DOI:
10.1039/D1SC04011A
(Perspective)
Chem. Sci., 2021,
12, 12866-12873
Transition metal-catalyzed organic reactions in undivided electrochemical cells
Received
22nd July 2021
, Accepted 24th August 2021
First published on 24th August 2021
Abstract
Transition metal-catalyzed organic electrochemistry is a rapidly growing research area owing in part to the ability of metal catalysts to alter the selectivity of a given transformation. This conversion mainly focuses on transition metal-catalyzed anodic oxidation and cathodic reduction and great progress has been achieved in both areas. Typically, only one of the half-cell reactions is involved in the organic reaction while a sacrificial reaction occurs at the counter electrode, which is inherently wasteful since one electrode is not being used productively. Recently, transition metal-catalyzed paired electrolysis that makes use of both anodic oxidation and cathodic reduction has attracted much attention. This perspective highlights the recent progress of each type of electrochemical reaction and relatively focuses on the transition metal-catalyzed paired electrolysis, showcasing that electrochemical reactions involving transition metal catalysis have advantages over conventional reactions in terms of controlling the reaction activity and selectivity and figuring out that transition metal-catalyzed paired electrolysis is an important direction of organic electrochemistry in the future and offers numerous opportunities for new and improved organic reaction methods.
1. Introduction
Synthetic methodologists are perpetually motivated to develop useful reactions that are also maximally sustainable. The past decade has witnessed a rapid increase in the use of electrochemical methods to accomplish new transformations of small organic molecules. Seminal electrochemical synthetic advances since the advent of the battery (in the year 1800)1 include the Kolbe electrolysis (1847),2 the Tafel rearrangement (1907),3 the Shono oxidation (1984),4 the Simons fluorination (1949),5a and the Baizer adiponitrile synthesis (1964).5b The culmination of the development of electrochemical techniques, methods, and equipment has allowed the current electrochemical organic synthesis renaissance.
The intersection of electrocatalysis and transition metal catalysis has resulted in a rapidly growing subdiscipline that seeks to unite the sustainable nature of electrochemistry and the chemoselective, regioselective, and stereoselective hallmarks of organotransition metal-catalyzed synthetic methods. The merger of these disciplines has proven successful in that the use of stoichiometric redox additives can be avoided and other limitations of more conventional methods have been overcome.6 Typically, though, only one of the two half-cell electrochemical reactions is used productively while a sacrificial reaction occurs at the counter electrode, which is wasteful. Paired electrolysis has garnered much attention since it could make use of both cathode and anode and thereby maximize the energy efficiency so long as the innate redox properties of the cathodic and anodic reactions are properly matched. Fortunately, the redox properties of an organotransition metal complex could be tuned via the modification of the organic ligand. Ultimately, paired electrolysis could permit simultaneous anodic oxidation and cathodic reduction of different catalytic intermediates en route to a target, and hence paired electrolysis could provide improved faradaic efficiency without diminishing atom economy.
Paired electrolysis has been successfully used for the industrial synthesis of nylon-6,6,7 gluconic acid, sorbitol,8 methyl ethyl ketone9 and others.10 There are several impressive reviews encompassing the advances of paired electrolysis,11 but these reviews do not focus on transition metal-catalyzed paired electrolysis since the development of such methods is in its infancy. Although there have been many reviews on transition metal-catalyzed organic electrochemistry,6,12,13 there is no review focusing on highlighting the anodic oxidation, cathodic reduction, and paired electrolysis which have advantages over thermochemical reactions. Herein, this perspective highlights the latest progress including our own progress in these areas, showcasing that paired electrolysis has great advantages in improving the atom and energy economy of electrosynthesis. Thus transition metal-catalyzed paired electrolysis is an important direction of organic electrochemistry in the future and offers numerous opportunities for new and improved organic reaction methods. This perspective introduces the transition metal-catalyzed paired electrolysis relatively in more detail for there have been many reviews on transition metal-catalyzed electrochemistry6,12,13 and both anodic oxidation and cathodic reduction are familiar to us. It is organized into three parts based on the productive (non-sacrificial) electrode: (1) anodic oxidation; (2) cathodic reduction; and (3) paired electrolysis. And we choose the most representative chemical transformations to introduce each type.
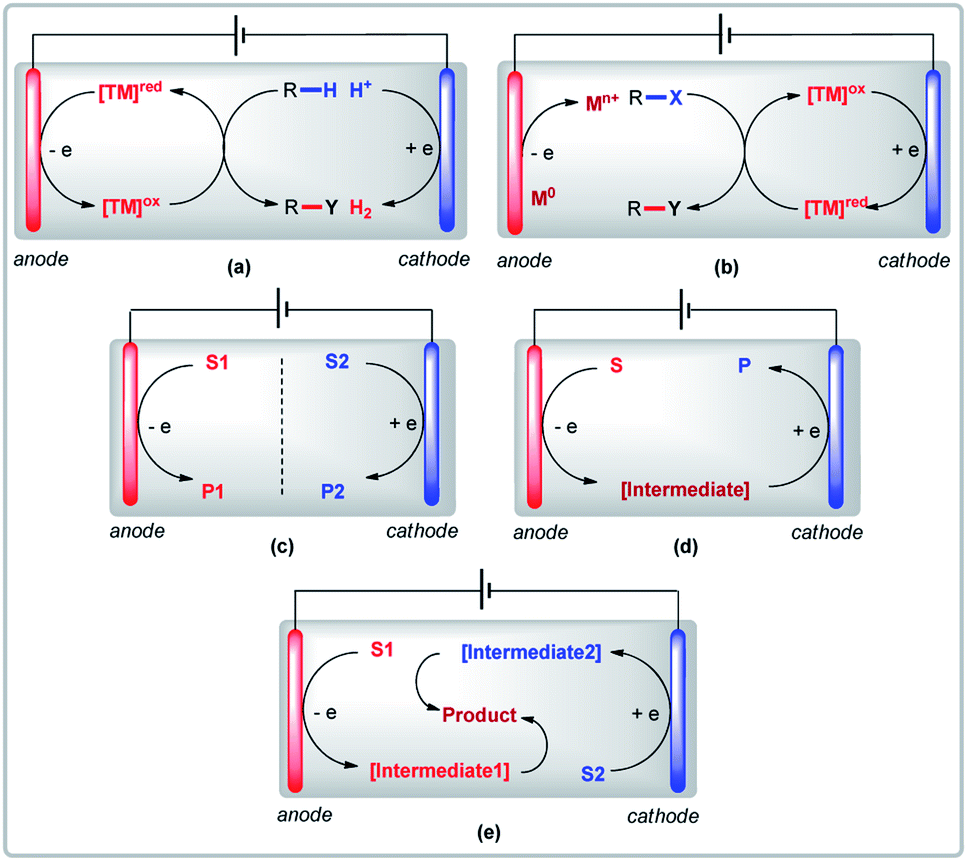
A visual glossary of electrolysis modes in transition metal-catalyzed organic reactions is provided below to illustrate the key differences between anodic oxidation, cathodic reduction, and variants of paired electrolysis (parallel, sequential, and convergent). With the exclusive use of either anodic oxidation or cathodic reduction to obtain target products, sacrificial reactions at the counter electrode are inevitable (Fig. 1a and b). Paired electrolysis has the potential to overcome this problem and use anodic oxidation and cathodic reduction synergistically. There are three types of paired electrolysis: (a) parallel paired electrolysis (Fig. 1c); (b) sequential paired electrolysis (Fig. 1d); and (c) convergent paired electrolysis (Fig. 1e). For clarity, only reactions involving undivided cells (UCs) will be discussed in this perspective.
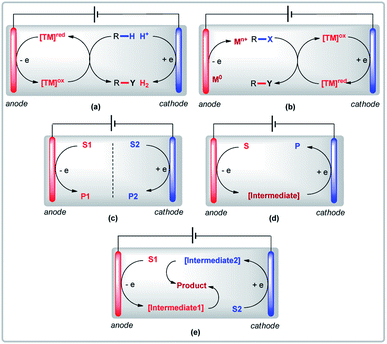 |
| | Fig. 1 (a) Anodic oxidation, (b) cathodic reduction, (c) parallel paired electrolysis, (d) sequential paired electrolysis, and (e) convergent paired electrolysis. | |
2. Transition metal-catalyzed reactions mediated by anodic oxidation
Anodic oxidation has special advantages in regulating the metal valence and producing free radicals. Recently, transition metal catalyzed anodic oxidation is mainly studied in the fields of C–H functionalization or radical reactions and has been developed and studied by the groups of Kakiuchi,6a Mei,6b Lin,6c Ackermann,6d Lei,6e Xu,14 and others,15 as a means for regenerating a transition metal catalyst or for generating a high-valent metal intermediate, in both cases replacing toxic (and often expensive) stoichiometric chemical oxidants. Herein, we highlight several representative studies of C–H functionalization and radical reactions which have shown that electrochemical reactions involving transition metal catalysis have advantages over conventional reactions in terms of controlling the reaction activity and selectivity.16 These transformations engage transition metal catalysts to regulate the chemo-, regio- and stereo-selectivity of reactions wherein such selectivity was traditionally dictated by the substrate.
2.1 Transition metal-catalyzed C–H bond functionalization mediated by anodic oxidation
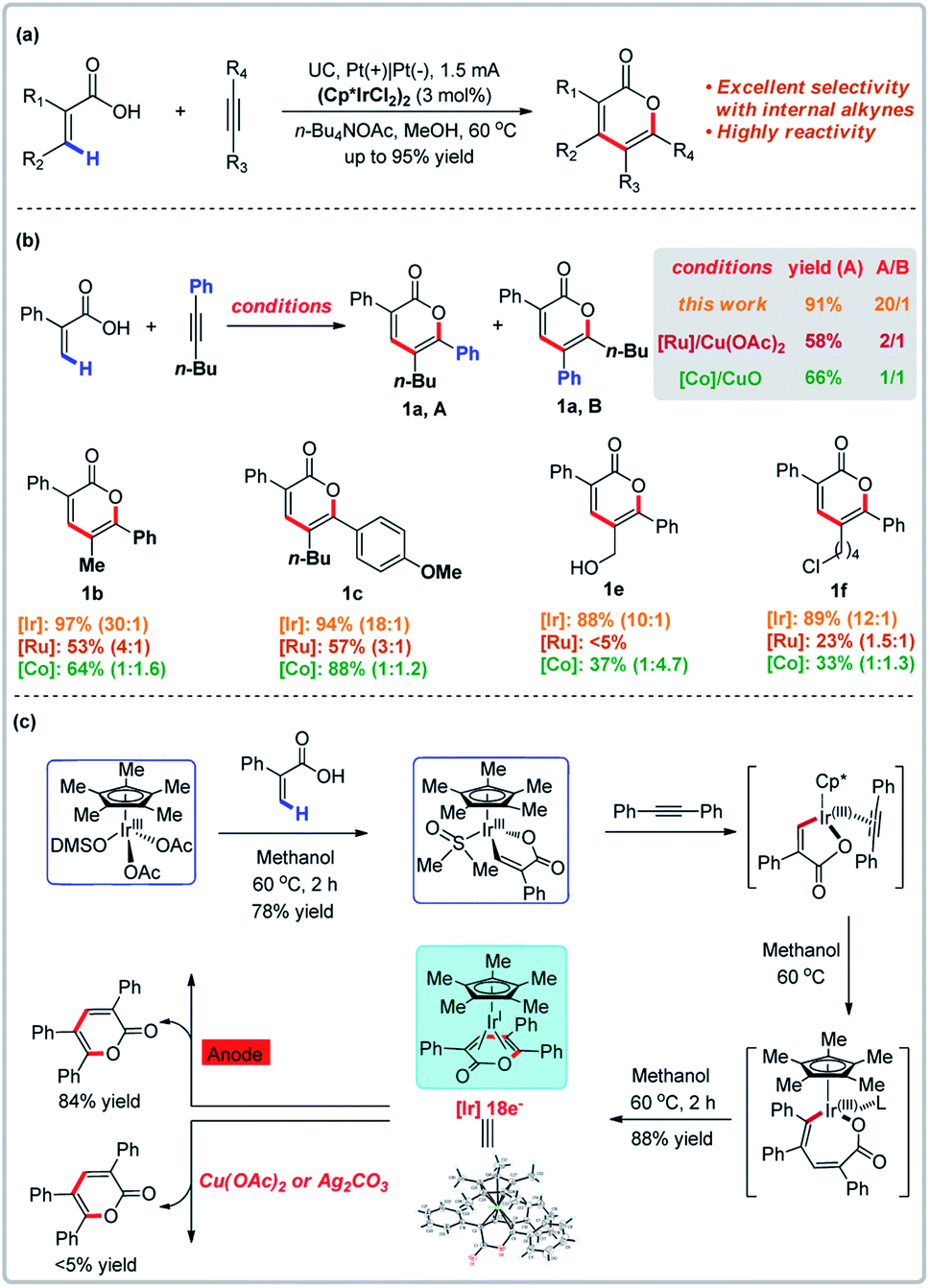
Transition metal-catalyzed direct C–H bond functionalization has witnessed an explosive development in recent decades and a variety of useful methods are now available to directly convert C–H bonds into various functional groups.17 Transition metal-catalyzed vinylic C–H annulations of acrylic acids with alkynes have long been established,18 but the regioselectivity of metal-catalyzed intramolecular [4 + 2] cyclization of unsymmetrical alkenes and alkynes is typically limited. In 2019, Mei and co-workers demonstrated the first example of Ir-catalyzed electrochemical vinylic C–H annulation of acrylic acids with alkynes (Fig. 2).19 This electrochemical variant was directly compared to established Co- or Ru-catalyzed annulations of acrylic acids with unsymmetrical internal alkynes and proved to be superior in terms of regioselectivity and functional group tolerance (for selected examples, see Fig. 2b). The Ir-catalyzed electrochemical annulation affords regioselectivites of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 30:1 and yields up to 95%. In contrast, the Co- or Ru-catalyzed annulation with chemical oxidants only affords regioselectivites of 1:1 to 1:4.7 and 1:1 to 4:1, respectively.18a,d Mechanistic experiments reported by Mei and co-workers revealed that anodic oxidation was crucial for reoxidation. X-ray crystallography revealed that the Ir(I) intermediate in the catalytic cycle has a saturated coordination sphere, which resists oxidation to the corresponding Ir(III) by a chemical oxidant. In contrast, the Ir(I) complex is easily oxidized to Ir(III) at the anode to give the product with a yield of 84%.
:1 to 30:1 and yields up to 95%. In contrast, the Co- or Ru-catalyzed annulation with chemical oxidants only affords regioselectivites of 1:1 to 1:4.7 and 1:1 to 4:1, respectively.18a,d Mechanistic experiments reported by Mei and co-workers revealed that anodic oxidation was crucial for reoxidation. X-ray crystallography revealed that the Ir(I) intermediate in the catalytic cycle has a saturated coordination sphere, which resists oxidation to the corresponding Ir(III) by a chemical oxidant. In contrast, the Ir(I) complex is easily oxidized to Ir(III) at the anode to give the product with a yield of 84%.
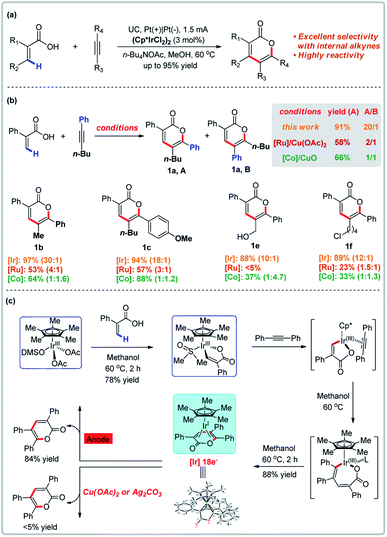 |
| | Fig. 2 C–H bond functionalization promoted by anodic oxidation. (a) Ir-Catalyzed electrochemical vinylic C–H annulation. (b) Selected examples compared to the Ru/Co system. (c) Proposed iridium redox catalysis under electrochemical conditions. | |
2.2 Transition metal-catalyzed radical reactions mediated by anodic oxidation
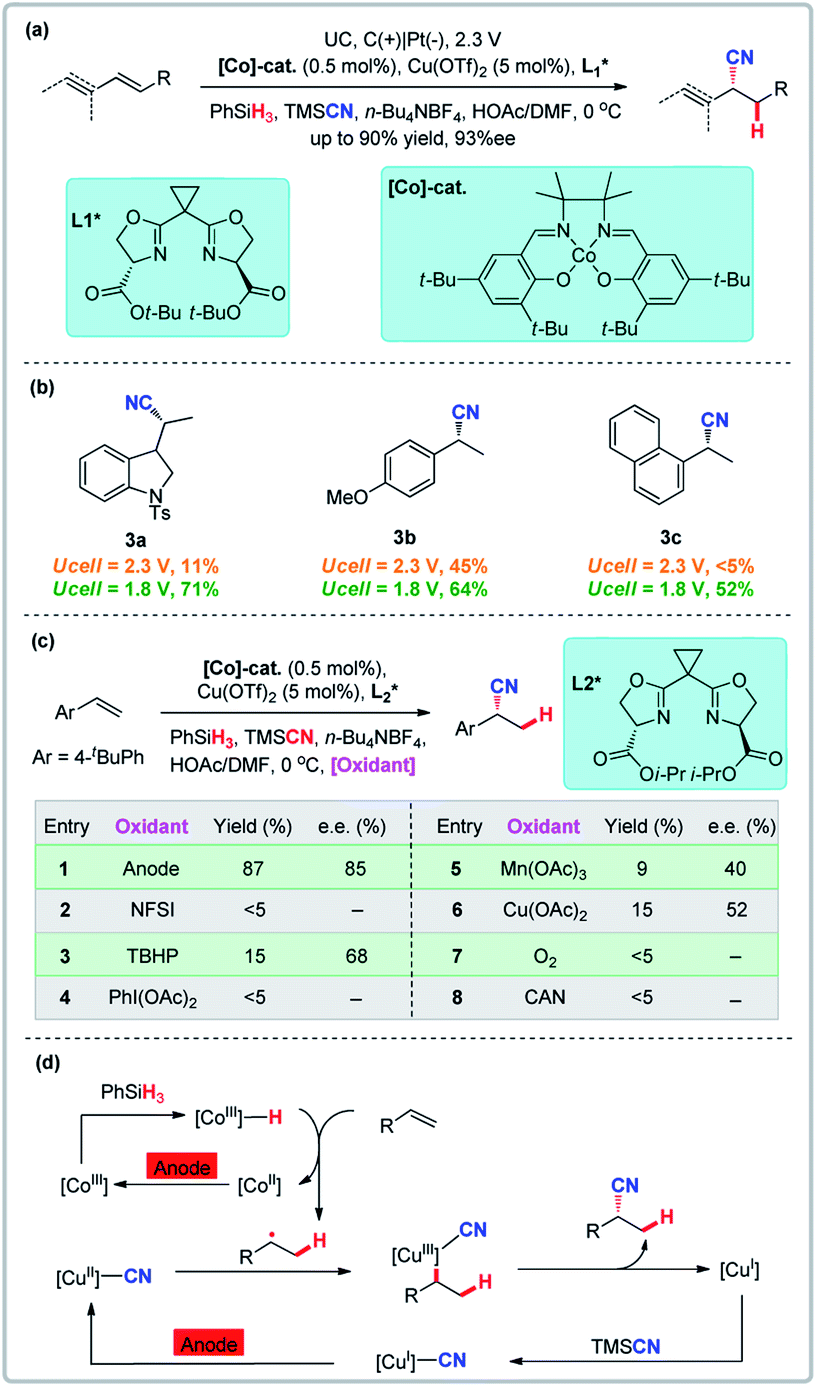
Radical reactions are useful for the difunctionalization of alkenes, with the atom transfer radical addition (ATRA) method being particularly noteworthy. Known radical cyanation examples are primarily limited to the intramolecular cyclization or reactions of activated alkenes.20 Reactive nickel hydride intermediates limit the scope and are likely responsible for the sensitivity of reaction yield and selectivity toward electronic properties of alkenes.21 In addition, enantioselective variants remain elusive.22 Early studies of electrochemical transition metal-catalyzed hydrofunctionalization of alkenes are also primarily limited to intramolecular cyclization or reactions of activated alkenes with few examples of intermolecular reactions of unactivated alkenes.23 Lin and co-workers developed the first novel dual electrocatalysis for enantioselective hydrocyanation of conjugated alkenes which is powered by a Co and Cu dual electrocatalytic process in which two canonical radical reactions (cobalt-catalyzed hydrogen-atom transfer and copper-promoted radical cyanation) are combined (Fig. 3).24 This approach demonstrates the feasibility of the electrochemical hydrofunctionalization of alkenes that has a broad scope, high functional group tolerance, high enantioselectivity, and electrochemical control of chemoselectivity. Compared with chemical oxidants which have their own unique oxidation potential, electrochemistry can regulate the hydrocyanation chemoselectivity as a function of the electrode potential input. Over-oxidation was successfully suppressed by lowering the external potential from 2.3 V to 1.8 V (Eanode ≈ 0.08 V) (Fig. 3b). A direct comparison to traditional thermochemical (non-electrochemical) reactions shows that none of the chemical oxidants surveyed promoted the reaction with efficiencies or enantioselectivities comparable to the electrochemical variant, thus clearly indicating that electrochemistry can circumvent problems with stoichiometric chemical oxidation (Fig. 3c).
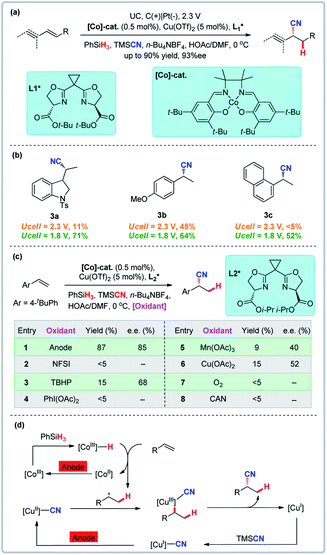 |
| | Fig. 3 Radical reactions promoted by anodic oxidation. (a) Enantioselective hydrocyanation of conjugated alkenes. (b) Electrochemical tuning of reaction chemoselectivity. (c) Comparison with chemical methods. (d) Proposed catalytic cycles for alkene hydrocyanation. | |
3. Transition metal-catalyzed reactions mediated by cathodic reduction
Compared to electrochemical reactions mediated by anodic oxidation, there are relatively few reports of reactions promoted by cathodic reduction.25 Thermochemical reduction, including reductive coupling reactions, provides a wide variety of C–C bond formation reactions, but traditionally relies on the presence of stoichiometric active metal reducing agents (Mn, Zn, and others). Thus, cathode reduction in transition metal catalysis could avoid the need for a metal reducing agent, thereby improving the atom economy of the reaction.
3.1 Reductive relay cross-coupling of alkyl halides to aryl halides mediated by cathodic reduction
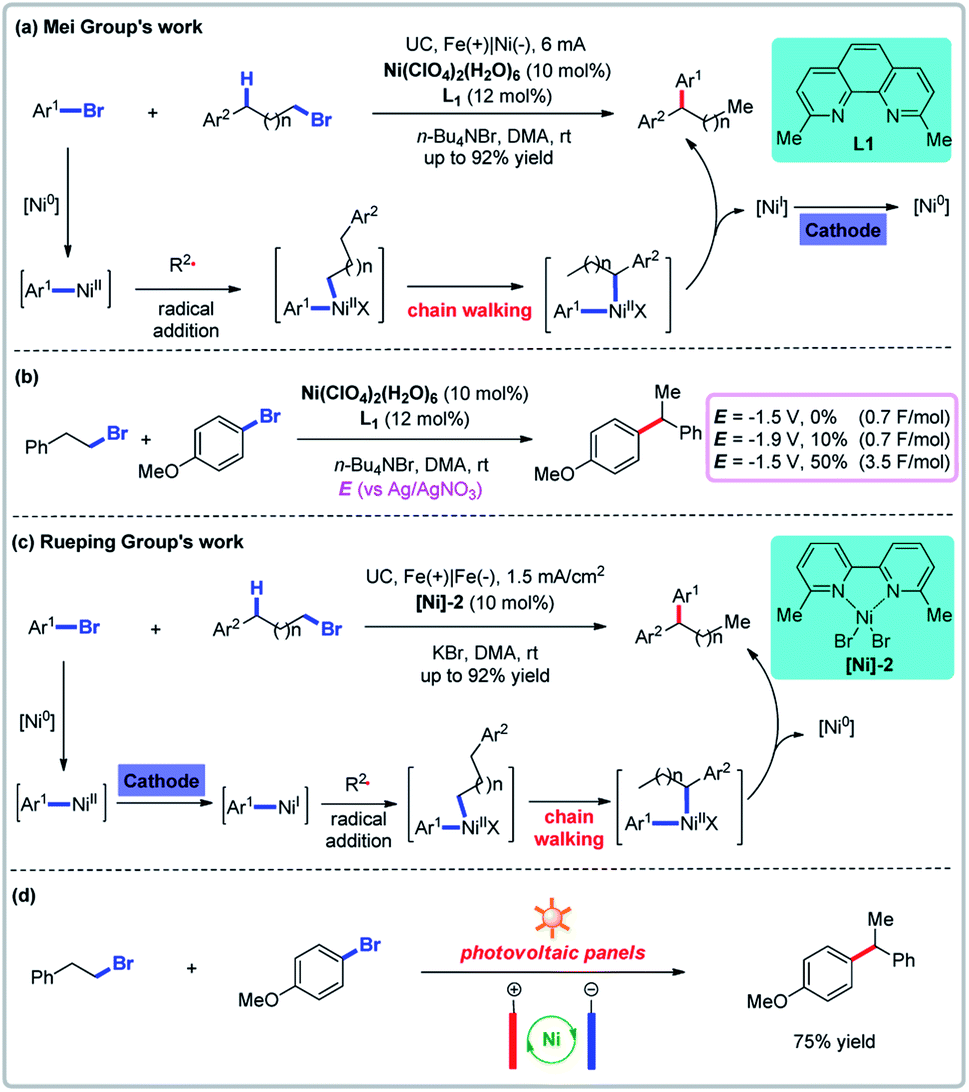
Nickel-catalyzed electrochemical reductive coupling reactions have been extensively developed as a powerful means to couple two electrophiles. This strategy has been employed to both homo-coupling and cross-coupling reactions. The groups of Mei and Rueping reported relay cross-couplings of aryl bromides (or chlorides) and aryl-substituted alkyl bromides simultaneously, notably avoiding the need to add a metal reducing agent (Fig. 4).26 Cyclic voltammetric analyses show that when the nickel catalyst and the ligand are both present, there are two quasireversible reductive peaks at −1.82 V and −1.29 V (vs. Ag/AgNO3) which Mei and co-workers attribute to the reductive potential of Ni(I)/Ni(0) and Ni(II)/Ni(I).26a The desired product could not be obtained by controlled potential electrolysis at −1.5 V vs. Ag/AgNO3 (0.7 F mol−1) but a lower potential of −1.9 V (0.7 F mol−1) could provide the product in 10% yield. The yield could be increased to 50% if five times the amount of electricity was used. Similar results were obtained by Rueping and co-workers. These results provide suggestive evidence for the mechanism of the catalytic cycle. In addition, Rueping and co-workers employed a photovoltaic panel as the source of electricity (Fig. 4d).26b
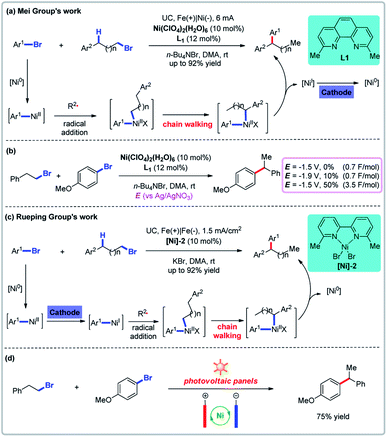 |
| | Fig. 4 Electrochemical reductive relay cross-coupling. (a) Mei group's nickel-catalyzed relay cross-couplings. (b) Controlled potential electrolysis. (c) Rueping group's nickel-catalyzed chain walking cross-couplings. (d) The use of solar energy for the reductive cross-coupling. | |
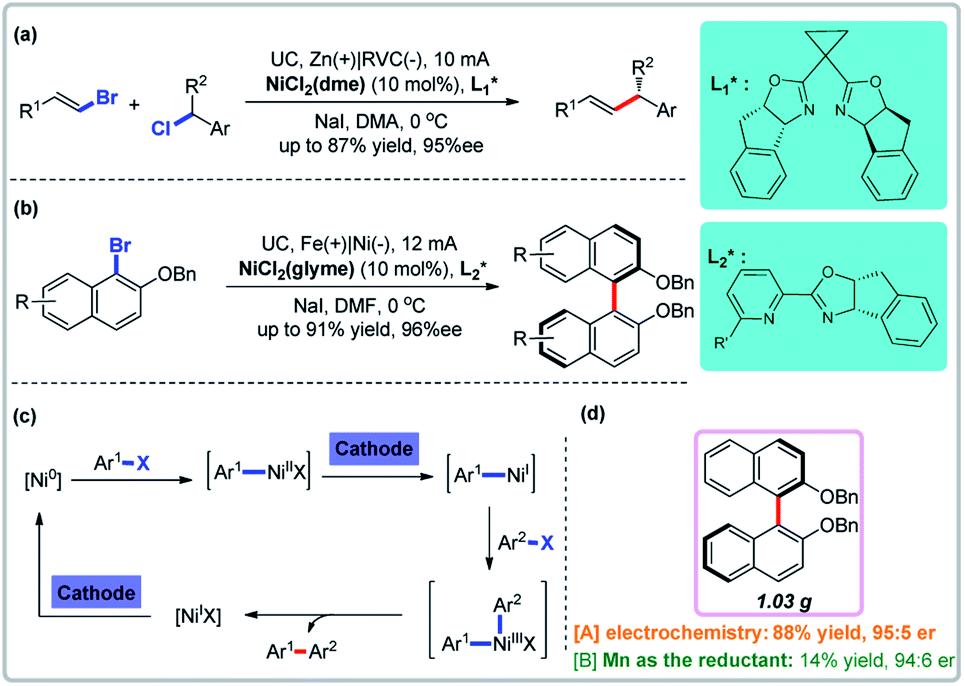
3.2 Reductive enantioselective coupling reactions mediated by cathodic reduction
Ni-Catalyzed reductive coupling reactions avoid the use of organometal nucleophiles and have been extensively studied using Mn, Zn, or Mg powders as reducing agents. Metal-catalyzed electrochemical reductive coupling reactions, especially asymmetric ones, are rare.27 Reisman and co-workers developed the first such reaction using cathodic reduction and nickel catalysis (Fig. 5a).28a Recently, Mei and co-workers disclosed the first nickel-catalyzed electrochemical reductive homocoupling reaction of aryl bromides (Fig. 5b).28b A direct comparison to non-electrochemical variants showed that the use of a stoichiometric amount of Mn powder resulted in lower yields in the absence of electrical current, underscoring the unique utility of electrochemistry (Fig. 5d). Both Reisman and Mei groups showed through control experiments that both the nickel catalyst and electrical current are essential for the reaction. There are two cathodic reductive steps in the plausible catalytic cycle of these nickel-catalyzed electrochemical reductive coupling reactions (Fig. 5c).
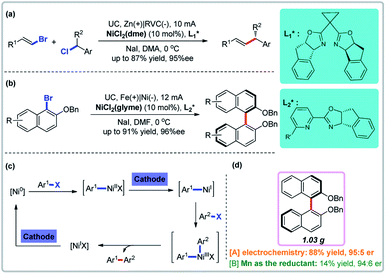 |
| | Fig. 5 Electrochemical reductive enantioselective coupling reactions. (a) Enantioselective cross-coupling of alkenyl and benzyl halides. (b) Enantioselective synthesis of biaryl atropisomers. (c) Proposed mechanism. (d) Direct comparison with homocoupling. | |
4. Transition metal-catalyzed reactions mediated by paired electrolysis
In paired electrolysis reactions, both the anode and cathode are involved in the transformation of a reactant or intermediate (neither electrode is used sacrificially). Paired electrolysis has recently attracted significant attention although most reports involve non-catalytic reactions.11 In 2019, Ye and co-workers developed metal-free convergent paired electrolysis for the direct arylation of α-amino C(sp3)–H bonds.29 Jensen and co-workers reported convergent paired microfluidic electrochemical decarboxylative arylation, α-amino C–H arylation, and deboronative arylation.30 Findlater and co-workers developed an alcohol arylation reaction using paired electrolysis.31 Dong and co-workers developed an efficient synthesis of vicinal dibromides and dichlorides via paired electrolysis.32 There are many other interesting studies in this area33 that have been reviewed previously.11 Since organotransition metal catalysis can tune chemoselectivity, regioselectivity, and stereoselectivity through ligand modification, synergy between transition metal catalysis and paired electrolysis has drawn more and more attention of late. In 2019, Chen and co-workers reported a manganese-catalyzed electrochemical oxychlorination of styrenes via paired electrolysis.34 Jiao and co-workers developed a nickel-catalyzed oxygenation of sulfides with water using paired electrolysis.35 Shimakoshi and co-workers reported an interesting one-pot synthesis of tertiary amides from organic trichlorides using a B12 derivative as the catalyst by paired electrolysis.36 Compared to the reported metal-free ones, transition metal catalyzed paired electrolysis is relatively rare.37 Some representative instances on sequential and convergent paired electrolysis will be discussed in detail.
4.1 Transition metal-catalyzed reactions mediated by sequential paired electrolysis
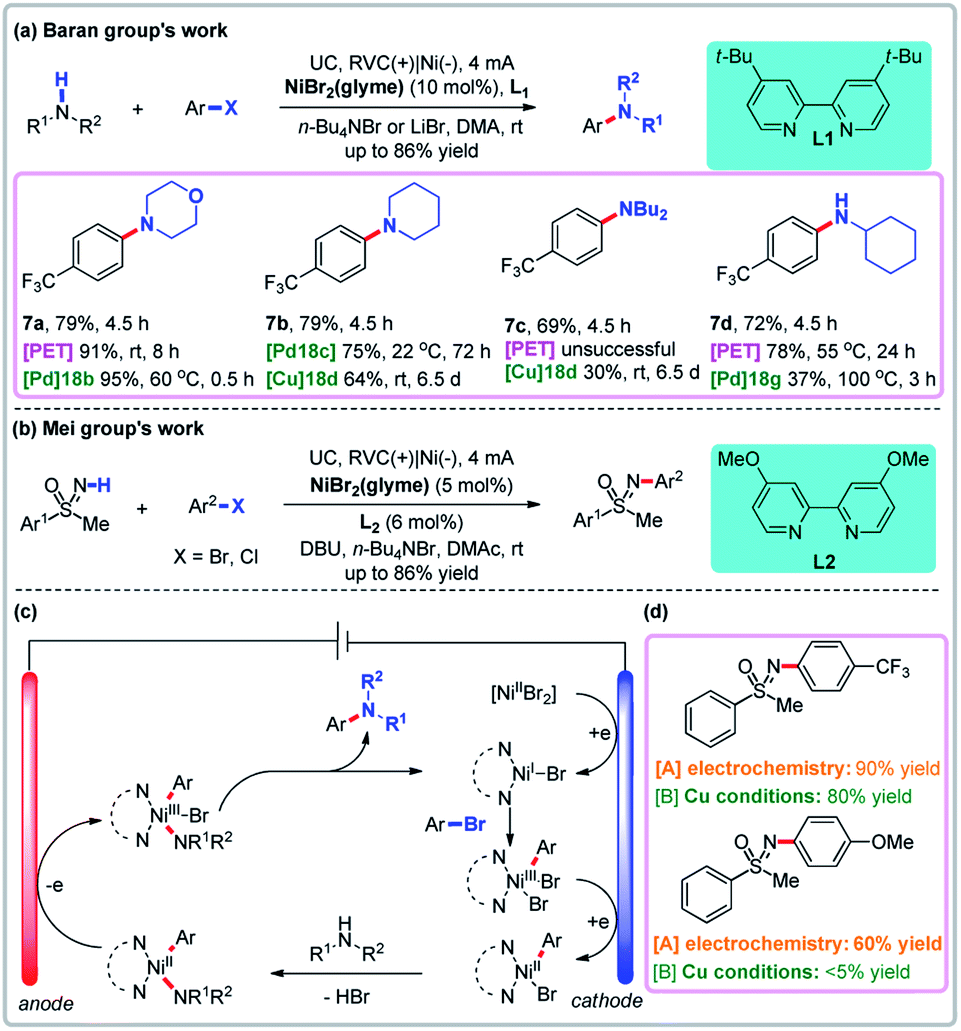
In transition metal-catalyzed reactions involving sequential paired electrolysis, a metal intermediate is oxidized and reduced (or vice versa) sequentially. Thermochemical nickel-catalyzed C–N coupling reactions are well established, but often employ air-sensitive nickel(0) catalysts, high temperatures, and harsh alkoxide bases.38 To skirt these issues, Baran and co-workers developed an electrochemical amination of aryl halides using nickel catalysis and sequential anodic oxidation and cathodic reduction to generate active catalyst species (Fig. 6a).39a Compared with thermochemical methods, the electrochemical reaction conditions are mild and no external base is needed. Although arene aminations combining nickel and photoredox catalysis afforded higher yield, such reactions did not work for some substrates. Through mechanistic studies, Baran and co-workers found that both the cathode and anode play critical roles in the catalytic cycle of NiI/NiIII (Fig. 6c).39b Specifically, the NiI intermediate that undergoes an oxidative addition to give the active NiIII intermediate is itself generated from the reduction of a NiII precatalyst on the cathode. The NiIII intermediate is then reduced on the cathode to give the stable NiII complex, which undergoes ligand exchange with the amine. The resultant NiII complex is oxidized on the anode to generate the highly active NiIII intermediate, which undergoes a facile reductive elimination process to give the arylamine product. This strategy expands the substrate scope of C–N coupling reactions to allow access to natural products and sugars that are difficult to access using non-electrochemical approaches such as the Buchwald–Hartwig reaction. Then the NiII complex was oxidized to generate the highly active NiIII intermediate which underwent a rapid reductive elimination process to give the arylamine product and the NiI intermediate on the anode. Recently, Mei and co-workers developed a nickel-catalyzed N-arylation of NH-sulfoximines using aryl halides via paired electrolysis under very mild conditions (Fig. 6b).40 In comparison, non-electrochemical copper-catalyzed reactions involving aryl chlorides are slow, although aryl bromides perform similarly. These reports show that the electrochemical reaction has great advantages compared to the traditional reactions.
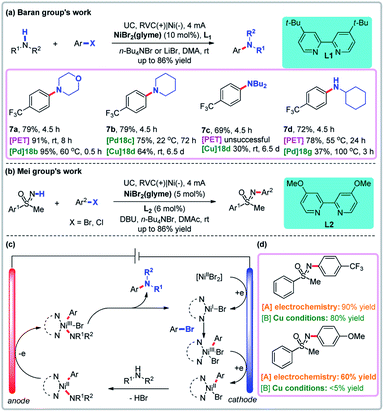 |
| | Fig. 6 Sequential paired electrolysis with Ni as the catalyst. (a) Baran group's amination via paired electrolysis with Ni as the catalyst. (b) Mei group's N-arylation via paired electrolysis with Ni as the catalyst. (c) Proposed mechanism. (d) Selective comparison. | |
4.2 Transition metal-catalyzed reactions mediated by convergent paired electrolysis
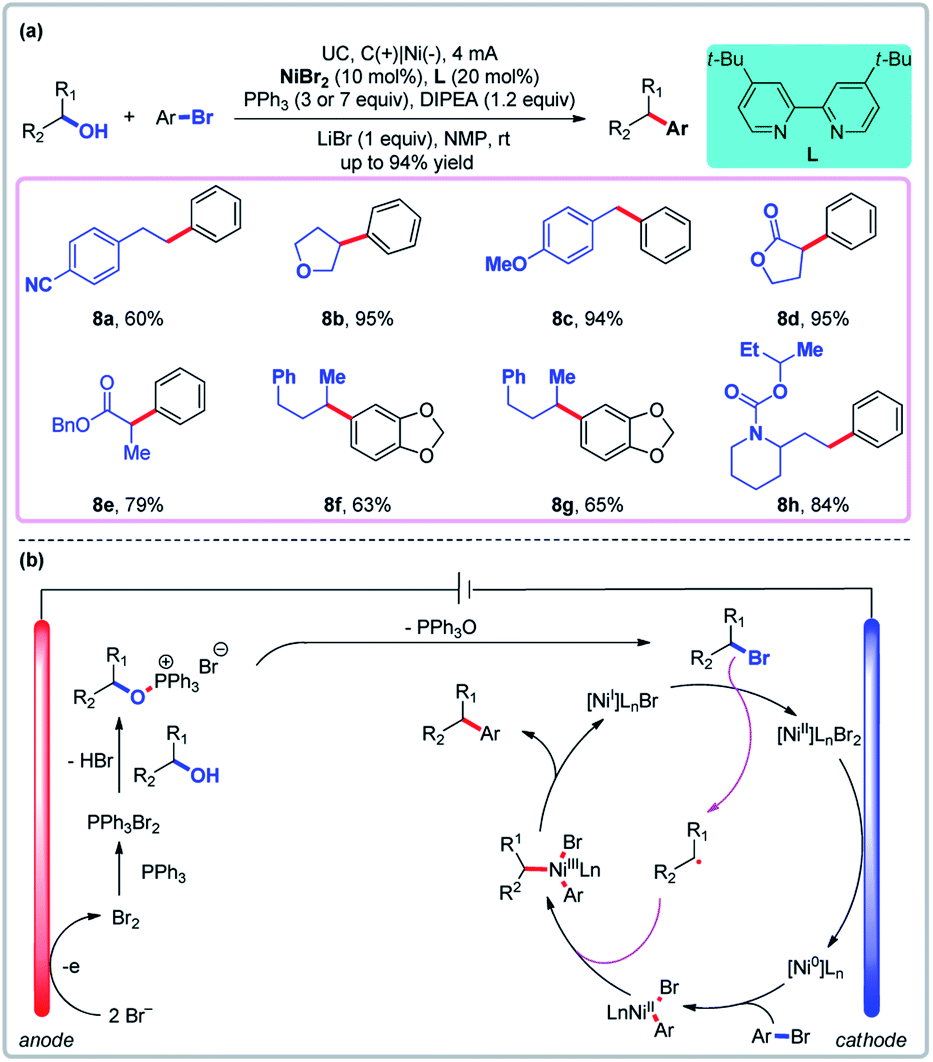
In convergent paired electrolysis, intermediates produced by anodic and cathodic processes react with each other to yield the product. Although alcohol derivatives have been widely used as cross-coupling partners,41 the direct use of unprotected aliphatic alcohols as C(sp3) synthons is rare.42 Recently, Li and co-workers reported a novel direct arylation of readily available free alcohols through a one-step paired electrolysis which elegantly pairs an anodic Appel reaction and a cathodic nickel-catalyzed cross-electrophile coupling (Fig. 7).43 The use of stoichiometric hazardous CBr4 or Br2 in the thermochemical Appel reaction and the use of stoichiometric Zn or Mn in traditional reductive cross-couplings are avoided using this procedure (LiBr serves as the source of bromine atoms). Cyclic voltammetry has shown that the bromide ion has lower oxidative potential than both PPh3 and alcohols in NMP (oxidative potential: 1.1, 1.7, and >2.0 V, respectively, for LiBr, PPh3, and alcohol vs. Ag/AgCl). The bromide ion is oxidized to afford Br2 on the anode, which can react with PPh3 to generate PPh3Br2. Then the PPh3Br2 is coupled to an alcohol to afford alkoxy triphenylphosphonium bromide, which can then undergo reductive cross-coupling (Fig. 7b). Again, the ability to use free alcohols as catalytic cross-coupling substrates is a unique feature of this paired electrolysis.
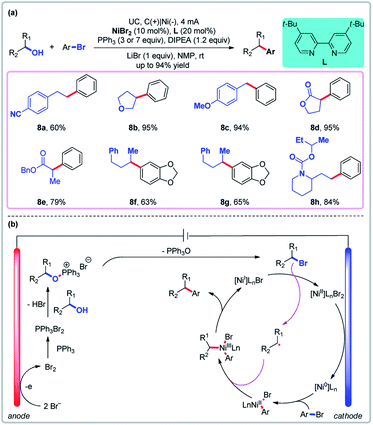 |
| | Fig. 7 Convergent paired electrolysis with Ni as the catalyst. (a) Dehydroxylative cross-coupling of alcohols with aryl halides. (b) Proposed mechanism. | |
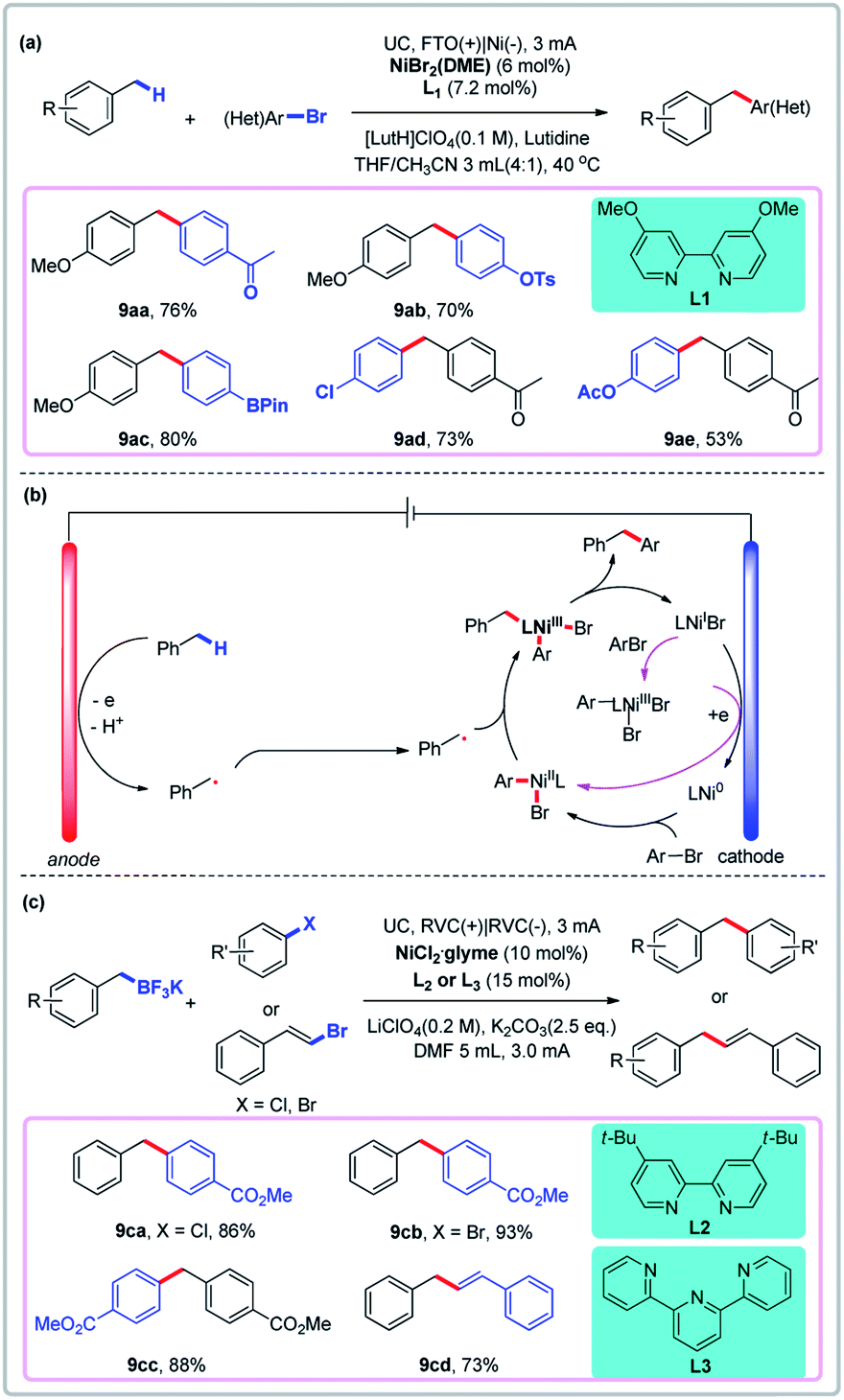
Ni-catalyzed electrochemical reductive coupling of aryl halides is well-established.44 For example, the synthesis of diarylmethanes via an in situ-generated benzylic cation and nucleophile has been developed by Yoshida and Waldvogel groups.45 More recently, Zhang and Hu reported the direct arylation of benzylic C–H bonds via convergent paired electrolysis using a nickel catalyst (Fig. 8a).46 In this process, toluene derivatives are oxidized to give benzyl radicals, which are then trapped by the nickel catalyst at the anode, thus avoiding sacrificial electrodes such as Zn and Mn. Linear sweep voltammetry (LSV) was used to gain more insight into the mechanism. They found that the benzylic substrate and the nickel catalyst have different redox properties on different electrodes. Fluorine-doped tin oxide (FTO) was shown to be a uniquely suitable anode for reactions in which 4-methylanisole and the nickel catalyst are oxidized at the anode (even though a carbon fiber anode could more easily oxidize the Ni catalyst than an FTO anode). In the putative mechanism, the toluene derivative is first oxidized to give the benzyl radical, which is then trapped by a Ni(II) species to give a Ni(III) intermediate, which undergoes reductive elimination to give the product (Fig. 8b). Very recently, Liu and co-workers developed an elegant method for Ni-catalyzed C(sp2)–C(sp3) cross-coupling reactions of benzyl trifluoroborate and organic halides via convergent paired electrolysis (Fig. 8c).47
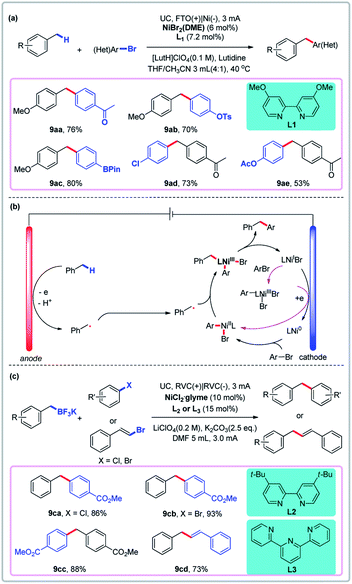 |
| | Fig. 8 Convergent paired electrolysis with Ni as the catalyst. (a) Direct arylation of benzylic C–H bonds. (b) Proposed mechanism. (c) Cross-coupling reactions of benzyl trifluoroborate and organic halides. | |
5. Conclusion and outlook
Organotransition metal-catalyzed electrochemical reactions have been rapidly advancing. Chemo-, regio-, and stereo-selectivity, redox potentials and coordination mode can all be tuned by the modification of organic ligands. The development of more efficient, more sustainable, and more broadly applicable transition metal-catalyzed electrochemical synthetic methods has virtually only just begun. The key foundational accomplishments described in this review and associated implication for future studies include (1) the transition metal catalyzed electrochemical functionalization of the C(sp3)–H bond and asymmetric variants should continue to be the focus; (2) a low catalyst turnover should be addressed by exploring catalyst-modified electrodes; (3) more easily recovered electrolytes are being developed and should be applied to these reactions; (4) paired electrolysis should be favored to avoid needless and wasteful sacrificial side reactions. The merger of organotransition metal catalysis and paired electrolysis has the potential to address each of these issues.
Author contributions
T.-S. M. conceived the idea of the perspective. C. M. and P. F. designed the structure of the perspective. C. M. wrote the initial manuscript. D. L., K.-J. J., P.-S. G. and H. Q. collected the papers related to the topic of the perspective. All the authors participated in the revision of the manuscript.
Conflicts of interest
There are no conflicts to declare.
References
- A. G. A. Volta, J. Nat. Philos. Chem. Arts, 1800, 4, 179 Search PubMed.
- H. Kolbe, Prakt. Chem., 1847, 41, 138 CrossRef.
- J. Tafel and H. Hahl, Ber. Dtsch. Chem. Ges., 1907, 40, 3312 CrossRef CAS.
- T. Shono, Tetrahedron, 1984, 40, 811 CrossRef CAS.
-
(a) J. H. Simons, J. Electrochem. Soc., 1949, 95, 47 CrossRef CAS;
(b) M. M. Baizer, J. Electrochem. Soc., 1964, 111, 215 CrossRef CAS.
-
(a) F. Kakiuchi and T. Kochi, Chem. Lett., 2020, 49, 1256 CrossRef CAS;
(b) K.-J. Jiao, Y.-K. Xing, Q.-L. Yang, H. Qiu and T.-S. Mei, Acc. Chem. Res., 2020, 53, 300 CrossRef CAS PubMed;
(c) J. C. Siu, N. Fu and S. Lin, Acc. Chem. Res., 2020, 53, 547 CrossRef CAS PubMed;
(d) L. Ackermann, Acc. Chem. Res., 2020, 53, 84 CrossRef CAS PubMed;
(e) Y. Yuan and A. Lei, Acc. Chem. Res., 2019, 52, 3309 CrossRef CAS PubMed;
(f) T. Wu, B. H. Nguyen, M. C. Daugherty and K. D. Moeller, Angew. Chem., Int. Ed., 2019, 58, 3562 CrossRef CAS PubMed;
(g) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230 CrossRef CAS PubMed.
- D. A. White and M. M. Baizer, J. Chem. Soc., Perkin Trans. 1, 1973, 2230 RSC.
- J. C. Yu, M. M. Baizer and K. Nobe, J. Electrochem. Soc., 1988, 135, 1400 CrossRef CAS.
- M. Baizer, T. Nonaka, K. Park, Y. Saito and K. Nobe, J. Appl. Electrochem., 1984, 14, 197 CrossRef CAS.
-
(a) K. Scott, Electrochim. Acta, 1991, 36, 1447 CrossRef CAS;
(b) A. Ansari and D. Nematollahi, Appl. Catal., B, 2020, 261, 118226 CrossRef CAS.
-
(a) N. Sbei, T. Hardwick and N. Ahmed, ACS Sustainable Chem. Eng., 2021, 9, 6148 CrossRef CAS;
(b) W. Zhang, N. Hong, L. Song and N. Fu, Chem. Rec., 2021 DOI:10.1002/tcr.202100025;
(c) G. Hilt, ChemElectroChem, 2020, 7, 395 CrossRef CAS;
(d) N. P. Martínez, M. Isaacs and K. K. Nanda, New J. Chem., 2020, 44, 5617 RSC;
(e) D. Pletcher, R. A. Green and R. C. Brown, Chem. Rev., 2018, 118, 4573 CrossRef CAS PubMed;
(f) J. G. Ibanez, B. A. Frontana-Uribe and R. Vasquez-Medrano, J. Mex. Chem. Soc., 2016, 60, 247 CAS;
(g) W. Li, T. Nonaka and T.-C. Chou, Electrochemistry, 1999, 67, 4 CrossRef CAS.
- C. Ma, P. Fang, Z.-R. Liu, S.-S. Xu, K. Xu, X. Cheng, A. Lei, H.-C. Xu, C. Zeng and T.-S. Mei, Sci. Bull., 2021 DOI:10.1016/j.scib.2021.07.011.
-
(a) Z. N. Gafurov, A. O. Kantyukov, A. A. Kagilev, O. G. Sinyashin and D. G. Yakhvarov, Coord. Chem. Rev., 2021, 442, 213986 CrossRef CAS;
(b) K. Yamamoto, M. Kuriyama and O. Onomura, Curr. Opin. Electrochem., 2021, 28, 100714 CrossRef CAS;
(c) J.-S. Zhong, Y. Yu, Z.-J. Shi and K.-Y. Ye, Org. Chem. Front., 2021, 8, 1315 RSC;
(d) J. Q. Lu, Y. K. Wang, T. McCallum and N. Fu, iScience, 2020, 23, 101796 CrossRef CAS PubMed;
(e) X. Chang, J. Zhang, Q. Zhang and C. Guo, Angew. Chem., Int. Ed., 2020, 59, 18500 CrossRef CAS PubMed;
(f) Y. Qin, J. Lu, Z. Zou, H. Hong, Y. Li, Y. Li, L. Chen, J. Hu and Y. Huang, Org. Chem. Front., 2020, 7, 1817 RSC;
(g) P. Gandeepan, L. H. Finger, T. H. Meyer and L. Ackermann, Chem. Soc. Rev., 2020, 49, 4254 RSC;
(h) T. H. Meyer, L. H. Finger, P. Gandeepan and L. Ackermann, Trends Chem., 2019, 1, 63 CrossRef CAS;
(i) J. B. Chen, S. D. Lv and S. Y. Tian, ChemSusChem, 2019, 12, 115 CrossRef CAS PubMed;
(j) F. Kakiuchi and T. Kochi, Chem. Rec., 2021, 21 DOI:10.1002/tcr.202100050.
-
(a) Z.-J. Wu, F. Su, W. Lin, J. Song, T.-B. Wen, H.-J. Zhang and H.-C. Xu, Angew. Chem., Int. Ed., 2019, 58, 16770 CrossRef CAS PubMed;
(b) F. Xu, Y.-J. Li, C. Huang and H.-C. Xu, ACS Catal., 2018, 8, 3820 CrossRef CAS.
-
(a) Z. Liu, Z.-C. Yin, W.-Q. Lu, C. Niu, M. Chen, S. Yang and G.-W. Wang, Org. Lett., 2021, 23, 4051 CrossRef CAS PubMed;
(b) Y. K. Au, H. Lyu, Y. Quan and Z. Xie, J. Am. Chem. Soc., 2020, 142, 6940 CrossRef CAS PubMed;
(c) J. Lai and M. A. Pericàs, Org. Lett., 2020, 22, 7338 CrossRef CAS PubMed;
(d) Z. Zhang, L. Zhang, Y. Cao, F. Li, G. Bai, G. Liu, Y. Yang and F. Mo, Org. Lett., 2019, 21, 762 CrossRef CAS PubMed;
(e) D.-Z. Lin, Y.-L. Lai and J.-M. Huang, ChemElectroChem, 2019, 6, 4188 CrossRef CAS;
(f) B. D. W. Allen, M. D. Hareram, A. C. Seastram, T. McBride, T. Wirth, D. L. Browne and L. C. Morrill, Org. Lett., 2019, 21, 9241 CrossRef CAS PubMed;
(g) X. Ye, P. Zhao, S. Zhang, Y. Zhang, Q. Wang, C. Shan, L. Wojtas, H. Guo, H. Chen and X. Shi, Angew. Chem., Int. Ed., 2019, 58, 17226 CrossRef CAS PubMed.
-
(a) L. Niu, C. Jiang, Y. Liang, D. Liu, F. Bu, R. Shi, H. Chen, A. D. Chowdhury and A. Lei, J. Am. Chem. Soc., 2020, 142, 17693 CrossRef CAS PubMed;
(b) L. Li, Y. Li, N. Fu, L. Zhang and S. Luo, Angew. Chem., Int. Ed., 2020, 59, 14347 CrossRef CAS PubMed;
(c) Z.-J. Wu, F. Su, W. Lin, J. Song, T.-B. Wen, H.-J. Zhang and H.-C. Xu, Angew. Chem., Int. Ed., 2019, 58, 16770 CrossRef CAS PubMed;
(d) Q.-L. Yang, X.-Y. Wang, J.-Y. Lu, L.-P. Zhang, P. Fang and T.-S. Mei, J. Am. Chem. Soc., 2018, 140, 11487 CrossRef CAS PubMed;
(e) S. Tang, D. Wang, Y. Liu, L. Zeng and A. Lei, Nat. Commun., 2018, 9, 798 CrossRef PubMed;
(f) N. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Science, 2017, 357, 575 CrossRef CAS PubMed.
-
(a) P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199 CrossRef CAS;
(b) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS PubMed;
(c) J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992 RSC;
(d) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed.
-
(a) R. Mandal and B. Sundararaju, Org. Lett., 2017, 19, 2544 CrossRef CAS PubMed;
(b) M. Sen, R. Mandal, A. Das, D. Kalsi and B. Sundararaju, Chem.–Eur. J., 2017, 23, 17454 CrossRef CAS PubMed;
(c) G. Duarah, P. P. Kaishap, T. Begum and S. Gogoi, Adv. Synth. Catal., 2019, 361, 654 CrossRef CAS;
(d) L. Ackermann, J. Pospech, K. Graczyk and K. Rauch, Org. Lett., 2012, 14, 930 CrossRef CAS PubMed;
(e) Y. Yang, K. Li, Y. Cheng, D. Wan, M. Li and J. You, Chem. Commun., 2016, 52, 2872 RSC;
(f) Y. Yu, L. Huang, W. Wu and H. Jiang, Org. Lett., 2014, 16, 2146 CrossRef CAS PubMed.
- Q.-L. Yang, Y.-K. Xing, X.-Y. Wang, H.-X. Ma, X.-J. Weng, X. Yang, H.-M. Guo and T.-S. Mei, J. Am. Chem. Soc., 2019, 141, 18970 CrossRef CAS PubMed.
-
(a) T. V. RajanBabu and A. L. Casalnuovo, J. Am. Chem. Soc., 1996, 118, 6325 CrossRef CAS;
(b) A. L. Casalnuovo, T. V. RajanBabu, T. A. Ayers and T. Warren, J. Am. Chem. Soc., 1994, 116, 9869 CrossRef CAS;
(c) T. V. RajanBabu and A. L. Casalnuovo, J. Am. Chem. Soc., 1992, 114, 6265 CrossRef CAS.
-
(a) A. W. Schuppe, G. M. Borrajo-Calleja and S. L. Buchwald, J. Am. Chem. Soc., 2019, 141, 18668 CrossRef CAS PubMed;
(b) X. Li, C. You, J. Yang, S. Li, D. Zhang, H. Lv and X. Zhang, Angew. Chem., Int. Ed., 2019, 58, 10928 CrossRef CAS PubMed;
(c) A. Falk, A. Cavalieri, G. S. Nichol, D. Vogt and H. G. Schmalz, Adv. Synth. Catal., 2015, 357, 3317 CrossRef CAS;
(d) A. Falk, A. L. Göderz and H. G. Schmalz, Angew. Chem., Int. Ed., 2013, 52, 1576 CrossRef CAS PubMed;
(e) J. Wilting, M. Janssen, C. Müller and D. Vogt, J. Am. Chem. Soc., 2006, 128, 11374 CrossRef CAS PubMed;
(f) B. Saha and T. V. RajanBabu, Org. Lett., 2006, 8, 4657 CrossRef CAS PubMed.
-
(a) Q. Lin, L. Li and S. Luo, Chem.–Eur. J., 2019, 25, 10033 CrossRef CAS PubMed;
(b) B. H. Nguyen, A. Redden and K. D. Moeller, Green Chem., 2014, 16, 69 RSC;
(c) S. Torii, P. Liu and H. Tanaka, Chem. Lett., 1995, 24, 319 CrossRef.
-
(a) L. Zhu, P. Xiong, Z.-Y. Mao, Y.-H. Wang, X. Yan, X. Lu and H.-C. Xu, Angew. Chem., Int. Ed., 2016, 55, 2226 CrossRef CAS PubMed;
(b) J. A. Miranda, C. J. Wade and R. D. Little, J. Org. Chem., 2005, 70, 8017 CrossRef CAS PubMed;
(c) T. Shono, S. Kashimura, Y. Mori, T. Hayashi, T. Soejima and Y. Yamaguchi, J. Org. Chem., 1989, 54, 6001 CrossRef CAS.
- S. Lin, N. Fu, B. G. Ernst, W. H. Lee, M. O. Frederick, R. A. J. DiStasio and S. Lin, Nat. Chem., 2020, 12, 747 CrossRef CAS PubMed.
-
(a) Y. Yuan and A. Lei, Nat. Commun., 2020, 11, 802 CrossRef CAS PubMed;
(b) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594 CrossRef CAS PubMed.
-
(a) K.-J. Jiao, D. Liu, H.-X. Ma, H. Qiu, P. Fang and T.-S. Mei, Angew. Chem., Int. Ed., 2020, 59, 6520 CrossRef CAS PubMed;
(b) S. K. Gadde, A. Peshkov, A. Brzozowska, P. Nikolaienko, C. Zhu and M. Rueping, Angew. Chem., Int. Ed., 2020, 59, 6513 CrossRef PubMed.
-
(a) X. Wang, Y. Dai and H. Gong, Top. Curr. Chem., 2016, 374, 43 CrossRef PubMed;
(b) D. J. Weix, Acc. Chem. Res., 2015, 48, 1767 CrossRef CAS PubMed;
(c) C. E. I. Knappke, S. Grupe, D. Gärtner, M. Corpet, C. Gosmini and A. J. von Wangelin, Chem.–Eur. J., 2014, 20, 6828 CrossRef CAS PubMed.
-
(a) T. J. DeLano and S. E. Reisman, ACS Catal., 2019, 9, 6751 CrossRef CAS PubMed;
(b) H. Qiu, B. Shuai, Y.-Z. Wang, D. Liu, Y.-G. Chen, P.-S. Gao, H.-X. Ma, S. Chen and T.-S. Mei, J. Am. Chem. Soc., 2020, 142, 9872 CrossRef CAS PubMed.
- Y. Ma, X. Yao, L. Zhang, P. Ni, R. Cheng and J. Ye, Angew. Chem., Int. Ed., 2019, 58, 16548 CrossRef CAS PubMed.
- Y. Mo, Z. Lu, G. Rughoobur, P. Patil, N. Gershenfeld, A. I. Akinwande, S. L. Buchwald and K. F. Jensen, Science, 2020, 368, 1352 CrossRef CAS PubMed.
- S. Zhang, L. Li, J. Li, J. Shi, K. Xu, W. Gao, L. Zong, G. Li and M. Findlater, Angew. Chem., Int. Ed., 2021, 60, 7275 CrossRef CAS PubMed.
- X. Dong, J. L. Roeckl, S. R. Waldvogel and B. Morandi, Science, 2021, 371, 507 CrossRef CAS PubMed.
-
(a) T. Wu and K. D. Moeller, Angew. Chem., Int. Ed., 2021, 60, 12883 CrossRef CAS PubMed;
(b) M. S. J. Strehl, M. L. Abraham and G. Hilt, Angew. Chem., Int. Ed., 2021, 60, 9996 CrossRef PubMed;
(c) S. Momeni and D. Nematollahi, J. Electroanal. Chem., 2020, 857, 113746 CrossRef CAS;
(d) M. Jamshidi, D. Nematollahi, F. Taheri and H. Alizadeh, ACS Sustainable Chem. Eng., 2019, 7, 1956 CrossRef CAS;
(e) Y. Jiang, G. Dou, L. Zhang, K. Xu, R. D. Little and C. Zeng, Adv. Synth. Catal., 2019, 361, 5170 CrossRef CAS;
(f) W. Jud, C. O. Kappe and D. Cantillo, Chem.–Eur. J., 2018, 24, 17234 CrossRef CAS PubMed;
(g) H.-B. Zhao, P. Xu, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2018, 57, 15153 CrossRef CAS PubMed;
(h) M. Sharafi-Kolkeshvandi, D. Nematollahi and F. Nikpour, Synthesis, 2017, 49, 1555 CAS;
(i) L.-S. Kang, M.-H. Luo, C. M. Lam, L.-M. Hu, R. D. Little and C. Zeng, Green Chem., 2016, 18, 3767 RSC;
(j) R. Matthessen, J. Fransaer, K. Binnemans and D. E. De Vos, ChemElectroChem, 2015, 2, 73 CrossRef CAS.
- S. Tian, X. Jia, L. Wang, B. Li, S. Liu, L. Ma, W. Gao, Y. Wei and J. Chen, Chem. Commun., 2019, 55, 12104 RSC.
- Y. Liang, S.-H. Shi, R. Jin, X. Qiu, J. Wei, H. Tan, X. Jiang, X. Shi, S. Song and N. Jiao, Nat. Catal., 2021, 4, 116 CrossRef.
- Z.-L. Luo, K. J. Imamura, Y. Shiota, K. Yoshizawa, Y. Hisaeda and H. Shimakoshi, J. Org. Chem., 2021, 86, 5983 CrossRef CAS PubMed.
-
(a) L. Wei, Z.-H. Wang, K.-J. Jiao, D. Liu, C. Ma, P. Fang and T.-S. Mei, J. Org. Chem., 2021 DOI:10.1021/acs.joc.1c00204;
(b) C. Zhu, H. Yue, P. Nikolaienko and M. Rueping, CCS Chem., 2020, 2, 179 CrossRef CAS;
(c) D. Liu, H. X. Ma, P. Fang and T. S. Mei, Angew. Chem., Int. Ed., 2019, 58(15), 5033 CrossRef CAS PubMed;
(d) J. Strehl and G. Hilt, Org. Lett., 2019, 21, 5259 CrossRef CAS PubMed;
(e) L. Zhang, Z. Zha, Z. Zhang, Y. Li and Z. Wang, Chem. Commun., 2010, 46, 7196 RSC;
(f) G. Hilt, Angew. Chem., Int. Ed., 2003, 42, 1720 CrossRef CAS PubMed.
-
(a) V. Bizet, R. Kowalczyk and C. Bolm, Chem. Soc. Rev., 2014, 43, 2426 RSC;
(b) S. Ge, R. A. Green and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 1617 CrossRef CAS PubMed;
(c) N. H. Park, G. Teverovskiy and S. L. Buchwald, Org. Lett., 2014, 16, 220 CrossRef CAS PubMed;
(d) N. F. Fine Nathel, J. Kim, L. Hie, X. Jiang and N. K. Garg, ACS Catal., 2014, 4, 3289 CrossRef CAS PubMed;
(e) G. Manolikakes, A. Gavryushin and P. Knochel, J. Org. Chem., 2008, 73, 1429 CrossRef CAS PubMed.
-
(a) C. Li, Y. Kawamata, H. Nakamura, J. C. Vantourout, Z. Liu, Q. Hou, D. Bao, J. T. Starr, J. Chen, M. Yan and P. S. Baran, Angew. Chem., Int. Ed., 2017, 56, 13088 CrossRef CAS PubMed;
(b) Y. Kawamata, J. C. Vantourout, D. P. Hickey, P. Bai, L. Chen, Q. Hou, W. Qiao, K. Barman, M. A. Edwards, A. F. Garrido-Castro, J. N. deGruyter, H. Nakamura, K. Knouse, C. Qin, K. J. Clay, D. Bao, C. Li, J. T. Starr, C. Garcia-Irizarry, N. Sach, H. S. White, M. Neurock, S. D. Minteer and P. S. Baran, J. Am. Chem. Soc., 2019, 141, 6392 CrossRef CAS PubMed.
- D. Liu, Z.-R. Liu, C. Ma, K.-J. Jiao, B. Sun, L. Wei, J. Lefranc, S. Herbert and T.-S. Mei, Angew. Chem., Int. Ed., 2021, 60, 9444 CrossRef CAS PubMed.
-
(a) X.-B. Yan, C.-L. Li, W.-J. Jin, P. Guo and X.-Z. Shu, Chem. Sci., 2018, 9, 4529 RSC;
(b) L. K. G. Ackerman, L. L. Anka-Lufford, M. Naodovic and D. J. Weix, Chem. Sci., 2015, 6, 1115 RSC;
(c) J.-H. Liu, C.-T. Yang, X.-Y. Lu, Z.-Q. Zhang, L. Xu, M. Cui, X. Lu, B. Xiao, Y. Fu and L. Liu, Chem.–Eur. J., 2014, 20, 15334 CrossRef CAS PubMed;
(d) L. L. Anka-Lufford, M. R. Prinsell and D. J. Weix, J. Org. Chem., 2012, 77, 9989 CrossRef CAS PubMed.
-
(a) D. H. Lee, K.-H. Kwon and C. S. Yi, Science, 2011, 333, 1613 CrossRef CAS PubMed;
(b) D.-H. Lee, K.-H. Kwon and C. S. Yi, J. Am. Chem. Soc., 2012, 134, 7325 CrossRef CAS PubMed.
- Z. Li, W. Sun, X. Wang, L. Li, Y. Zhang and C. Li, J. Am. Chem. Soc., 2021, 143, 3536 CrossRef CAS PubMed.
-
(a) T. Koyanagi, A. Herath, A. Chong, M. Ratnikov, A. Valiere, J. Chang, V. Molteni and J. Loren, Org. Lett., 2019, 21, 816 CrossRef CAS PubMed;
(b) H. Li, C. P. Breen, H. Seo, T. F. Jamison, Y.-Q. Fang and M. M. Bio, Org. Lett., 2018, 20, 1338 CrossRef CAS PubMed;
(c) R. J. Perkins, D. J. Pedro and E. C. Hansen, Org. Lett., 2017, 19, 3755 CrossRef CAS PubMed;
(d) C. Amatore, A. Jutand and L. Mottier, J. Electroanal. Chem., 1991, 306, 125 CrossRef CAS;
(e) C. Amatore and A. Jutand, J. Am. Chem. Soc., 1991, 113, 2819 CrossRef CAS.
-
(a) Y. Imada, J. L. Röckl, A. Wiebe, T. Gieshoff, D. Schollmeyer, K. Chiba, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 12136 CrossRef CAS PubMed;
(b) R. Hayashi, A. Shimizu and J.-i. Yoshida, J. Am. Chem. Soc., 2016, 138, 8400 CrossRef CAS PubMed.
- L. Zhang and X. Hu, Chem. Sci., 2020, 11, 10786 RSC.
- J. Luo, B. Hu, W. Wu, M. Hu and T. L. Liu, Angew. Chem., Int. Ed., 2021, 60, 6107 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*