
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improvement of the antimicrobial potency, pharmacokinetic and pharmacodynamic properties of albicidin by incorporation of nitrogen atoms†
Lieby
Zborovsky‡
a,
Leonardo
Kleebauer‡
 a,
Maria
Seidel
a,
Arseni
Kostenko
a,
Leonard
von Eckardstein
a,
Frank Otto
Gombert
b,
John
Weston
a and
Roderich D.
Süssmuth
*a
a,
Maria
Seidel
a,
Arseni
Kostenko
a,
Leonard
von Eckardstein
a,
Frank Otto
Gombert
b,
John
Weston
a and
Roderich D.
Süssmuth
*a
aInstitut für Organische Chemie, Technische Universität Berlin, Straße des 17. Juni 124, 10623 Berlin, Germany. E-mail: suessmuth@chem.tu-berlin.de
bGombert Pharma Research Solutions (GPRS), Dornacherstrasse 120, CH 4053 Basel, Switzerland
First published on 19th October 2021
Abstract
The worrisome development and spread of multidrug-resistant bacteria demands new antibacterial agents with strong bioactivities particularly against Gram-negative bacteria. Albicidins were recently structurally characterized as highly active antibacterial natural products from the bacterium Xanthomonas albilineans. Albicidin, which effectively targets the bacterial DNA-gyrase, is a lipophilic hexapeptide mostly consisting of para amino benzoic acid units and only one α-amino acid. In this study, we report on the design and synthesis of new albicidins, containing N-atoms on each of the 5 different phenyl rings. We systematically introduced N-atoms into the aromatic backbone to monitor intramolecular H-bonds and for one derivative correlated them with a significant enhancement of the antibacterial activity and activity spectrum, particularly also towards Gram-positive bacteria. In parallel we conducted DFT calculations to find the most stable conformation of each derivative. A drastic angle-change was observed for the lead compound and shows a preferred planarity through H-bonding with the introduced N-atom at the D-fragment of albicidin. Finally, we went to the next level and conducted the first in vivo experiments with an albicidin analogue. Our lead compound was evaluated in two different mouse experiments: In the first we show a promising PK profile and the absence of toxicity and in the second very good efficiency and reduction of the bacterial titre in an E. coli infection model with FQ-resistant clinically relevant strains. These results qualify albicidins as active antibacterial substances with the potential to be developed as a drug for treatment of infections caused by Gram-negative and Gram-positive bacteria.
Introduction
Antibiotic resistance is one of the biggest public health threats today.1 Worldwide, 17 million people die each year from bacterial infections. In the United States, approximately two million people are infected annually with bacteria resistant to antibiotics, and 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 of these infections are fatal. In Europe, 25000 people die from drug-resistant bacteria every year.2
000 of these infections are fatal. In Europe, 25000 people die from drug-resistant bacteria every year.2
The current need for novel antibiotics with new modes of action is especially critical for drug-resistant Gram-negative pathogens.3,4 Gram-negative microorganisms have a highly restrictive permeability barrier, which limits the penetration of most antibiotics.5,6 Only few antibacterial molecule classes can cross this barrier. The quinolones developed in the 1960s are the last big class of antibiotics acting against Gram-negative bacteria.4,7 Since then, discovery has been largely limited to narrow-spectrum compounds.4,8 This lack of Gram-negative antibiotics is largely responsible for the current antimicrobial resistance crisis.9 Pathogens such as Escherichia coli, Klebsiella pneumoniae, Pseudomonas aeruginosa and Acinetobacter baumannii have acquired resistance to most, and in some cases to all, antibiotics currently available in clinics leading to a decreasing number of treatment options.10 The WHO has classified these drug resistant pathogens as a critical priority for global human health.11 Thus, there is an urgent need to provide new effective treatments to meet the increasing public health burden caused by multidrug-resistant Gram-negative bacteria. To meet this need, there is an ongoing search for novel antibiotics, involving novel molecular targets and modes of action. Many such compounds are derived from natural products made by soil microorganisms, primarily Actinomycetes, examples include aminoglycosides, tetracyclines and β-lactams.12 Exploration of natural products remains one of the most promising approaches for the discovery of novel antibiotics.
Albicidin (Fig. 1) is a phytotoxic oligoaromatic peptide synthesized by the Gram-negative sugarcane pathogen Xanthomonas albilineans that causes leaf scald disease in sugarcane plants.13 Structurally related compounds, the cystobactamids and coralmycins have also been identified and are under ongoing development.14,15 Albicidin exhibits antibacterial activity at nanomolar concentrations against both Gram-positive and Gram-negative microorganisms and is active against resistant strains. The molecular target is DNA gyrase16 with a half maximum inhibitory concentration (IC50) of 40 nM (compared to 80 nM of cystobactamid),17 thus exhibiting an inhibitory activity similar to that of DNA gyrase-inhibiting quinolones and coumarins.16 Hence, albicidin represents a promising lead structure in the search for a new class of antibiotics.
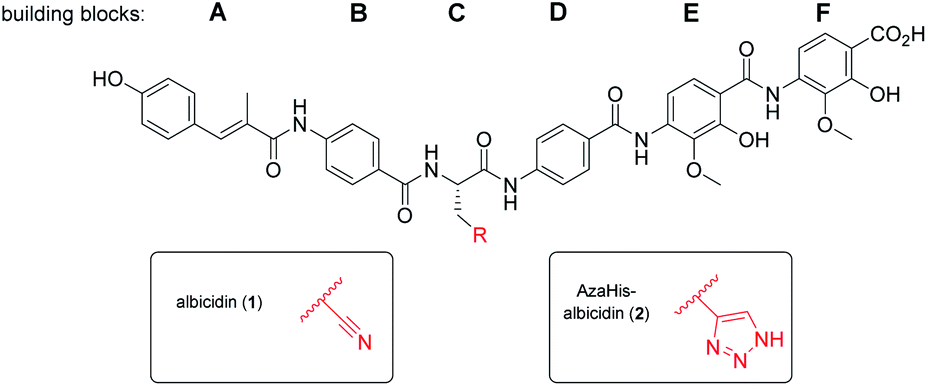 | ||
| Fig. 1 Molecular structure of albicidin (1) and AzaHis-albicidin (2). | ||
With regard to its biosynthesis, albicidin is assembled by a hybrid polyketide synthase/non-ribosomal peptide synthetase (PKS/NRPS) complex.18,19 The structure of albicidin is composed of six building blocks, namely A–F. The N-terminal part is a cinnamoyl residue (building block A) and a para-aminobenzoic acid (pABA; building block B). The C terminal part consists of a dipeptidic moiety of pABA, each decorated with adjacent methoxy and hydroxy groups (building blocks E and F) and an undecorated pABA moiety (building block D).18 Both, N-terminal and C-terminal parts are linked by a central L-cyanoalanine (building block C) which is the only stereo-centre of the molecule, forming a hinge-like structure (Fig. 1).
Previously, various resistance factors against albicidin have been characterized. The two main resistant factors are the albicidin binding protein AlbA20,21 and the serine protease AlbD.22
To maximize the bioactivity of albicidin in the quest for a potent novel antimicrobial drug, structure–activity relationship (SAR) studies are key. The first total synthesis of albicidin established by our group employed an allyl protecting group strategy23 and paved the way for initial SAR studies. This included many variations of the cinnamoyl residue (building block A)24,25 and the incorporation of various α-amino acids at building block C.26,27 A recent study demonstrated the significance and influence of the hydroxy and methoxy substituents on the E and F building blocks on the antibacterial activity.28 Furthermore, with an azahistidine we have established a new important modification in the central building block C, which resulted in improved antibacterial activities and prevented hydrolysis.28
In our ongoing studies we have systematically investigated the effect of the variation of the functional groups on each building block on the antibacterial activity. For the selection of a lead structure however, the compound further needs to display optimised balance between physicochemical, pharmacokinetic (PK) and target related pharmacodynamic (PD) properties. This includes the determination of solubility, plasma stability, plasma protein binding as key parameters and the subsequent in vivo profiling in an animal model as the ultimate proof of the drug ability of an antibacterial compound.
Heterocycles play an important role in medicinal chemistry optimisation studies. It is not surprising that the phenyl to pyridine exchange is among the most common and beneficial in medicinal chemistry and more than 60 pyridine-containing drugs have been approved by the FDA.29 Pyridine is an electron-deficient aromatic heterocycle containing a ring nitrogen atom, which is present in many natural products including vitamins, coenzymes and alkaloids.29 The ring nitrogen is more electronegative than the ring carbons, naturally making the remaining ring carbons more electropositive in comparison to the ring carbons of benzene. The replacement of a CH group with an N atom in aromatic ring systems can have many important effects on molecular and physicochemical properties such as increased basicity which leads to decreased lipophilicity and improved water solubility. Additionally, the introduction of the heterocyclic nitrogen can result in the formation of intra- and intermolecular interactions such as hydrogen bonding (acceptor) and more efficient π-stacking. This may lead to improved pharmacological properties such as active site binding, elevated cell permeability and improved antimicrobial activity. All these properties make pyridine an excellent bioisostere for the phenyl fragment.
As the albicidin antibiotic contains five phenyl rings in its core structure, a SAR study involving a phenyl to pyridyl exchange could lead to new and more potent and pharmacokinetically improved antibiotic compounds. This idea has been probed before by our group, when the hydroxy group of the E and/or F building blocks was replaced by a heterocyclic nitrogen.28 However, a systematic study describing the phenyl to pyridyl replacement (pyridine scan) on a full length albicidin as well as its cystobactamid and coralmycin analogues is lacking.
In this work, we report the systematic phenyl to pyridyl building block replacement along the albicidin sequence followed by a thorough SAR testing. We found that substituting the phenyl ring of the D building block with a pyridyl has the highest beneficial impact on the antimicrobial activity leading to a novel albicidin derivative with unprecedented potency. We show, using DFT quantum mechanical calculations that the phenyl to pyridyl replacement leads to preferable planarization of the albicidin backbone, caused by intramolecular hydrogen bonding, which we believe is beneficial for the albicidin's in vivo activity. In addition, we report the investigation of the pharmacokinetic and pharmacodynamic properties of this new albicidin derivative. The experimental outcome of an E. coli sepsis model proves exposure of the antibacterial by reducing the bacterial burden by several orders of magnitude thus turning albicidin into a promising lead structure for a future antibiotic agent.
Results
Synthesis of N-aryl substituted albicidins
As was previously reported, the substitution of the central cyanoalanine group in building block C with a 2-amino-3-(1H-1,2,3-triazol-4-yl)propanoic acid (azahistidine) moiety AzaHis-albicidin 2, significantly improved the antibacterial activities against various host strains and overcame stability issues of the cyano group which is prone to hydrolysis and thus loss of activity.28 For this reason, we chose 2 as a template structure for our pyridine scan/SAR studies aiming for increased, bioactivity and solubility. Since we have previously found that it is possible to replace the methoxy and hydroxy substituted pABA rings with unsubstituted phenyls, whilst retaining comparable antimicrobial activities,28 for the simplicity and ease of synthesis, we replaced the phenyl rings in E and F with an unsubstituted pyridyl. For each pyridyl building block we aimed for the synthesis of two isomers, the picolinic acid and nicotinic acid derivatives, thus yielding compounds 3–10, 12 and 13 (Fig. 2) and compound 15 missing the C-terminal carboxy group.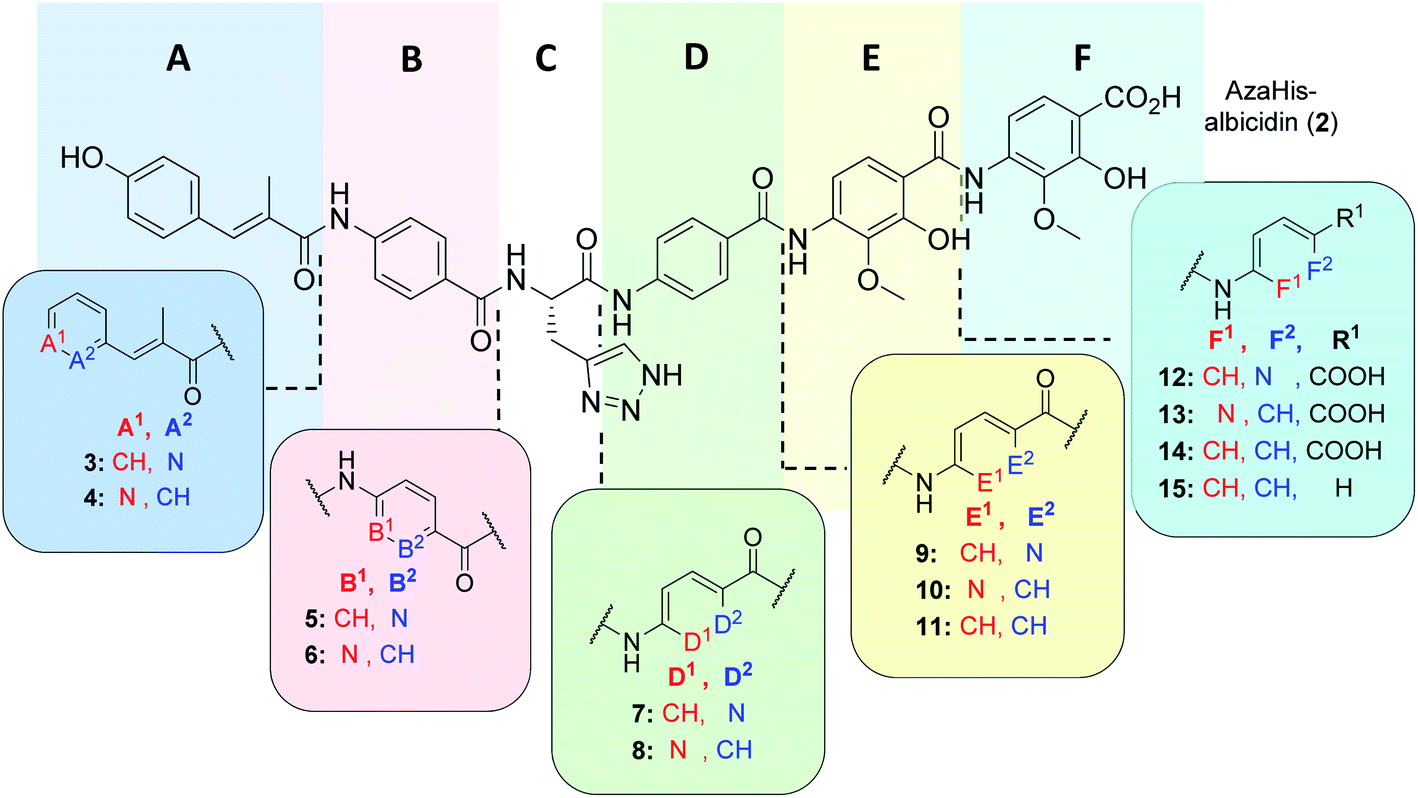 | ||
| Fig. 2 Molecular structure of albicidin derivatives 2,2711 & 14 (ref. 28) and aimed derivatives 3–10, 12, 13 and 15. | ||
To rapidly generate derivatives of albicidin, we adhered to our previously established convergent synthesis strategy26 and modified or improved it when necessary. We were able to modify each of the five different phenyl rings (Fig. 2). Starting with the A-building block from the commercially available aldehydes 16a and 16b (Scheme 1), we used a Horner–Wadsworth–Emmons reaction to give the desired coumaric acid ester isomers with exclusive (E)-selectivity, which were directly saponified to the acids 18a and 18b in good yields. The further steps towards the AB-PCP ester (22a–b, Scheme 2) were carried out in analogy with the synthesis variations of the B-fragment (see ESI†): there, acetyl-protected coumaric acid 19 (see ESI†) was coupled via the acid chlorides with the amine 20c or 20d. After removal of both protecting groups, the methyl ester and the acetyl group, the hydroxy group was again re-protected with Boc to obtain 21c and 21d, respectively. To avoid possible by-products in the further coupling which has been observed by using HATU as coupling reagent we prepared the active esters 22c and 22d instead.
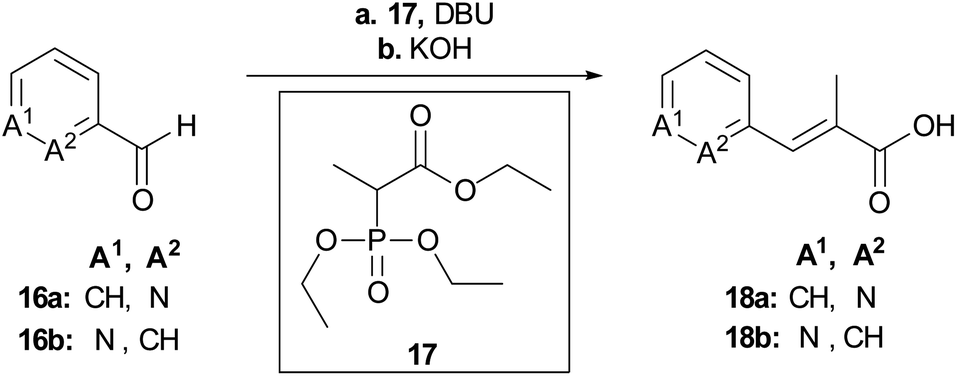 | ||
| Scheme 1 Synthesis of the key building blocks for A-derivatives. Conditions: a LiCl, DBU, MeCN, 25 °C, b aq. KOH, THF, 25 °C, 71% (18a), 80% (18b). | ||
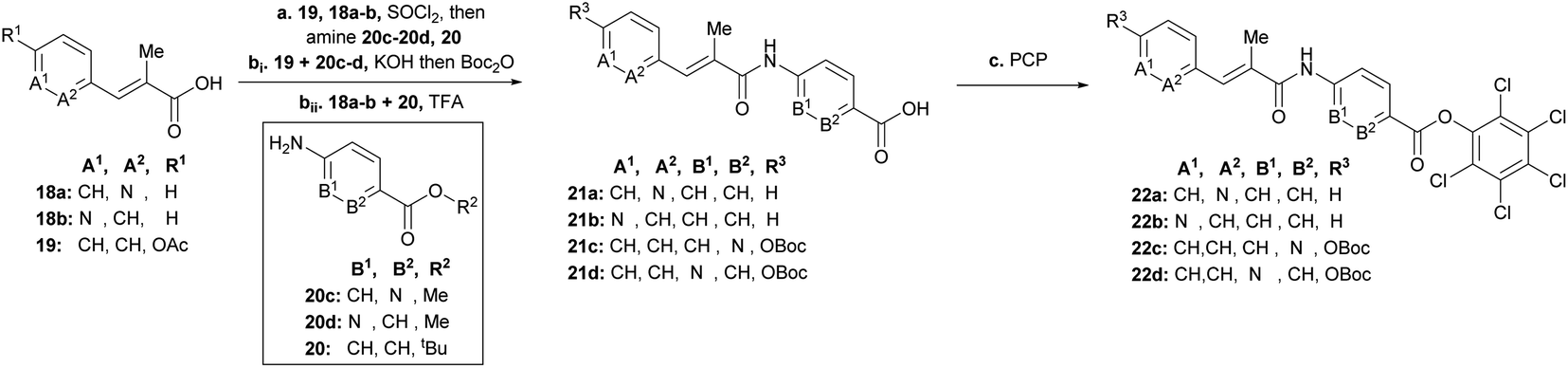 | ||
| Scheme 2 Synthesis of AB-fragments with modified A or B building block. Conditions: a19, 18a–b, SOCl2, 95 °C, then (20c or 20d to 19) or (20 to 18a or 18b) and NEt3 or Na2CO3 (for 20d), THF, 0 °C → 25 °C; bi product of 19 and 20c or 20d, KOH, THF, 25 °C, then Boc2O, DMAP, THF, 25 °C, 75% (21c, 3 steps), 59% (21d, 3 steps); bii product of 20 and 19a or 19b, TFA/CH2Cl2 (1:1), 0 °C, 57% (21a), 68% (21b), c [(21a or 21b), SOCl2, 95 °C] or [(21c or 21d), DIPEA, DMAP, EDC, THF], then pentachlorophenol (PCP), THF, 25 °C, 51% (22a), 51% (22b), 12% (22c), 18% (22d). | ||
Having the N-variations of the AB fragments in hand for the final coupling with an already synthetically established C–D–E–F fragment, we focused on preparing the N-substituted EF variations. One of our key building blocks, compound 23 (Scheme 3), was synthesized in 6 steps23 and then coupled via the acid chloride with heterocycles 24a and 24b, respectively, to obtain dipeptides with a nitroaryl group, which were subsequently reduced to 25a and 25b (Scheme 3). While the reduction to dipeptide 25b was uncomplicated, a by-product was observed during reduction of the picolinic acid dipeptide: the methyl ester was reduced to the alcohol 26 yielding a 1:1 ratio, with a yield of 30% of the desired product (25a). We could enhance the yields by changing the solvent from ethanol to chloroform yielding 25a with a yield of 96%.
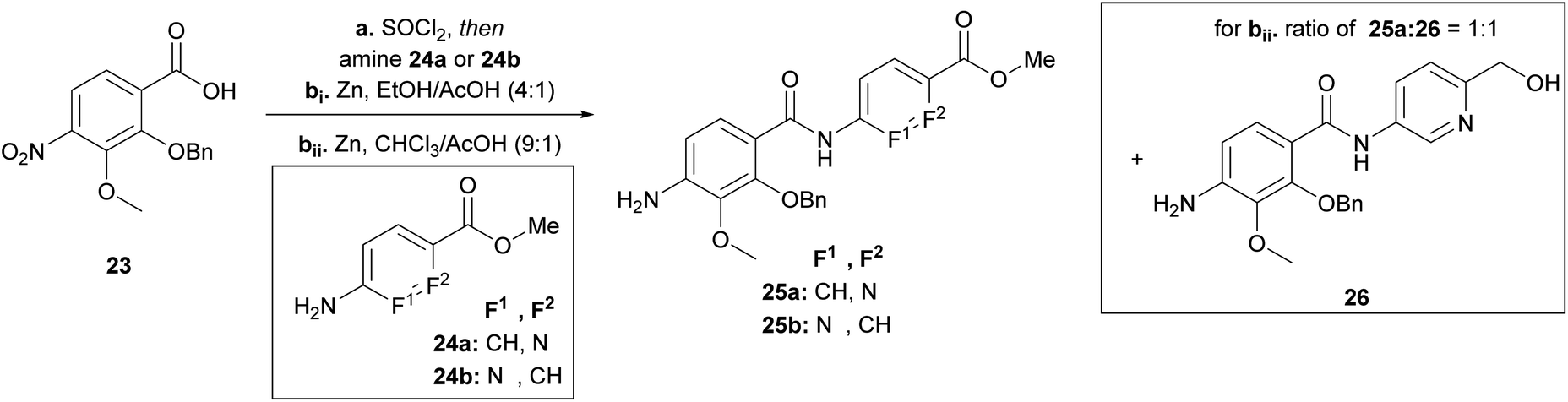 | ||
| Scheme 3 Synthesis of the EF-building block for F-variations. Conditions: a SOCl2, 95 °C, then amine 25a (80%) or 25b (79%), NEt3, THF, 0 °C → 25 °C; bi Zn, EtOH/AcOH (4:1), 0 °C → 25 °C, 25a (30%), 25b (96%), 26 (32%); bii Zn, CHCl3/AcOH (9:1), 0 °C → 25 °C, 25a (96%). | ||
Next, we focused on the variation of the E-building block (Scheme 4). For the picolinic acid 27a the transformation to the acid chloride worked well. This was followed by coupling with 28a and reduction to 33a. However, coupling with nicotinic acid 27b decreased the yield of 29b dramatically, due to the formation of the by-product 30 coming from a nucleophilic substitution of the nitro group by a chlorine. Previously, similar reactions have been reported in literature (see ESI†).30 We decided at this point for an alternative approach with a Boc-protected amine group (31) replacing the nitro group (27b). Knowing that the Boc group would be cleaved under harsh acidic activation conditions (SOCl2), we switched to oxalyl chloride in a ratio of 1:1.2 (31:(COCl)2) at 0 °C, to obtain dipeptide 32. Removal of the Boc group with hydrochloric acid and trifluoro acetic acid also cleaved the benzyl ester to give corresponding carboxylic acid 34 (see ESI†), whereas TBAF rendered the desired product 33b.
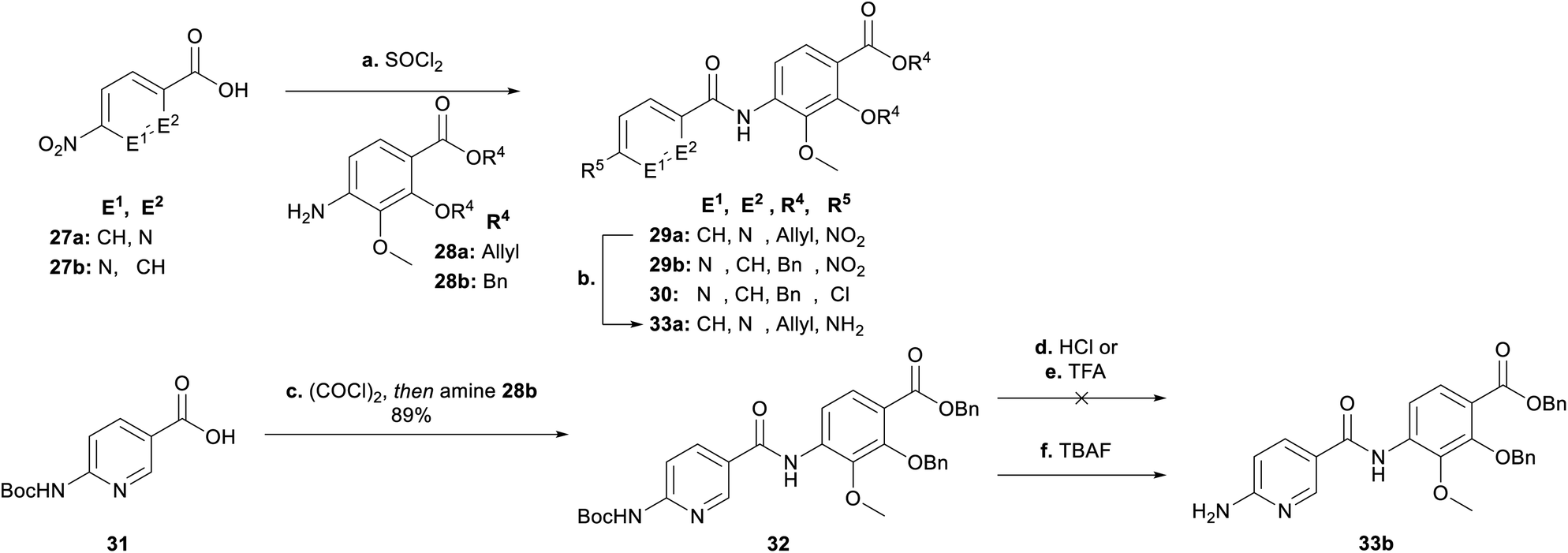 | ||
| Scheme 4 Synthesis of the EF-building block for E-variations. Conditions: ai27a, SOCl2, 95 °C, then 28a, NEt3, THF, 0 °C → 25 °C, 89% (29a); aii27b, SOCl2, 95 °C, then 28b, NEt3, THF, 0 °C → 25 °C, 11% (29b), 25% (30); b Zn, AcOH/EtOH (1:4), 0 °C, 92% (33a); c (COCl)2, THF, cat. DMF, 0 °C → 25 °C, then 28b, NEt3, THF, 0 °C → 25 °C, 89%; d HCl 4 N in dioxane; 0 °C; e TFA/CH2Cl2 (1:4), 0 °C; f TBAF, THF, 120 °C, 79% (33b). | ||
After having all variations of EF building blocks (25a–b, 33a–b, 35a (for synthesis of compound 15, see ESI†), 35b (ref. 23)) in hand (Scheme 5), the assembly of the tripeptides was carried out: this was performed either by coupling with benzoyl chloride (27c) or picolinic acid (27a)/SOCl2. The nitroaryl peptides were reduced with zinc to yield 36a–f.
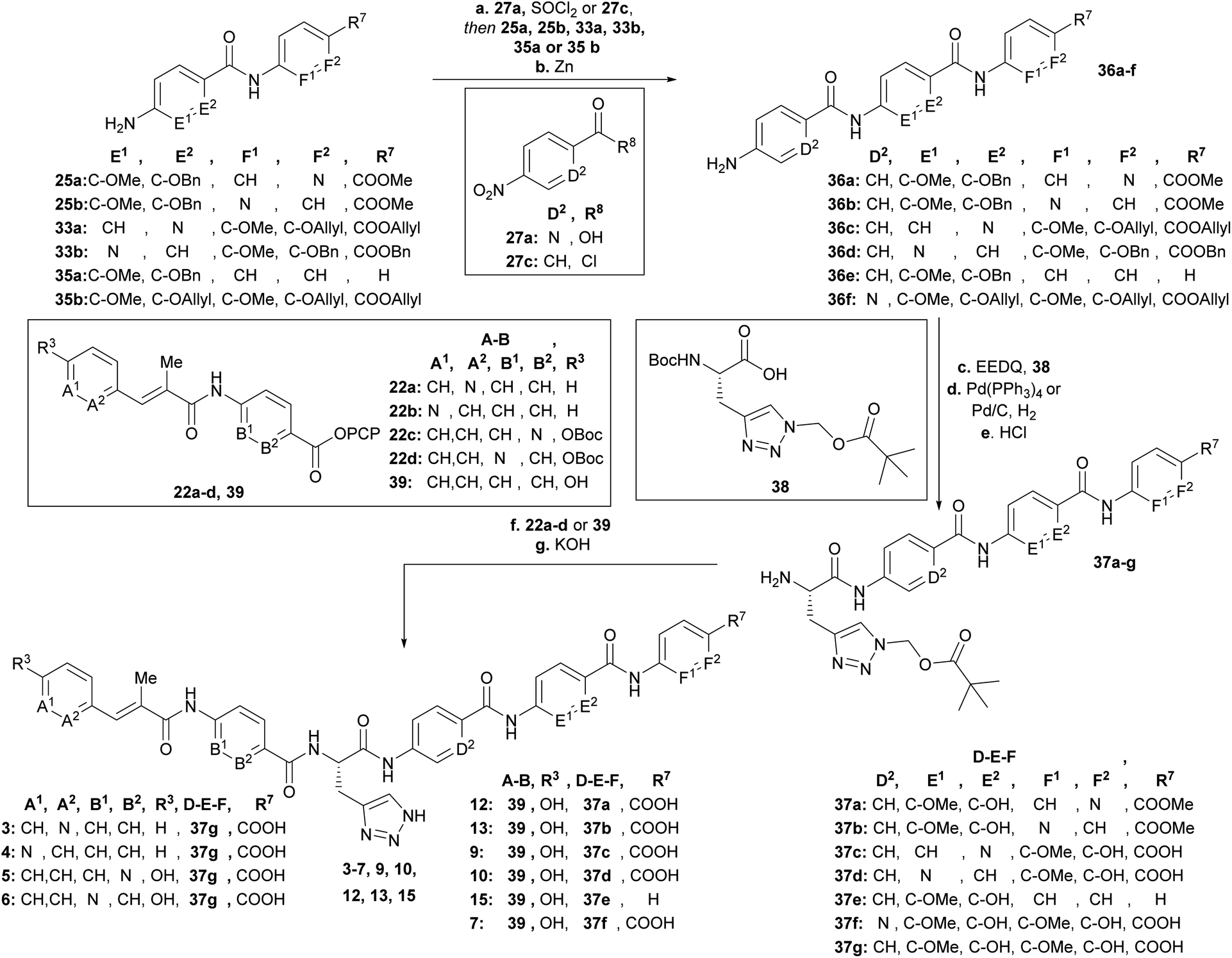 | ||
| Scheme 5 Synthesis of final derivatives by assembling of the building blocks. Conditions: a (SOCl2, 90 °C 27a then 35b, DIPEA) or [27c and (25a, 25b, 33a, 33b or 35a), NEt3], THF, 0 °C → 25 °C; b Zn, EtOH/AcOH (4:1) or CHCl3/AcOH (9:1), 0 °C, 2 steps: 82% (36a), 54% (36b), 41% (36c), 32% (36d), 76% (36e), 75% (36f); c EEDQ, 38, THF, 25 °C; d Pd(PPh3)4, morpholine, THF or Pd/C, H2, EtOH/THF/MeOH (1:1:1); e 4N HCl, dioxane, 25 °C, 3 steps: 65% (37a), 66% (37b), 20% (37c), 53% (37d), 51% (37e), 37% (37f); f (37a–f, 39) or (37g, 22a–b) or (first 22a or 22b, TFA/CH2Cl2 (1:1), 0 °C, then 37g), NEt3, DMF, 25 °C, g 3N KOH, DMF, 25 °C, 2–3 steps: 15% (3), 25% (4), 8% (5), 12% (6), 23% (7), 16% (9), 12% (10), 5% (12), 8% (13), 15% (15). | ||
Next the AzaHis building block C (38)27 was coupled with the tripeptides 36a–f (Scheme 5) and subsequently the allyl (36c, 36f)− or benzyl (36a, 36b, 36d, 36e) protecting groups were removed with tetrakis (triphenylphosphine)palladium(0), or Pd/C respectively. The remaining Boc groups of all tetrapeptides were then cleaved under acidic conditions to obtain the tetrapeptides 37a–f, ready for the final coupling with the AB building block. Finally, we used either AB-PCP ester 39 (ref. 28) or PCP-esters 22a–d (Scheme 2) for coupling with the tetrapeptides 36a–f. Literature-known tetrapeptide 37g,28 was prepared in analogy to the C–D–E–F fragments 37a–f, for coupling with the different AB-PCP esters 22a–d. The pivaloyloxymethyl (POM) protecting group on the C part was removed under basic conditions (KOH, DMF/H2O (1:1)) to obtain the full-length derivatives 3–7, 9, 10, 12, 13 and 15.
Having managed to overcome several synthetic issues, we then tackled the synthesis of C-terminal tripeptide analogues of compound 8 (Fig. 2). The coupling with the para-amino nicotinic acid in building block D was extremely challenging. The formation of the corresponding tri- and tetra-peptide was achieved in only low yields. The coupling to give the final derivative was highly inefficient and additionally the final derivative (8) was found to be chemically unstable during the purification process. Thus, further attempts on the synthesis and antibacterial profiling of 8 were not continued.
Antibacterial activity and physicochemical characterization of N-aryl substituted albicidins
Compounds 3 and 4 represent the N-terminal part of the pyridine-N-scan, namely building block A. As it has been previously shown for variations in building block A24 aromaticity is important to retain antibacterial activity, otherwise a number of structural variations were allowed. Results from the antibacterial assays showed that compounds 3 and 4 (Table 1) with pyridine replacing the hydroxy-phenyl in the cinnamoyl fragment, exhibited similar antimicrobial activities regardless of the position of the heterocyclic nitrogen. Both compounds however showed lower activities compared to the parent compound AzaHis-albicidin 2. While the activities of 3 and 4 against Gram-negative strains are only an order of magnitude lower, there is extremely low activity against Gram-positive strains. This observation is in line with the previously shown importance of the para-hydroxy substitution on the building block A of albicidin and the observation that variations of A basically led to no increase in bioactivity.24 The phenyl to pyridyl exchange in building block B leading to compounds 5 (picolinic acid) and 6 (nicotinic acid), showed that both compounds have very good antibacterial activity, which is fairly comparable to the activity of compound 2 (Table 1).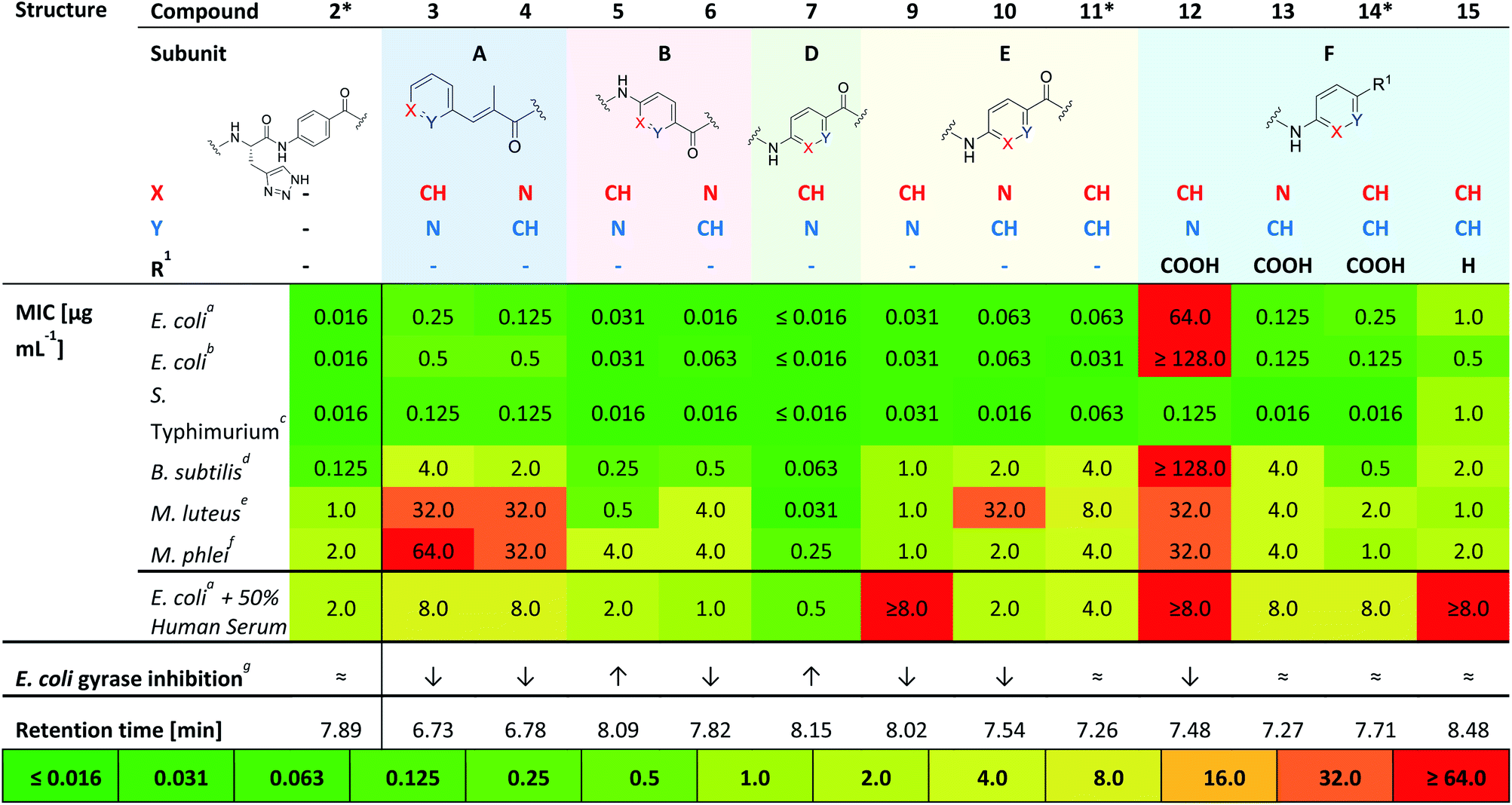
The next test set of compounds comprises pyridyl variations in building blocks E and F: interestingly, compound 9, with the picolinic acid in building block E shows very good bioactivity against all Gram-negative strains. This observation can be compared with activities for compound 10 (nicotinic acid), for which there is however very poor activity against one bacterial strain (M. luteus 32 μg mL−1). Comparison of 9 and 10 to compound 11 (ref. 28) (with pABA in building block E) shows that the N-substitutions lead to a slight but measurable enhancement of antibacterial activity against Gram-positive bacteria, especially in the case of derivative 9.
Next, we investigated variations in building block F. As we had found out in our previous work,28 replacing building block F with unsubstituted pABA (compound 14), resulted in an antibacterial activity that was comparable to compound 2. Removal of the C-terminal carboxy group gave 15 which only displayed moderate antimicrobial activities against Gram-negative and Gram-positive organisms. This points to the significance of this functional group for potent inhibitory activity. Replacing the F building block with para-amino picolinic acid (compound 12) and para-amino nicotinic acid (compound 13) gives compounds, which show strikingly different antimicrobial properties (Table 1). For compound 12 the activity loss is nearly total with MIC values of >32 μg mL−1 for both Gram-negative (except for S. typhimurium) and Gram-positive strains. Compound 13 shows better MIC values (0.125 μg mL−1) against the Gram-negative strains and a slightly better activity (4 μg mL−1) against the Gram-positive strains. Overall, the bioactivity of 13 is comparable to that of the previously reported compound 14 (Table 1) with a loss of activity for Gram positive strains.28 Interestingly, it is noticeable that the change of the position of the heterocyclic nitrogen from position 2 to position 3 in building block E has a smaller impact on MICs than the analogous change in building block F.
Finally, with compound 7 building block D was assessed: This derivative with the pyridyl in building-block D (nitrogen ortho to carboxylic acid) demonstrates an excellent antimicrobial activity against all tested Gram-negative and also Gram-positive strains including M. luteus (0.031 μg mL−1) and Mycobacterium phlei (0.25 μg mL−1), which is more than two orders of magnitude superior to that of the parent compound 2 and all other tested derivatives. Interestingly, the MIC data against E. coli are mostly paralleled by in vitro inhibition data on E. coli gyrase (Table 1 and ESI Fig. S1;† relative comparison to albicidin 1) which particularly confirms the better performance of compound 7 with regard to antibacterial activity.
Compound 2 was representatively probed for development of bacterial resistance on a narrow selection of bacterial strains. The formation of resistance (FoR) observed for the tested organisms were low (10−10 to 10−11 range at eight times MIC (see ESI Table S3†). The value of 10−11 for E. coli is lower compared to representative literature values obtained for the reference drug ciprofloxacin (10−9 at 16 times MIC), and points to a desirably low development of resistance by mutations.31
In summary, from this initial antibacterial profiling we have found that the phenyl to pyridyl exchange plays a more significant role for the C-terminal half of the molecule. Moreover, the position of the heterocyclic nitrogen in the C-terminal building blocks F, E and most significantly, building block D plays a key role in the bioactivity of the albicidin derivatives. Thus, we identified compound 7 as the most active in our primary screens.
Therefore, 7 was subjected to a follow-up antibacterial profiling against a panel of clinically relevant and ciprofloxacin (CIP)-resistant pathogens (Table 2).
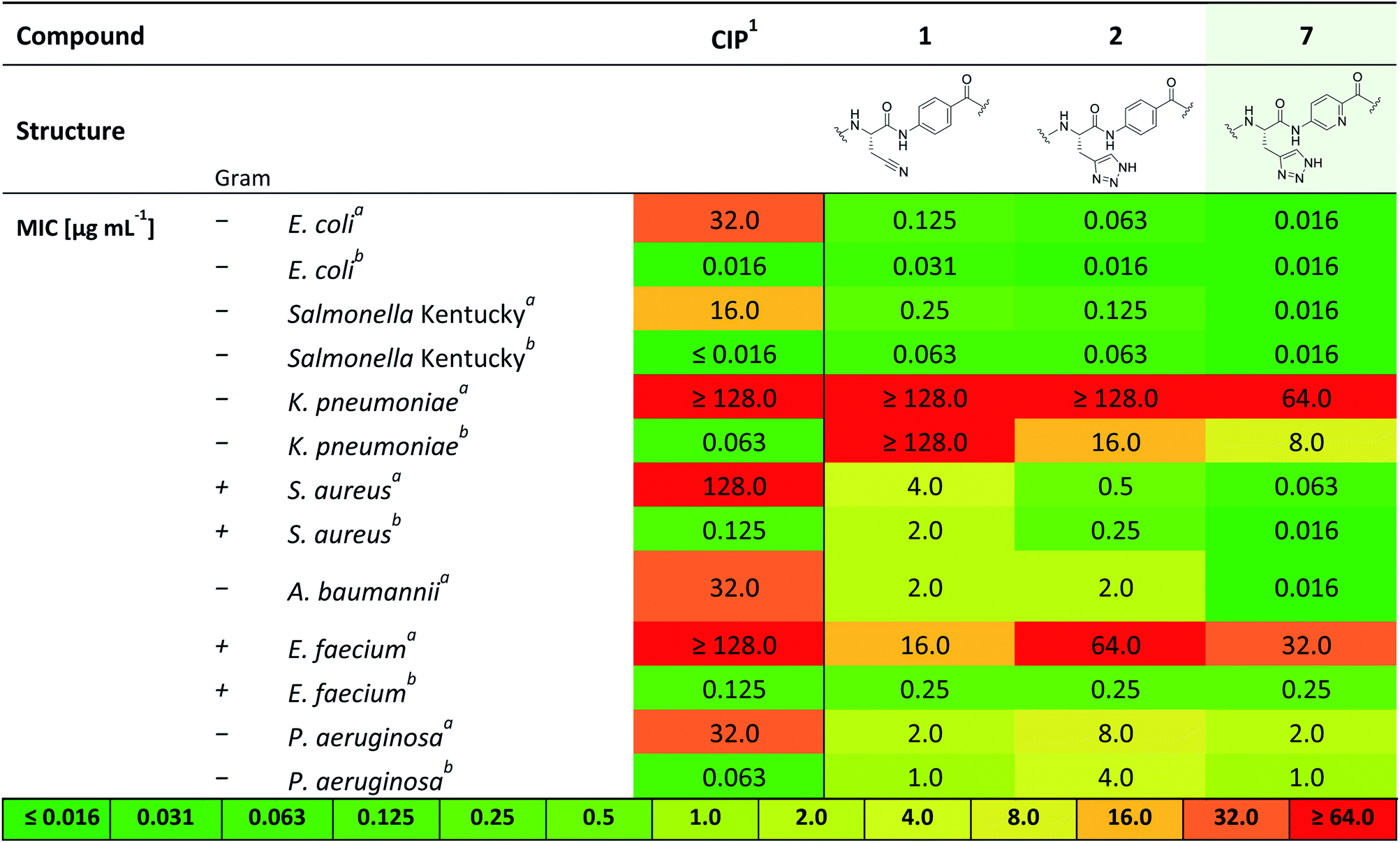
Compound 7 shows excellent activity against CIP-resistant strains of E. coli (4 times higher compared to 2), Salmonella Kentucky (8 times higher compared to 2), S. aureus (8 times higher compared to 2) and A. baumannii (125 times higher compared to 2), as well as the CIP-sensitive strains of these bacteria. Moreover, 7 shows improved, yet moderate activities against CIP sensitive strains of K. pneumonia and P. aeruginosa (8 and 4 times higher than 2, respectively). We further investigated the biological activity of 7 against a panel of gyrase- and topoisomerase-deficient isolates of E. coli GK571 and against selected E. coli knockout mutants from the KEIO collection (Table S2†). The potent antimicrobial activity of compound 7 against E. coli was not affected by quinolone resistance-mediating mutations (0.031–0.016 μg mL−1, 10-fold higher than 1 and 2). In comparison, the antimicrobial activity of cystobactamid analogue CYS-22 (Fig. S1, SI†) was previously reported to be 0.5 μg mL−1 on gyrase-deficient E. coli WT-3-1 [gyrA (S83L, D87G)].32 In addition, we observed a weak effect of the TolC (efflux transporter) knockout mutation on the antimicrobial activity of 7. The efficacy increased by a factor of 4 from 0.063 to ≤0.016 μg mL−1 presumably due to the impaired efflux mechanisms of the strain. In contrast, the knockout mutation of the tsx gene resulted in a 10-fold decrease in activity caused by a diminished compound uptake by the bacterial cells (Table S2†).33,34
This investigation was completed by assessment of the antibacterial activity in presence of human plasma (50%) (Table 1), since high plasma protein binding can severely impair antibacterial activity and thus efficacy. Remarkably, with an MIC 0.5 μg mL−1 against E. coli DSM 1116 compound 7 also showed the strongest activity in presence of plasma, which is superior to all other candidate molecules.
With this strong antibacterial profile in hand, we determined the kinetic solubility and logD values for 7 in comparison to albicidin 1 and AzaHis-albicidin 2. Interestingly, with a solubility of 14.2 μg mL−1 for 7 and 14.8 μg mL−1 for 2, both synthetic compounds are roughly three times more soluble in phosphate buffer (pH 7.5, DMSO f.c.: 5%) than albicidin (5.53 μg mL−1) (Table S3†). The logD7.4 1.63 of compound 7 is higher than that for the parent compound 2 (logD7.4 1.06) but still in a reasonable range <5. For the last step of in vitro profiling, we examined plasma half-life in both human and mouse plasma. We found a half-life (t1/2) for 1 of 60 min in human and 30 min in mouse plasma, respectively. Half-life was found increased to >240 min in human plasma and 95 min in mouse plasma for 2 and 7 which is sufficient for the subsequent progression into an animal model. Hemolysis and cell proliferation determined with HepG2 cells were in support of the conductance of in vivo experiments (see ESI, Table S3 and Fig. S2†).
Computational analysis
Intrigued by the striking bioactivity differences observed in the various pyridine substituted derivatives, we performed a computational structural analysis with and without incorporation of heterocyclic nitrogen into the albicidin structure at various sites. Some molecular dynamics (MD) calculations have been previously applied to the truncated cystobactamids35 and to a full cystobactamid derivative.36 However, structural investigation of albicidin or its analogues using density functional theory (DFT) has never been reported. We aimed at conducting systematic structural investigations using state of the art DFT methods. For this purpose, the GFN-xTB tight-binding method was used for initial conformational searching in water employing the GBSA solvation model. The structures were fully optimized at the B3LYP CPCM (water) level of theory, with the BJ-damped DFT-D3 dispersion correction and 6-311+G (d,p) basis set (see ESI† for details). Analytical frequencies were calculated to verify the optimized geometries as minima (with no imaginary frequency).Interestingly, the optimized geometry of 2 (ESI Fig. S3†) reveals a hairpin-like folded structure of the albicidin core. The molecule is folded via a twist around the sp3 carbon of the central AzaHis amino acid (building block C) and forms a cis-amide bond between C and D, resulting in the C-terminal and N-terminal “arms” facing each other. A similar structure was described for cystobactamid by Kirschning and co-workers using MD simulations.36 Hairpin-like structures of similar type have been reported in naturally derived (e.g. gramicidins37) and peptidomimetic drugs such as plantazolicin A38 and protegrin I39 and others.40 Furthermore, aromatic oligoamide foldamers have been constructed using a rigid turn unit into β-sheet hairpin-like structures stabilized by π-stacking interactions.41 For our part, we did not find experimental evidence by NOESY NMR spectroscopy for these structures, and conclude that the proposed hair-pin structure has to be taken with some reservation and should be studied in more detail in the future.
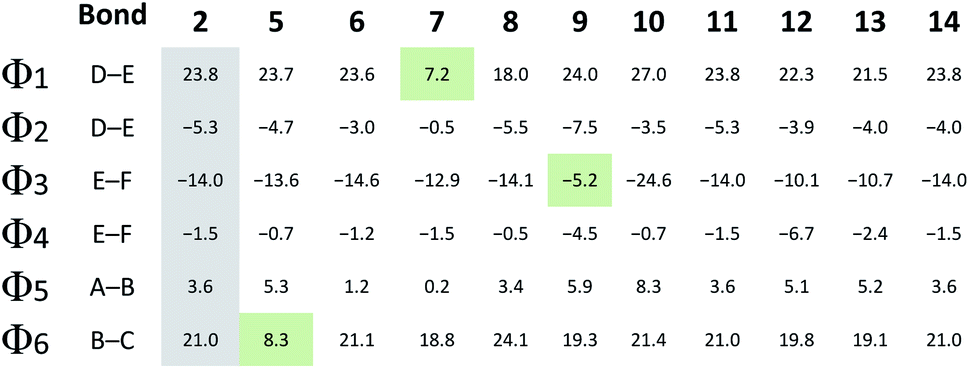
As the D–E–F fragment of the C-terminal part of albicidin shares its main structural features with aromatic oligoamides (AOA),42 it is to be expected that albicidin might show coordination patterns similar with AOA and its geometrical structure can be analysed in the same way. It has been shown that the incorporation of hydrogen acceptors, such as alkoxy substituents or a heterocyclic nitrogen into the aromatic rings of the AOA scaffold results in the formation of intramolecular hydrogen bonds (IMHB), which are responsible for conformational restrictions leading to a number of molecular architectures.43 Especially the ether oxygen of the lipophilic methoxy groups as H bond acceptor is important for the overall geometry. As expected, the calculated geometry of the D-E–F fragment of 2 (Fig. 3a reveals IMHB between the oxygen of the aryl methoxy substituents of building blocks F and E and the neighbouring amide hydrogens (shown with black dashed lines), with a distance of 2.1 Å which is in the common range of O–H hydrogen bonding. The corresponding MeO–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–N–H dihedral angles of 2 (Fig. 3a) are essentially planar with Φ2 = −4.0° and Φ4 = −1.8°.
C–N–H dihedral angles of 2 (Fig. 3a) are essentially planar with Φ2 = −4.0° and Φ4 = −1.8°.
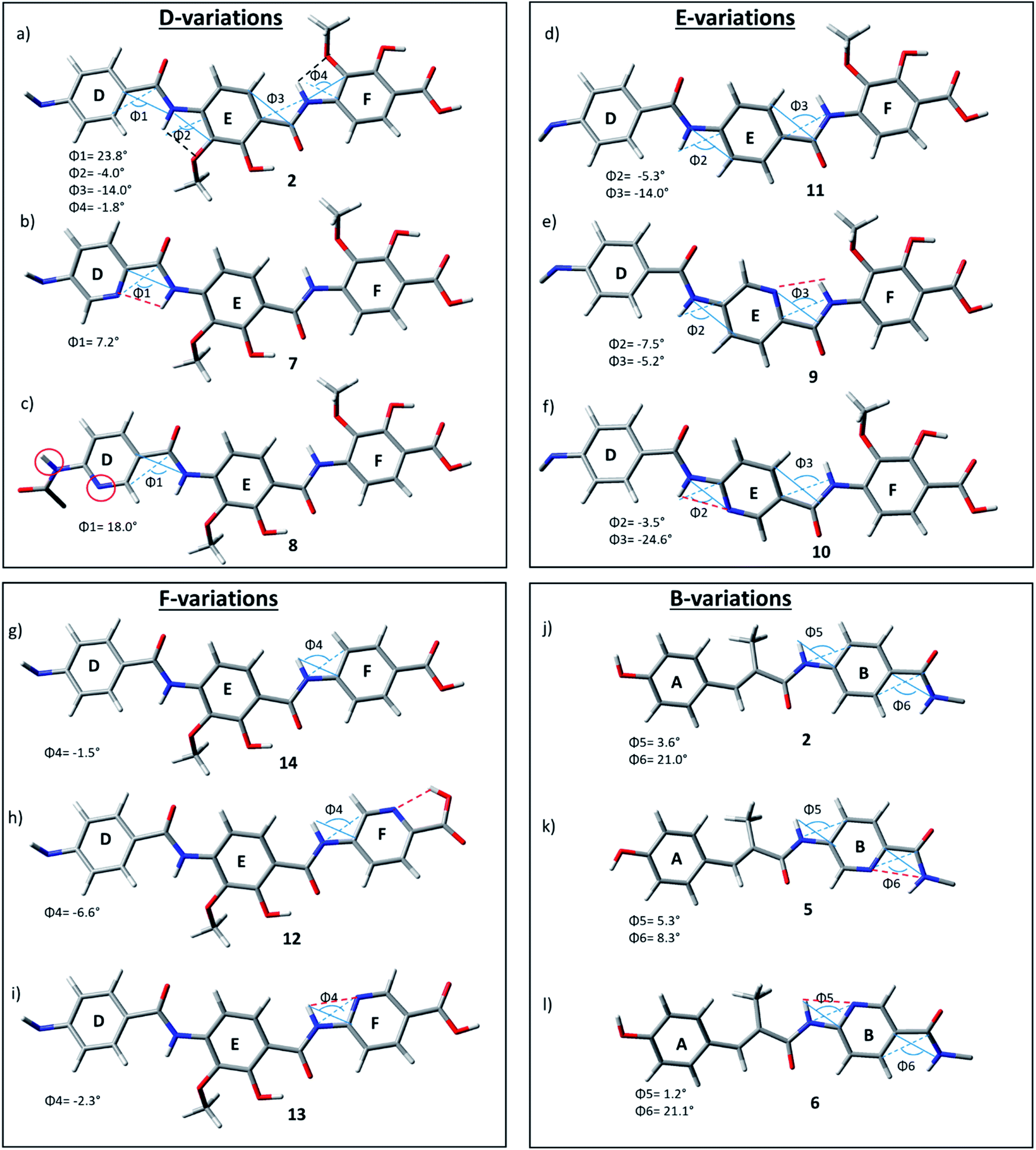 | ||
| Fig. 3 D–F fragments of derivatives 2 (a), 7–14 (b–i) and A–B fragments of derivatives 2 (j), 5 (k), 6 (l), extracted from optimized geometries of the corresponding full albicidin derivatives. | ||
In contrast to the N-terminal dihedral angles, the C-terminal angles of 2 (Fig. 3a, Φ1 and Φ3) are substantially twisted (23.8° and −14.0° respectively). Introduction of picolinic acid in building block D (derivative 7) provides an additional IMHB between the ring nitrogen and the amide hydrogen (Fig. 3b, red dashed line), similar to the IMHB observed for the truncated (tri-peptide) cystobactamid 507 analogues.35 The formation of the additional N–H hydrogen bond in 7 results in a dihedral angle Φ1 = 7.2° (Fig. 3b) which is 16.6° smaller compared to the analogous angle in 2 (Φ1 = 23.8°) and leads to a planarization of the C-terminal part. In the resulting structure, the three aromatic rings of building blocks D, E and F are aligned in a slightly distorted linear structure (Fig. 3b). (Table 3)
 ), shown in Fig. 3 angle decrease/increase >5° (
), shown in Fig. 3 angle decrease/increase >5° ( )
)
|
For the derivatives containing pyridine in building block B (5, 6), we observe an adverse effect. For compound 5, the formation of an additional IMHB results in a significantly smaller dihedral angle Φ6 = 8.3° (Fig. 3k) versus Φ6 = 21.0° (parent compound 2, Fig. 3j). This local geometrical change, however, has no significant effect on the planarity of the entire molecule since it is located next to the sp3 amino acid of building block C and is not connected directly with the C-terminal arm. For compound 6 the change in angle is much smaller with Φ5 = 1.2° (Fig. 3l), which is only 2.4° narrower than the corresponding angle in 2 (Fig. 3j, Φ5).
Since albicidin (1) and AzaHis-albicidin 2 contain substituted pABA in building blocks E and F, it would be more accurate to compare the geometry of the pyridine substituted compounds in E (9, 10) and F (12, 13) with the pABA derivative in E (11) and pABA derivative in F (14) respectively. Curiously, when replacing building block E with pABA (compound 11, Fig. 3) the dihedral angle Φ2 is only slightly affected (Φ2 = −5.3°) implying that the planarity of the DEF fragment is not determined by the MeO substitution in building block E. The same is true for the compound with pABA in F (14, Fig. 3g) where angle Φ4 is essentially not changed upon removal of the MeO substituent (Φ4 = −1.5° in 14 (Fig. 3g) vs. Φ4 = −1.8° in 2 (Fig. 3a)). A similar trend is observed for compounds 12 and 13, where the dihedral angle of parent compound 14 (Φ4 = −1.5) is already small (Fig. 3g–i). The picolinic acid derivative 12 stands out in our range of pyridine containing derivatives since the heterocyclic nitrogen located at the C-terminal position is not suitable for the formation of IMHB with the amide hydrogens. Compound 12 is even more intriguing concerning its complete loss of antimicrobial activity compared with compounds 2 and 14 (Table 1). The heterocyclic nitrogen in the picolinic acid unit coordinates with the acidic hydrogen of the carboxylic acid (Fig. 3h, red dashed line) with the distance of 1.94 Å between the heterocyclic nitrogen and the acidic hydrogen thus altering the C-terminal properties of the albicidin molecule. Picolinic acid is more acidic than benzoic acid by 3.2 pKa units. The increased acidity might play an additional role in the loss of activity in 12 compared with 14.
In summary, using DFT methods we have shown that the oligoaromatic albicidin scaffold is strongly impacted by IMHB through the introduction of heterocyclic nitrogens. In this context, the placement of picolinic acid renders a much stronger planarization effect through IMHB formation than in the nicotinic acid derivatives. This effect is the largest for building block D (compound 7) resulting in the planar alignment of the C-terminal tripeptide fragment and thus possibly influencing the antimicrobial potency of 7. This information is of high importance, since it enables us to rationally design further albicidin analogues with a local incorporation of IMHBs which should improve the SAR profile. Once the mode of action and the molecular binding site of albicidin are elucidated, these data can become a game changer in the design and synthesis of novel potent alibicidin derivatives.
Pharmacokinetics and pharmacodynamics of albicidin analogues 2 and 7
Encouraged by the antimicrobial in vitro results, the in vivo profile of compound 2 and 7 was then examined. The optimized compound formulation for in vivo experiments was established: From this work, a clear solution of analogues 2 was observed for a formulation mixture of DMSO/PEG400 (10:90) (Table S3†), which later was also applied for compound 7. In a first step, a tolerability study was conducted with albicidin analogue 2 in adult male CD-1 mice. Doses of 12.5, 25 and 50 mg kg−1 were administered twice at t = 0 h and t = 12 h by intravenous (i.v.) application route with a volume of 5 mL kg−1 (n = 3 per dose). No adverse effects and signs indicating pain were noticed throughout 24 hours. Blood cell count and macroscopic necropsy of inner organs did not show any pathological modifications (Table S4†). We then subjected both compounds 2 and 7 to a pharmacokinetic analysis. For each compound, a single dose of 50 mg kg−1 was administered i.v. in 5 mL kg−1 to three male CD-1 mice. Samples of plasma were collected at designated time points and analysed for the compound concentration levels by HPLC-MS/MS (Fig. 4, Table S7†). Subsequently, PK parameters were calculated using a two-compartment model (Table 4).
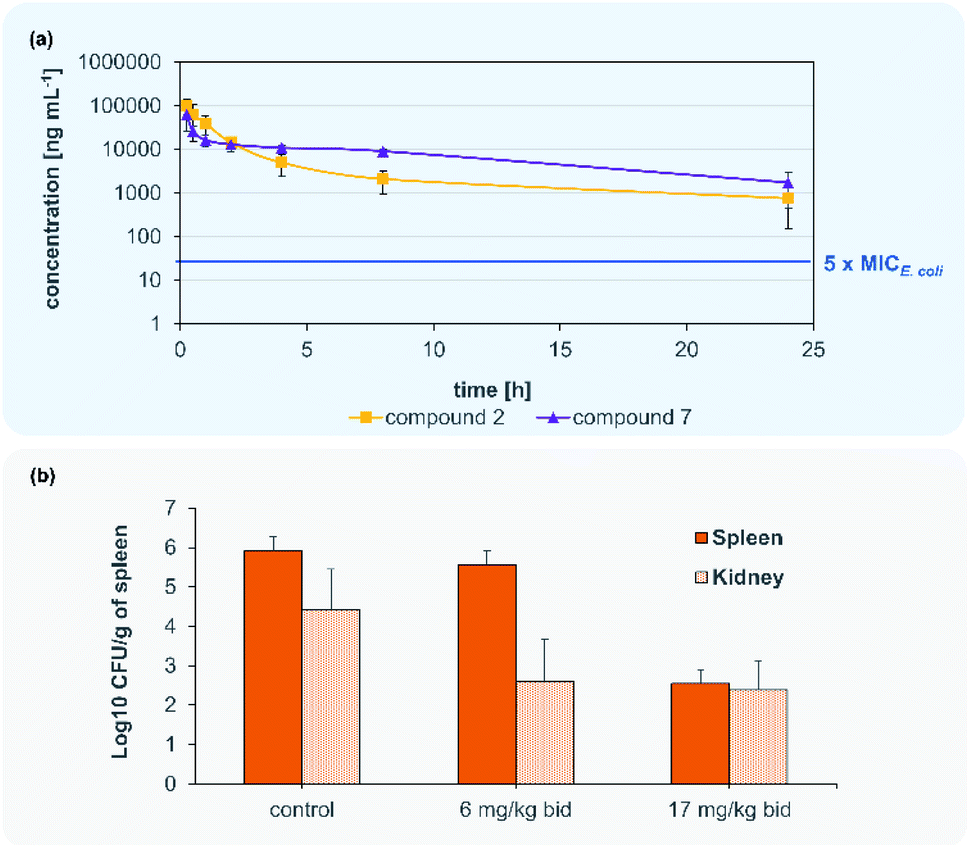 | ||
| Fig. 4 Blood concentration of 2 and 7 after 50 mg kg−1 i.v. administration (a). Murine septicaemia model for the i.v. administration of 7 to CD-1 mice infected with a fluoroquinolone-resistant (FQR) E. coli isolate infected (b). | ||
| PK parameter | 2 | 7 | CYS-22 |
|---|---|---|---|
| 50 mg kg−1 | 50 mg kg−1 | 5 mg kg−1 | |
| AUC [ng mL h−1] | 153320.0 ± 47239.0 |
224852.0 ± 49029.0 |
2087.0 ± 573.1 |
| T 1/2 [h] | 1.2 ± 0.6 | 1.373 ± 1.5 | 0.98 ± 0.1 |
| V Z [L kg−1] | 4.9 ± 5.2 | 2.7 ± 1.1 | 3.5 ± 1.2 |
| CL [mL h−1 kg−1] | 352.4 ± 128.6 | 229.2 ± 47.5 | 2490 ± 684 |
Accordingly, compounds 2 and 7 attained plasma levels in amounts higher than 5-times MIC for E. coli (=16 ng mL−1 for compound 7) even after 24 h (Fig. 4). By comparing the plasma level after 8 h of our lead compound 7 (i.v. 50 mg kg−1) with the lead compound CYS-22 from Testolin et al.32 (i.v. 5 mg kg−1, 8 h) to our delight we found a much higher (about 2000-fold) plasma concentrations of100 μg mL−1 for 7 compared to 0.05 μg mL−1 for CYS-22), even taking into account a 10-fold higher dosing of 7 compared to CYS-22. This much higher plasma concentrations resulting mainly from the 7- to 10-fold lower clearance rate compared to compound CYS-22 leads to >10-fold higher plasma concentration after 24 h above the microbiological important 5-fold MIC-value. This overcomes the need for repeated and complicated dosing regimen to achieve satisfactory plasma levels over the anticipated whole therapeutic treatment period. Importantly, no adverse effects were observed on the tested mice at the used dose of 50 mg kg−1. The full set of pharmacokinetic parameters are found in the ESI (Table S8†).
Following the encouraging pharmacokinetic results, we then proceeded to an in vivo efficacy study in a murine septicaemia model. We decided to further follow compound 7 in the in vivo infection model, due to the better antibacterial activity in presence of plasma and the pharmacological parameters allowing a bi-daily dosing of the test compound. From a panel of clinical ESKAPE isolates one fluoroquinolone (FQR)-resistant E. coli strain was selected for the preparation of the infection model (Table S3†). A solution of 7 was administered intravenously to 6 weeks old CD-1 male mice infected with a ciprofloxacin resistant E. coli clinical isolate (MIC = 0.031 μg kg−1) in two doses of 6 mg kg−1 (12 mg per kg per day) and 17 mg kg−1 (34 mg per kg per day) at t = 2 h and t = 12 h post infection. All mice infected with the lethal dose of FQR resistant bacteria survived after 24 h while treated with doses of 6 and 17 mg kg−1 injected after 2 h and 10 h. Bacterial payload (CFU) was measured from spleens and kidneys and compared to untreated control animals. Compound 7 showed significant reduction of bacterial counts (>3log10 CFU g−1) in kidneys in both tested doses (Fig. 4). The dose of 17 mg kg−1 of compound 7 indicates a main elimination of the unmetabolized compound via the kidney.
Conclusion
In summary, we have performed a systematic synthesis and SAR study for the replacement of the phenyl by pyridines in the aromatic building blocks of albicidin. Such replacements commonly are performed in regions which are not necessarily pharmacophore regions of the molecule. Following our study, we succeeded in obtaining a novel albicidin derivative 7 with a pyridine as building block D, of unprecedented antibacterial activity against a microbial panel of clinically relevant multi-resistant bacteria clearly showing the broad spectrum of activity of this new analogue of albicidin. The best antibacterial potency was observed towards Gram-negative E. coli and Gram-positive S. aureus FQR isolates. We are delighted to report a breakthrough regarding the activity in presence of plasma, and in vitro plasma stability. The tolerability study in mice by intravenous injection of a dose of 50 mg kg−1 of compound (7) did not show any adverse effects or histopathological observations which provides evidence very promising safety profile. The pharmacokinetic data obtained after intravenous injection with a safe dose of 50 mg kg−1 in mice revealed a bi-phasic elimination of compound 2 and 7 suggesting a 2-compartmental distribution in the body. In the in vivo proof-of-concept experiment, treating animals infected with a lethal dose of FQR clinical isolate (BSL2) E. coli at a dose of 17 mg kg−1 compound 7 applied twice, all mice survived. The CFU reduction in the infected isolated organs was more than 3-log units in spleen and 2-log units in kidney. In summary, the pharmacokinetic and pharmacodynamics testing of albicidin analogue 7 has clearly demonstrated efficacy and revealed 7 as a lead structure for a novel antibiotic drug. After this successful target validation in a rodent animal model the broader anti-bacterial testing towards WHO priority 1 and 2 clinical isolates of these new albicidin analogues will set the stage for a full pre-clinical profiling. This will be paralleled by MOA studies and a further thorough understanding of albicidin resistance mechanisms as a prerequisite for the pre-clinical and clinical development of new antibiotic therapies addressing AMR.Data availability
Experimental procedures and analytical data are availiable in the ESI.†Author contributions
LZ and LK designed and synthesized albicidins analyzed the data and wrote the manuscript. LvE designed and synthesized albicidins (5, 6). MS was responsible for biological testing and wrote the manuscript. AK developed the theory and performed the computations. FOG and JW aided in interpreting the results and worked on the manuscript. RDS supervised the project. All authors provided critical feedback and helped shape the research, analysis and manuscript.Conflicts of interest
FOG is employee of Gombert Pharma Research Solutions (GPRS) and shareholder of Selmod LLC. The other authors declare no conflict of interest. Patents are filed.Acknowledgements
All experimental procedures were approved by and conducted in accordance with the regulations of local Animal Welfare authorities (Landesamt für Gesundheit und Verbraucherschutz, Abteilung Lebensmittel-und Veterinärwesen, Saarbrücken, Germany). The authors acknowledge financial support granted by the Deutsche Forschungsgemeinschaft (DFG, SU 239/11-1, PE 2600/1-1 and RTG 2473 “Bioactive Peptides” (LK), the Alexander-von-Humboldt-Foundation (LZ) and the BMBF (VIP grant: 03VP00030). The authors also thank Dr Andi Mainz for valuable discussions as well as Kay Hommernick, Marcello Spinczyk and Marcel Kulike for their contribution in the laboratory. Furthermore, the authors thank Marc Griffel and Dr Maria Schlangen for HRLC-MS measurements as well as Samantha Voges and Dr Sebastian Kemper for NMR measurements. Lastly, the authors thank Anika Christoffel as Team Manager of Pharmacelsus for conducting the in vivo experiments.References
- R. Laxminarayan, Science, 2014, 345, 1299–1301 CrossRef CAS PubMed.
- E. Martens and A. L. Demain, J. Antibiot., 2017, 70, 520–526 CrossRef CAS PubMed.
- D. J. Payne, M. N. Gwynn, D. J. Holmes and D. L. Pompliano, Nat. Rev. Drug Discovery, 2007, 6, 29–40 CrossRef CAS PubMed.
- K. Lewis, Nat. Rev. Drug Discovery, 2013, 12, 371–387 CrossRef CAS PubMed.
- S. J. Baker, D. J. Payne, R. Rappuoli and E. De Gregorio, Proc. Natl. Acad. Sci., 2018, 115, 12887–12895 CrossRef CAS PubMed.
- R. Tommasi, R. Iyer and A. A. Miller, ACS Infect. Dis., 2018, 4, 686–695 CrossRef CAS PubMed.
- M. I. Hutchings, A. W. Truman and B. Wilkinson, Curr. Opin. Microbiol., 2019, 51, 72–80 CrossRef CAS PubMed.
- E. D. Brown and G. D. Wright, Nature, 2016, 529, 336–343 CrossRef CAS PubMed.
- P. Hunter, EMBO Rep., 2020, 21 Search PubMed.
- W. H. Organization, Global antimicrobial resistance and use surveillance system, (GLASS) report, 2020 Search PubMed.
- E. Tacconelli, E. Carrara, A. Savoldi, S. Harbarth, M. Mendelson, D. L. Monnet, C. Pulcini, G. Kahlmeter, J. Kluytmans, Y. Carmeli, M. Ouellette, K. Outterson, J. Patel, M. Cavaleri, E. M. Cox, C. R. Houchens, M. L. Grayson, P. Hansen, N. Singh, U. Theuretzbacher, N. Magrini, A. O. Aboderin, S. S. Al-Abri, N. Awang Jalil, N. Benzonana, S. Bhattacharya, A. J. Brink, F. R. Burkert, O. Cars, G. Cornaglia, O. J. Dyar, A. W. Friedrich, A. C. Gales, S. Gandra, C. G. Giske, D. A. Goff, H. Goossens, T. Gottlieb, M. Guzman Blanco, W. Hryniewicz, D. Kattula, T. Jinks, S. S. Kanj, L. Kerr, M.-P. Kieny, Y. S. Kim, R. S. Kozlov, J. Labarca, R. Laxminarayan, K. Leder, L. Leibovici, G. Levy-Hara, J. Littman, S. Malhotra-Kumar, V. Manchanda, L. Moja, B. Ndoye, A. Pan, D. L. Paterson, M. Paul, H. Qiu, P. Ramon-Pardo, J. Rodríguez-Baño, M. Sanguinetti, S. Sengupta, M. Sharland, M. Si-Mehand, L. L. Silver, W. Song, M. Steinbakk, J. Thomsen, G. E. Thwaites, J. W. M. van der Meer, N. van Kinh, S. Vega, M. V. Villegas, A. Wechsler-Fördös, H. F. L. Wertheim, E. Wesangula, N. Woodford, F. O. Yilmaz and A. Zorzet, Lancet Infect. Dis., 2018, 18, 318–327 CrossRef PubMed.
- P. Beyer and S. Paulin, ACS Infect. Dis., 2020, 6, 1289–1291 CrossRef CAS.
- R. G. Birch and S. S. Patil, Microbiology, 1985, 131, 1069–1075 CAS.
- S. Baumann, J. Herrmann, R. Raju, H. Steinmetz, K. I. Mohr, S. Hüttel, K. Harmrolfs, M. Stadler and R. Müller, Angew. Chem., Int. Ed., 2014, 53, 14605–14609 CrossRef CAS PubMed.
- Y. J. Kim, H.-J. Kim, G.-W. Kim, K. Cho, S. Takahashi, H. Koshino and W.-G. Kim, J. Nat. Prod., 2016, 79, 2223–2228 CrossRef CAS PubMed.
- S. M. Hashimi, M. K. Wall, A. B. Smith, A. Maxwell and R. G. Birch, Antimicrob. Agents Chemother., 2007, 51, 181–187 CrossRef CAS PubMed.
- S. Baumann, J. Herrmann, R. Raju, H. Steinmetz, K. I. Mohr, S. Hüttel, K. Harmrolfs, M. Stadler and R. Müller, Angew. Chem., Int. Ed., 2014, 53, 14605–14609 CrossRef CAS PubMed.
- S. Cociancich, A. Pesic, D. Petras, S. Uhlmann, J. Kretz, V. Schubert, L. Vieweg, S. Duplan, M. Marguerettaz, J. Noëll, I. Pieretti, M. Hügelland, S. Kemper, A. Mainz, P. Rott, M. Royer and R. D. Süssmuth, Nat. Chem. Biol., 2015, 11, 195–197 CrossRef CAS PubMed.
- M. Royer, L. Costet, E. Vivien, M. Bes, A. Cousin, A. Damais, I. Pieretti, A. Savin, S. Megessier, M. Viard, R. Frutos, D. W. Gabriel and P. C. Rott, Mol. Plant-Microbe Interact., 2004, 17, 414–427 CrossRef CAS PubMed.
- L. Rostock, R. Driller, S. Grätz, D. Kerwat, L. v. Eckardstein, D. Petras, M. Kunert, C. Alings, F.-J. Schmitt, T. Friedrich, M. C. Wahl, B. Loll, A. Mainz and R. D. Süssmuth, Nat. Commun., 2018, 9, 3095 CrossRef PubMed.
- A. Sikandar, K. Cirnski, G. Testolin, C. Volz, M. Brönstrup, O. V. Kalinina, R. Müller and J. Koehnke, J. Am. Chem. Soc., 2018, 140, 16641–16649 CrossRef CAS PubMed.
- L. Vieweg, J. Kretz, A. Pesic, D. Kerwat, S. Grätz, M. Royer, S. Cociancich, A. Mainz and R. D. Süssmuth, J. Am. Chem. Soc., 2015, 137, 7608–7611 CrossRef CAS PubMed.
- J. Kretz, D. Kerwat, V. Schubert, S. Grätz, A. Pesic, S. Semsary, S. Cociancich, M. Royer and R. D. Süssmuth, Angew. Chem., Int. Ed., 2014, 54, 1969–1973 CrossRef PubMed.
- D. Kerwat, S. Grätz, J. Kretz, M. Seidel, M. Kunert, J. B. Weston and R. D. Süssmuth, ChemMedChem, 2016, 11, 1899–1903 CrossRef CAS PubMed.
- I. Behroz, L. Kleebauer, K. Hommernick, M. Seidel, S. Grätz, A. Mainz, J. B. Weston and R. Süssmuth, Chem.–Eur. J., 2021, 27, 9077–9086 CrossRef CAS PubMed.
- L. von Eckardstein, D. Petras, T. Dang, S. Cociancich, S. Sabri, S. Grätz, D. Kerwat, M. Seidel, A. Pesic, P. C. Dorrestein, M. Royer, J. B. Weston and R. D. Süssmuth, Chem.–Eur. J., 2017, 23, 15316–15321 CrossRef CAS PubMed.
- S. Grätz, D. Kerwat, J. Kretz, L. von Eckardstein, S. Semsary, M. Seidel, M. Kunert, J. B. Weston and R. D. Süssmuth, ChemMedChem, 2016, 11, 1499–1502 CrossRef PubMed.
- I. Behroz, P. Durkin, S. Grätz, M. Seidel, L. Rostock, M. Spinczyk, J. B. Weston and R. D. Süssmuth, Chemistry, 2019, 25, 16538–16543 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- Y. A. Azev, G. A. Mokrushina and I. Y. Postovskii, Chem. Heterocycl. Compd., 1974, 10, 687–690 CrossRef.
- D. I. Andersson, D. Hughes and J. Z. Kubicek-Sutherland, Drug Resist. Updates, 2016, 26, 43–57 CrossRef CAS PubMed.
- G. Testolin, K. Cirnski, K. Rox, H. Prochnow, V. Fetz, C. Grandclaudon, T. Mollner, A. Baiyoumy, A. Ritter, C. Leitner, J. Krull, J. Van Den Heuvel, A. Vassort, S. Sordello, M. M. Hamed, W. A. M. Elgaher, J. Herrmann, R. W. Hartmann, R. Müller and M. Brönstrup, Chem. Sci., 2020, 11, 1316–1334 RSC.
- R. G. Birch, J. M. Pemberton and W. V. S. Basnayake, J. Gen. Microbiol., 1990, 136, 51–58 CrossRef CAS PubMed.
- J. Ye and B. Van Den Berg, EMBO J., 2004, 23, 3187–3195 CrossRef CAS PubMed.
- W. A. M. Elgaher, M. M. Hamed, S. Baumann, J. Herrmann, L. Siebenbürger, J. Krull, K. Cirnski, A. Kirschning, M. Brönstrup, R. Müller and R. W. Hartmann, Chem.–Eur. J., 2020, 26, 7219–7225 CrossRef CAS PubMed.
- T. Planke, M. Moreno, S. Hüttel, J. Fohrer, F. Gille, M. D. Norris, M. Siebke, L. Wang, R. Müller and A. Kirschning, Org. Lett., 2019, 21, 1359–1363 CrossRef CAS PubMed.
- B. F. Erlanger and L. Goode, Nature, 1954, 174, 840–841 CrossRef CAS PubMed.
- H. Wada, H. E. L. Williams and C. J. Moody, Angew. Chem., Int. Ed., 2015, 54, 15147–15151 CrossRef CAS PubMed.
- S. C. Shankaramma, Z. Athanassiou, O. Zerbe, K. Moehle, C. Mouton, F. Bernardini, J. W. Vrijbloed, D. Obrecht and J. A. Robinson, ChemBioChem, 2002, 3, 1126–1133 CrossRef CAS PubMed.
- J. A. Robinson, Acc. Chem. Res., 2008, 41, 1278–1288 CrossRef CAS PubMed.
- L. Sebaoun, V. Maurizot, T. Granier, B. Kauffmann and I. Huc, J. Am. Chem. Soc., 2014, 136, 2168–2174 CrossRef CAS PubMed.
- T. Seedorf, A. Kirschning and D. Solga, Chem.–Eur. J., 2021, 27, 7321–7339 CrossRef CAS PubMed.
- D.-W. Zhang, X. Zhao, J.-L. Hou and Z.-T. Li, Chem. Rev., 2012, 112, 5271–5316 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc04019g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |