
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of an enolate alkynylation approach towards the synthesis of the taiwanschirin natural products†
Maxwell B.
Haughey
a,
Kirsten E.
Christensen‡
a,
Darren L.
Poole
 b and
Timothy J.
Donohoe
*a
b and
Timothy J.
Donohoe
*a
aDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: timothy.donohoe@chem.ox.ac.uk
bGlaxoSmithKline Medicines Research Centre, Stevenage, SG1 2NY, UK
First published on 20th September 2021
Abstract
Through the use of model studies, an approach was conceived towards the synthesis of the taiwanschirin family of natural products. These are structurally complex compounds which represent highly challenging and biologically active targets for total synthesis. This work describes a successful synthesis of the complex taiwanschirin fused [8,6,5] core through a novel alkynylation reaction coupled with an intramolecular Heck reaction used to construct the 8-membered ring.
In nature, pyruvic acids and their derivatives are crucial to a myriad of metabolic pathways, including the synthesis of amino acids.1 Seminal work by Krebs and Johnson disclosed the central role of pyruvic acid in the citric acid (Krebs) cycle, responsible for the supply of energy to living cells through the aerobic oxidation of acetyl-CoA.2 In stark contrast to the ease by which Nature uses pyruvates and alpha-keto acid derivatives, there are few mild synthetic methods available for the manipulation of pyruvates. Accordingly, a recent program of research within our group has led to the development of novel methodologies for the alpha-arylation of oxabicyclo[2.2.2]octyl (OBO) ortho-ester protected pyruvate equivalents.3 Work by Johnson has also developed a complementary method for the alpha-arylation of tert-butyl pyruvates.4 Subsequent chemistry was later realised to assemble a wide variety of alpha-acyl substituted heterocycles,3b resulting in the bioinspired syntheses of lamellarins D and Q in 7 steps from pyruvic acid.3c This chemistry demonstrated the application of the pyruvate alpha-arylation methodology in a more complex setting. Our continued interest in masked pyruvate equivalents led us to identify the taiwanschirin family of natural products as suitable targets for total synthesis (Fig. 1).
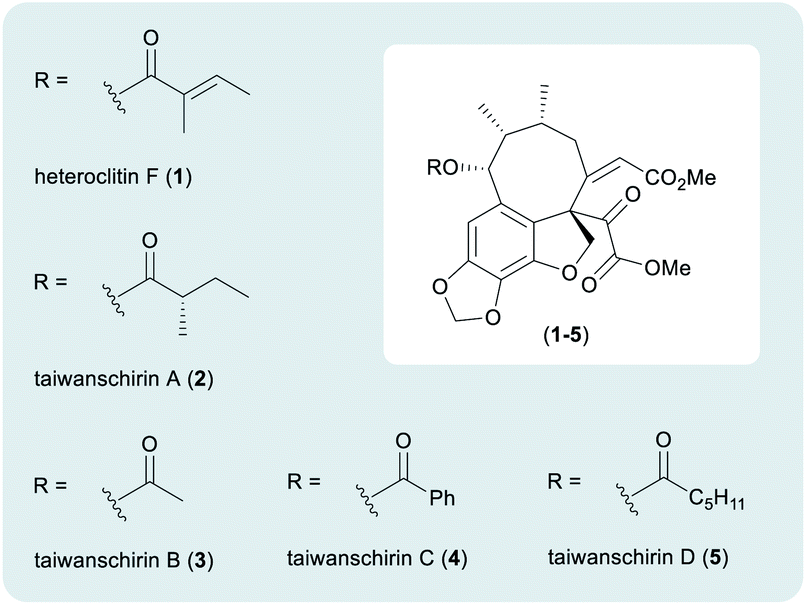 | ||
| Fig. 1 The taiwanschirin family of natural products. | ||
Isolated from the stems of Kadsura heteroclita, heteroclitin F (1) was the first member of the taiwanschirin family to be discovered (Fig. 1).5 In 1999, the isolation of taiwanschirins A (2), B (3) and C (4) from Schisandra arisanensis was reported,6 and the final member of the family to be discovered was taiwanschirin D (5), isolated from Kadsura matsudai.7 The therapeutic properties of the Schisandraceae family have long been applied in traditional Chinese medicine.8,9 The stems of Kadsura heteroclita have been used for the treatment of many diseases and activity against human type B hepatitis has been demonstrated by taiwanschirins C (4) and D (5).6,7
The taiwanschirins are highly structurally complex natural products containing an extremely rare ortho-peri-fused [8,6,5] cyclooctabenzofuran skeleton, together with a fully substituted pyruvate unit. A synthesis of this class of natural product must overcome multiple synthetic challenges including formation of the thermodynamically unstable (Z)-enone and the peri-fused 8-membered ring featuring three contiguous stereogenic centres including one quaternary stereogenic centre. There is little literature precedent regarding approaches to these compounds,10 but during the preparation of this manuscript Lumb reported an elegant biomimetically inspired synthesis of several related lignan natural products.11 In this work a redox-neutral photocatalytic approach was used to assemble the key structural fragments.
Our retrosynthesis began with the acylation of alcohol 7 (Scheme 1) which would in turn be obtained by a diastereoselective reduction of ketone 8.12 A double ozonolysis of alkene 9 (containing a sulphur ylide masked pyruvate) would result in concomitant formation of the ketone and pyruvate functionality. We decided to install the pyruvate group at a late-stage due to its general instability and sensitivity to chemical manipulations. Next, a key exo-selective Heck cyclisation would lead to the formation of the 8-membered ring, thereby forming the carbon skeleton. The precursor for the Heck cyclisation would be obtained from the conjugate addition of an organocuprate derived from iodide 12 into alkyne 11. This alkyne intermediate was a key component of our retrosynthesis and we proposed that it could be synthesised via an alkynylation reaction of an enolate derived from cyanoketothiane 13 with beta-haloalkyne 14.
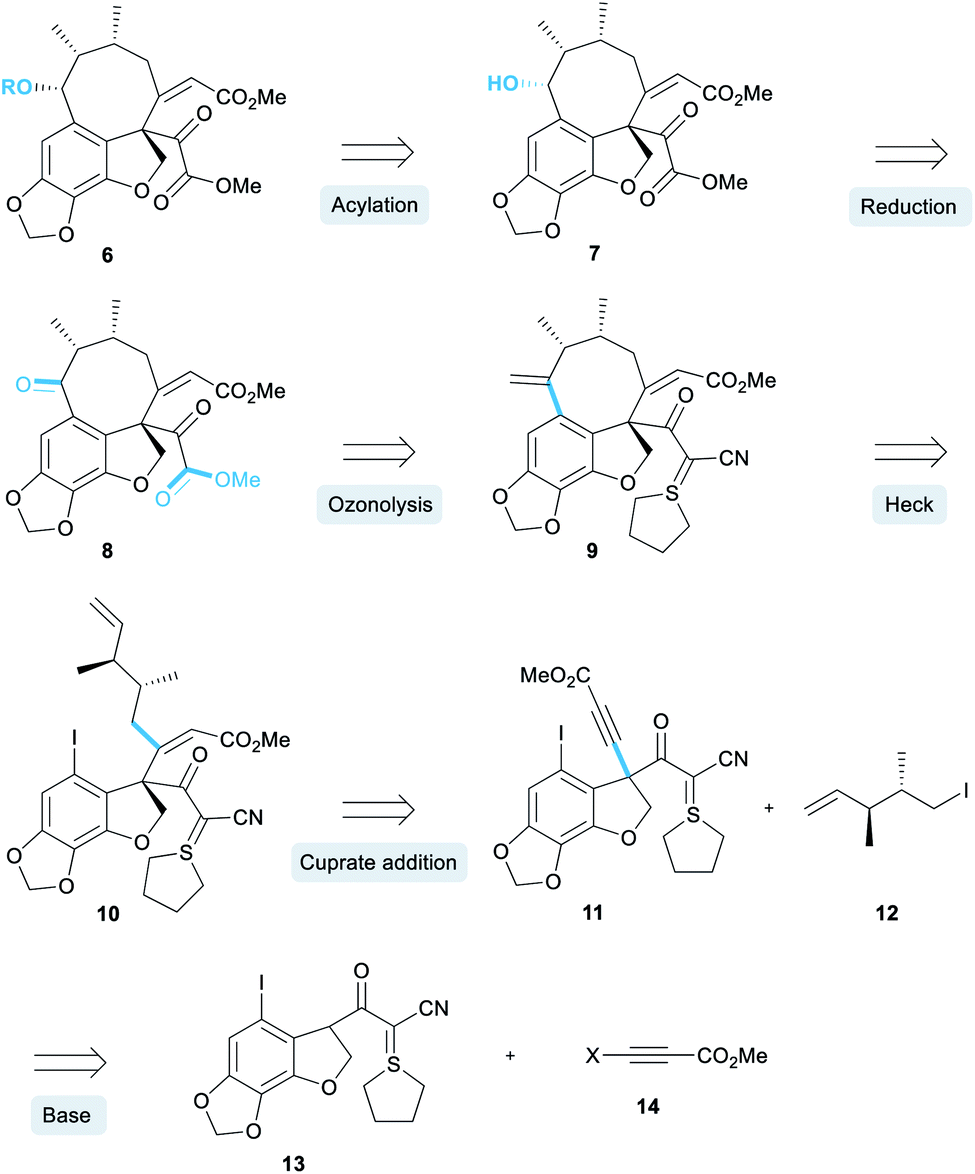 | ||
| Scheme 1 Proposed retrosynthesis. | ||
Our inspiration for the alkynylation reaction derived from work by Jørgensen detailing the enantioselective alpha-alkynylation of cyclic beta-ketoesters.13 Here, beta-haloalkynes incorporating an electron withdrawing group underwent a conjugate addition–elimination reaction with cyclic beta-ketoesters under asymmetric phase-transfer conditions. Earlier workers have developed methodologies for the reaction of haloalkynes with ester enolates,14 and as partners in cycloaddition with alkenes.15 There are also reports of acetylenic substitution employing alternative classes of reagents, including notable work on hypervalent iodine reagents by Waser16 and lead triacetate reagents.17 To this end, we sought to further develop this general reaction to enable the reaction of haloacetylene 14 with enolates derived from a wide range of carbonyl compounds. We anticipated that a powerful alkynylation methodology could be developed that would not only enable the synthesis of alkyne 11, and aid our synthesis of the taiwanschirins, but would also be highly useful in its own right.
Owing to the extended synthesis of cyanoketothiane 13, we looked towards a model compound to test our synthetic strategy. Consequently, dihydrobenzofuran 17 was prepared in two steps from 2-hydroxybenzaldehyde 15 following a report by Hossain (Scheme 2a).18 Following this protocol, 2-hydroxybenzaldehyde 15 was reacted with ethyl diazoacetate and tetrafluoroboric acid. After treatment with concentrated sulfuric acid, benzofuran 16 was obtained in 85% yield. The reduction of benzofuran 16 using magnesium turnings in MeOH then afforded the model dihydrobenzofuran 17 in 70% yield.19 Transesterification of the ester from ethyl to methyl was also observed in this reaction.
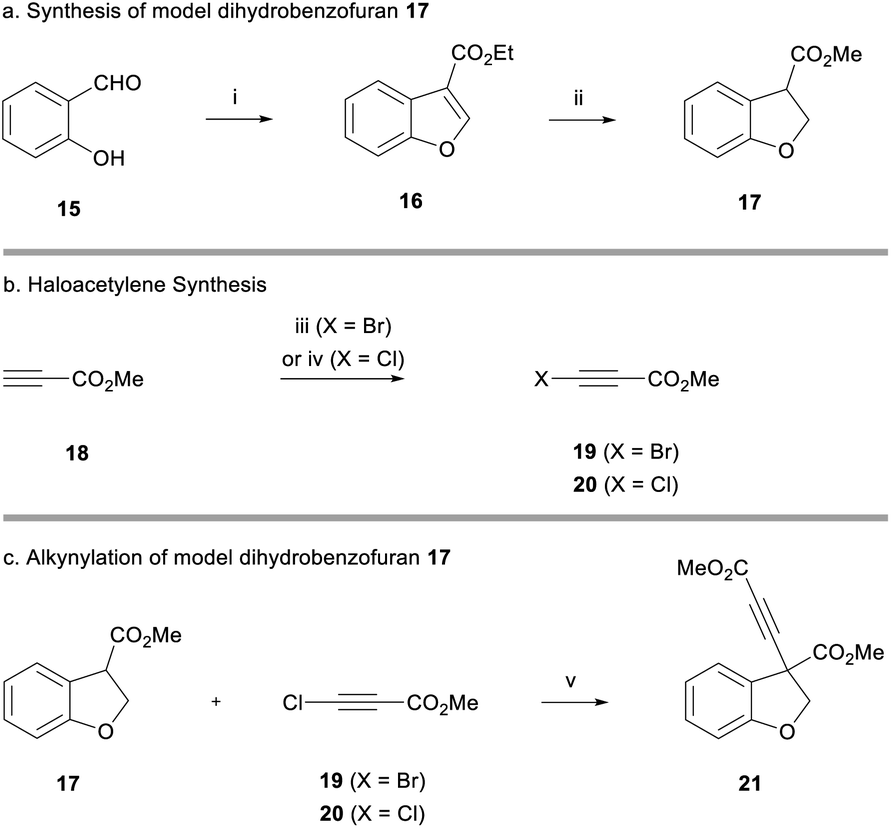 | ||
| Scheme 2 (a) Synthesis of model dihydrobenzofuran 17. Reagents and conditions: (i) ethyl diazoacetate, HBF4·Et2O, CH2Cl2, rt, 30 min, then H2SO4, 30 min, 85%; (ii) Mg, MeOH, rt, 1 h, 70%. (b) Haloacetylene synthesis. Reagents and conditions: (iii) N-bromosuccinimide, acetone, rt, 30 min, 76%; (iv) tBuOCl, tBuOK, tBuOH, rt, 30 min, 60%. (c) Alkynylation of model dihydrobenzofuran 17. Reagents and conditions: (v) LiHMDS, THF, −78 °C, 5 min, then 19, −78 °C, 30 min, 38%, or 20, 94%. | ||
Bromoacetylene 19 was prepared in 76% yield through reaction of N-bromosuccinimide with methylpropiolate (Scheme 2b).20 Chloroacetylene 20 was synthesised in 60% yield according to Jørgensen's procedure. We were now in a position to examine the alkynylation of dihydrobenzofuran 17.21 Early work had shown that the enolate derived from 17 was only stable at low temperatures (i.e. −78 °C) because it underwent rapid beta-elimination and we observed by-products derived from opening of the heterocyclic ring. Deprotonation with weaker alkoxide bases was unsatisfactory, and thus we found it optimal to use a strong lithium amide base to deprotonate the ester at −78 °C. Pleasingly, after deprotonation with LiHMDS, dihydrobenzofuran 17 formed an enolate that went on to react with bromoacetylene 19 (reaction maintained at low temperature) to afford alkyne 21 in 38% yield (Scheme 2c). The use of chloroacetylene 20 gave a much cleaner reaction profile and a considerably higher yield of 94%. It has been suggested that the chloroalkyne electrophile is less likely to act as a halogenating agent towards the nucleophile, compared to the bromoalkyne, which may explain the significantly improved yield.13a
In addition to developing a general route to the taiwanschirins, we were keen to explore the general applicability of this dihydrobenzofuran enolate alkynylation methodology. Therefore, the scope of this reaction was demonstrated using a wide range of substituted acyl dihydrobenzofurans. A summary of the different products of this new reaction is presented below (21–29, Scheme 3). As shown in Scheme 3, the alkynylation reaction afforded good yields for ester substrates featuring electron donating groups including methylenedioxy (22-as found in the target natural products) and trimethylsilyl (23) affording the product alkynes in 68% and 69% yields respectively. The highest yields, however, were obtained from dihydrobenzofuran esters that were not substituted on the aromatic ring, for example esters 21 and 24 were obtained in 94% and 96% yields respectively. It was also found that the alkynylation reaction could be performed with tert-butyl chloroacetylene 30 to afford ester 25 and amide 26 in 100% and 85% yield.
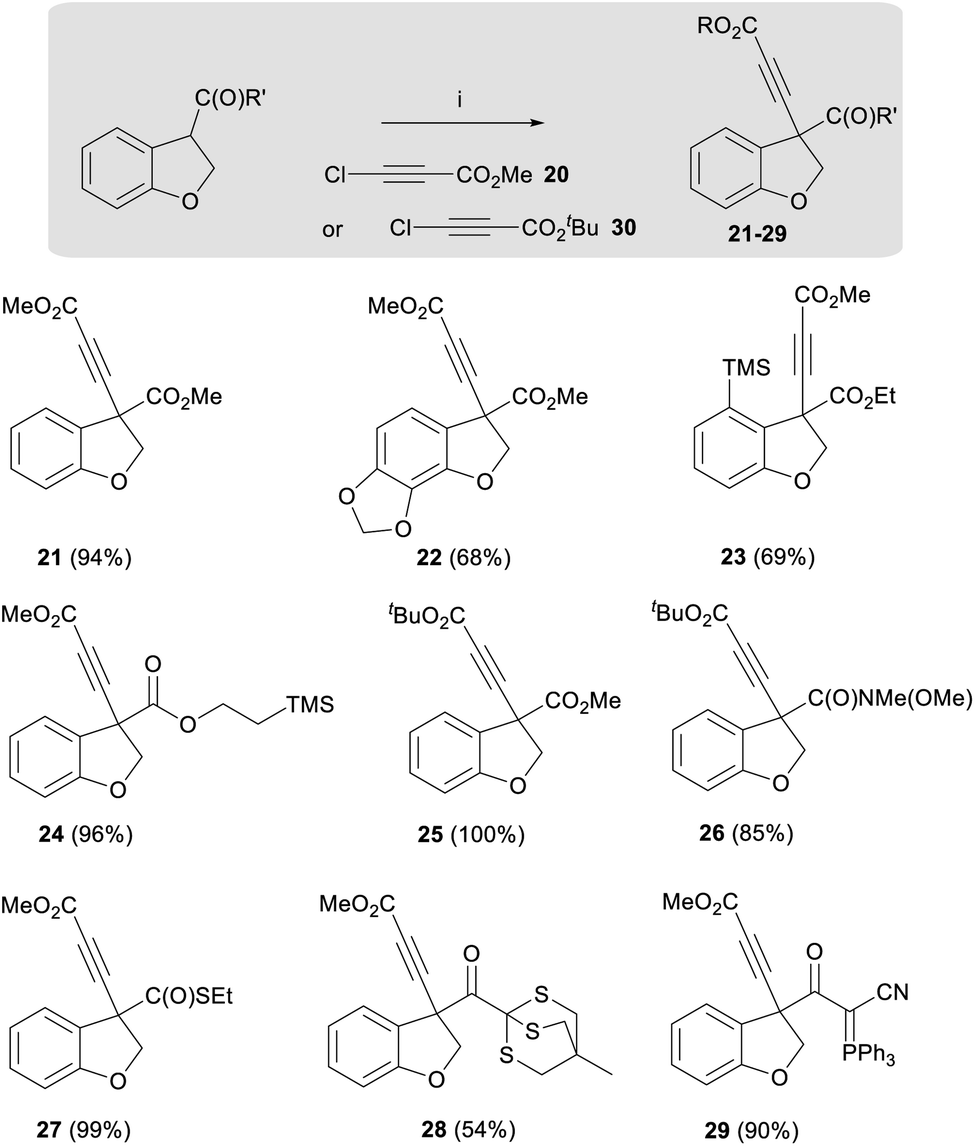 | ||
| Scheme 3 Alkynylation substrate scope. Reagents and conditions: (i) LiHMDS, THF, −78 °C, 5 min, then 20 or 30, −78 °C, 30 min. | ||
Notably, a thioester analogue was also found to display high reactivity, affording alkyne 27 in near quantitative yield. Further unprecedented reactivity was obtained with a thioether, leading to the isolation of alkyne 28 in 54% yield. The alkynylation reaction could also be applied to cyanoketophosphoranes enabling the synthesis of alkyne 29 in 90% yield.
The successful development of the alkynylation methodology meant that we could next explore methods to functionalise the alkyne leading to trisubstituted olefins as found in the target. The addition of organocuprate reagents to ynones has been described as a method of accessing trisubstituted enones with overall cis-addition.22 Pleasingly, we found that the organocuprate reagent derived from MeLi reacted with alkyne 21 to afford the desired (Z)-enone 31 exclusively as a single diastereoisomer in 92% yield (Scheme 4). This process was shown to be general, and organocuprate reagents derived from various organolithium reagents were tolerated, for example nBuLi and iBuLi afforded enones 32 and 33 in yields of 89% and 91% respectively.
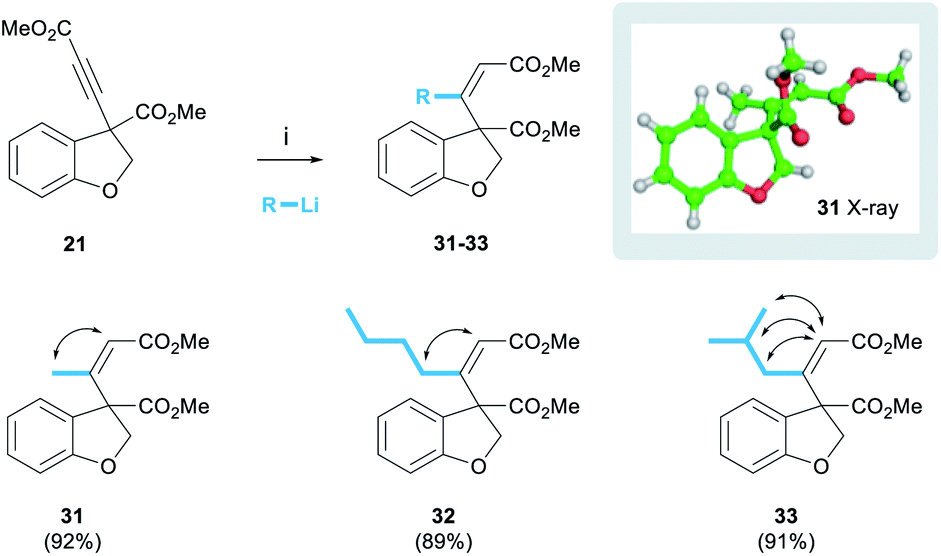 | ||
| Scheme 4 (Z)-Enone incorporation. Reagents and conditions: (i) 4 eq. RLi, 2 eq. CuI, THF, −78–0 °C, 1 h, then −78 °C, 21, 30 min. | ||
The alkene stereochemistry of these products was proven unequivocally. NOESY spectra were obtained for the three enones below (cross-peaks represented by double-headed arrows) and the (Z)-configuration was proven in each case (see ESI† for spectra). Furthermore, a crystal of enone 31 was grown and the structure proven by single crystal X-ray analysis.23 We also observed that the 1H NMR chemical shift of the olefinic proton was diagnostic of enone stereochemistry. In the case of the desired (Z)-enones, the olefinic proton was typically in the range of 5.86–5.93 ppm. However, in preliminary work using the same procedure but quenching the reaction with aqueous NH4Cl, rather than MeOH, we isolated quantities (approximately 20%) of the undesired (E)-enone whose olefinic protons appeared in a lower range of 5.62–5.63 ppm (see ESI†). We speculate that the reasons behind this difference in selectivity lie in the rate of reaction quenching versus temperature as the reaction warms to room temperature.
Next, we turned our attention towards the fused 8-membered ring of the taiwanschirin target. In order to test the intramolecular Heck cyclisation approach, it was necessary to incorporate a halogen functional group at the peri-position of the benzofuran. To this end, 3-bromophenol 34 was deprotonated with NaH and reacted with diethylcarbamoyl chloride to form a carbamate (Scheme 5). With this protecting group in place we performed a directed deprotonation using LDA in THF at −78 °C. Following reaction with DMF and subsequent deprotection under acidic conditions, aldehyde 36 was formed in 69% yield over 3 steps. As before, aldehyde 36 was then reacted with ethyl diazoacetate to afford peri-bromobenzofuran 37 in 84% yield.
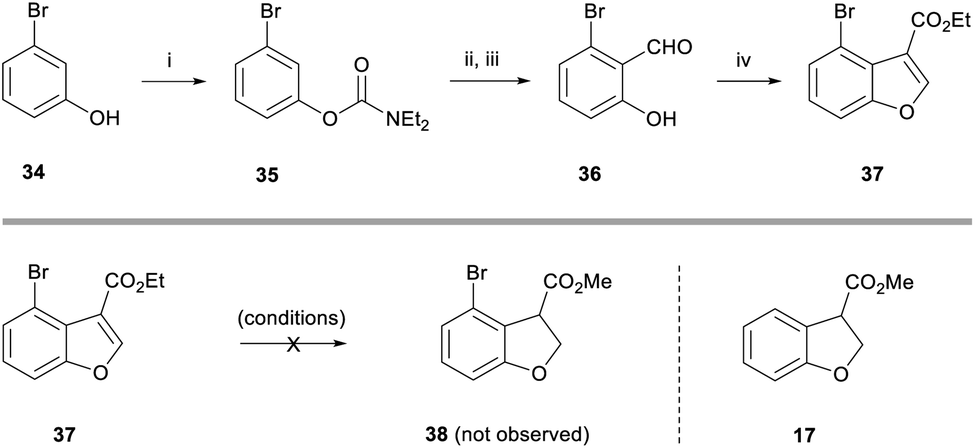 | ||
| Scheme 5 Synthesis of benzofuran 37 and attempted reduction. Reagents and conditions: (i) NaH, diethylcarbamoyl chloride, THF, rt, 16 h; (ii) LDA, THF, −78 °C, 30 min, then DMF, −78 °C, 30 min; (iii) 2 M HCl (aq.), 0 °C, 20 min, 69% over 3 steps; (iv) ethyl diazoacetate, HBF4·OEt2, rt, 30 min, then H2SO4, rt, 30 min, 84%. | ||
We then subjected benzofuran 37 to a range of reducing conditions in order to form the corresponding dihydrobenzofuran 38. Disappointingly, the magnesium-promoted reduction conditions used previously were unsuccessful (Scheme 5). Dihydrobenzofuran 38 was not observed in the reaction mixture by 1H NMR spectroscopy, however the de-brominated dihydrobenzofuran 17 was isolated in 23% yield from a complex mixture. Conducting the reaction at 0 °C resulted in only starting material being observed by 1H NMR spectroscopy.
A variety of different conditions for the partial reduction of benzofuran 37 were then investigated as detailed below. Surprisingly, the use of palladium-catalysed hydrogenation conditions failed to afford the desired product; the de-brominated benzofuran was not observed.24 Both triethylsilane/trifluoracetic acid (TFA),25 and samarium(II) iodide also resulted in only starting material being observed by 1H NMR spectroscopy of the crude reaction mixtures.26
In order to overcome this problem, a palladium-catalysed stannylation reaction was proposed to convert the bromobenzofuran 37 to the corresponding tributylstannane 40. We postulated that the aryl stannane may be reduced to the dihydrobenzofuran under the magnesium-promoted reduction, and the stannane then exchanged for a halogen, thereby enabling the key intramolecular Heck cyclisation. Initially, only starting material was observed when Pd(PPh3)4 was used as the catalyst for the stannylation of 37 (Scheme 6, entry 1).27 However, a change of catalyst to PdCl2(PhCN)2, resulted in a 33% yield of the desired product (entry 2).28 Pleasingly, the use of 2.5 mol% Pd2(dba)3 in combination with 15 mol% JohnPhos resulted in 65% yield (entry 3).
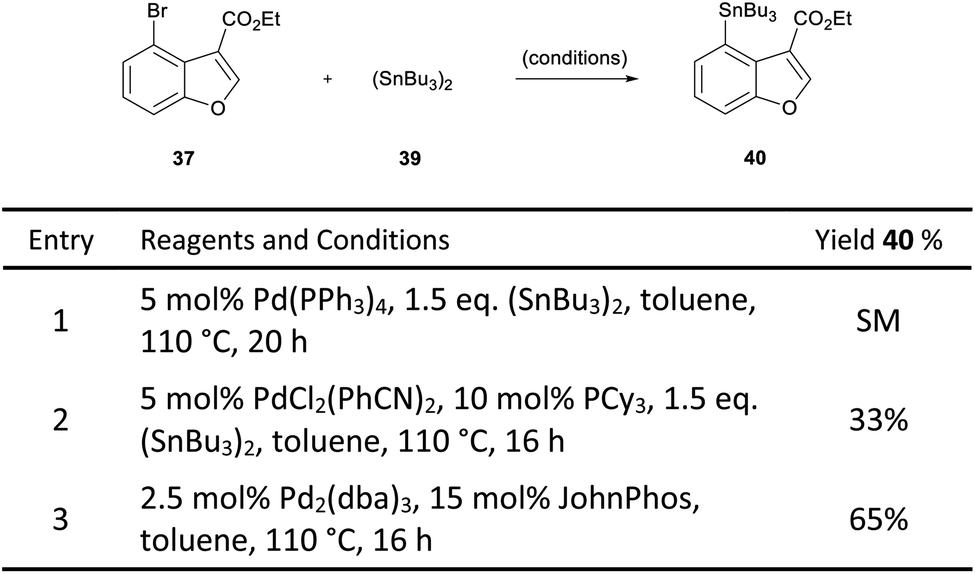 | ||
| Scheme 6 Stannylation of benzofuran 37. | ||
With benzofuran 40 in hand, we were able to test the magnesium-promoted reduction step and observed the formation of stannane 41 in 92% yield (Scheme 7). Furthermore, a comparable yield of 84% was achieved when the reaction was performed on a 1.6 g scale. Crucially, heating at 70 °C was necessary for complete transesterification to form the methyl ester.
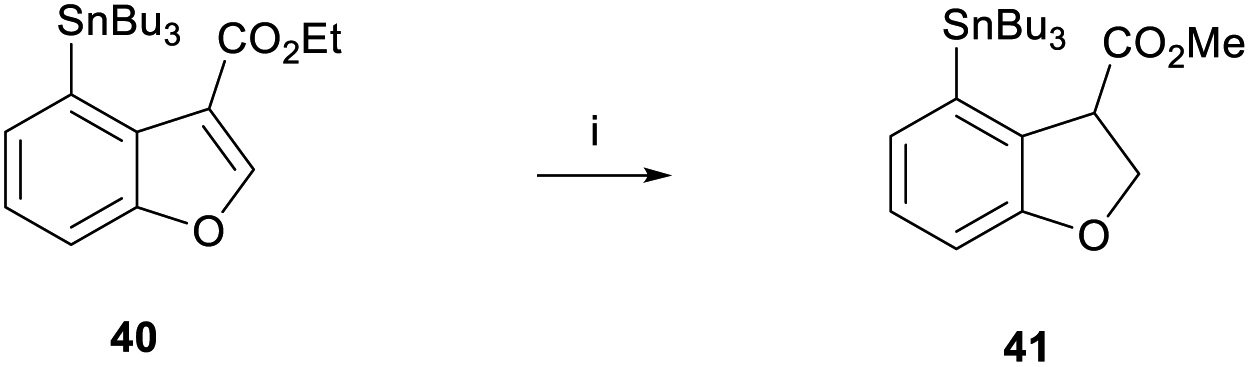 | ||
| Scheme 7 Magnesium reduction of benzofuran 40. Reagents and conditions: (i) Mg, MeOH, rt, 4 h then 70 °C, 1 h, 92%. | ||
Stannane 41 was then subjected to the newly-developed alkynylation reaction conditions to form alkyne 42 in 48% yield (Scheme 8). Tin-iodine exchange transformed stannane 42 to iodide 43 in 87% yield. The following cuprate addition also progressed cleanly to afford alkene 45 in 45% yield as the single (desired) (Z)-diastereomer. This assignment was made by analogy with the previous examples (Scheme 4); additional data included the presence of the olefinic proton resonance at 5.90 ppm, comfortably within the range found for (Z)-enones of 5.86–5.93 ppm.
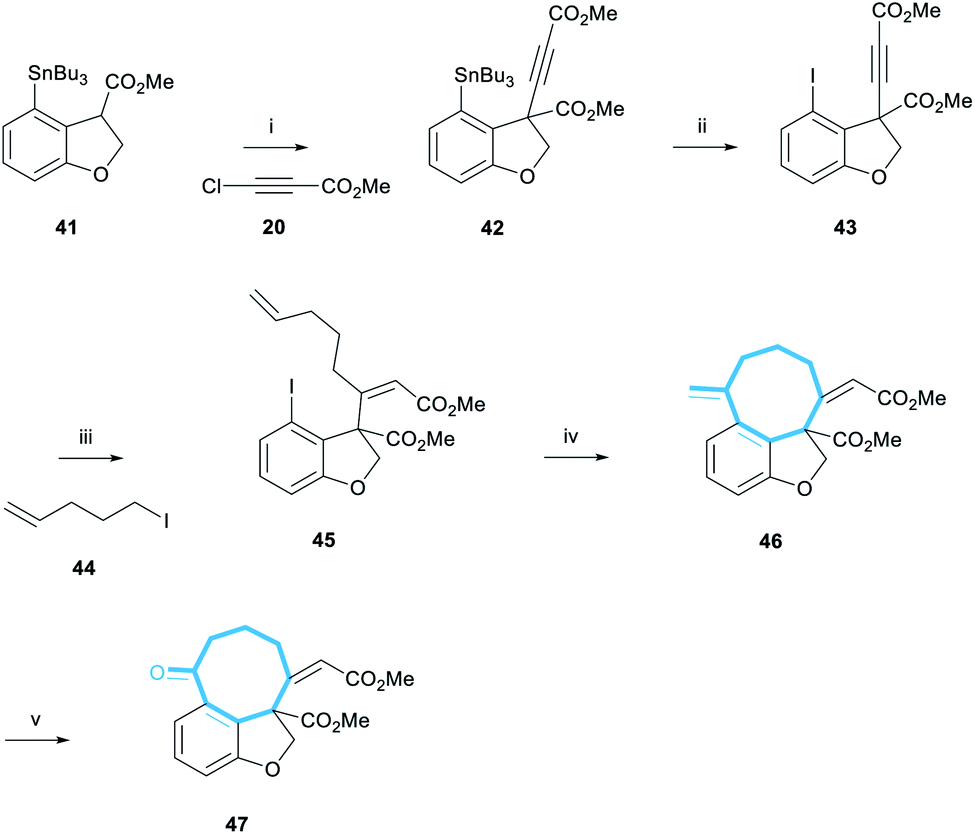 | ||
Scheme 8 Synthesis of iodide 45 and subsequent Heck cyclisation and dihydroxylation-periodate cleavage to afford alkene 47. Reagents and conditions: (i) LiHMDS, 41, THF, −78 °C, 10 min, then 20, −78 °C, 1 h, 48%; (ii) I2, CHCl3, rt, 1 h, 87%; (iii) 44, tBuLi, pentane/Et2O 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, −78 °C, 10 min, CuI, then 0 °C, 1 h, then −78 °C, 43, 30 min, 45%; (iv) 10 mol% Pd(OAc)2, 40 mol% PPh3, 10 eq. Et3N, MeCN, 80 °C, 1 h, 81%; (v) 5 mol% OsO4, 5 eq. NaIO4, THF/H2O 2:1, rt, 2 h, 69%. :2, −78 °C, 10 min, CuI, then 0 °C, 1 h, then −78 °C, 43, 30 min, 45%; (iv) 10 mol% Pd(OAc)2, 40 mol% PPh3, 10 eq. Et3N, MeCN, 80 °C, 1 h, 81%; (v) 5 mol% OsO4, 5 eq. NaIO4, THF/H2O 2:1, rt, 2 h, 69%. | ||
With the pendant alkene installed, we could now test the key Heck-cyclisation reaction. We were delighted to observe that a slight modification of literature conditions resulted in an 81% yield of the desired alkene 46 (Scheme 8).29 Furthermore, the reaction was regioselective, affording the 8-exo-regioisomer exclusively, and the undesired 9-endo-cyclisation product was not observed in the 1H NMR spectrum of the crude reaction mixture. The selectivity of this reaction is noteworthy given previous reports detailing the low selectivity of intramolecular Heck cyclisation reactions during the formation of medium-sized rings.30a We note that in this reaction, the tether between the aryl iodide and the alkene contains a (Z)-alkene, 3 trigonal carbons and a single quaternary-substituted carbon. Thus, we suggest that the high exo-selectivity of this reaction is a consequence of having a relatively restricted and sterically encumbered framework which is unable to accommodate the larger steric requirements of an endo cyclication to form a 9-membered ring.30b,c
Finally, alkene 46 was reacted with catalytic OsO4 in the presence of NaIO4.31 This dihydroxylation-periodate cleavage reaction afforded ketone 47 in 69% yield, representing the most advanced compound en-route to the taiwanschirin family synthesised. Overall, ketone 47 was synthesised in 4% yield over 11 steps from 3-bromophenol 34. With the carbon skeleton of the taiwanschirin family now nearly complete, the final challenge to be solved was incorporation of the pyruvate group.
Installing the pyruvate proved to be the most challenging part of this project. A wide range of strategies were explored before we examined the use of the Bode salt 49 as a pyruvate equivalent, itself prepared in a single step from tetrahydrothiophene and bromoacetonitrile (Scheme 9).32 Dihydrobenzofuran 17 was subjected to ester hydrolysis conditions to afford carboxylic acid 48. Pleasingly, the reaction of 48 and 49 in the presence of EDC·HCl provided cyanoketothiane 50 in 78% yield. Cyanoketothiane 50 was subsequently deprotonated with LDA and reacted with chloroacetylene 20 to afford alkyne 51 in 60% yield. Here, LDA base was found to afford optimum yields of the desired product. The addition of an organocuprate reagent derived from iBuLi to alkyne 51 afforded (Z)-enone 52 in 79% yield. The stereochemical assignment of 52 was made by analogy with the previous examples of this reaction (Scheme 4) and the 1H NMR chemical shift of the olefinic proton of alkene 52 was observed at 5.90 ppm. We were delighted to observe that enone 52 reacted with ozone at −78 °C in the presence of methanol, to form the desired pyruvate 53 in 34% yield.33 The crude 1H NMR spectrum of this reaction showed complete conversion of enone 52 to pyruvate 53, with almost no other contaminants; the modest yield was attributed to the small scale of this model reaction.
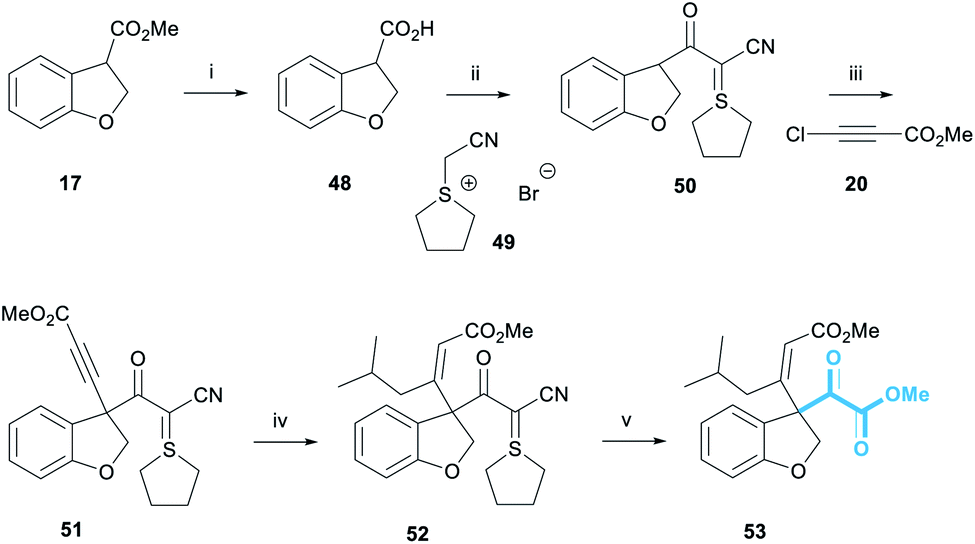 | ||
| Scheme 9 Pyruvate incorporation. Reagents and conditions: (i) Na2CO3, THF/H2O 1:1, rt, 16 h, 54% (ii) EDC·HCl, DIPEA, DMAP, CH2Cl2, rt, 16 h, 78%; (iii) LDA, 50, THF, −78 °C, 10 min, then 20, −78 °C, 30 min, 60%; (iv) iBuLi, CuI, THF, −78–0 °C, 1 h, then −78 °C, 51, 5 min, 79%; (v) O3, CH2Cl2/MeOH 4:1, −78 °C, 10 min, 34%. | ||
Thus, we have demonstrated a comprehensive approach towards the taiwanschirin family of natural products that incorporate the key structural fragments identified at the outset. Future work will combine these elements to complete the synthesis of these targets and also address the issue of an asymmetric alkynylation reaction using controlling factors such as chiral auxiliaries, chiral lithium amides or asymmetric catalysis.34
Conclusions
This work describes model studies developed towards the structural core of the taiwanschirin family of natural products. An approach was conceived to enable installation of the (Z)-enone as a single diastereoisomer. A novel alkynylation reaction has been successfully applied to a wide substrate scope including esters, amides, ketones and thioesters. Furthermore, these studies have led to the assembly of an 8-membered ring in high yield through the development of a regioselective Heck cross-coupling reaction. Under palladium-catalysed conditions, we observed the formation of the desired alkene as a single (exo)-regioisomer in 81% yield. Through the use of Bode salt, the pyruvate functional group could also be accessed, and this approach enabled the assembly of a model compound that incorporated both the pyruvate group and the (Z)-enone.Data availability
Full experimental and characterisation data are provided as part of the ESI.†Author contributions
MBH performed the experiments. KEC performed the X-ray crystallography experiments. All authors contributed to the discussion and prepared the manuscript. TJD directed the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC and GlaxoSmithKline for funding.Notes and references
- H. Yoo, J. R. Widhalm, Y. Qian, H. Maeda, B. R. Cooper, A. S. Jannasch, I. Gonda, E. Lewinsohn, D. Rhodes and N. Dudareva, Nat. Commun., 2013, 4, 2833 CrossRef PubMed.
- H. A. Krebs and W. A. Johnson, Biochem. J., 1937, 31, 645 CrossRef CAS PubMed.
- (a) C. H. Alves Esteves, C. J. J. Hall, P. D. Smith and T. J. Donohoe, Org. Lett., 2017, 19, 5248 CrossRef CAS PubMed; (b) C. H. Alves Esteves, M. Koyioni, K. E. Christensen, P. D. Smith and T. J. Donohoe, Org. Lett., 2018, 20, 4048 CrossRef CAS PubMed; (c) H. S. Shirley, M. Koyioni, F. Muncan and T. J. Donohoe, Chem. Sci., 2019, 10, 4334 RSC.
- B. P. Zavesky, S. L. Bartlett and J. S. Johnson, Org. Lett., 2017, 19, 2126 CrossRef CAS PubMed.
- X.-W. Yang, H. Miyashiro, M. Hattori, T. Namba, Y. Tezuka, T. Kikuchi, D.-F. Chen, G.-J. Xu, T. Hori, M. Extine and H. Mizuno, Chem. Pharm. Bull., 1992, 40, 1510 CrossRef CAS PubMed.
- Y.-H. Kuo, H.-C. Huang, L. Y. Kuo and C.-F. Chen, J. Org. Chem., 1999, 64, 7023 CrossRef CAS.
- S.-Y. Li, M.-D. Wu, C.-W. Wang, Y.-H. Kuo, R.-L. Huang and K.-H. Lee, Chem. Pharm. Bull., 2000, 48, 1992 CrossRef CAS PubMed.
- (a) N. Ookawa, Y. Ikeya, H. Taguchi and I. Yosiokam, Chem. Pharm. Bull., 1981, 29, 123 CrossRef CAS; (b) Y.-C. Shen, Y.-C. Lin, Y.-B. Cheng, Y.-H. Kuo and C.-C. Liaw, Org. Lett., 2005, 23, 5297 CrossRef PubMed.
- L.-J. Xu, H.-T. Liu, Y. Peng and P.-G. Xiao, J. Syst. Evol., 2008, 46, 692 Search PubMed.
- For an approach to the spirocyclic cyclohexadienone core of the related schiarisanrin family of natural products see: R. S. Coleman, J. M. Guernon and J. T. Roland, Org. Lett., 2000, 2, 277 CrossRef CAS PubMed.
- Z. Huang and J.-P. Lumb, Nat. Chem., 2021, 13, 24 CrossRef CAS PubMed.
- W. Gong and T. V. Rajanbabu, Chem. Sci., 2013, 4, 3979 RSC.
- (a) T. B. Poulsen, L. Bernardi, J. Alemá, J. Overgaard and K. A. Jørgensen, J. Am. Chem. Soc., 2007, 129, 441 CrossRef CAS PubMed; (b) T. B. Poulsen, L. Bernardi, M. Bell and K. A. Jørgensen, Angew. Chem., Int. Ed., 2006, 45, 6551 CrossRef CAS PubMed.
- A. S. Kende, P. Fludzinski, J. H. Hill, W. Swenson and J. Clardy, J. Am. Chem. Soc., 1984, 106, 3551 CrossRef CAS.
- (a) B. B. Snider, D. M. Roush, D. J. Rodini, D. Gonzalez and D. Spindell, J. Org. Chem., 1980, 45, 2773 CrossRef CAS; (b) K. Villeneuve, N. Riddell, R. W. Jordan, G. C. Tsui and W. Tam, Org. Lett., 2004, 6, 4543 CrossRef CAS PubMed.
- (a) J. P. Brand and J. Waser, Chem. Soc. Rev., 2012, 41, 4165 RSC; (b) F. González, J. P. Brand and J. Waser, Chem.–Eur. J., 2010, 16, 9457 CrossRef PubMed.
- M. G. Moloney, J. T. Pinhey and E. G. Roche, J. Chem. Soc., Perkin Trans. 1, 1989, 333 RSC.
- M. E. Dudley, M. M. Morshed and M. M. Hossain, Synthesis, 2006, 1711 CAS.
- P. Bongen, J. Pietruszka and R. C. Simon, Chem.–Eur. J., 2012, 18, 11063 CrossRef CAS PubMed.
- W. Lewis, C. J. Moody, E. Packard, A. T. Murray, G. Jones, D. Hamza and A. Nortcliffe, Eur. J. Org. Chem., 2017, 138 CAS.
- For literature examples of the alkylation of related dihydrobenzofuran ester enolates see: (a) Z. Gu and A. Zakarian, Org. Lett., 2011, 13, 1080 CrossRef CAS PubMed; (b) P. Lu, A. Mailyan, Z. Gu, D. M. Guptill, H. Wang, H. M. L. Davies and A. Zakarian, J. Am. Chem. Soc., 2014, 136, 17738 CrossRef CAS PubMed; (c) S. Antus, Á. Gottsegen, P. Kolonits and H. Wagner, Liebigs Ann. Chem., 1989, 6, 593 CrossRef.
- E. J. Corey and J. A. Katzenellenbogen, J. Am. Chem. Soc., 1969, 91, 1851 CrossRef CAS.
- Single crystal X-ray diffraction data were collected using a (Rigaku) Oxford Diffraction SuperNova diffractometer and CrysAlisPro. Crystallographic data have been deposited with the Cambridge Crystallographic Data Centre (CCDC 2072549).† For details concerning solving and refining these structures, see: (a) L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786 CrossRef CAS; (b) P. Parois, R. I. Cooper and A. L. Thompson, Chem. Cent. J., 2015, 9, 30 CrossRef PubMed; (c) R. I. Cooper, A. L. Thompson and D. J. Watkin, J. Appl. Crystallogr., 2010, 43, 1100 CrossRef CAS.
- A. Mori, T. Mizusaki, Y. Miyakawa, E. Ohashi, T. Haga, T. Maegawa, Y. Monguchi and H. Sajiki, Tetrahedron, 2006, 62, 11925 CrossRef CAS.
- J. Zhang, J. Zhang, Y. Kang, J. Shi and C. Yao, Synlett, 2016, 27, 1587 CrossRef CAS.
- L. Guo, F. Zhang, W. Hu, L. Li and Y. Jia, Chem. Commun., 2014, 50, 3299 RSC.
- J.-L. Li, H.-W. Zhao, X. Qin, J. Cui, S. Su, H.-L. Li, Y.-Y. Yue and X.-Q. Song, Synth. Commun., 2013, 43, 3175 CrossRef CAS.
- E. B. Corcoran, A. B. Williams and R. N. Hanson, Org. Lett., 2012, 14, 4630 CrossRef CAS PubMed.
- J.-M. Gaudin, Tetrahedron Lett., 1991, 32, 6113 CrossRef CAS.
- (a) L. F. Tietze, K. M. Sommer, G. Schneider, P. Tapolcsányi, J. Wölfling, P. Müller, M. Noltemeyer and H. Terlau, Synlett, 2003, 1494 CrossRef CAS; (b) for a comprehensive review see: J. T. Link, Org. React., 2002, 60, 157 CAS; (c) K. C. Majumdar and B. Chattopadhyay, Synlett, 2008, 979 CrossRef CAS.
- P. Wipf, C. R. J. Stephenson and M. A. A. Walczak, Org. Lett., 2004, 6, 3009 CrossRef CAS PubMed.
- J. Lei, A. R. Lippert and J. W. Bode, J. Am. Chem. Soc., 2008, 10, 4253 Search PubMed.
- H. H. Wasserman and A. K. Petersen, Tetrahedron Lett., 1997, 38, 953 CrossRef CAS.
- For a review of methods for the enantioselective construction of quaternary stereocentres see: J. Christoffers and A. Mann, Angew. Chem., Int. Ed., 2001, 40, 4591 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2072549. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04247e |
| ‡ Chemical Crystallography, Chemistry Research Laboratory, Oxford. |
| This journal is © The Royal Society of Chemistry 2021 |