
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Azine-N-oxides as effective controlling groups for Rh-catalysed intermolecular alkyne hydroacylation†
Daniel F.
Moseley
,
Jagadeesh
Kalepu
and
Michael C.
Willis
 *
*
Chemistry Research Laboratory, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: michael.willis@chem.ox.ac.uk
First published on 14th September 2021
Abstract
Heterocycle-derived aldehydes are challenging substrates in metal-catalysed hydroacylation chemistry. We show that by using azine N-oxide substituted aldehydes, good reactivity can be achieved, and that they are highly effective substrates for the intermolecular hydroacylation of alkynes. Employing a Rh(I)-catalyst, we achieve a mild and scalable aldehyde C–H activation, that permits the coupling with unactivated terminal alkynes, in good yields and with high regioselectivities (up to >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 l:b). Both substrates can tolerate a broad variety of functional groups. The reaction can also be applied to diazine aldehydes that contain a free N-lone pair. We demonstrate conversion of the hydroacylation products to the corresponding azine, through a one-pot hydroacylation/deoxygenation sequence. A one-pot hydroacylation/cyclisation, using N-Boc propargylamine, additionally leads to the synthesis of a bidentate pyrrolyl ligand.
:1 l:b). Both substrates can tolerate a broad variety of functional groups. The reaction can also be applied to diazine aldehydes that contain a free N-lone pair. We demonstrate conversion of the hydroacylation products to the corresponding azine, through a one-pot hydroacylation/deoxygenation sequence. A one-pot hydroacylation/cyclisation, using N-Boc propargylamine, additionally leads to the synthesis of a bidentate pyrrolyl ligand.
Introduction
C(2)-Substituted azines are becoming increasingly prevalent in a wide selection of pharmaceuticals and agrochemicals1 (Fig. 1). Incorporation of azines and other N-heterocyclic motifs into drug candidates can lead to a plethora of benefits, such as adjusted target specificity/potency, lipophilicity and aqueous solubility.2 Thus, their controlled functionalisation is of paramount importance in medicinal chemistry.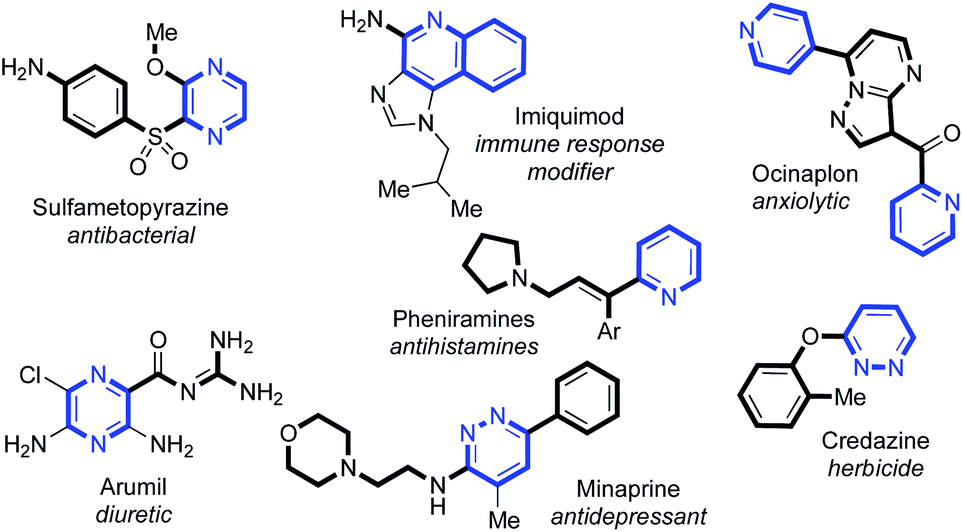 | ||
| Fig. 1 A selection of C(2)-substituted azines present in biologically important compounds. | ||
The utility of N-oxides has propelled the field of catalytic azine C–H functionalisation.3 From an atom-economy perspective, N-oxides are effective directing groups as their removal comprises the loss of a single ‘O’ atom in a straightforward redox process.4 This can sometimes be incorporated into a reaction's catalytic cycle, either through direct deoxygenation5 or O-atom transfer,6 thus relinquishing the need for external oxidants or subsequent reduction steps. A wealth of catalytic reactions exploit the enhanced reactivity that the N-oxide provides to the C(2)-position, priming the azine substrate for C–H bond cleavage.7 A variety of PdII/AgI/Ni0/CuI/II/RhIII-catalysed couplings, towards C–C,8,4b C–O,9 C–S10 and C–N11 functionalised products have been achieved using this strategy. Prior work on azine C(2)-functionalisation has suggested that a metal-coordinated N-oxide species is not necessarily an active catalytic intermediate.4b However, a RhI-catalysed alkenylation of quinoline-N-oxide by Shibata12 was the first method to demonstrate how the N-oxide could formally direct a C–H bond activation, through the generation of a rhodacycle intermediate, and deliver exclusive C(8)-H regioselectivity. This reactivity has been applied to a range of quinoline-N-oxide reactions, yielding C–C,6a,c,d,12,13 C–N14 and C–I14 functionalised products using RhIII/IrIII/PdII and CoIII-catalysts. Despite these advances, methods that utilise N-oxides as a formal directing group remain in their infancy. The success of Shibata's chemistry prompted us to consider the use of N-oxides in intermolecular hydroacylation.15 Azine aldehydes are challenging substrates in hydroacylation as the pyridyl nitrogen can prevent the formation of chelated rhodacycle intermediates, which are key for reaction progression in many hydroacylation systems.16,17 Catalyst inhibition can also be problematic. Jun and Lee previously established an intermolecular alkene hydroacylation with 2-pyridyl aldehydes, via the in situ formation of an aldimine intermediate (Scheme 1A).18 Respectable yields could only be obtained on a single alkene example by hindering the disfavoured N-coordination of the 2-pyridyl group, either through the use of a Zr-additive or with a sterically obstructing ortho-methyl group. The utility of N-oxides as directing groups for aldehyde C–H activations in hydroacylation remains unexplored, and would provide a simple and attractive solution towards the challenges faced when using azines in hydroacylation. Herein, we report the first N-oxide directed hydroacylation of unactivated terminal alkynes using a range of azine aldehydes (Scheme 1B).
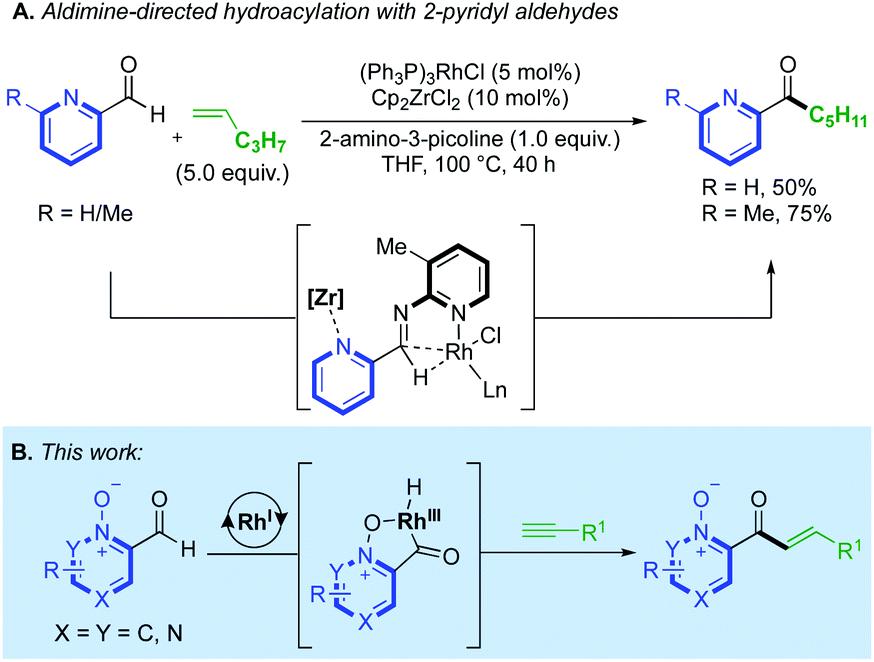 | ||
| Scheme 1 (A) Challenges faced with pyridyl groups in intermolecular hydroacylation; (B) azine-N-oxides as a solution to this compromised reactivity. | ||
Results and discussion
We began our study by investigating the reaction between aldehyde 1a and 1-octyne using Rh(I) catalysts. Our initial ligand evaluation showed that three ligands could produce promising yields: rac-BINAP, dppf and DPEPhos (Scheme 2A). The high yield produced by the dppf-derived catalyst is significant, as this ligand has previously promoted hydroacylation directed by a salicylaldehyde phenolate-anion,19 which is a coordinating group iso-electronic to the pyridine-N-oxide substrate 1a. We decided to further study the result obtained using the DPEPhos-derived catalyst,20 as it delivered the highest linear:branched (l:b) selectivity. Many reactions from the ligand assessment revealed full consumption of aldehyde 1a after 18 h (see ESI, Section 3†), however, yields remained moderate. Pleasingly, increasing the loading of 1-octyne to 2.0 equivalents made a significant improvement in product yield to 67% (Scheme 2B). From this result, a higher yield could then be obtained by making sequential adjustments in scale, reaction concentration (M), and reaction time (h). We additionally investigated the use of alternative solvents; however, despite our efforts, no other solvent system provided an appropriate balance between yield and selectivity (see ESI, Section 3†). Control reactions established that some decomposition of both the aldehyde substrate, and enone products, was possible under the reaction conditions (see ESI, Section 6†). Rh(III)-derived catalysts were not effective in the present system, with N–O reduction products dominating.16d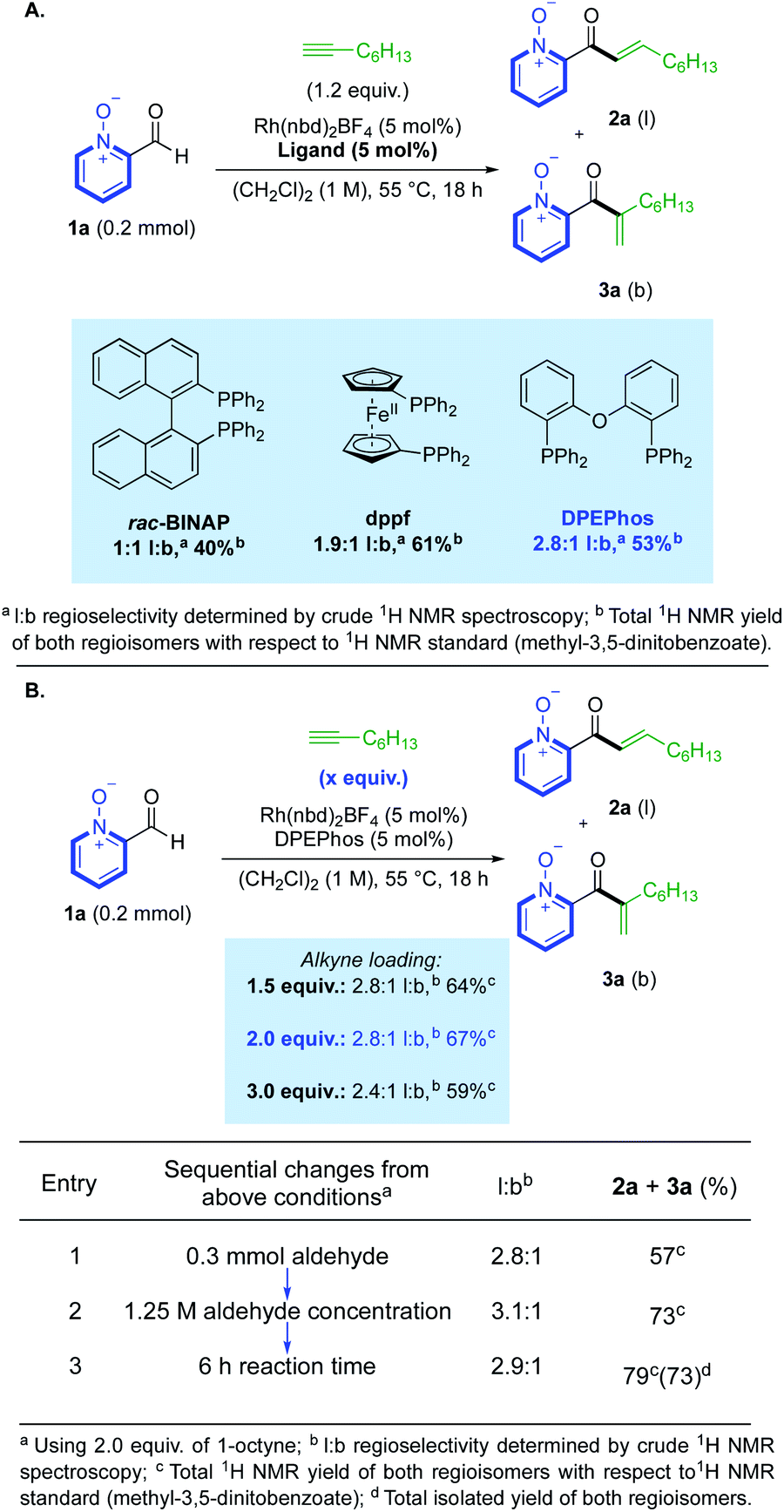 | ||
| Scheme 2 (A) Initial ligand evaluation for reactivity. (B) Selected optimisation of hydroacylation reactivity. | ||
With optimised conditions established, these were then used to explore the reactivity of different alkyne substrates (Scheme 3). Although a return to the longer reaction duration was necessary for more sterically hindered aliphatic alkynes, we were delighted to observe a significant increase in linear regioselectivity, up to >20:1 l:b, in examples 2b–d. TMS-acetylene reacted in a similar fashion to t-Bu-acetylene, delivering linear hydroacylation product 2e in 74% and a >20:1 l:b. ratio.
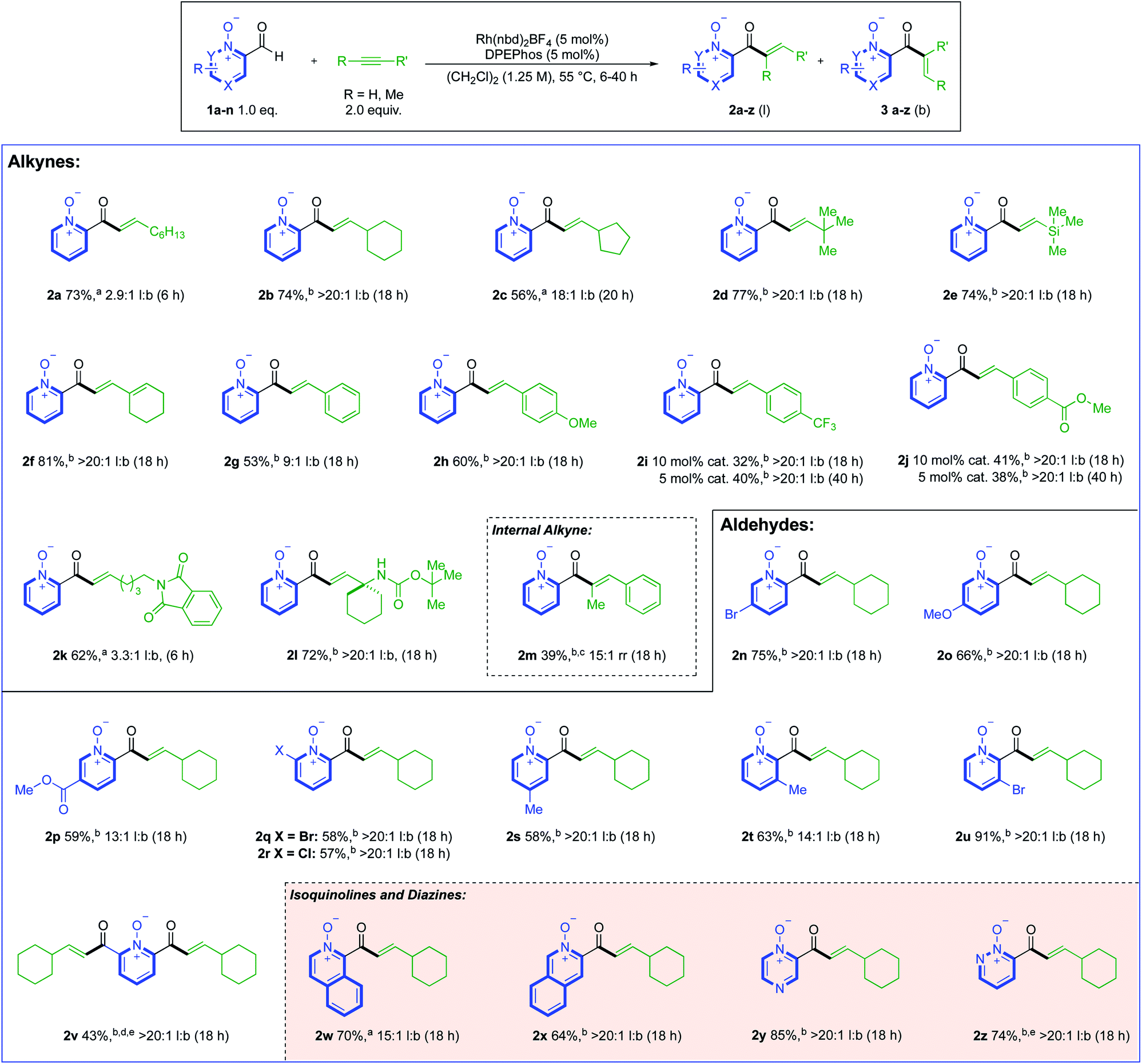 | ||
| Scheme 3 Reaction scope of both alkyne and aldehyde components using 0.3 mmol of aldehyde 1. aYield corresponds to the combined yield of both regioisomers. bYield corresponds to the isolated yield of a single regioisomer. cReaction temperature was 80 °C. d Using 4.0 equiv. alkyne. eReactions were performed on a 0.2 mmol scale. | ||
Cyclohexenyl substituted linear product 2f provided the highest yield of 81%, with >20:1 l:b selectivity. Phenylacetylene proved to be a challenging substrate (2g); however, we could obtain >20:1 l:b selectivity by altering the substrate electronics (2h–j). For the electron-poor phenylacetylene substrates, full conversion to products 2i and 2j could only be achieved with a higher catalyst loading, or longer reaction duration (40 h). We next evaluated the efficacy of nitrogen-bearing alkynes as substrates, with a phthalimide-tethered alkyne (2k), and a cyclohexyl-substituted propargyl amine (2l) both working well. Internal alkynes were generally poor substrates, with both 3-octyne, and diphenylacetylene delivering only trace products. However, employing 1-propynylbenzene as substrate and reacting at 80 °C allowed 39% of trisubstituted-enone 2m to be isolated (with 15:1 rr). Alkene substrates were unreactive (see ESI, Section 6†). We then turned our focus towards the scope of the aldehyde component, where ethynylcyclohexane was used as the alkyne coupling partner (Scheme 3). Substituents at the 5-position of the pyridine aldehyde provided excellent reactivity, with 5-bromo (2n), 5-methoxy (2o), and nicotinate (2p) groups providing the expected products in good yields. More sterically encumbered 6-halo-substituted aldehydes could also be used, and delivered highly selective reactions (2q and 2r). 4- and 3-Me-substituted examples 2s and 2t were obtained in similar yields to 5- and 6-substituted products, and a 3-bromo-substituted aldehyde gave linear enone 2u in excellent yield and selectivity. A double-hydroacylation to deliver 2,6-bis-functionalised product 2v, was achieved in 43%, with >20:1 l:b selectivity. The low reaction conversion for this example was attributed to the poor solubility of the 2,6-bis-aldehyde precursor. Moving away from the pyridine core, we found that 3-formyl-2-isoquinoline-oxide and 4-formyl-4-isoquinoline-oxide both delivered good reactivity, providing enones 2w and 2x, respectively. The aldehyde scope was expanded to include diazines, with pyrazine (2y) and pyridazine (2z) derived N-oxides working well. These two results are significant, as related free azine aldehydes have previously been reported to be poorly reactive in hydroacylation chemistry.18,21
With an effective hydroacylation using azine N-oxide aldehydes achieved, we then set out to confirm we could access the corresponding free 2-pyridyl motifs, through product derivatisation (Scheme 4). Deoxygenation of the N–O bond in hydroacylation product 2b was achieved using PCl3, delivering free pyridyl enone 4 in 72% yield. Previous literature accounts have reported various conditions that permit this deoxygenation in tandem with a non-catalytic C–H functionalization at the 6-position of the pyridine ring.22 Inspired by this work, we performed a deoxyamination reaction on enone 2b to form 6-aminopyridylic enone 5 in a respectable 56% yield. Accessing the free 2-pyridyl enone directly from the starting N-oxide aldehyde, via an in situ reduction, was also achieved through a one-pot hydroacylation/deoxygenation sequence (Scheme 4). This used aldehyde 1a and ethynylcyclohexane, and yielded pyridyl enone 4 in 46%, >20:1 l:b. A control reaction between 2-formylpyridine 1o and ethynylcyclohexane, confirmed no reactivity, thus reinforcing the necessity for the N-oxide group in these transformations.
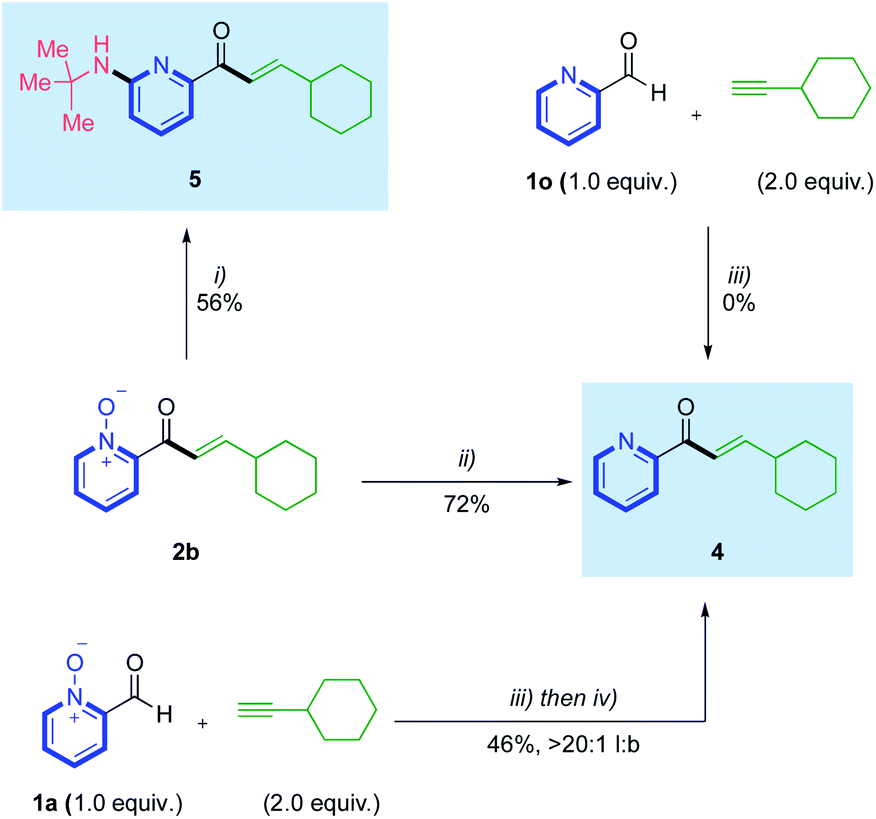 | ||
| Scheme 4 Access to free 2-pyridylic enones 4 and 5 from N-oxide deoxygenations; conditions: (i) Ts2O (3.5 equiv.), t-BuNH2 (8.0 equiv.), 2.5:1 PhCF3:CH2Cl2, 30 °C, 18 h; (ii) PCl3 (1.2 equiv.), toluene (0.2 M), rt, 15 min; (iii) Rh(nbd)2BF4 (5 mol%), DPEPhos (5 mol%), (CH2Cl)2 (1.25 M), 55 °C, 18 h; (iv) PCl3 (1.2 equiv.), (CH2Cl)2 (0.2 M), rt, 15 min. | ||
We also demonstrated how hydroacylation using N-Boc-propargylamine could be performed in tandem with subsequent cyclisation, achieved with stoichiometric p-TSA,23 to generate pyrrolyl ligand 6, from a single reaction pot, in 39% yield and >20:1 l:b (Scheme 5A). Similar products can be synthesised through a pyridine-N-oxide-directed C(2)-H heteroarylation; however, our strategy prevents the formation of 2-/3-pyrrolyl-coupled regioisomeric mixtures which were observed by Tzschucke.24 We additionally conducted a hydroacylation reaction on a larger 1.0 mmol scale, which provided clean conversion to t-Bu-substituted enone 2d in 83%, >20:1 l:b (Scheme 5B).
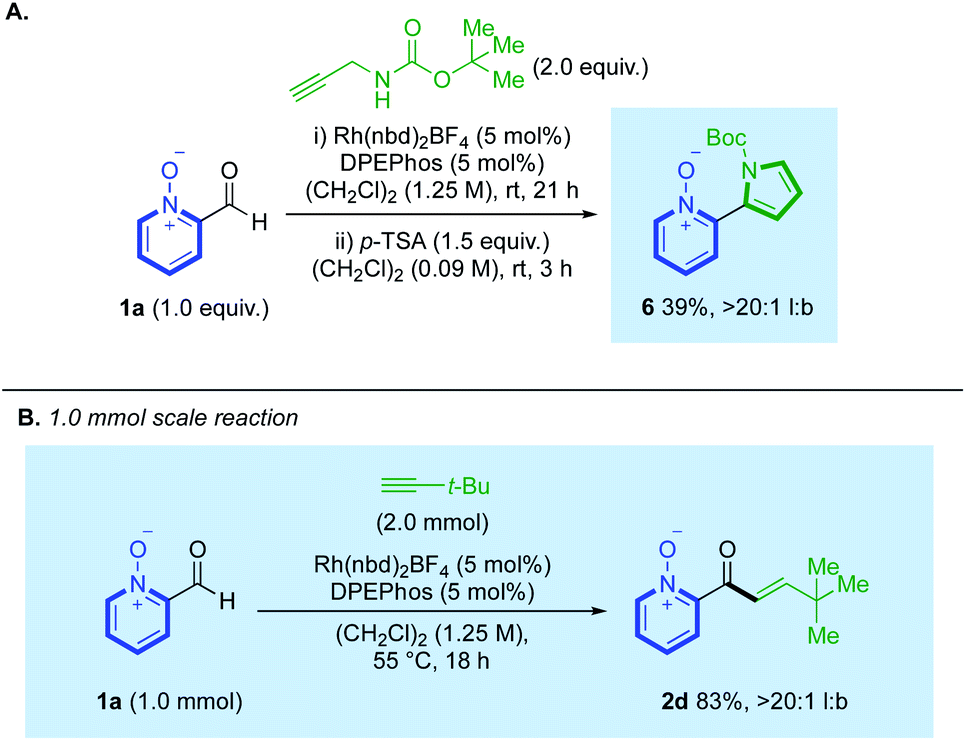 | ||
| Scheme 5 (A) A two-step one-pot hydroacylation/cyclisation towards pyrrolyl ligand 6; (B) employment of aldehyde 1a on a 1.0 mmol scale. | ||
Conclusions
We have demonstrated that azine aldehyde N-oxides can be used as suitable substrates in intermolecular rhodium-catalysed hydroacylation reactions. Using milder temperatures than other equivalent RhI/III-catalysed quinoline-N-oxide C(8)-H functionalisation reactions, this hydroacylation enables the coupling with unactivated terminal alkynes, generating the desired linear enone products in high regioselectivity. The ability for this methodology to tolerate diazines bearing a free nitrogen lone-pair, without the need for other additives, steric modifications or more complex directing groups, is testament to the method's potential synthetic application.Data availability
Full experimental and characterisation data are provided as part of the ESI.†Author contributions
D. F. M. and J. K. performed the experiments and analysed the data. All authors contributed to the discussion and prepared the manuscript. MCW directed the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
DFM is grateful to the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine (EP/L015838/1) for a studentship, and the generous support by GlaxoSmithKline, Vertex, AstraZeneca, Diamond Light Source, Defence Science and Technology Laboratory, Evotec, Janssen, Novartis, Pfizer, Syngenta, Takeda, and UCB is gratefully acknowledged.Notes and references
- (a) I. Mohd and A. Mohammad, Russ. J. Bioorg. Chem., 2020, 46, 745–767 CrossRef; (b) M. M. Heravi and V. Zadsirjan, RSC Adv., 2020, 10, 44247–44311 RSC; (c) V. Nosálová, K. Drábiková, V. Jančinová, R. Nosál, T. MačIčková, J. Pečivová, J. Nedelčevová and R. Sotníková, Inflammation Res., 2009, 58, 68–69 CrossRef PubMed.
- A. Gomtsyan, Chem. Heterocycl. Compd., 2012, 48, 7–10 CrossRef CAS.
- (a) C.-c. Arylations, X. A. F. Cook, A. D. Gombert, J. McKnight, R. E. Pantaine and M. C. Willis, Angew. Chem., Int. Ed., 2021, 60, 11068–11091 CrossRef PubMed; (b) E. N. Da Silva Júnior, G. A. M. Jardim, R. S. Gomes, Y. F. Liang and L. Ackermann, Chem. Commun., 2018, 54, 7398–7411 RSC; (c) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107–1295 RSC; (d) G. Yan, A. J. Borah and M. Yang, Adv. Synth. Catal., 2014, 356, 2375–2394 CrossRef CAS.
- (a) L. C. Campeau, D. R. Stuart, J. P. Leclerc, M. Bertrand-Laperle, E. Villemure, H. Y. Sun, S. Lasserre, N. Guimond, M. Lecavallier and K. Fagnou, J. Am. Chem. Soc., 2009, 131, 3291–3306 CrossRef CAS PubMed; (b) H. C. Seung, J. H. Seung and S. Chang, J. Am. Chem. Soc., 2008, 130, 9254–9256 CrossRef PubMed.
- J. Wu, X. Cui, L. Chen, G. Jiang and Y. Wu, J. Am. Chem. Soc., 2009, 131, 13888–13889 CrossRef CAS PubMed.
- (a) N. Barsu, M. Sen, J. R. Premkumar and B. Sundararaju, Chem. Commun., 2016, 52, 1338–1341 RSC; (b) R. Odani, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2015, 80, 2384–2391 CrossRef CAS PubMed; (c) X. Zhang, Z. Qi and X. Li, Angew. Chem., Int. Ed., 2014, 53, 10794–10798 CrossRef CAS PubMed; (d) U. Sharma, Y. Park and S. Chang, J. Org. Chem., 2014, 79, 9899–9906 CrossRef CAS PubMed.
- (a) Y. Tan, F. Barrios-Landeros and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 3683–3686 CrossRef CAS PubMed; (b) H. Y. Sun, S. I. Gorelsky, D. R. Stuart, L. C. Campeau and K. Fagnou, J. Org. Chem., 2010, 75, 8180–8189 CrossRef CAS PubMed.
- (a) O. V. Larionov, D. Stephens, A. Mfuh and G. Chavez, Org. Lett., 2014, 16, 864–867 CrossRef CAS PubMed; (b) Z. Wu, C. Pi, X. Cui, J. Bai and Y. Wu, Adv. Synth. Catal., 2013, 355, 1971–1976 CrossRef CAS; (c) W. Mai, J. Yuan, Z. Li, G. Sun and L. Qu, Synlett, 2012, 145–149 CrossRef CAS; (d) F. Gosselin, S. J. Savage, N. Blaquiere and S. T. Staben, Org. Lett., 2012, 14, 862–865 CrossRef CAS PubMed; (e) M. P. Huestis and K. Fagnou, Org. Lett., 2009, 11, 1357–1360 CrossRef CAS PubMed; (f) H. Q. Do, R. M. K. Khan and O. Daugulis, J. Am. Chem. Soc., 2008, 130, 15185–15192 CrossRef CAS PubMed; (g) K. S. Kanyiva, Y. Nakao and T. Hiyama, Angew. Chem., Int. Ed., 2007, 46, 8872–8874 CrossRef CAS PubMed; (h) L. C. Campeau, S. Rousseaux and K. Fagnou, J. Am. Chem. Soc., 2005, 127, 18020–18021 CrossRef CAS PubMed; (i) J. R. Huckins, E. A. Bercot, O. R. Thiel, T. L. Hwang and M. M. Bio, J. Am. Chem. Soc., 2013, 135, 14492–14495 CrossRef CAS PubMed.
- X. Chen, C. Zhu, X. Cui and Y. Wu, Chem. Commun., 2013, 49, 6900–6902 RSC.
- Z. Wu, H. Song, X. Cui, C. Pi, W. Du and Y. Wu, Org. Lett., 2013, 15, 1270–1273 CrossRef CAS PubMed.
- (a) G. Li, C. Jia and K. Sun, Org. Lett., 2013, 15, 5198–5201 CrossRef CAS PubMed; (b) C. Zhu, M. Yi, D. Wei, X. Chen, Y. Wu and X. Cui, Org. Lett., 2014, 16, 1840–1843 CrossRef CAS PubMed.
- T. Shibata and Y. Matsuo, Adv. Synth. Catal., 2014, 356, 1516–1520 CrossRef CAS.
- (a) J. Jeong, P. Patel, H. Hwang and S. Chang, Org. Lett., 2014, 16, 4598–4601 CrossRef CAS PubMed; (b) K. Shin, S. W. Park and S. Chang, J. Am. Chem. Soc., 2015, 137, 8584–8592 CrossRef CAS PubMed; (c) D. E. Stephens, J. Lakey-Beitia, A. C. Atesin, T. A. Ateşin, G. Chavez, H. D. Arman and O. V. Larionov, ACS Catal., 2015, 5, 167–175 CrossRef CAS PubMed; (d) D. E. Stephens, J. Lakey-Beitia, G. Chavez, C. Ilie, H. D. Arman and O. V. Larionov, Chem. Commun., 2015, 51, 9507–9510 RSC; (e) D. Kalsi, R. A. Laskar, N. Barsu, J. R. Premkumar and B. Sundararaju, Org. Lett., 2016, 18, 4198–4201 CrossRef CAS PubMed; (f) B. Wang, C. Li and H. Liu, Adv. Synth. Catal., 2017, 359, 3029–3034 CrossRef CAS; (g) C. You, C. Pi, Y. Wu and X. Cui, Adv. Synth. Catal., 2018, 360, 4068–4072 CrossRef CAS; (h) C. M. R. Volla, R. K. Shukla, A. M. Nair and S. Khan, Angew. Chem., Int. Ed., 2020, 59, 2–9 CrossRef.
- H. Hwang, J. Kim, J. Jeong and S. Chang, J. Am. Chem. Soc., 2014, 136, 10770–10776 CrossRef CAS PubMed.
- (a) R. Guo and G. Zhang, Synlett, 2018, 29, 1801–1806 CrossRef CAS; (b) W. W. Chen and M. H. Xu, Org. Biomol. Chem., 2017, 15, 1029–1050 RSC; (c) A. Ghosh, K. F. Johnson, K. L. Vickerman, J. A. Walker and L. M. Stanley, Org. Chem. Front., 2016, 3, 639–644 RSC; (d) J. C. Leung and M. J. Krische, Chem. Sci., 2012, 3, 2202–2209 RSC; (e) G. Rousseau and B. Breit, Angew. Chem., Int. Ed., 2011, 50, 2450–2494 CrossRef CAS PubMed; (f) M. C. Willis, Chem. Rev., 2010, 110, 725–748 CrossRef CAS PubMed; (g) C. H. Jun, E. A. Jo and J. W. Park, Eur. J. Org. Chem., 2007, 1869–1881 CrossRef CAS; (h) R. T. Davison, E. L. Kuker and V. M. Dong, Acc. Chem. Res., 2021, 54, 1236–1250 CrossRef CAS PubMed.
- (a) H. Lee and C. H. Jun, Bull. Korean Chem. Soc., 1995, 16, 66–68 CAS; (b) T. J. Coxon, M. Fernandez, J. Barwick-Silk, A. I. McKay, L. E. Britton, A. S. Weller and M. C. Willis, J. Am. Chem. Soc., 2017, 139, 10142–10149 CrossRef CAS PubMed; (c) M. Castaing, S. L. Wason, B. Estepa, J. F. Hooper and M. C. Willis, Angew. Chem., Int. Ed., 2013, 52, 13280–13283 CrossRef CAS PubMed; (d) T. Zhang, Z. Qi, X. Zhang, L. Wu and X. Li, Chem.–Eur. J., 2014, 20, 3283–3287 CrossRef CAS PubMed; (e) R. N. Straker, M. Formica, J. D. Lupton, J. Niu and M. C. Willis, Tetrahedron, 2018, 74, 5408–5414 CrossRef CAS; (f) C.-H. Jun, D.-Y. Lee, H. Lee and J.-B. Hong, Angew. Chem., Int. Ed., 2000, 39, 3070–3072 CrossRef CAS; (g) J. F. Hooper, S. Seo, F. R. Truscott, J. D. Neuhaus and M. C. Willis, J. Am. Chem. Soc., 2016, 138, 1630–1634 CrossRef CAS PubMed; (h) A. B. Chaplin, J. F. Hooper, A. S. Weller and M. C. Willis, J. Am. Chem. Soc., 2012, 134, 4885–4897 CrossRef CAS PubMed.
- For an example of a Ru-catalysed hydroesterification process that uses a pyridyl-directing group, see: Y. Na, S. Ko, L. K. Hwang and S. Chang, Tetrahedron Lett., 2003, 44, 4475–4478 CrossRef CAS.
- C. H. Jun, D. Y. Lee and J. B. Hong, Tetrahedron Lett., 1997, 38, 6673–6676 CrossRef CAS.
- (a) X. W. Du and L. M. Stanley, Org. Lett., 2015, 17, 3276–3279 CrossRef CAS PubMed; (b) K. Kokubo, K. Matsumasa, Y. Nishinaka, M. Miura and M. Nomura, Bull. Chem. Soc. Jpn., 1999, 72, 303–311 CrossRef CAS; (c) K. Kokubo, K. Matsumasa, M. Miura and M. Nomura, J. Org. Chem., 1997, 62, 4564–4565 CrossRef CAS; (d) M. von Delius, C. M. Le and V. M. Dong, J. Am. Chem. Soc., 2012, 134, 15022–15032 CrossRef CAS PubMed.
- G. L. Moxham, H. E. Randell-Sly, S. K. Brayshaw, R. L. Woodward, A. S. Weller and M. C. Willis, Angew. Chem., Int. Ed., 2006, 45, 7618–7622 CrossRef CAS PubMed.
- Although the N-oxide from quinoline-8-carboxaldehyde was unreactive in the present chemistry (it would require a 6-membered chelate), the parent aldehyde has been used in a stiochiometric Rh-promoted hydroacylation: J. W. Suggs, J. Am. Chem. Soc., 1978, 100, 640–641 CrossRef CAS.
- A. V. Kutasevich, V. P. Perevalov and V. S. Mityanov, Eur. J. Org. Chem., 2021, 2021, 357–373 CrossRef CAS.
- M. K. Majhail, P. M. Ylioja and M. C. Willis, Chem.–Eur. J., 2016, 22, 7879–7884 CrossRef CAS PubMed.
- S. Liu and C. C. Tzschucke, Eur. J. Org. Chem., 2016, 3509–3513 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03915f |
| This journal is © The Royal Society of Chemistry 2021 |