
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic asymmetric transformations of racemic α-borylmethyl-(E)-crotylboronate via kinetic resolution or enantioconvergent reaction pathways†
Shang
Gao‡
 ,
Jiaming
Liu‡
and
Ming
Chen
*
,
Jiaming
Liu‡
and
Ming
Chen
*
Department of Chemistry and Biochemistry, Auburn University, Auburn, AL 36849, USA. E-mail: mzc0102@auburn.edu
First published on 20th September 2021
Abstract
We report herein catalytic asymmetric transformations of racemic α-borylmethyl-(E)-crotylboronate. The Brønsted acid-catalyzed kinetic resolution–allylboration reaction sequence of the racemic reagent gave (Z)-δ-hydroxymethyl-anti-homoallylic alcohols with high Z-selectivities and enantioselectivities upon oxidative workup. In parallel, enantioconvergent pathways were utilized to synthesize chiral nonracemic 1,5-diols and α,β-unsaturated aldehydes with excellent optical purity.
Introduction
Enantioenriched molecules are indispensable components in organic chemistry and modern drug discovery.1 In the past twenty years, asymmetric catalysis has been the most adopted approach to synthesize such compounds.2 Other strategies, however, also play important roles in different research settings. For instance, by taking advantage of the different reaction rates of each enantiomer of a racemate with a chiral, nonracemic reagent or catalyst, kinetic resolution enables access to a variety of highly enantiomerically enriched molecules.3 On the other hand, enantioconvergent processes operate in a way such that both enantiomers of the racemate are converted into the same enantiomer of the product.4(Z)-2-Methyl-3-pentene-1,5-diols and their reduced forms (highlighted in red and blue in Fig. 1) are common structural motifs in numerous bioactive natural products.5 Methods that allows for the access to such structural entities mainly rely on a multistep reaction sequence.6 For example, in the synthesis of a fragment of discodermolide reported by the Cossy group, enantioenriched homoallylic alcohol I was converted into lactone II in two steps. DIBAL reduction of II gave a lactol intermediate, which was reduced by NaBH4 to give (Z)-2-methyl-3-pentene-1,5-diol III (Scheme 1a).6a In the total synthesis of dictyostatin, Curran and co-workers transformed chiral nonracemic propargylic mesylate IV into (Z)-vinyl iodide V in four steps. Li-halogen exchange of V and addition of the resulting vinyl lithium to an aldehyde gave VI as a mixture of two diastereomers (Scheme 1b).6b While these methods can deliver the desired alcohol products with a meaningful quantity, streamlined synthesis of these molecules via catalytic asymmetric transformations would also be valuable. As part of our ongoing research program in acyclic stereochemical control via novel organoboron compounds,7 we describe herein asymmetric synthesis of (Z)-2-methyl-3-pentene-1,5-diols 4via chiral phosphoric acid-catalyzed kinetic resolution–allylation using racemic α-borylmethyl-(E)-crotylboronate (±)-3 (Scheme 1). Moreover, enantioconvergent syntheses of diols 6 and aldehydes 9 were accomplished from the same racemic boron reagent (±)-3, where both enantiomers of the racemate were converted into 6 or 9 with high enantioselectivities.
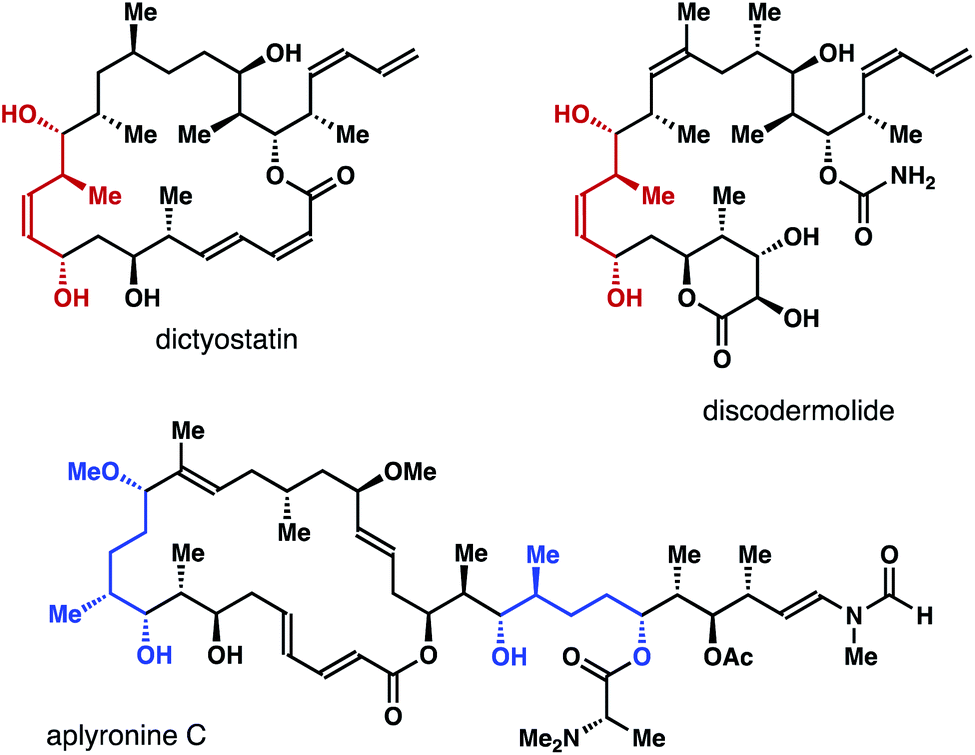 | ||
| Fig. 1 Selected natural products that contain (Z)-2-methyl-3-pentene-1,5-diols or their reduced forms. | ||
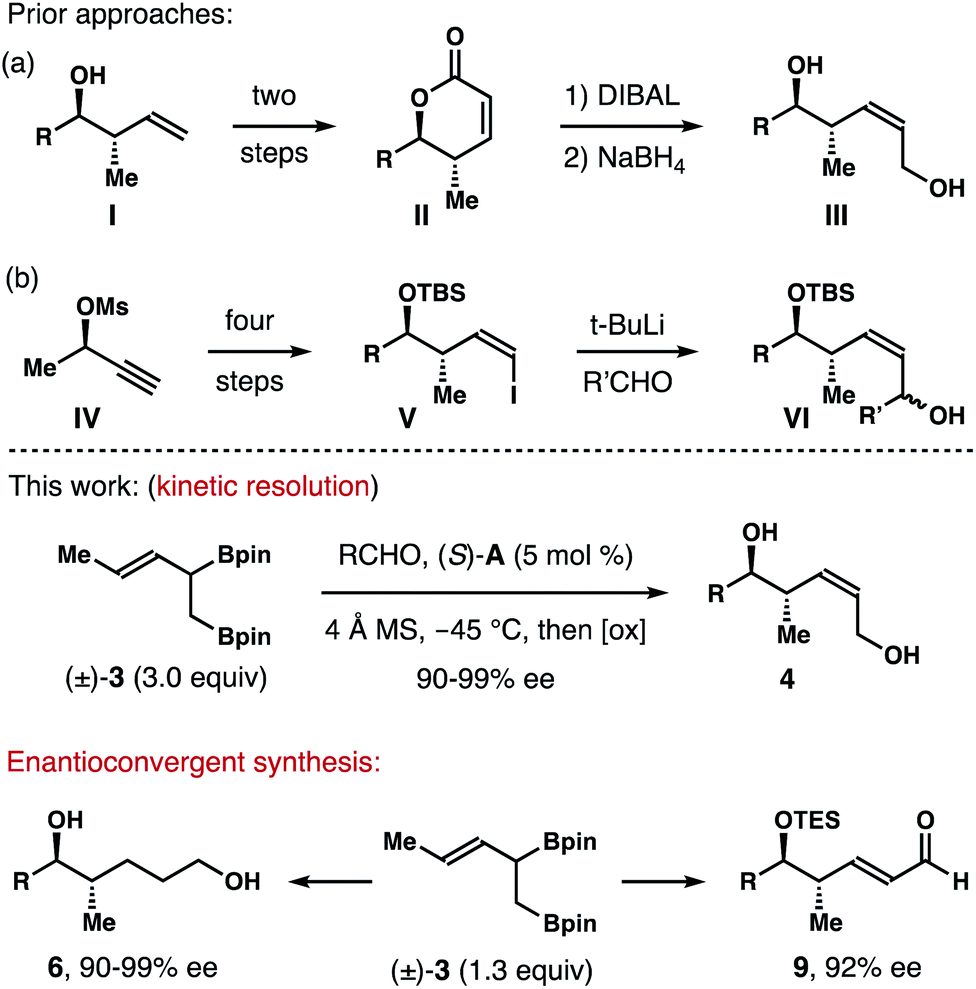 | ||
| Scheme 1 Approaches to (Z)-2-methyl-3-pentene-1,5-diols. | ||
Results and discussion
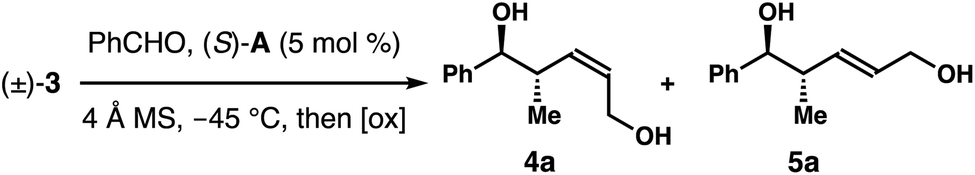
To evaluate the feasibility of proposed kinetic resolution approach to synthesize (Z)-2-methyl-3-pentene-1,5-diols 4, we obtained racemic boronate (±)-3 according to the reported protocols.8 In the absence of any catalyst, the reaction of (±)-3 with benzaldehyde gave a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of two products upon oxidative workup, favouring the Z-isomer, (±)-4a. The results indicate the pseudo axial preference (3:1) of the α-CH2Bpin group of reagent (±)-3 in the reaction transition state. On the basis of recent studies on Z-selective allylboration catalyzed by chiral phosphoric acids,9,10 we suspect that the Z-selectivity could be enhanced by using a proper phosphoric acid catalyst. However, a more pertinent question is whether the catalyst could differentiate the two enantiomers of racemate (±)-3 in reactions with aldehydes such that Z-isomer 4 could be obtained with high enantioselectivities.
:1 mixture of two products upon oxidative workup, favouring the Z-isomer, (±)-4a. The results indicate the pseudo axial preference (3:1) of the α-CH2Bpin group of reagent (±)-3 in the reaction transition state. On the basis of recent studies on Z-selective allylboration catalyzed by chiral phosphoric acids,9,10 we suspect that the Z-selectivity could be enhanced by using a proper phosphoric acid catalyst. However, a more pertinent question is whether the catalyst could differentiate the two enantiomers of racemate (±)-3 in reactions with aldehydes such that Z-isomer 4 could be obtained with high enantioselectivities.
To gather more experimental data to answer this question, we prepared enantioenriched boron reagents (S)-3 and (R)-3 (94% ee, see ESI† for detailed procedure). And allylation studies with the single enantiomer reagents in the presence of chiral phosphoric acid catalyst (S)-A were conducted. As shown in Scheme 2, the reaction between (S)-3 (1.2 equiv.) and benzaldehyde with 5 mol% (S)-A as the catalyst gave product 4a in 89% yield with >50:1 Z-selectivity and >99% ee. Intriguingly, the reaction with the enantiomeric reagent (R)-3 in the presence of the same catalyst (S)-A generated E-isomer 5a in 83% yield with 15:1 E-selectivity and >99% ee. The data described here bear several important implications. First, it is apparent that the two enantiomers of 3, (S)-3 and (R)-3, reacted with benzaldehyde in the presence of the same acid catalyst (S)-A formed products 4a and 5a that are not enantiomers, which is reminiscent of pathways for enantiodivergent reactions. Second, both 4a and 5a have the same relative and absolute configurations at the stereocenters with a hydroxyl group and a methyl group, only differing in the olefin geometry. Lastly, perhaps the most significant aspect, the enantiomeric excesses of 4a and 5a are amplified (>99% ee vs. 94% ee of (S)-3 and (R)-3). It is well-established that the reactions of aldehydes with chiral nonracemic α-substituted crotylboronates proceed via chirality transfer; and the optical purity of the starting allylboron reagents will be retained in the reaction products. In another word, we would expect 94% ee for products 4a and 5a, given the enantiomeric excess of starting allylboron reagents (S)-3 and (R)-3 is 94% ee. The amplification of enantiopurity and high Z-selectivity in the reaction with (S)-3 indicate that the minor component (R)-3 in the starting boron reagent (3% based on 94% ee of (S)-3) likely did not participate in the reaction with benzaldehyde. Otherwise, formation of the E-isomer 5a would be observed. With (S)-A as the catalyst, only (S)-3 (97% based on 94% ee of (S)-3) reacted with benzaldehyde to deliver 4a with outstanding Z-selectivity and enantioselectivity. On the other hand, in the reaction of (R)-3 with the same acid (S)-A as the catalyst, the selectivity (15:1) is inferior to the one from the reaction with (S)-3 (>50:1). Therefore, we infer that the reaction of (S)-3 with aldehydes in the presence of catalyst (S)-A is stereochemically matched. The reaction with the (R)-3/(S)-A pairing is likely mismatched. These results form the basis of a potential kinetic resolution of racemic reagent (±)-3 using single enantiomer acid catalyst (S)-A and would eliminate the need to synthesize enantioenriched boron reagents (e.g. (S)-3) for use in allylboration reactions.
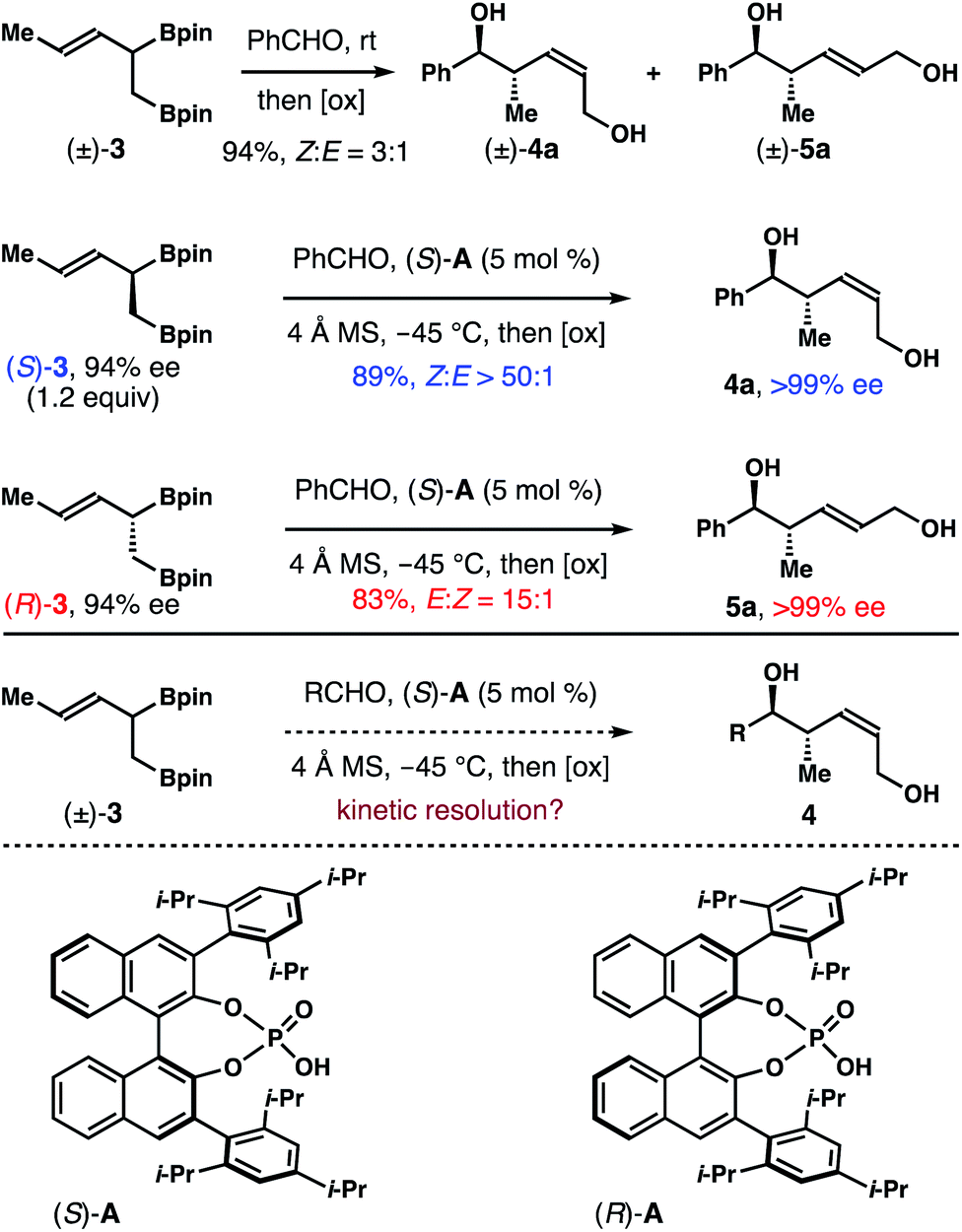 | ||
| Scheme 2 Initial studies with single enantiomer reagents. | ||
To identify the optimal conditions for the kinetic resolution process, the reactions of benzaldehyde with various amounts of reagent (±)-3 in the presence of 5 mol% catalyst (S)-A were conducted. As shown in Table 1, the reaction with 1.3 equiv. of reagent (±)-3 formed a 3:1 mixture of 4a and 5a in a combined 89% yield. The enantiomeric excess of 4a is 96% ee, and 99% ee for 5a. Excellent enantiopurity was also observed for the recovered reagent 3. With the aim to improve the Z-selectivity, we increased the amount of starting reagent (±)-3. When 2 equiv. of (±)-3 was used, the Z-selectivity was enhanced to 5:1 (entry 2). Further improvement of the Z-selectivity (7:1) was observed with 2.5 equiv. of reagent (±)-3 (entry 3). Finally, 10:1 Z-selectivity was achieved when 3.0 equiv. of (±)-3 was used in the reaction (entry 4). In all cases, enantiopurities of both 4a and 5a remained excellent; the optical purity of recovered reagent 3 decreased as anticipated (96% to 28% ee).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Equiv. (3) |
4a:5a |
ee (4a/5a) | ee (recovered 3) | Yield (4a + 5a) |
| a Reaction conditions: allylboronate (±)-3, (S)-A (5 mol%), PhCHO (0.1 mmol, 1.0 equiv.), toluene (0.3 mL), −45 °C, 12 h. b The ratios of 4a and 5a were determined by 1H NMR analysis of the crude reaction products. c Yields of isolated products are listed. d Enantiomeric excesses were determined by HPLC analysis. e Yield of 4a is listed. | |||||
| 1 | 1.3 | 3:1 |
96/99 | 96 | 89 |
| 2 | 2.0 | 5:1 |
98/99 | 58 | 94 |
| 3 | 2.5 | 7:1 |
98/99 | 39 | 90 |
| 4 | 3.0 | 10:1 |
99/99 | 28 | 83e |
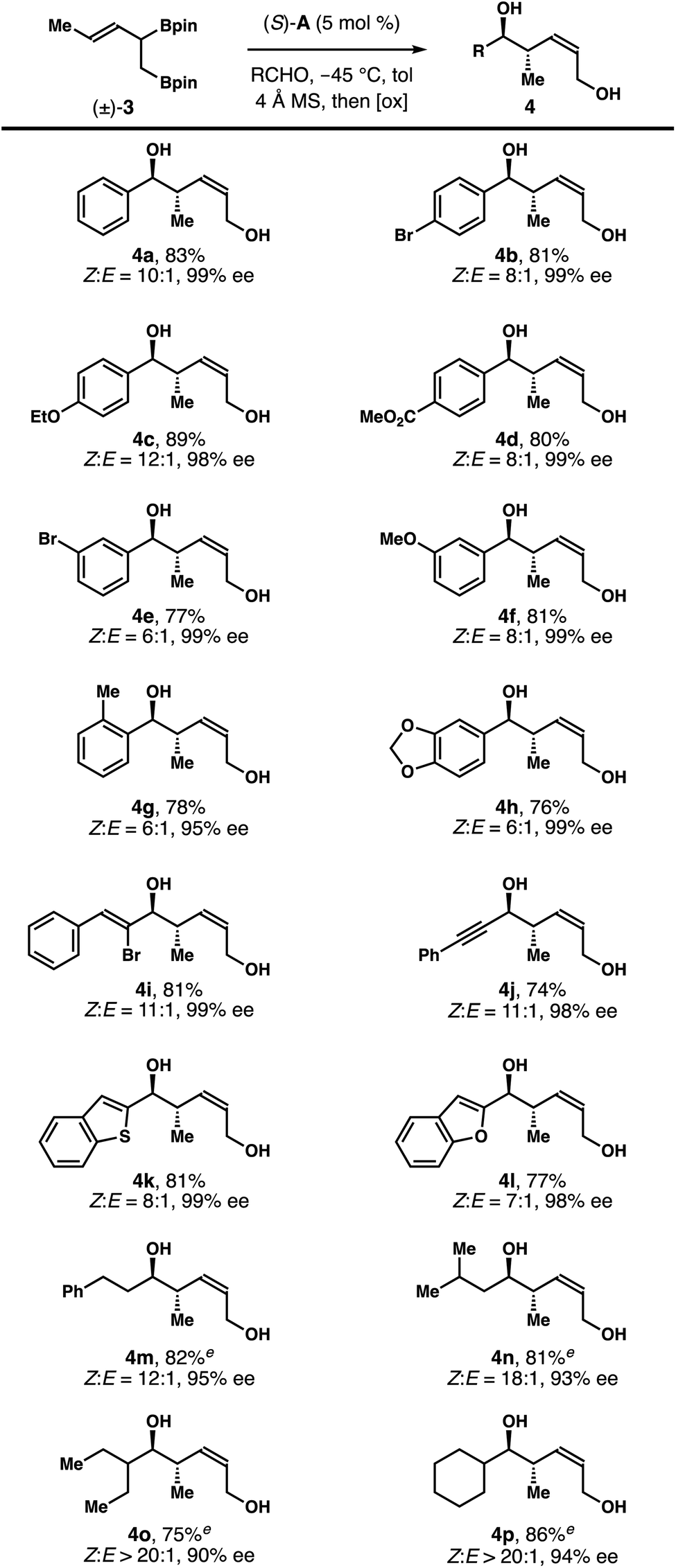
Although further enhancement of the Z-selectivity might be achievable, we decided to explore the reaction scope with 3.0 equiv. of (±)-3 to balance the amount of (±)-3 used in the reaction and the Z-selectivity of products 4. As summarized in Table 2, under the developed conditions, the reactions worked reasonably well for a broad range of aldehyde substrates. For instance, reactions of (±)-3 with aromatic aldehydes with a substituent at the para-position gave products 4b–4d with (8–12):1 Z-selectivity and 98–99% ee. Reactions of aromatic aldehydes with other substitution patterns proceeded to generate diols 4e–4h with (6–8):1 Z-selectivity and 95–99% ee. Reactions with α,β-unsaturated aldehydes occurred smoothly to form products 4i and 4j with 11:1 Z-selectivity and 98–99% ee. Heteroaromatic aldehydes are tolerated under the reaction conditions, affording diols 4k and 4l with (8 and 7):1 Z-selectivity and 98–99% ee. Importantly, aliphatic aldehydes also participated in the reactions with (±)-3 to deliver products 4m–4p with (12–20):1 Z-selectivity and 90–95% ee. It should be noted that the reactions with aliphatic aldehydes were slow with 5 mol% of the acid catalyst. Optimal reactions rates and selectivities were achieved by increasing the catalyst loading to 10 mol%.
| a Reaction conditions: allylboronate (±)-3 (0.3 mmol, 3.0 equiv.), aldehyde (0.1 mmol, 1.0 equiv.), (S)-A (5 mol%), 4 Å molecular sieves, toluene, −45 °C; then NaOH, H2O2, 0 °C. b Z/E-selectivities were determined by 1H NMR analysis of the crude reaction products. c Yields of isolated products 4 are listed. d Enantiopurities of 4 were determined by HPLC analysis. e The reactions were conducted with 10 mol% of (S)-A. |
|---|
|
As shown in Table 1, both products 4a and 5a generated from racemic boron reagent (±)-3 have identical relative and absolute configuration at the stereocenters bearing a hydroxyl group and a methyl group. The only difference is the geometry of the alkene unit. The results imply that the chiral phosphoric acid catalyst (S)-A controls the face selective addition to the aldehyde substrates, regardless of the absolute configuration of reagent 3, (R) or (S). The Z or E olefin geometry of 4a or 5a reflects the pseudo axial or equatorial orientation of the α-CH2Bpin group in the reaction transition states. As shown in Fig. 1, the reduced form (e.g.6, Scheme 3) of 2-methyl-3-pentene-1,5-diol is also a prevalent structural motif in a myriad of biologically relevant molecules. We envisioned that reduction of the alkene units of diols 4a and 5a should converge both compounds into the exact same product 6a. Therefore, the overall reaction could proceed through an enantioconvergent pathway such that both enantiomers of racemic boron reagent (±)-3 can be utilized to form enantioenriched 1,5-diol products 6, and large excess of reagent (±)-3 will not be required as it is in the kinetic resolution pathway.
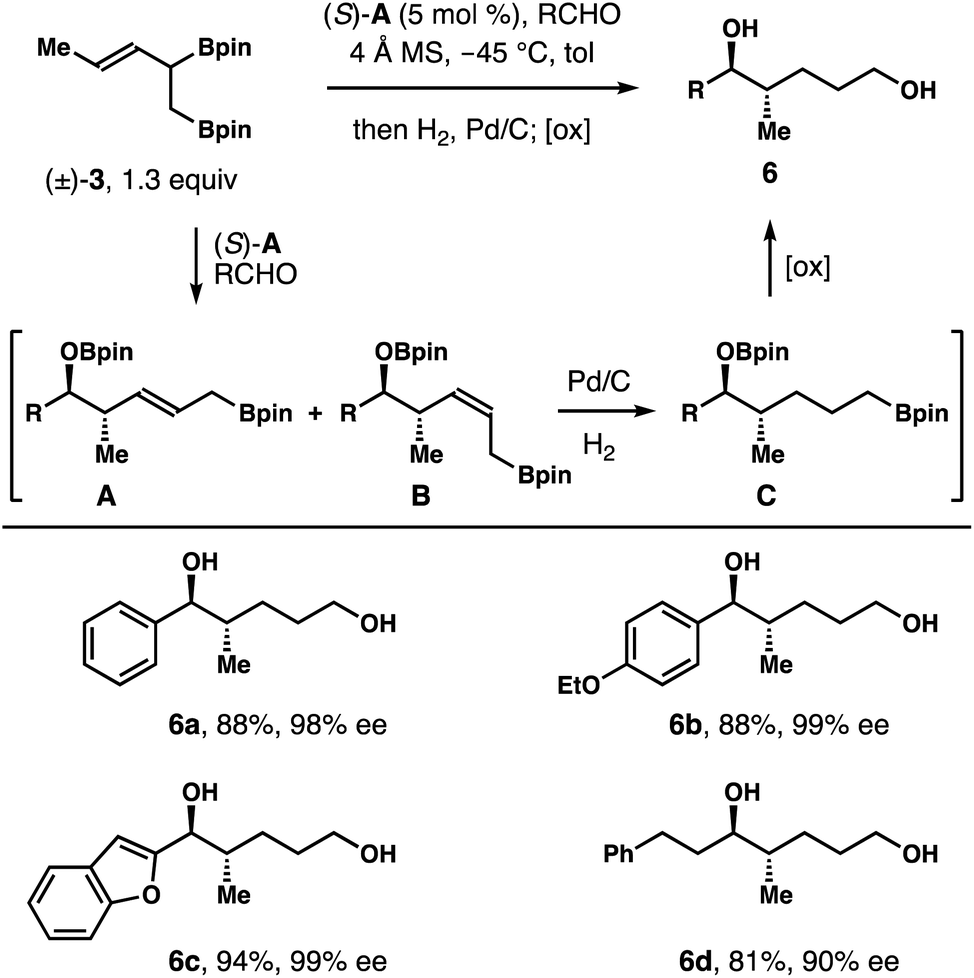 | ||
| Scheme 3 Enantioconvergent synthesis of diols 6. | ||
To validate our hypothesis, reactions of several aldehydes with reagent (±)-3 (1.3 equiv.) were conducted in the presence of the catalyst (S)-A. After completion of the allylation, the resulting mixture, presumably intermediates A and B (Scheme 3), was subjected to the standard hydrogenation conditions in the same reaction vessel. Upon full reduction of the alkene unit, the resulting intermediate (e.g., C) was treated with oxidative workup conditions (NaOH, H2O2) to give diol product 6. As summarized in Scheme 3, several representative aldehydes, including aromatic, heteroaromatic and aliphatic aldehydes, reacted under these conditions to give diols 6a–6d in 81–94% yields with 90–99% ee. In the case of 6d, 10 mol% of catalyst (S)-A was employed for an optimal reaction rate. This process is highly valuable because only a slight excess of racemic reagent (±)-3 is required to generate highly enantioenriched diol products 6 and only a catalytic amount of chiral phosphoric acid catalyst (S)-A is needed as the source of asymmetric induction for the reactions.
As shown in Scheme 4, an enantioconvergent synthesis of aldehyde 9 was also developed. The racemic boron reagent (±)-3 (1.3 equiv.) was treated with hydrocinnamic aldehyde under the standard asymmetric allylation conditions first. After completion of the reaction, the secondary alcohol group was protected in situ as a TES ether. Final oxidative workup gave a 3:1 mixture of allylic alcohols 7 and 8 in a combined 72% yield. Dess–Martin oxidation of the mixture of 7 and 8 followed by treatment of the resulting crude aldehydes with DMAP gave aldehyde 9 in 78% yield with 92% ee. The aldehyde group in 9 can now serve as a handle for further chain elongation reactions. For example, chiral phosphoric acid (S)-A-catalyzed asymmetric crotylation of aldehyde 9 with crotylboron reagent 10 gave alcohol 11 in 87% yield with >20:1 dr.11,12 When acid (R)-A was employed as the catalyst, the reaction of 9 with reagent 10 formed alcohol 12 in 77% yield again with >20:1 dr.
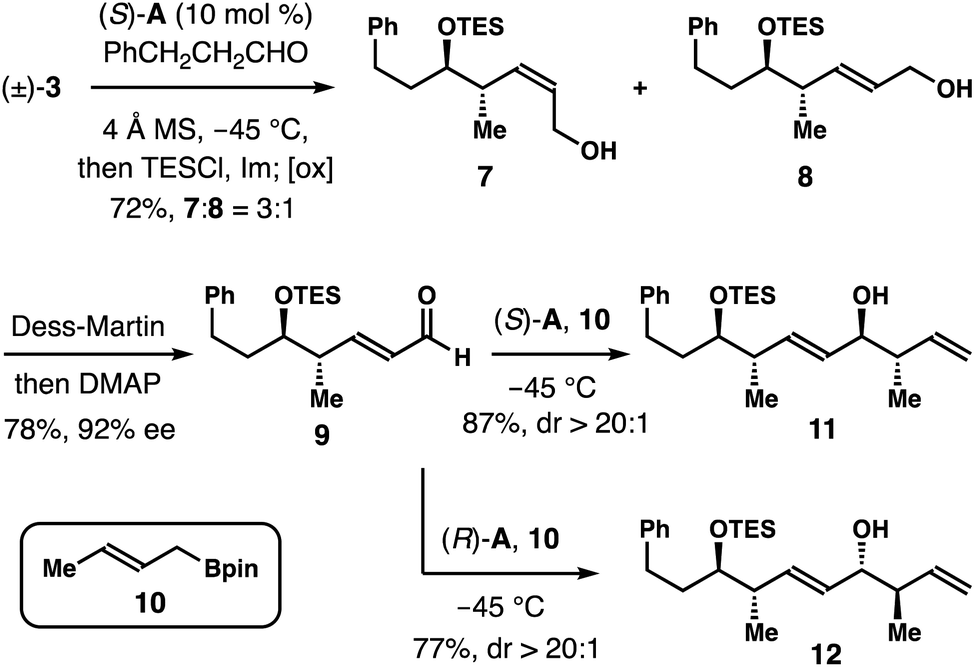 | ||
| Scheme 4 Enantioconvergent synthesis of aldehyde 9. | ||
We and other groups have documented that chiral biaryl phosphoric acid-catalyzed reactions of α-substituted achiral allylboronates with aldehydes form homoallylic alcohols with excellent Z-selectivity. And the origins of observed Z-selectivity were further explored by extensive computational studies.10 These reports provide foundation for us to rationalize the Z-selectivity in chiral phosphoric acid (S)-A-catalyzed reactions with boronate (±)-3 (Scheme 5). In Scheme 2, we showed that the reaction of an aldehyde with the (S)-enantiomer of (±)-3 is stereochemically matched when acid catalyst (S)-A is used. Therefore, the reaction of (R)-enantiomer of (±)-3, (R)-3, with (S)-A as the catalyst is disfavoured under the kinetic resolution pathway. In the reaction of (S)-3 and an aldehyde substrate with (S)-A as the catalyst, two competing transition states, TS-1 and TS-2, will lead to the Z-product 4 and E-product 5 respectively. As depicted in Scheme 5, TS-2 involves the addition to the re-face of the aldehyde substrate. This mode of addition is opposite to the sense of asymmetric induction of the acid catalyst, (S)-A.9 Consequently, unfavourable steric interactions between the pinacol group on boron and the aromatic moiety of the acid catalyst will be developed (shown with a red arrow in TS-2). In addition, the pseudo equatorially positioned CH2Bpin group in TS-2 will have gauche interactions with the pinacol group on boron, which further destabilizes TS-2. By contrast, the si-face addition to the aldehyde substrate in TS-1 is consistent with the sense of asymmetric induction of catalyst (S)-A, which eliminates the steric interactions between the pinacol group on boron and the catalyst. Moreover, the CH2Bpin group is oriented in a pseudo axial position. And the unfavourable gauche steric interactions are absent in TS-1. Therefore, the reaction proceeded via the favoured transition state TS-1 to give Z-isomer 4 as the major product.
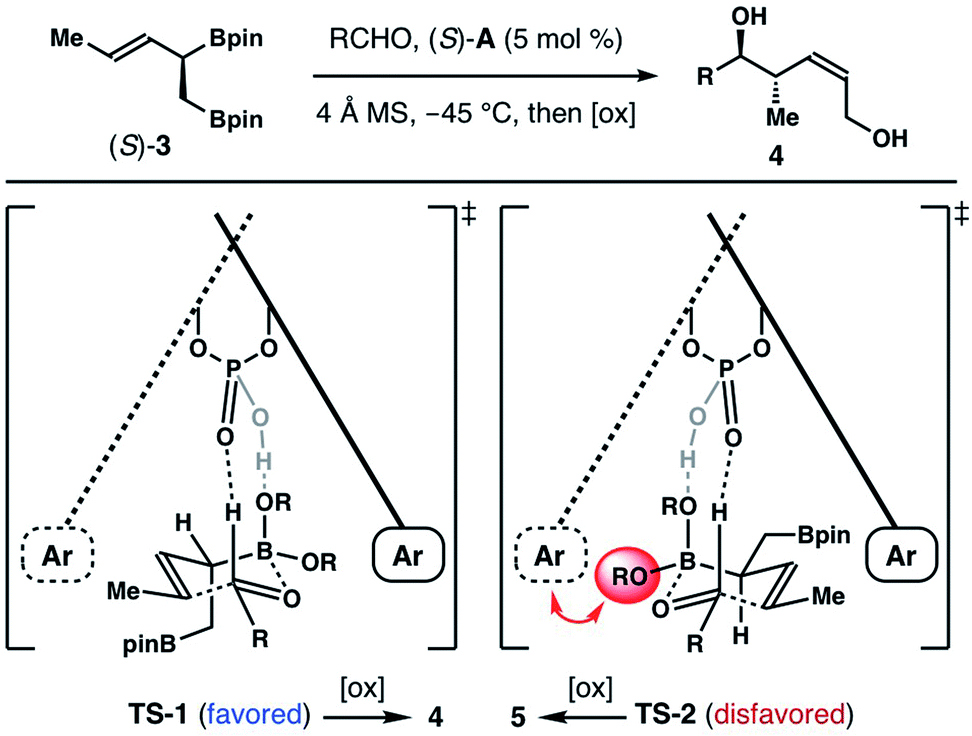 | ||
| Scheme 5 Rationale for the observed Z-selectivity. | ||
Conclusions
In summary, we developed asymmetric transformations of racemic α-borylmethyl-(E)-crotylboronate (±)-3via either a kinetic resolution or enantioconvergent reaction pathways. In the presence of a catalytic amount of chiral phosphoric acid (S)-A, kinetic resolution–allylation of reagent (±)-3 produces (Z)-2-methyl-3-pentene-1,5-diols 4 with high Z-selectivities and excellent enantioselectivities. In the enantioconvergent reaction manifold, both enantiomers of racemic reagent (±)-3 were converted into highly enantioenriched 1,5-diols 6 or aldehyde 9. Importantly, these compounds can serve as synthetically useful intermediates for stereochemically complex natural product synthesis. It is worth noting that the enantioconvergent processes do not demand a large excess of racemic reagent (±)-3; and only a catalytic amount of chiral phosphoric acid (S)-A is required as the source of asymmetric induction to obtain products with excellent optical purities. Synthetic applications of this method will be reported in due course.Data availability
All the data have been included in the ESI.†Author contributions
Shang Gao and Jiaming Liu contributed equally to the manuscript by conducting experiments in Tables 1, 2, and Schemes 3 and 4.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support provided by Auburn University and National Science Foundation (CAREER Award CHE-2042353) is gratefully acknowledged.References
- (a) J. M. Hawkins and T. J. N. Watson, Angew. Chem., Int. Ed., 2004, 43, 3224 CrossRef CAS PubMed; (b) V. Farina, J. T. Reeves, C. H. Senanayake and J. J. Song, Chem. Rev., 2006, 106, 2734 CrossRef CAS PubMed.
- (a) E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Comprehensive Asymmetric Catalysis, Springer, New York, 1999, vol. I–III, suppl. I–II Search PubMed; (b) I. Ojima, Catalytic Asymmetric Synthesis, Wiley VCH, New York, 2nd edn, 2000 CrossRef; (c) E. M. Carreira and H. Yamamoto, Comprehensive Chirality, Elsevier Science, Amsterdam, 2012, vol. 1–9 Search PubMed.
- For selected reviews on kinetic resolution: (a) H. B. Kagan and J. C. Fiaud, in Topics in Stereochemistry, ed. A. L. Allinger and E. Eliel, Wiley, New York, 1988, vol. 18, p. 249 Search PubMed; (b) E. Vedejs and M. Jure, Angew. Chem., Int. Ed., 2005, 44, 3974 CrossRef CAS PubMed.
- (a) S. L. Bartlett and J. S. Johnson, Acc. Chem. Res., 2017, 50, 2284 CrossRef CAS PubMed; (b) H. Zhou, Z.-L. Li, Q.-S. Gu and X.-Y. Liu, ACS Catal., 2021, 11, 7978 CrossRef CAS . For selected recent examples: ; (c) M. Chen and W. R. Roush, J. Am. Chem. Soc., 2011, 133, 5744 CrossRef CAS PubMed; (d) M. Terada, Y. Ota, F. Li, Y. Toda and A. Kondoh, J. Am. Chem. Soc., 2016, 138, 11038 CrossRef CAS PubMed; (e) A. Bartoszewicz, C. D. Matier and G. C. Fu, J. Am. Chem. Soc., 2019, 141, 14864 CrossRef CAS PubMed; (f) H. Yin and G. C. Fu, J. Am. Chem. Soc., 2019, 141, 15433 CrossRef CAS PubMed; (g) S. J. He, J. W. Wang, Y. Li, Z. Y. Xu, X. X. Wang, X. Lu and Y. Fu, J. Am. Chem. Soc., 2020, 142, 214 CrossRef CAS PubMed; (h) S.-P. Jiang, X.-Y. Dong, Q.-S. Gu, L. Ye, Z.-L. Li and X.-Y. Liu, J. Am. Chem. Soc., 2020, 142, 19652 CrossRef CAS PubMed; (i) H. Huo, B. J. Gorsline and G. C. Fu, Science, 2020, 367, 559 CrossRef CAS PubMed; (j) Z.-P. Yang, D. J. Freas and G. C. Fu, J. Am. Chem. Soc., 2021, 143, 2930 CrossRef CAS PubMed.
- For selected reviews: (a) J. Rohr, Angew. Chem., Int. Ed., 2000, 39, 2847 CrossRef CAS; (b) Macrolide Antibiotics: Chemistry, Biology, and Practice, ed. S. Omura, Academic Press, New York, 2nd edn, 2002 Search PubMed. For selected examples: (c) S. P. Gunasekera, M. Gunasekera and R. E. Longley, J. Org. Chem., 1990, 55, 4912 CrossRef CAS; (d) K. Yamada, M. Ojika, T. Ishigaki, Y. Yoshida, H. Ekimoto and M. Arakawa, J. Am. Chem. Soc., 1993, 115, 11020 CrossRef CAS; (e) G. R. Pettit, Z. A. Cichacz, F. Gao, M. R. Boyd and J. M. Schmidt, J. Chem. Soc., Chem. Commun., 1994, 30, 1111 RSC.
- (a) S. BouzBouz and J. Cossy, Org. Lett., 2003, 5, 3029 CrossRef CAS PubMed; (b) W. Zhu, M. Jiménez, W.-H. Jung, D. P. Camarco, R. Balachandran, A. Vogt, B. W. Day and D. P. Curran, J. Am. Chem. Soc., 2010, 132, 9175 CrossRef CAS PubMed; (c) H. Kigoshi, M. Ojika, T. Ishigaki, K. Suenaga, T. Mutou, A. Sakakura, T. Ogawa and K. Yamada, J. Am. Chem. Soc., 1994, 116, 7443 CrossRef CAS; (d) M. Kita, H. Oka, A. Usui, T. Ishitsuka, Y. Mogi, H. Watanabe, M. Tsunoda and H. Kigoshi, Angew. Chem., Int. Ed., 2015, 54, 14174 CrossRef CAS PubMed.
- (a) S. Gao, M. Wang and M. Chen, Org. Lett., 2018, 20, 7921 CrossRef CAS PubMed; (b) S. Gao, J. Chen and M. Chen, Chem. Sci., 2019, 10, 3637 RSC; (c) S. Gao and M. Chen, Chem. Sci., 2019, 10, 7554 RSC; (d) S. Gao and M. Chen, Chem. Commun., 2019, 55, 11199 RSC; (e) M. Wang, S. Gao and M. Chen, Org. Lett., 2019, 21, 2151 CrossRef CAS PubMed; (f) J. Chen, S. Gao, J. D. Gorden and M. Chen, Org. Lett., 2019, 21, 4638 CrossRef CAS PubMed; (g) J. Chen, S. Gao and M. Chen, Org. Lett., 2019, 21, 8800 CrossRef CAS PubMed; (h) J. Chen, S. Gao and M. Chen, Org. Lett., 2019, 21, 9893 CrossRef CAS PubMed; (i) J. Liu, S. Gao and M. Chen, Org. Process Res. Dev., 2019, 23, 1659 CrossRef CAS; (j) J. Liu, X. Tong and M. Chen, J. Org. Chem., 2020, 85, 5193 CrossRef CAS PubMed; (k) J. Chen, E. Miliordos and M. Chen, Angew. Chem., Int. Ed., 2021, 60, 840 CrossRef CAS PubMed; (l) J. Liu, B. Su and M. Chen, Org. Lett., 2021, 23, 6035 CrossRef CAS PubMed.
- (a) T. Ishiyama, M. Yamamoto and N. Miyaura, Chem. Commun., 1997, 33, 689 RSC; (b) E. Davenport and E. Fernández, Chem. Commun., 2018, 54, 10104 RSC.
- For selected reviews, see: (a) T. Akiyama, Chem. Rev., 2007, 107, 5744 CrossRef CAS PubMed; (b) M. Terada, Chem. Commun., 2008, 44, 4097 RSC; (c) T. Akiyama and K. Mori, Chem. Rev., 2015, 115, 9277 CrossRef CAS PubMed . For pioneering allylboration studies: ; (d) P. Jain and J. C. Antilla, J. Am. Chem. Soc., 2010, 132, 11884 CrossRef CAS PubMed . For computational studies: ; (e) M. N. Grayson, S. C. Pellegrinet and J. M. Goodman, J. Am. Chem. Soc., 2012, 134, 2716 CrossRef CAS PubMed; (f) H. Wang, P. Jain, J. C. Antilla and K. N. Houk, J. Org. Chem., 2013, 78, 1208 CrossRef CAS PubMed.
- (a) T. Miura, J. Nakahashi, W. Zhou, Y. Shiratori, S. G. Stewart and M. Murakami, J. Am. Chem. Soc., 2017, 139, 10903 CrossRef CAS PubMed; (b) S. Gao, M. Duan, K. N. Houk and M. Chen, Angew. Chem., Int. Ed., 2020, 59, 10540 CrossRef CAS PubMed; (c) S. Gao, M. Duan, Q. Shao, K. N. Houk and M. Chen, J. Am. Chem. Soc., 2020, 142, 18355 CrossRef CAS PubMed; (d) J. Chen and M. Chen, Org. Lett., 2020, 22, 7321 CrossRef CAS PubMed; (e) T. Miura, N. Oku, Y. Shiratori, Y. Nagata and M. Murakami, Chem.–Eur. J., 2021, 27, 3861 CrossRef CAS PubMed.
- (a) T. Miura, Y. Nishida, M. Morimoto and M. Murakami, J. Am. Chem. Soc., 2013, 135, 11497 CrossRef CAS PubMed; (b) C. A. Incerti-Pradillos, M. A. Kabeshov and A. V. Malkov, Angew. Chem., Int. Ed., 2013, 52, 5338 CrossRef CAS PubMed; (c) Y. Huang, X. Yang, Z. Lv, C. Cai, C. Kai, Y. Pei and Y. Feng, Angew. Chem., Int. Ed., 2015, 54, 7299 CrossRef CAS PubMed; (d) P. Barrio, E. Rodriguez, K. Saito, S. Fustero and T. Akiyama, Chem. Commun., 2015, 51, 5246 RSC; (e) T. Miura, J. Nakahashi and M. Murakami, Angew. Chem., Int. Ed., 2017, 56, 6989 CrossRef CAS PubMed; (f) S. Gao and M. Chen, Org. Lett., 2018, 20, 6174 CrossRef CAS PubMed; (g) B. E. Hetzler, G. Volpin, E. Vignoni, A. P. Petrovic, G. Proni, T. H. Chunhua and D. Trauner, Angew. Chem., Int. Ed., 2018, 57, 14276 CrossRef CAS PubMed; (h) T. Miura, N. Oku and M. Murakami, Angew. Chem., Int. Ed., 2019, 58, 14620 CrossRef CAS PubMed; (i) S. Gao and M. Chen, Org. Lett., 2020, 22, 400 CrossRef CAS PubMed; (j) J. Liu and M. Chen, Org. Lett., 2020, 22, 8967 CrossRef CAS PubMed; (k) J. Park, Y. Jung, J. Kim, E. Lee, S. Y. Lee and S. H. Cho, Adv. Synth. Catal., 2021, 363, 2371 CrossRef CAS.
- (a) P. Jain, H. Wang, K. N. Houk and J. C. Antilla, Angew. Chem., Int. Ed., 2012, 51, 1391 CrossRef CAS PubMed; (b) L. R. Reddy, Org. Lett., 2012, 14, 1142 CrossRef CAS PubMed; (c) M. Chen and W. R. Roush, J. Am. Chem. Soc., 2012, 134, 10947 CrossRef CAS PubMed; (d) A. S. Tsai, M. Chen and W. R. Roush, Org. Lett., 2013, 15, 1568 CrossRef CAS PubMed; (e) M. Wang, S. Khan, E. Miliordos and M. Chen, Org. Lett., 2018, 20, 3810 CrossRef CAS PubMed; (f) L. R. Reddy, Chem. Commun., 2012, 48, 9189 RSC; (g) M. Wang, S. Khan, E. Miliordos and M. Chen, Adv. Synth. Catal., 2018, 360, 4634 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc04047b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |