
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDecreasing the coordinated N atoms in a single-atom Cu catalyst to achieve selective transfer hydrogenation of alkynes†
Xuge
Zhang‡
ab,
He
Lin‡
c,
Jian
Zhang‡
*b,
Yajun
Qiu
a,
Zedong
Zhang
a,
Qi
Xu
a,
Ge
Meng
b,
Wensheng
Yan
 d,
Lin
Gu
e,
Lirong
Zheng
f,
Dingsheng
Wang
*a and
Yadong
Li
a
d,
Lin
Gu
e,
Lirong
Zheng
f,
Dingsheng
Wang
*a and
Yadong
Li
a
aDepartment of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: wangdingsheng@mail.tsinghua.edu.cn
bCollege of Chemistry and Materials Engineering, Wenzhou University, Wenzhou, Zhejiang 325035, China. E-mail: zhangjian@wzu.edu.cn
cState Key Laboratory of Chemistry and Utilization of Carbon Based Energy Resources, Key Laboratory of Advanced Functional Materials, Autonomous Region, Institute of Applied Chemistry, College of Chemistry, Xinjiang University, Urumqi, 830046, Xinjiang, China
dNational Synchrotron Radiation Laboratory, CAS Center for Excellence in Nanoscience, University of Science and Technology of China, Hefei 230029, China
eBeijing National Laboratory for Condensed Matter Physics, Institute of Physics, Chinese Academy of Sciences, Beijing 100190, China
fBeijing Synchrotron Radiation Facility, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China
First published on 19th October 2021
Abstract
Single-atom (SA) catalysts have attracted broad attention due to their distinctive catalytic properties in diverse reactions. Increasing the unsaturated coordination sites of active centers is a valid and challenging approach to improve the performance of such catalysts. Herein, we report an oxide compounding strategy to decrease the N coordination number of a SA Cu catalyst by reducing the thickness of the N-doped carbon carrier with a lower density of N atoms. The SA Cu catalyst with a more unsaturated N coordination structure can achieve transfer hydrogenation of alkynes with good activity and selectivity, which is disabled over the common N coordinated SA Cu catalyst on pure CN. It is found that individual Cu centers coordinated by fewer N atoms can accelerate the hydrogen transfer from ammonia–borane and still leave proper adsorption sites for alkynes to realize the entire hydrogenation reaction. This work will open up new opportunities to modulate the unsaturated coordination structure of SA catalysts for creating better-performing heterogeneous catalysts.
Introduction
Single-atom (SA) catalysts have recently attracted much attention1–4 due to their unique performance in numerous fields such as electrochemistry,5–8 organic synthesis9–12 and industrial catalysis.13–15 The specific properties of such catalysts derive from the low coordination of metal centers, which affords a peculiar electronic structure and favorable adsorption of reactants.16–18 For now, SA catalysts on N-doped carbon (CN) have emerged as versatile and classic catalysts, and are usually obtained from the pyrolysis of organic polymers19,20 or metal–organic frameworks.21,22 It is noteworthy that the general metal centers in such catalysts inevitably suffer from saturated coordination by abundant N atoms in pure CN due to the better stability, which restricts their catalytic behavior in many cases.23,24 Thus, an effective strategy to regulate the coordination environment of metal centers for increasing unsaturated sites is crucial to enhance their catalytic performance.25–27 So far, a limited number of approaches have been reported to tailor the coordination number of metal centers in CN supported SA catalysts like changing pyrolysis temperature,28,29 choosing different precursors30,31 and altering the immobilization of metal.32–34 Therefore, it is urgent but tough to develop new pathways to modulate the unsaturated coordination structures of SA catalysts on CN and further disclose their impact on the catalytic activity.Semihydrogenation of alkynes represents the most convenient and straightforward method to produce olefins, the important intermediates35–37 and raw chemical material38–40 in organic synthesis. Benefitting from the superior reusability, a few heterogeneous catalysts have been applied in this transformation.41–45 However, these heterogeneous catalysts are mainly based on a noble metal with selectivity issues or suffering from harsh reaction conditions (high temperature and high pressure of H2). Thus, it is highly desirable to develop more abundant non-noble metal based heterogeneous catalysts for such reaction with good selectivity under mild conditions. For this purpose, heterogeneous base metal catalyzed transfer hydrogenation of alkynes provides a potential approach for avoiding the high pressure of H2.46,47 In this context, active metal centers need to serve as the station to transfer hydrogen species from the hydrogen source to alkynes, which demands enough adsorption sites for the hydrogen source and alkynes.48 Thus, it is essential to fabricate targeted base metal heterogeneous catalysts with more unsaturated coordination sites.
Herein, we demonstrate an oxide compounding CN strategy to reduce coordinated N atoms in a SA Cu catalyst for realizing transfer semihydrogenation of alkynes. Owing to the CN on an Al2O3 matrix being thinner with decreased doped N atoms, the general Cu–N3 and Cu–N4 features of a SA Cu catalyst on pure CN can be tuned to the more unsaturated Cu–N2 feature. The obtained Cu–N2 structure makes the SA Cu catalyst succeed in achieving transfer hydrogenation of alkynes, which cannot occur over Cu–N3 and Cu–N4 structures. The optimized SA Cu catalyst exhibits good activity and selectivity to produce a variety of olefins. It is clarified that the lower-coordination state of the Cu–N2 structure benefits hydrogen species transferring from ammonia–borane (AB) to Cu centers, and simultaneously affords the appropriate adsorption site for alkynes to complete the hydrogenation reaction smoothly.
Results and discussion
The SA Cu catalyst supported on pure CN (denoted as Cu1/CN) is prepared through pyrolyzing melamine–formaldehyde (MF) resin with loading of Cu (Fig. S1†).49 To increase the unsaturated coordination sites of the SA Cu catalyst, the MF resin loaded Cu is generated on an Al2O3 matrix via the in situ polymerization process followed by the same pyrolysis treatment (denoted as Cu1/CN/Al2O3) (Fig. S1†). The irregular thick nanosheet morphology of Cu1/CN can be observed in high-resolution transmission electron microscopy (HR-TEM) and scanning transmission electron microscopy (STEM) images (Fig. 1a and S2†). Meanwhile, for Cu1/CN/Al2O3, a thin CN coating layer can be distinguished from the Al2O3 matrix (Fig. 1b), which is formed from the coated MF resin as revealed by HR-TEM and Fourier transform infrared (FT-IR) analyses (Fig. S3 and S4†). No visible Cu or its derivative nanoparticles can be found in either of the two catalysts by HR-TEM, STEM or X-ray diffraction (XRD) analysis (Fig. 1a and b, S2, S5 and S6†). The homogeneous distribution of Cu and N species on the two catalysts is confirmed through energy dispersive X-ray (EDX) elemental mapping experiments (Fig. 1c and d). The presence of SA Cu species is verified by aberration corrected high-angle annular dark-field scanning transmission electron microscopy (AC HAADF-STEM) measurements which show a number of bright dots assigned to independent Cu species on Cu1/CN and Cu1/CN/Al2O3 (Fig. 1e and f). The loading content of Cu is determined to be 1.65 and 0.98 wt% for Cu1/CN and Cu1/CN/Al2O3 according to inductively coupled plasma optical emission spectrometry (ICP-OES) tests.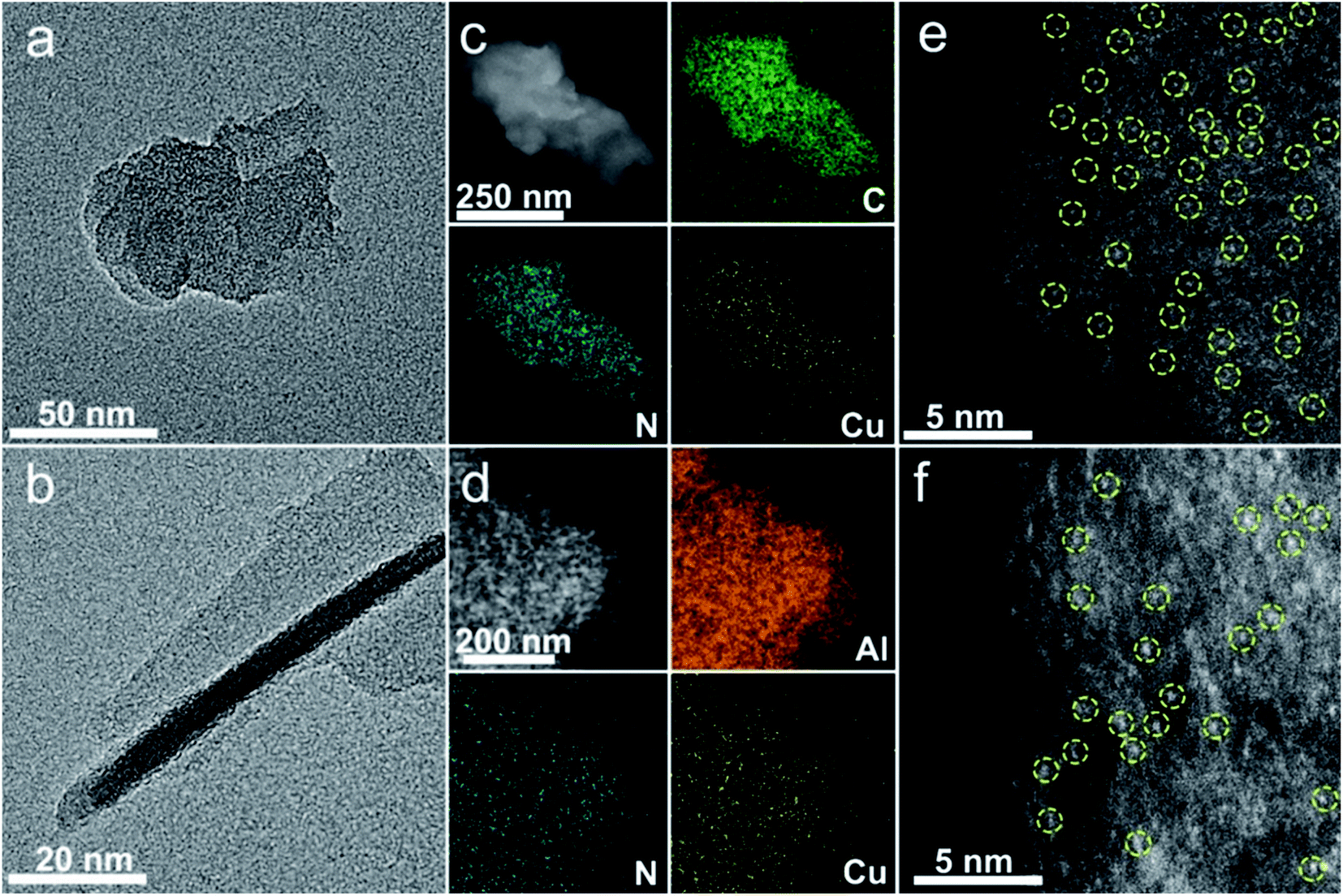 | ||
| Fig. 1 Characterization of Cu1/CN and Cu1/CN/Al2O3. (a) HR-TEM image of Cu1/CN. (b) HR-TEM image of Cu1/CN/Al2O3. (c) EDX elemental mapping analysis of Cu1/CN. (d) EDX elemental mapping analysis of Cu1/CN/Al2O3. (e) Representative AC HAADF-STEM image of Cu1/CN. (f) Representative AC HAADF-STEM image of Cu1/CN/Al2O3. The yellow circles in e and f are drawn around partial SA Cu species. | ||
The local atomic and electronic structures of the two catalysts are investigated by X-ray absorption spectroscopy (XAS) techniques. As shown in Fourier transformed extended X-ray absorption fine structure (FT-EXAFS) spectra at the Cu K-edge (Fig. 2a, for EXAFS in k-space see Fig. S7†), there is only one primary peak attributed to the Cu–N bond (∼1.5 Å) but no other peaks of the Cu–Cu bond (∼2.1 Å) are observed for Cu1/CN or Cu1/CN/Al2O3, which provides more proof of SA Cu species in them.50–52 It is noteworthy that the oxidation states of the two SA Cu species are quite different from those of each other as indicated by their X-ray absorption near-edge structure (XANES) spectra (Fig. 2b). The adsorption peak ascribed to the dipole-allowed 1s → 4p transition of Cu is close to that of Cu2O for Cu1/CN (ca. 8982.5 eV) while it is closer to that of CuO for Cu1/CN/Al2O3 (ca. 8986.1 eV) with differently intense white line peaks (ca. ∼8996.0 eV), indicating the higher oxidation state of Cu species in Cu1/CN/Al2O3. This conclusion can also be drawn from X-ray photoelectron spectroscopy (XPS) spectra in the Cu 2p region. The deconvoluted Cu 2p3/2 XPS peak indicates that Cu1/CN involves one major Cu species at a binding energy of 932.0 eV with the other minor Cu species at a binding energy of 934.3 eV. Meanwhile, for Cu1/CN/Al2O3, a solely dominant peak at 933.0 eV can be deconvoluted from its Cu 2p3/2 XPS spectrum, illustrating the higher oxidation state of its Cu species compared to most Cu species in Cu1/CN (Fig. 2c). Moreover, electron paramagnetic resonance (EPR) studies found that Cu1/CN/Al2O3 exhibits a notable Cu(II) signal (g = 2.045), while Cu1/CN just displays a relatively weak Cu(II) signal at a different g value of 1.997 (Fig. 2d).53–55 Given Cu(I) species is silent in EPR, it is apparent that Cu1/CN contains a majority of Cu(I) species and so leads to its lower oxidation state of Cu in contrast to Cu1/CN/Al2O3 which contains predominant Cu(II) species.54
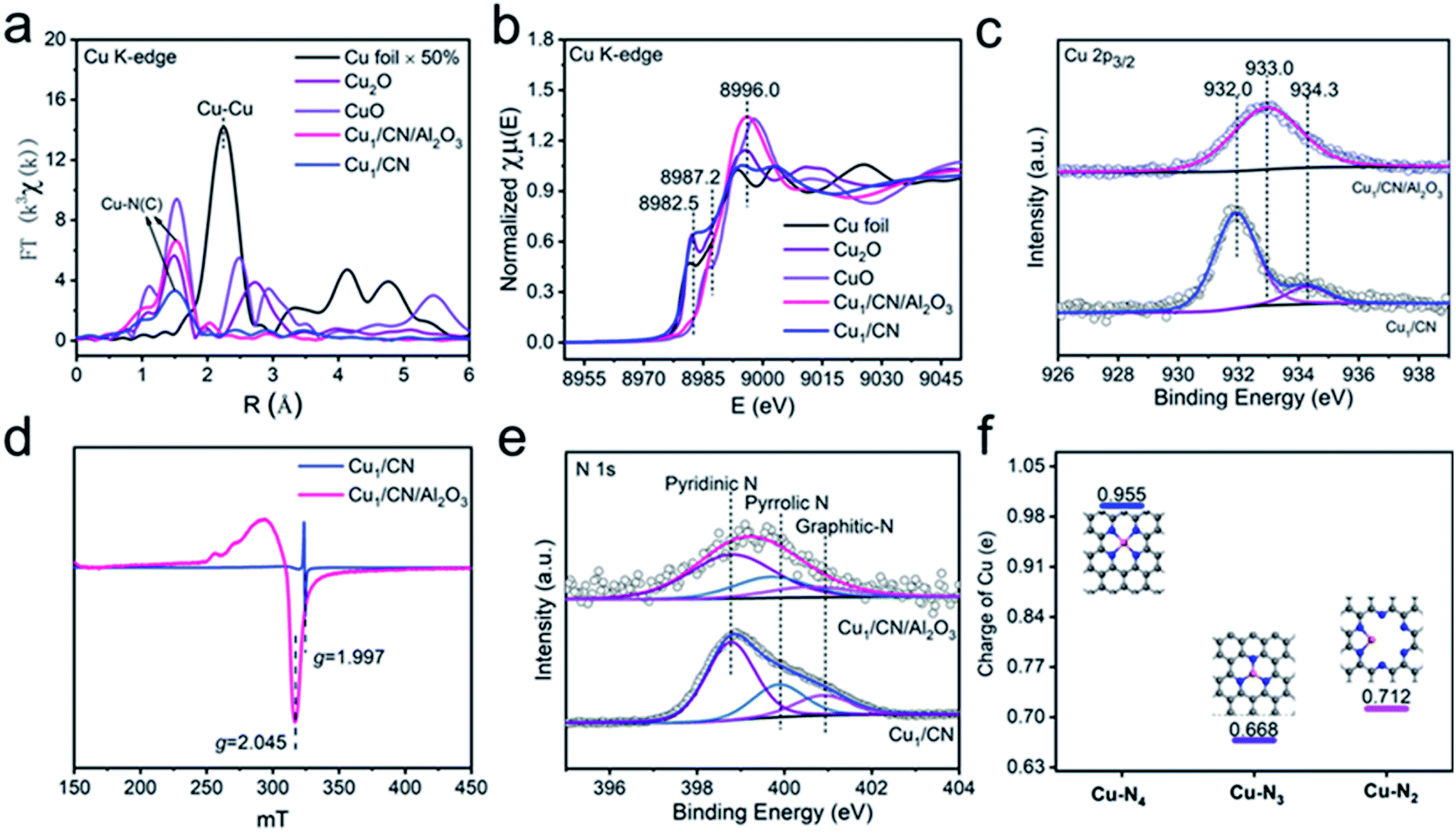 | ||
| Fig. 2 Structural analyses of Cu1/CN and Cu1/CN/Al2O3. (a) FT-EXAFS spectra of Cu1/CN and Cu1/CN/Al2O3 with CuO, Cu2O and Cu foil as references at the Cu K-edge. (b) XANES spectra of Cu1/CN and Cu1/CN/Al2O3 with CuO, Cu2O and Cu foil as references at the Cu K-edge. (c) Deconvoluted XPS spectra of Cu1/CN and Cu1/CN/Al2O3 in the Cu 2p3/2 region. (d) EPR spectra of Cu1/CN and Cu1/CN/Al2O3. (e) N 1s XPS fitting analysis of Cu1/CN and Cu1/CN/Al2O3. (f) DFT calculation studies on the oxidation state of SA Cu species in different coordination structures. | ||
The character of the CN carrier is next probed on the two catalysts. The CN carriers in Cu1/CN/Al2O3 and Cu1/CN both display pyridinic-N (398.7 eV) as the major doped N species with minor amounts of pyrrolic-N (399.8 eV) and graphitic-N (400.9 eV), disclosed by deconvoluted N 1s XPS spectra (Fig. 2e, for fitting parameters see Table S1†).54 However, the content of N species in the CN of Cu1/CN/Al2O3 decreases a lot compared to that of Cu1/CN according to the significantly lower intensity of the pyridinic-N signal for Cu1/CN/Al2O3 in soft X-ray absorption spectroscopy (sXAS) spectra (Fig. S8†). This result is also proved by elemental analyses which indicate that the content of N in Cu1/CN/Al2O3 is just 1.04 wt% which is much less than that of Cu1/CN (30.77 wt%) (Table S2†). The more sparse N atoms result from the CN carrier being thin enough in Cu1/CN/Al2O3 instead of the thick nanosheet morphology in Cu1/CN. It should be noted that, owing to the decrease of N atoms, the ratio of N to Cu reduced a lot from 18.66 for Cu1/CN to 1.07 for Cu1/CN/Al2O3 (Table S2†). It is predictable that fewer N atoms can lead to a smaller N coordination number for Cu centers in Cu1/CN/Al2O3. This can be revealed by the EXAFS fitting analysis where the N coordination number of Cu is 3.2 for Cu1/CN while it is just 1.9 for Cu1/CN/Al2O3 (Fig. S9†).
To confirm the lower-coordination structure, density functional theory (DFT) calculations are carried out to analyze the feature of SA Cu centers on the CN carrier with different coordination structures (Fig. S10 and Table S3†). Considering that N atoms in pure CN are abundant, Cu centers in Cu1/CN can exist as Cu–N3 and Cu–N4 structures as commonly reported in previous studies of CN supported SA Cu catalysts.56–58 For increasing unsaturated coordination sites, the Cu–N2 structure of Cu centers may be present in Cu1/CN/Al2O3. Therefore, since the main N species in the CN of the two catalysts is pyridinic-N, the potential Cu–N4, Cu–N3 and Cu–N2 structures with pyridinic-N coordination are simulated to explore their differences (Fig. 2f). It turns out that the charge of Cu in Cu–N2 (0.712e) lies right between that in Cu–N3 (0.668e) and Cu–N4 (0.955e). This conclusion is consistent with the observation from Cu 2p3/2 XPS spectra that the oxidation state of Cu in Cu1/CN/Al2O3 is between that of the two Cu species in Cu1/CN. This strongly verifies the lower coordinated Cu–N2 structure in Cu1/CN/Al2O3 instead of Cu–N3 and Cu–N4 structures in Cu1/CN.
The performance of Cu1/CN/Al2O3 and Cu1/CN for transfer hydrogenation of alkynes is evaluated to clarify the influence of distinct coordination structures. Delightfully, the hydrogenation of phenylacetylene proceeds smoothly over Cu1/CN/Al2O3 with AB as the hydrogen source (Fig. 3a). The product styrene is obtained in 94% yield and 99% selectivity without potential ethylbenzene by-product detected by gas chromatography-mass spectrometry (GC-MS) analysis (Fig. S11†). The turnover number (TON) is calculated to be 74 which is greater than that of reported heterogeneous non-noble metal based catalysts (Table S4†). Meanwhile, over Cu1/CN, this hydrogenation reaction fails to occur under the same conditions. A pure Al2O3 supported SA Cu catalyst (Cu1/Al2O3) is also prepared for comparison, and it possesses uniformly dispersed Cu species with a loading content of 0.5 wt% as revealed by STEM, EDX elemental mapping, XAS, and ICP-OES analyses (Fig. S12 and S13†). This Cu1/Al2O3 exhibits a very poor activity in contrast to Cu1/CN/Al2O3. Clean CN/Al2O3 is also inert for the reaction. Thus, it is concluded that the great performance of Cu1/CN/Al2O3 derives from the distinct structure of the SA Cu species on CN from that of Cu1/CN. A recycling test is conducted to evaluate the stability of Cu1/CN/Al2O3. It shows that Cu1/CN/Al2O3 can be reused at least five times without obvious loss of activity and selectivity at a relatively low conversion level (Fig. 3b). The loading content of Cu in the catalyst remains nearly the same as 0.97 wt% after the reaction as indicated by ICP-OES. STEM and EDX elemental mapping analyses manifest homogeneously dispersed Cu components and unchanged morphology in the recovered Cu1/CN/Al2O3 (Fig. S14†). This demonstrates the good stability of Cu1/CN/Al2O3 for such transfer hydrogenation reactions.
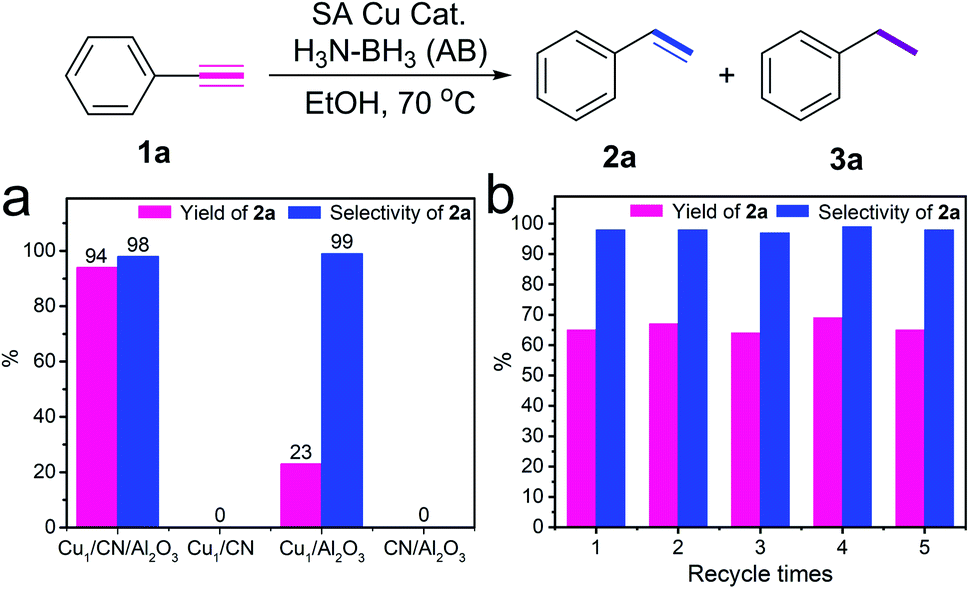 | ||
| Fig. 3 Performance evaluation for transfer hydrogenation of alkynes. (a) Activity evaluation of SA Cu catalysts for transfer hydrogenation of phenylacetylene. Standard reaction conditions: phenylacetylene (1a, 0.2 mmol), AB (1.0 mmol) in EtOH (4.0 mL) at 70 °C for 8 h with the catalyst: Cu1/CN/Al2O3 (16.9 mg, Cu = 1.0 mol%), Cu1/CN (9.8 mg, Cu = 1.0 mol%), Cu1/Al2O3 (25.6 mg Cu = 1.0 mol%), CN/Al2O3 (16.9 mg). Yields are determined by gas chromatography (GC) analysis with dodecane as the internal standard. Selectivities are determined by GC-MS analysis. (b) Recycling test of Cu1/CN/Al2O3 for transfer hydrogenation of phenylacetylene. | ||
We next explore the impact of substrate categories on the catalytic performance of Cu1/CN/Al2O3. As displayed in Fig. 4, phenylacetylene derivatives with electron-donating functional groups (MeO−, tBu−, and Me−) can be transformed into alkenes in good yields and selectivities (2b–2d), while those with electron-withdrawing functional groups (Br− and Cl−) suffer from relatively low conversions (2e and 2f) for longer reaction time. This discloses that alkyne substrates have an electronic effect on the catalytic behavior of Cu1/CN/Al2O3, where electron-poor alkynes can decrease the catalytic efficiency. The meta-methyl and ortho-methyl substituted phenylacetylenes are both hydrogenated into the corresponding alkenes (2h and 2i) with high conversions and selectivities, but the more steric naphthyl acetylene lowers the hydrogenation efficiency with just 83% conversion (2i), indicating the steric effect of substrates on the performance of Cu1/CN/Al2O3. In addition, the hydrogenation of internal aryl alkynes like diphenylacetylene and 1-phenyl-1-propyne also proceeds over Cu1/CN/Al2O3 with decreased conversions of 89% and 87% to provide alkene products with quantitative selectivities (2j and 2k). As for aliphatic alkynes such as cyclohexylacetylene, Cu1/CN/Al2O3 exhibits an unchanged catalytic efficiency to convert it into the target cyclohexylethylene (2l) with 99% conversion and 98% selectivity.
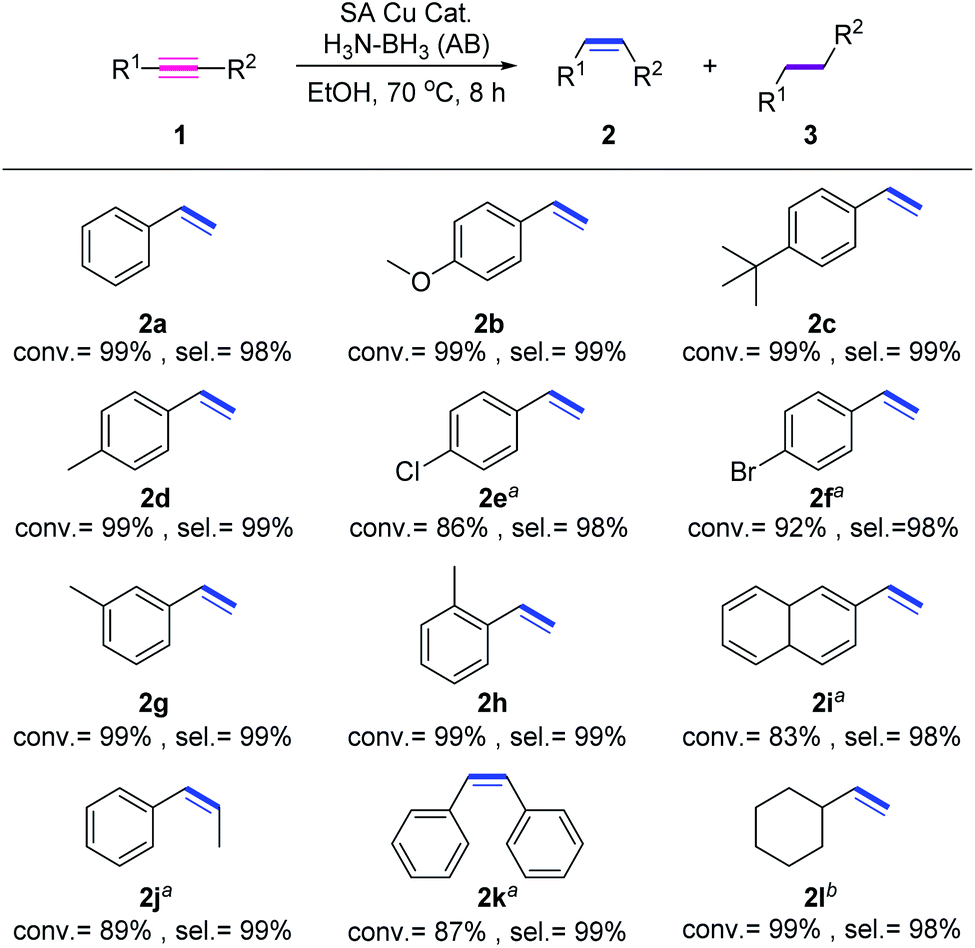 | ||
| Fig. 4 Substrate scope of transfer hydrogenation of alkynes over Cu1/CN/Al2O3. Standard reaction conditions: substrate 1 (0.20 mmol), AB (1.0 mmol), Cu1/CN/Al2O3 (16.9 mg, Cu = 1.0 mol%) in EtOH (4.0 mL) at 70 °C for 8 h. Conversion of 1 (conv.) is determined by GC analysis with dodecane as the internal standard. Selectivity of 2 (sel.) is determined by GC-MS analysis. aReaction time is prolonged to 12 h. bReaction time is shortened to 6 h. | ||
The prominent performance of Cu1/CN/Al2O3 with respect to Cu1/CN can be attributed to the more unsaturated coordination of Cu centers caused by the Al2O3 compounding CN strategy. To gain more insight into the relationship between the performance and unsaturated coordination structures, DFT calculations are performed to analyze the transfer hydrogenation pathway over the two catalysts. Based on characterization results, the Cu–N2 and Cu–N3 models are constructed to simulate Cu1/CN/Al2O3 and Cu1/CN. The adsorption performance of AB molecules on the two SA Cu catalysts is firstly investigated for the reason that the initial step of the transfer hydrogenation is a hydrogen species shift from AB molecules to active Cu centers.59,60 The energy barrier of this process is 2.07 eV on the Cu–N2 structure and 2.69 eV higher on the Cu–N3 structure, suggesting a more favorable hydrogen transfer on the former (Fig. 5a and b). Over hydrogenated Cu–N2 and Cu–N3 structures, the following transfer of hydrogen species to alkynes is next investigated. The phenylacetylene can adsorb on the hydrogenated Cu center in the Cu–N2 structure, and the energy barrier of the hydrogen species shift to phenylacetylene is as low as 0.24 eV (Fig. 5c). Meanwhile, for the Cu–N3 structure, since the hydrogenated Cu center is coordination-saturated, the adsorption of phenylacetylene is hindered. In order to achieve the hydrogenation process, a spillover hydrogenation procedure may be involved.61 The hydrogen species shifts from the Cu center to the ortho carbon to provide the adsorption site for phenylacetylene (Fig. S15†), and then transfers back to realize the hydrogenation reaction (Fig. 5d). Even so, the total energy barrier of such procedures comes to 2.62 eV which is significantly higher than that of the hydrogenation process on the Cu–N2 structure, demonstrating the dramatic difficulty in the transfer hydrogenation reaction.
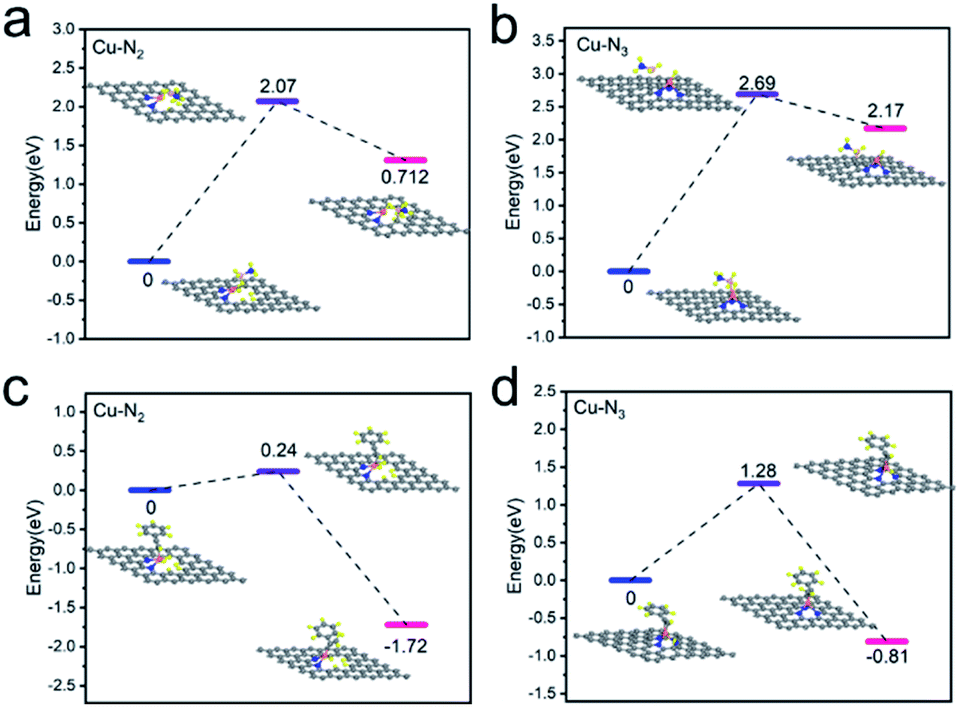 | ||
| Fig. 5 Investigations on the source of the improved activity. (a) DFT calculation studies on the hydrogen transfer from AB molecule to Cu1/CN/Al2O3. (b) DFT calculation studies on the hydrogen transfer from AB molecule to Cu1/CN. (c) DFT calculation studies on the transfer hydrogenation of phenylacetylene over Cu1/CN/Al2O3. (d) DFT calculation studies on the transfer hydrogenation of phenylacetylene over Cu1/CN. | ||
Conclusions
In summary, we report that reducing the coordination sites of a SA Cu catalyst on CN can achieve remarkable performance for selective transfer hydrogenation of alkynes. Through compounding with Al2O3, the CN carrier becomes very thin with fewer N atoms, leading to a more unsaturated Cu–N2 coordination structure of independent Cu centers. Unlike the pure CN supported SA Cu catalyst with common Cu–N3 and Cu–N4 structures disabling the transfer hydrogenation of alkynes, the SA Cu catalyst anchored by decreased N atoms exhibits great activity for the hydrogenation reaction, furnishing various alkenes with good activity and selectivity. The decrease of coordinated N atoms makes SA Cu sites superior for abstracting hydrogen from ammonia–borane and adsorbing alkyne substrates to reach the transfer hydrogenation process, which causes the outstanding catalytic behavior of the catalyst. This work confirms that increasing the unsaturated coordination sites of SA catalysts via precise regulation of their coordination environment can readily and efficiently change the catalytic performance, providing new opportunities to develop better-performing SA catalysts for heterogeneous catalysis.Data availability
Data associated with this article, including synthesis, characterization, and experimental procedures, are available in the ESI.†Author contributions
X. Z. performed the experiments, collected and analyzed the data, and wrote the paper. H. L. conducted the density functional theory calculation and analysis. Y. Q., Z. Z., Q. X. and G. M. assisted in HR-TEM, STEM, XAS and EDX elemental mapping characterizations. W. Y. helped with the sXAS analysis. L. G. assisted in the AC HAADF-STEM characterization. J. Z., D. W. and Y. L. conceived the experiments, planned the synthesis, analyzed the results, and wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2018YFA0702003), the National Natural Science Foundation of China (22102119, 21890383, 21871159 and 52002249), the Guangdong Basic and Applied Basic Research Foundation (2019A1515110025), the National Postdoctoral Program for Innovative Talents (BX20190167), the Shuimu Tsinghua Scholar Program, and the China Postdoctoral Science Foundation (2020M670283). We thank the 1W1B station of the Beijing Synchrotron Radiation Facility (BSRF) and BL12B station of the National Synchrotron Radiation Laboratory (NRSL) in Hefei for XAS and sXAS measurements.Notes and references
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- X. Cui, W. Li, P. Ryabchuk, K. Junge and M. Beller, Nat. Catal., 2018, 1, 385–397 CrossRef CAS.
- J. Yang, W. Li, S. Tan, K. Xu, Y. Wang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 19085–19091 CrossRef CAS PubMed.
- J. Yang, W. Li, D. Wang and Y. Li, Small Struct., 2021, 2, 20051 Search PubMed.
- Y. Zhang, L. Guo, L. Tao, Y. Lu and S. Wang, Small Methods, 2019, 3, 1800406 CrossRef.
- L. Fan, P. F. Liu, X. Yan, L. Gu, Z. Z. Yang, H. G. Yang, S. Qiu and X. Yao, Nat. Commun., 2016, 7, 10667 CrossRef CAS PubMed.
- Z.-Y. Wu, M. Karamad, X. Yong, Q. Huang, D. A. Cullen, P. Zhu, C. Xia, Q. Xiao, M. Shakouri, F.-Y. Chen, J. Y. Kim, Y. Xia, K. Heck, Y. Hu, M. S. Wong, Q. Li, I. Gates, S. Siahrostami and H. Wang, Nat. Commun., 2021, 12, 2870 CrossRef CAS PubMed.
- Q. Qu, S. Ji, Y. Chen, D. Wang and Y. Li, Chem. Sci., 2021, 12, 4201–4215 RSC.
- L. Wang, E. Guan, J. Zhang, J. Yang, Y. Zhu, Y. Han, M. Yang, C. Cen, G. Fu, B. C. Gates and F.-S. Xiao, Nat. Commun., 2018, 9, 1362 CrossRef PubMed.
- J. Zhang, Z. Wang, W. Chen, Y. Xiong, W.-C. Cheong, L. Zheng, W. Yan, L. Gu, C. Chen, Q. Peng, P. Hu, D. Wang and Y. Li, Chem, 2020, 6, 725–737 CAS.
- Z. Chen, E. Vorobyeva, S. Mitchell, E. Fako, M. A. Ortuño, N. López, S. M. Collins, P. A. Midgley, S. Richard, G. Vilé and J. Pérez-Ramírez, Nat. Nanotechnol., 2018, 13, 702–707 CrossRef CAS PubMed.
- Y. Xiong, W. Sun, Y. Han, P. Xin, X. Zheng, W. Yan, J. Dong, J. Zhang, D. Wang and Y. Li, Nano Res., 2021, 14, 2418–2423 CrossRef CAS.
- X. Li, X. Huang, S. Xi, S. Miao, J. Ding, W. Cai, S. Liu, X. Yang, H. Yang, J. Gao, J. Wang, Y. Huang, T. Zhang and B. Liu, J. Am. Chem. Soc., 2018, 140, 12469–12475 CrossRef CAS PubMed.
- G. Meng, K. Ji, W. Zhang, Y. Kang, Y. Wang, P. Zhang, Y. G. Wang, J. Li, T. Cui, X. Sun, T. Tan, D. Wang and Y. Li, Chem. Sci., 2021, 12, 4139–4146 RSC.
- S. Bai, F. Liu, B. Huang, F. Li, H. Lin, T. Wu, M. Sun, J. Wu, Q. Shao, Y. Xu and X. Huang, Nat. Commun., 2020, 11, 954 CrossRef CAS PubMed.
- X. Liu, Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2019, 141, 9664–9672 CrossRef CAS PubMed.
- T. Zhang, X. Nie, W. Yu, X. Guo, C. Song, R. Si, Y. Liu and Z. Zhao, iScience, 2019, 22, 97–108 CrossRef CAS PubMed.
- W. Liu, L. Zhang, X. Liu, X. Liu, X. Yang, S. Miao, W. Wang, A. Wang and T. Zhang, J. Am. Chem. Soc., 2017, 139, 10790–10798 CrossRef CAS PubMed.
- L. Han, S. Song, M. Liu, S. Yao, Z. Liang, H. Cheng, Z. Ren, W. Liu, R. Lin, G. Qi, X. Liu, Q. Wu, J. Luo and H. L. Xin, J. Am. Chem. Soc., 2020, 142, 12563–12567 CrossRef CAS PubMed.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W.-C. Cheong, Y. Wang, L. Zheng, J. Luo, Y. Lin, Y. Liu, C. Liu, J. Li, Q. Lu, X. Chen, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed.
- X. Hu, G. Luo, Q. Zhao, D. Wu, T. Yang, J. Wen, R. Wang, C. Xu and N. Hu, J. Am. Chem. Soc., 2020, 142, 16776–16786 CrossRef CAS PubMed.
- J. Li, L. Jiao, E. Wegener, L. L. Richard, E. Liu, A. Zitolo, M. T. Sougrati, S. Mukerjee, Z. Zhao, Y. Huang, F. Yang, S. Zhong, H. Xu, A. J. Kropf, F. Jaouen, D. J. Myers and Q. Jia, J. Am. Chem. Soc., 2020, 142, 1417–1423 CrossRef CAS PubMed.
- L. Zhang, A. Wang, W. Wang, Y. Huang, X. Liu, S. Miao, J. Liu and T. Zhang, ACS Catal., 2015, 5, 6563–6572 CrossRef CAS.
- S. Chen, Y. Li, Z. Bu, F. Yang, J. Luo, Q. An, Z. Zeng, J. Wang and S. Deng, J. Mater. Chem. A, 2021, 9, 1705–1712 RSC.
- D. Deng, X. Chen, L. Yu, X. Wu, Q. Liu, Y. Liu, H. Yang, H. Tian, Y. Hu, P. Du, R. Si, J. Wang, X. Cui, H. Li, J. Xiao, T. Xu, J. Deng, F. Yang, P. N. Duchesne, P. Zhang, J. Zhou, L. Sun, J. Li, X. Pan and X. Bao, Sci. Adv., 2015, 1, e1500462 CrossRef PubMed.
- H. Wu, H. Li, X. Zhao, Q. Liu, J. Wang, J. Xiao, S. Xie, R. Si, F. Yang, S. Miao, X. Guo, G. Wang and X. Bao, Energy Environ. Sci., 2016, 9, 3736–3745 RSC.
- Y. Chen, R. Gao, S. Ji, J. Li, K. Tang, P. Jiang, H. Hu, Z. Zhang, H. Hao, Q. Qu, X. Liang, W. Chen, J. Dong, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 3212–3221 CrossRef CAS PubMed.
- Y. Pan, Y. Chen, K. Wu, Z. Chen, S. Liu, X. Cao, W.-C. Cheong, T. Meng, J. Luo, L. Zheng, C. Liu, D. Wang, Q. Peng, J. Li and C. Chen, Nat. Commun., 2019, 10, 4290 CrossRef PubMed.
- Z. Geng, Y. Cao, W. Chen, X. Kong, Y. Liu, T. Yao and Y. Lin, Appl. Catal., B, 2019, 240, 234–240 CrossRef CAS.
- T. Sun, S. Mitchell, J. Li, P. Lyu, X. Wu, J. Perez-Ramirez and J. Lu, Adv. Mater., 2021, 33, e2003075 CrossRef PubMed.
- H. Zhang, J. Li, S. Xi, Y. Du, X. Hai, J. Wang, H. Xu, G. Wu, J. Zhang, J. Lu and J. Wang, Angew. Chem., Int. Ed., 2019, 58, 14871–14876 CrossRef CAS PubMed.
- Y. Zhang, L. Jiao, W. Yang, C. Xie and H.-L. Jiang, Angew. Chem., Int. Ed., 2021, 60, 7607–7611 CrossRef CAS PubMed.
- Y. Wang, G. Jia, X. Cui, X. Zhao, Q. Zhang, L. Gu, L. Zheng, L. H. Li, Q. Wu, D. J. Singh, D. Matsumura, T. Tsuji, Y.-T. Cui, J. Zhao and W. Zheng, Chem, 2021, 7, 436–449 CAS.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang, W. Huang, J. Yang, X. Hong, S. Wei, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS PubMed.
- A. Henrick, Tetrahedron, 1977, 33, 1845–1889 CrossRef.
- G. C. Tron, T. Pirali, G. Sorba, F. Pagliai, S. Busacca and A. A. Genazzani, J. Med. Chem., 2006, 49, 3033–3044 CrossRef CAS PubMed.
- T. Brown, H. Holt Jr and M. Lee, in Heterocyclic Antitumor Antibiotics, ed. M. Lee, Springer Berlin Heidelberg, Berlin, Heidelberg, 2006, pp. 1–51, DOI: DOI:10.1007/7081_003.
- G. M. Richardson, I. Douair, S. A. Cameron, J. Bracegirdle, R. A. Keyzers, M. S. Hill, L. Maron and M. D. Anker, Nat. Commun., 2021, 12, 3147 CrossRef CAS PubMed.
- X. Lu, B. Xiao, Z. Zhang, T. Gong, W. Su, J. Yi, Y. Fu and L. Liu, Nat. Commun., 2016, 7, 11129 CrossRef PubMed.
- K. Dong, X. Fang, S. Gülak, R. Franke, A. Spannenberg, H. Neumann, R. Jackstell and M. Beller, Nat. Commun., 2017, 8, 14117 CrossRef PubMed.
- T. Mitsudome, M. Yamamoto, Z. Maeno, T. Mizugaki, K. Jitsukawa and K. Kaneda, J. Am. Chem. Soc., 2015, 137, 13452–13455 CrossRef CAS PubMed.
- S. P. Desai, J. Ye, J. Zheng, M. S. Ferrandon, T. E. Webber, A. E. Platero-Prats, J. Duan, P. Garcia-Holley, D. M. Camaioni, K. W. Chapman, M. Delferro, O. K. Farha, J. L. Fulton, L. Gagliardi, J. A. Lercher, R. L. Penn, A. Stein and C. C. Lu, J. Am. Chem. Soc., 2018, 140, 15309–15318 CrossRef CAS PubMed.
- S. Wang, Z. Xin, X. Huang, W. Yu, S. Niu and L. Shao, Phys. Chem. Chem. Phys., 2017, 19, 6164–6168 RSC.
- R. Guo, Q. Chen, X. Li, Y. Liu, C. Wang, W. Bi, C. Zhao, y. Guo and M. Jin, J. Mater. Chem. A, 2019, 7, 4714–4720 RSC.
- J. Hori, K. Murata, T. Sugai, H. Shinohara, R. Noyori, N. Arai, N. Kurono and T. Ohkuma, Adv. Synth. Catal., 2009, 351, 3143–3149 CrossRef CAS.
- X. Wen, X. Shi, X. Qiao, Z. Wu and G. Bai, Chem. Commun., 2017, 53, 5372–5375 RSC.
- V. Polshettiwar, B. Baruwati and R. S. Varma, Green Chem., 2009, 11, 127–131 RSC.
- S. Fu, N. Y. Chen, X. Liu, Z. Shao, S. P. Luo and Q. Liu, J. Am. Chem. Soc., 2016, 138, 8588–8594 CrossRef CAS PubMed.
- S. Mou, Y. Lu and Y. Jiang, Appl. Surf. Sci., 2016, 384, 258–262 CrossRef CAS.
- M. Abdel-Mageed, B. Rungtaweevoranit, M. Parlinska-Wojtan, X. Pei, O. M. Yaghi and R. J. Behm, J. Am. Chem. Soc., 2019, 141, 5201–5210 CrossRef PubMed.
- F. Li, G.-F. Han, H.-J. Noh, S.-J. Kim, Y. Lu, H. Y. Jeong, Z. Fu and J.-B. Baek, Energy Environ. Sci., 2018, 11, 2263–2269 RSC.
- P. Ren, Q. Li, T. Song and Y. Yang, ACS Appl. Mat. Interfaces, 2020, 12, 27210–27218 CrossRef CAS PubMed.
- E. Crusson-Blouet, A. Aboukais, C. F. Aissi and M. Guelton, Chem. Mater., 1992, 4, 1129–1131 CrossRef CAS.
- T. Zhang, D. Zhang, X. Han, T. Dong, X. Guo, C. Song, R. Si, W. Liu, Y. Liu and Z. Zhao, J. Am. Chem. Soc., 2018, 140, 16936–16940 CrossRef CAS PubMed.
- R. Kydd, W. Y. Teoh, K. Wong, Y. Wang, J. Scott, Q.-H. Zeng, A.-B. Yu, J. Zou and R. Amal, Adv. Funct. Mater., 2009, 19, 369–377 CrossRef CAS.
- M. Tong, F. Sun, Y. Xie, Y. Wang, Y. Yang, C. Tian, L. Wang and H. Fu, Angew. Chem., Int. Ed., 2021, 60, 14005–14012 CrossRef CAS PubMed.
- C. Huang, L. Zheng, W. Feng, A. Guo, X. Gao, Z. Long and X. Qiu, ACS Sustainable Chem. Eng., 2020, 8, 14030–14038 CrossRef CAS.
- X. Lu, S. Gao, H. Lin, L. Yu, Y. Han, P. Zhu, W. Bao, H. Yao, Y. Chen and J. Shi, Adv. Mater., 2020, 32, 2002246 CrossRef CAS PubMed.
- G. Jaiswal, V. G. Landge, M. Subaramanian, R. G. Kadam, R. Zbořil, M. B. Gawande and E. Balaraman, ACS Sustainable Chem. Eng., 2020, 8, 11058–11068 CrossRef CAS.
- V. R. Bakuru, D. Samanta, T. K. Maji and S. B. Kalidindi, Dalton Trans., 2020, 49, 5024–5028 RSC.
- R. Prins, Chem. Rev., 2012, 112, 2714–2738 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc04344g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |