed by the studies.
Results and discussion
Initially, silicon-tethered substrates 4a–c were prepared to probe the feasibility of the anticipated HDDA reactions (Fig. 2). Upon heating in decalin, products 5a–c were obtained from the desired HDDA cascade reactions.‡ The silicon tether was then readily removed via desilylation to provide 7 in high yields. However, the HDDA reactions of 4a–c required heating at very high temperature (240–280 °C) over extended reaction times (ca. 20 h), which limited the selection of compatible trapping reagents. Encouraged by these preliminary results, we continued to explore additional silicon tethers and found alkoxysilanes 8 to be both efficient and more reactive (cyclizations at ∼100–150 °C lower temperature than 4) polyyne substrates (e.g., nearly full conversion at 140 °C after 20 h). We attribute this greater reactivity in the rate-limiting HDDA cycloisomerization for 8vs.5a to the shorter distance between the pair of proximal alkyne carbons9 in the former, which has longer bonds to only one third-row Si atom rather than two. Substrates 5b and 5c contain four-atom linkers and require formation of benzyne with a fused six-membered ring, here containing two silicon atoms. It is notable that other than these two examples, we know of only one other case in which a 1,3-diyne separated from a diynophile by four atoms gave rise to an intermediate HDDA-benzyne;10 in that instance the alkynes in the substrate were embedded within a macrocycle, so the HDDA reaction was intramolecular in nature. | ||
| Fig. 2 Preliminary results demonstrating that silicon-tethered HDDA cascades are feasible. | ||
Because of their greater reactivity as well as easier and more versatile routes of preparative access, tri- and tetrayne HDDA substrates based on the silyl ether linker motif as seen in 8 were used for the remainder of the studies reported here. As a bonus and as described below, these unsymmetrical substrates demonstrated intriguing (and complete) regioselectivity with respect to their direction of cycloisomerization, including the reversal of inherent reactivity in some instances.
Each of the triyne and tetrayne substrates 12a–g (Fig. 3, 5, and 9) contains a three-atom alkoxysilane tether. They can be conveniently prepared11 from dimethyl- or diisopropylchlorosilane (9a or 9b) in two steps and high yield. The diynylsilanes 10a–c were generated by reacting various diynyllithium species with 9a or 9b. In a subsequent one-pot process, each diynylsilane 10 was then converted, in situ, to its corresponding bromosilane 11, which was directly treated with a diynyl alcohol to provide the tetraynes 12a–d. Triyne substrates 12e–g were synthesized in a similar fashion (see ESI†).
 | ||
| Fig. 3 Preparation of tetraynes 12a–d. (PMP = p-methoxyphenyl). | ||
In our initial experiment, a solution of tetrayne 12a and 2,5-dimethylfuran in o-dichlorobenzene was heated at 150 °C for 3 h, and product 14a was produced in 90% yield (Fig. 4a). HDDA reactions of unsymmetrical tetrayne substrates such as 12a can give rise to two isomeric benzynes involving the same pair of proximal but opposite pairs of distal atoms of the two 1,3-diynes. In this instance, the second possibility would have led to 14a′. The assignment of structure 14a was supported by a significant nuclear Overhauser enhancement (NOE) observed between the methyl protons at 1.94 ppm and the methylene protons in the benzoxasilole.
 | ||
| Fig. 4 (a) Exclusive formation of 14a (via a single benzyne) from the tetrayne substrate 12a. (b) DFT calculations of the relative free energies (ΔG) of species on the potential energy surface for the reaction of the slightly simplified, model tetrayne 15a (at 413 K) [SMD(chloroform)/(U)B3LYP-D3BJ/6-311+G(d,p)//(U)M062X/6-311+G(d,p)]. | ||
This intriguing selectivity in the benzyne formation prompted us to conduct DFT calculations to gain additional insight to the energy barriers for the formation of each of the two isomeric benzynes. These were performed starting from the simplified model tetrayne 15a (Fig. 4b). Prior experimental studies12 have shown that the HDDA cycloisomerization can proceed by a stepwise mechanism via a diradical intermediate, at least for one class of HDDA polyyne substrate, although various computational investigations13 often indicate similar energies of activation for concerted vs. stepwise pathways over a range of different triyne or tetrayne substrates. In the present case, the energy barrier (TS1) leading to the diradical intermediate 15b was computed to be 23.6 kcal mol−1. This diradical, common to the paths leading to both 15c and 15d, was seen to then ring-close through the lower energy TS2 (23.9 kcal mol−1; a 5.2 kcal mol−1 barrier) compared to TS2′ (29.0 kcal mol−1; 10.3 kcal mol−1 barrier). Thus, the computations suggest that ring-closure significantly favors formation of benzyne 15c, consistent with the experimental isolation of only 14a from the trapping reaction of 12a. We were also able to identify concerted TSs for cycloaddition of 15a directly to 15c and 15d (top of Fig. 4b). While these also showed a strong preference for the former, both of these concerted pathways were, nonetheless, calculated to be considerably less favorable (higher Eacts) than that of the stepwise-diradical alternative.
Examination of the geometry computed for the intermediate diradical 15b allows us to offer a reason for the exclusive formation of constitutional isomer 14a in the cycloisomerization of 12a. In 15b the distal pair of atoms leading to 15c (blue line) are ca. 1 Å closer than those leading to 15d (red line). This is a consequence of the more pronounced bend in the propargylic radical in the bottom fragment compared to that in the top (150° vs. 184°, gray box in Fig. 4b). We further suggest that this difference in distortion is due to enhanced orbital overlap between the carbon–silicon sigma bond and the radical character at the adjacent sp-hybridized carbon atom in the more linear orientation. It is relevant that hyperconjugative stabilization of radicals by β-silyl substituents has been demonstrated.14
The reactions of these alkoxysilane-tethered tetraynes are quite general (Fig. 5). Products 14a–f were all produced from tetrayne 12a. These examples demonstrate trapping reactions through 4π-cycloaddition (14a–c), nucleophilic addition (with imidazole to 14d and acetic acid to 14e), and a phenol–ene reaction15 (14f). The last three examples, each involving an unsymmetrical nucleophilic trapping agent, show a preference for addition to Ca of the benzyne 16a. DFT calculation [M062X/6-311+G(d,p)] shows that this benzyne has a nearly symmetrical geometry; the internal bond angles at Ca and Cb are essentially the same (Δ∠b–∠a = 0.8°). This contrasts with many benzynes having five-membered rings comprising all second-row atoms in the same position of the ring fusion, which are significantly distorted, leading to selective nucleophilic addition to Cb. In the case of benzyne 16a the regioselectivity of attack by an external nucleophile is, therefore, likely governed more by steric rather than distortion factors; that is, the aryl substituent is effectively larger than the methylene group in the fused ring, allowing easier access to Ca. The dimethylsilyl-containing substrate 12b was also shown to be viable (viz. formation of 14g). The tetrayne substrate 12c gave rise to 14h–l through similar reactions. The naphthalene derivatives 14i and 14j were formed when an α-pyrone derivative was used as the trapping agent. The intermediate [4 + 2] adduct ejects carbon dioxide under the reaction conditions; the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of products presumably reflects a similar extent of steric hindrance in the two possible orientations by which the pyrone can engage the intermediate benzyne 16a. Substrate 12d contains a tethered TBS ether that serves to trap the benzyne intramolecularly, producing 14m. Finally, the triyne 12e was also a viable substrate; its conversion to the adduct 14n proceeded somewhat more slowly than that of analogous tetraynes, consistent with the accelerating effect of a bystander alkynyl substituent, present in each of 12a–d, arising from its greater radical stabilizing effect relative to a carboxy ester.11,16
:1 mixture of products presumably reflects a similar extent of steric hindrance in the two possible orientations by which the pyrone can engage the intermediate benzyne 16a. Substrate 12d contains a tethered TBS ether that serves to trap the benzyne intramolecularly, producing 14m. Finally, the triyne 12e was also a viable substrate; its conversion to the adduct 14n proceeded somewhat more slowly than that of analogous tetraynes, consistent with the accelerating effect of a bystander alkynyl substituent, present in each of 12a–d, arising from its greater radical stabilizing effect relative to a carboxy ester.11,16
 | ||
| Fig. 5 Products 14 from HDDA cascades using alkoxysilane-tethered tetraynes/triynes 12 and various trapping agents. (TPCPD = tetraphenylcyclopentadienone). | ||
We next sought to demonstrate ways of further functionalizing the various products from these HDDA cascade reactions. First, the oxabenzonorbornadiene products 14a, 14b, 14g, and 14n (cf.Fig. 5) could be readily converted to the corresponding naphthalene derivatives (17a–d, Fig. 6) by reductive deoxyaromatization with TMSI.17 In the case of the TMS-substituted terminal alkyne 14h, concomitant hydroiodination18 provided iodoalkene 17e.
 | ||
| Fig. 6 Synthesis of naphthalene derivatives 17via deoxygenation of benzoxanorbornadiene derivatives. | ||
Because of the considerable breadth and versatility of organosilicon chemistry—indeed, a significant subdiscipline of the entirety of organic chemistry—the C–Si bond in these products offers a host of opportunities for further transformations. We have demonstrated a few. For example, halo-desilylation reactions of the benzoxasilole in 14g were effective, giving 18a–b upon treatment with AgF and NIS or NBS (Fig. 7). Interestingly, the iodination of naphthalene 17b to give 18c was quite clean, but treatment with AgF/NBS gave rise to both the naphthyl bromide 18d and the brominated benzoisochromene 18e.
 | ||
| Fig. 7 Iodination and bromination of the C–Si bond (with concomitant cleavage of the tether). | ||
As demonstrated by the examples in Fig. 8, the aryl–silicon bond can be replaced by bonds to hydrogen, oxygen, or carbon substituents. Simple disposal by protodesilylation with TBAF is straightforward, giving, for example, 19 and 20 (Fig. 8a). If desired, the alkynyl trimethylsilyl group in 14h can be selectively cleaved by treatment with K2CO3 in MeOH/THF leaving the Ar–Si bond intact. Oxygenation of the Ar–Si bond is also feasible (Fig. 8b). Upon subjection to peracetic acid, 14g afforded the diol 22 with concomitant epoxide formation. On the other hand, the alkene in either 14g or 14a survived treatment of those substrates with basic hydrogen peroxide to give the substituted benzofuran 23. A cross-coupling reaction (Fig. 8c) was demonstrated in the case of the dimethylated oxasilole 17b using conditions like those effective for arylation of alkenyloxasiloles [PdCl2(PPh3)2, CuI, CsF, DMF, room temperature].19 Lastly, nucleophilic displacement of 14g with MeLi occurred smoothly to generate the ortho-TMS-substituted benzyl alcohol 25 (Fig. 8d).
 | ||
| Fig. 8 Synthetically useful transformations of benzoxasiloles (with concomitant cleavage of the tether). | ||
In the discussion above accompanying Fig. 4, we described the observation that 12a gave, exclusively, product 14a, indicating that the silicon substituent was serving as an unanticipated control element to guide the HDDA cycloisomerization in only one of two possible directions. Seeing this, we prepared substrate 12f to explore another possible unique directing effect by a silicon substituent (Fig. 9). There is ample evidence3,20 that triynes analogous to 12f but in which each of the linking atoms is C, O, N, and/or S will undergo highly preferential cycloisomerization via a tetradehydro-Diels–Alder (TDDA)1c,21 pathway (blue). In contrast, upon heating the silyl-containing triyne 12f, we observed a complete reversal; namely, none of the TDDA product 14o′ was detected and the furan-trapped HDDA adduct 14o was isolated in excellent yield (viz. red, not blue, Fig. 9a). This type of reactivity reversal was precedented in the silicon-linked triynes studied by the Shibata group.7 The silicon atom in the tether is clearly capable of redirecting the innate regioselectivity of dehydro-DA reactions for this type of triyne. Finally, the complementary triyne 12g was also examined. This produced only the TDDA product 14p and none of the benzyne trapped, HDDA product 14p′ (blue, not red, Fig. 9b). In all three instances of 12a, 12f, and 12g, the sp-carbon beta to the silicon atom (dot, top) closes to the terminus of the 4π-component (triangle, bottom), consistent with the rationale and calculations associated with Fig. 4b and explained by radical stabilization by the β-silyl substituent.
 | ||
| Fig. 9 Triyne substrates 12f and 12g show complementary modes of ring-closing selectivity, dictated by the relative position of diyne and monoyne relative to the silicon atom. (a) The first (12f) proceeds through HDDA cyclization. (b) The second (12g) proceeds through TDDA cyclization. In each case (as with 12a), the alkyne carbon beta to the silicon atom (•) participated in the closure to the new six-membered ring. | ||
Conclusions
We have discovered that hexadehydro-Diels–Alder substrates in which the diyne and diynophile are linked by a tether containing a silicon atom provides new strategic opportunities and practical versatility to HDDA chemistry. The cycloisomerization reactions are often quite efficient. The product benzoxasiloles can be erased through protodesilylation or functionalized by replacement with halide, hydroxyl, or aryl groups. In an intriguing outcome and of fundamental mechanistic interest, the presence of the silicon atom completely biases the direction of cyclization, providing a control element for overriding the inherent regioselectivity of some dehydro-Diels–Alder reactions. We suggest that this arises from the (DFT-computed) geometry of an initially formed diradical intermediate, in turn a manifestation of the stabilization of radical character by the β-silyl substituent. In sum, the presence of a silicon atom-containing tether provides elementary control of these polyyne cyclizations and the benzoxasilole products provide opportunity for further, versatile derivatization.Experimental
Typical procedure for the cyclization of a silicon-tethered substrate (here, 12a trapped with furan to give 14b, Fig. 5): A solution of tetrayne 12a (28 mg, 0.071 mmol) and furan (0.15 mL, 2.1 mmol) in ethanol-free CHCl3 (3.5 mL, 0.02 M) was placed in a threaded culture tube and the headspace was flushed with N2. The tube was fitted with an inert, Teflon®-lined cap, firmly sealed, and heated in an oil bath that had been pre-equilibrated to 140 °C; this mixture was heated for 3 h, cooled to rt and concentrated. The crude product was purified by passage through a small plug of silica gel (15% EtOAc/hexanes as eluent), and the residue was purified by medium pressure liquid chromatography (20% EtOAc/hexanes) to give compound 14b (28 mg, 85%) as a pale-yellow, crystalline solid.Author contributions
M. P. S. prepared and established the cyclizations of the initial substrates 4a–c. M. L. carried out the bulk of the experimentation and co-wrote the early drafts of the manuscript. All authors reviewed and edited the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The National Institute of General Medical Sciences of the U.S. Department of Health and Human Services (R35 GM127097) provided support for this research. A portion of the NMR spectral data was obtained using an instrument funded through the NIH shared Instrumentation Grant program (S10OD011952). Mass spectral data were collected at the Masonic Cancer Center, University of Minnesota in the Analytical Biochemistry Shared Resource laboratory; instrumentation there was partially funded by a Cancer Center Support Grant (CA-77598). M. P. S. was supported in part by a Heisig-Gleysteen Fellowship. Computational studies were performed using resources provided by the University of Minnesota Supercomputing Institute (MSI).Notes and references
- (a) A. Z. Bradley and R. P. Johnson, J. Am. Chem. Soc., 1997, 119, 9917–9918 CrossRef CAS; (b) K. Miyawaki, R. Suzuki, T. Kawano and I. Ueda, Tetrahedron Lett., 1997, 38, 3943–3946 CrossRef CAS; (c) T. R. Hoye, B. Baire, D. Niu, P. H. Willoughby and B. P. Woods, Nature, 2012, 490, 208–212 CrossRef CAS PubMed.
- (a) C. H. née Hall and M. F. Greaney, Angew. Chem., Int. Ed., 2014, 53, 5746–5749 CrossRef PubMed; (b) O. J. Diamond and T. B. Marder, Org. Chem. Front., 2017, 4, 891–910 RSC; (c) L. L. Fluegel and T. R. Hoye, Chem. Rev., 2021, 121, 2413–2444 CrossRef CAS PubMed.
- M. P. Smela and T. R. Hoye, Org. Lett., 2018, 20, 5502–5505 CrossRef CAS PubMed.
- (a) M. Bols and T. Skrydstrup, Chem. Rev., 1995, 95, 1253–1277 CrossRef CAS; (b) L. Fensterbank, M. Max and S. Sieburth, Synthesis, 1997, 7, 813–854 CrossRef; (c) D. R. Gauthier Jr, K. S. Zandi and K. J. Shea, Tetrahedron, 1998, 54, 2289–2338 CrossRef; (d) S. Bracegirdle and E. A. Anderson, Chem. Soc. Rev., 2010, 39, 4114–4129 RSC; (e) P. A. Evans, Temporary Silicon-Tethered Ring-Closing Metathesis Reactions in Natural Product Synthesis, in Metathesis in Natural Product Synthesis, ed. J. Cossy, S. Areniyadis and C. Meyer, Wiley-VCH, Weinheim, Germany, 2010, pp. 225–259 Search PubMed; (f) M. Parasram and V. Gevorgyan, Acc. Chem. Res., 2017, 50, 2038–2053 CrossRef CAS PubMed; (g) M. Usman, X.-W. Zhang and W.-B. Liu, Synthesis, 2019, 51, 1529–1544 CrossRef CAS.
- Y. Himeshima, T. Sonoda and H. Kobayashi, Chem. Lett., 1983, 1211–1214 CrossRef CAS.
- A. Nishii, H. Takikawa and K. Suzuki, Chem. Sci., 2019, 10, 3840–3845 RSC.
- A. Mitake, R. Nagai, A. Sekine, H. Takano, N. Sugimura, K. S. Kanyiva and T. Shibata, Chem. Sci., 2019, 10, 6715–6720 RSC.
- (a) T. Kitamura, Z. Meng and Y. Fujiwara, Tetrahedron Lett., 2000, 41, 6611–6614 CrossRef CAS; (b) T. Kitamura, M. Todaka and Y. Fujiwara, (Phenyl)[2-(trimethylsilyl)phenyl]iodonium Triflate. An Efficient and Mild Benzyne Precursor, in Organic Syntheses, ed. W. R. Roush, Wiley, New York, 2000, vol. 78, pp. 104–112 Search PubMed.
- B. P. Woods, B. Baire and T. R. Hoye, Org. Lett., 2014, 16, 4578–4581 CrossRef CAS PubMed.
- (a) S. Nobusue, H. Yamane, H. Miyoshi and Y. Tobe, Org. Lett., 2014, 16, 1940–1943 CrossRef CAS PubMed; (b) For a different outcome with several other 4-atom-tethered triynes see: X. Xiao, B. P. Woods, W. Xiu and T. R. Hoye, Angew. Chem., Int. Ed., 2018, 57, 9901–9905 CrossRef CAS PubMed.
- For the preparation of other R2Si(alkynyl)(OR′′) compounds, see: M. Petit, G. Chouraqui, C. Aubert and M. Malacria, Org. Lett., 2003, 5, 2037–2040 CrossRef CAS PubMed.
- T. Wang, D. Niu and T. R. Hoye, J. Am. Chem. Soc., 2016, 138, 7832–7835 CrossRef CAS PubMed.
- (a) R. P. Johnson, J. Phys. Org. Chem., 2010, 23, 283–292 CAS; (b) Y. Liang, X. Hong, P. Yu and K. N. Houk, Org. Lett., 2014, 16, 5702–5705 CrossRef CAS PubMed; (c) D. J. Marell, L. R. Furan, B. P. Woods, X. Lei, A. J. Bendelsmith, C. J. Cramer, T. R. Hoye and K. T. Kuwata, J. Org. Chem., 2015, 80, 11744–11754 CrossRef CAS PubMed; (d) S. L. Skraba-Joiner, R. P. Johnson and J. Agarwal, J. Org. Chem., 2015, 80, 11779–11787 CrossRef CAS PubMed; (e) M. Chen, C. Q. He and K. N. Houk, J. Org. Chem., 2019, 84, 1959–1963 CrossRef CAS PubMed.
- (a) T. Kawamura and J. K. Kochi, J. Am. Chem. Soc., 1972, 94, 648–650 CrossRef CAS; (b) R. A. Jackson, K. U. Ingold, D. Griller and A. S. Nazran, J. Am. Chem. Soc., 1985, 107, 208–211 CrossRef CAS; (c) N. Auner, R. Walsh and J. Westrup, J. Chem. Soc., Chem. Commun., 1986, 207–208 RSC; (d) I. M. T. Davidson, T. J. Barton, K. J. Hughes, S. Ijadi-Maghsoodi, A. Revis and G. C. Paul, Organometallics, 1987, 6, 644–646 CrossRef CAS; (e) M. R. Ibrahim and W. L. Jorgensen, J. Am. Chem. Soc., 1989, 111, 819–824 CrossRef CAS; (f) J. R. Hwu, K. Y. King, I.-F. Wu and G. H. Hakimelahi, Tetrahedron Lett., 1998, 39, 3721–3724 CrossRef CAS.
- J. Zhang, D. Niu, V. A. Brinker and T. R. Hoye, Org. Lett., 2016, 18, 5596–5599 CrossRef CAS PubMed.
- (a) H. J. Bernstein, Spectrochim. Acta, 1962, 18, 161–170 CrossRef CAS; (b) D. J. Pasto, R. Krasnansky and C. Zercher, J. Org. Chem., 1987, 52, 3062–3072 CrossRef CAS; (c) D. J. Henry, C. J. Parkinson, P. M. Mayer and L. Radom, J. Phys. Chem. A, 2001, 105, 6750–6756 CrossRef CAS; (d) H. Zipse, Top. Curr. Chem., 2006, 263, 163–189 CrossRef CAS.
- K. Jung and M. Koreeda, J. Org. Chem., 1989, 54, 5667–5675 CrossRef CAS.
- A. H. Sato, S. Mihara and T. Iwasawa, Tetrahedron Lett., 2012, 53, 3585–3589 CrossRef CAS.
- S. E. Denmark and T. Kobayashi, J. Org. Chem., 2003, 68, 5153–5159 CrossRef CAS PubMed.
- (a) D. Rodríguez, L. Castedo, D. Domínguez and C. Saá, Tetrahedron Lett., 1999, 40, 7701–7704 CrossRef; (b) T. Kawano, M. Suehiro and I. Ueda, Chem. Lett., 2006, 35, 58–59 CrossRef CAS; (c) L. Li, Q. Hu, P. Zhou, H. Xie, Z. Zhang, H. Zhang, H. Wang and Y. Hu, Synthesis, 2014, 46, 1547–1554 CrossRef; (d) T. Shibata, A. Sekine, A. Mitake and K. S. Kanyiva, Angew. Chem., Int. Ed., 2018, 57, 15862–15865 CrossRef CAS PubMed.
- Generic tetradehydro-Diels–Alder (TDDA) reaction of i to iii (via the strained allene ii), typically favoured over the possible HDDA reaction of the triyne:
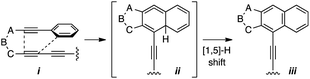 .
.

Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details for the preparation of new compounds; spectroscopic data for their characterization, including copies of 1H and 13C NMR spectra (PDF). See DOI: 10.1039/d1sc04082k |
| ‡ We note that benzoxadisiloles8a such as that in 5a, as well as 1,2-disilylated benzenes,8b (viz.5b–c) are known precursors to benzynes via derived iodonium species. |
| This journal is © The Royal Society of Chemistry 2021 |