
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilacyclization through palladium-catalyzed intermolecular silicon-based C(sp2)–C(sp3) cross-coupling†
Ying
Qin
,
Lianghui
Li
 ,
Jin-Yuan
Liang
,
Kailong
Li
and
Dongbing
Zhao
*
,
Jin-Yuan
Liang
,
Kailong
Li
and
Dongbing
Zhao
*
State Key Laboratory and Institute of Elemento-Organic Chemistry, Frontiers Science Center for New Organic Matter, College of Chemistry, Nankai University, Tianjin, 300071, China. E-mail: dongbing.chem@nankai.edu.cn
First published on 7th October 2021
Abstract
Silicon-based cross-coupling has been recognized as one of the most reliable alternatives for constructing carbon–carbon bonds. However, the employment of such reaction as an efficient ring expansion strategy for silacycle synthesis is comparatively little known. Herein, we develop the first intermolecular silacyclization strategy involving Pd-catalyzed silicon-based C(sp2)–C(sp3) cross-coupling. This method allows the modular assembly of a vast array of structurally novel and interesting sila-benzo[b]oxepines with good functional group tolerance. The key to success for this reaction is that silicon atoms have a stronger affinity for oxygen nucleophiles than carbon nucleophiles, and silacyclobutanes (SCBs) have inherent ring-strain-release Lewis acidity.
Introduction
Because of the high availability, low toxicity, and high-stability of organosilicon molecules, silicon-based cross-coupling reactions have been recognized as one of the most reliable alternatives for constructing carbon–carbon bonds.1 Although rapid progress in silicon-based cross-coupling has been achieved, the employment of Hiyama–Denmark cross-coupling as an efficient ring expansion strategy for silacycle synthesis is comparatively rare. Recently, we described the first intramolecular strain-release silicon-based cross-coupling reaction, which constitutes an efficient ring expansion route for the synthesis of diversified silacycles.2 However, the necessity of pre-synthesized starting substrates in this intramolecular method limits the full utilization of this elegant method. Further expanding this concept to intermolecular reactions would represent a significant and important step in organosilicon-based cross-coupling for silacycle synthesis.However, in this effort, we may face some challenges: (1) the use of alkylsilanes for construction of the Csp2–Csp3 bond in silicon-based coupling has the problem that the alkyl palladium intermediates formed by the transmetallation step normally tend to undergo β-hydride elimination; (2) a competitive reaction involving C–Si bond formation, in which the C-nucleophile attacks the silicon atom on SCBs, may occur during the reaction as the Pd-catalyzed cross-coupling of simple aryl iodides with SCBs has been reported to deliver a mixture of allylsilanes (Scheme 1a).3 Inspired by the mode of activation of the Si–C bonds in silicon-based cross-coupling reaction presented by Hiyama (Scheme 1b)4 and Denmark,5 involving coordination of the oxygen atom to the silicon atom, and the ample body of reactions utilizing SCBs as synthetic linchpins,6 we designed a new intermolecular cascade reaction between 2-halophenols and SCBs involving Si–O bond formation by an inter-reaction between the hydroxyl moiety of 2-halophenols and the silicon atom of SCBs, followed by Hiyama–Denmark C(sp2)–C(sp3) cross-coupling (Scheme 1c). If successful, the reaction is expected to provide facile access to sila-benzo[b]oxepines from readily available substrates. Notably, benzo[b]oxepine scaffolds are prevalent in various bioactive molecules (Scheme 1d).7 Incorporation of silicon into benzo[b]oxepines is of great significance because sila-congeners generally possess significantly altered physicochemical properties with a lack of element-associated toxicity,8 but remains a great synthetic challenge.9 Herein, we describe the implementation of the first intermolecular silicon-based C(sp2)–C(sp3) cross-coupling between 2-iodophenols and SCBs. This strategy is essentially the state of the art in terms of practicality and versatility for the synthesis of sila-benzo[b]oxepines and allows efficient late-stage modification of natural products.
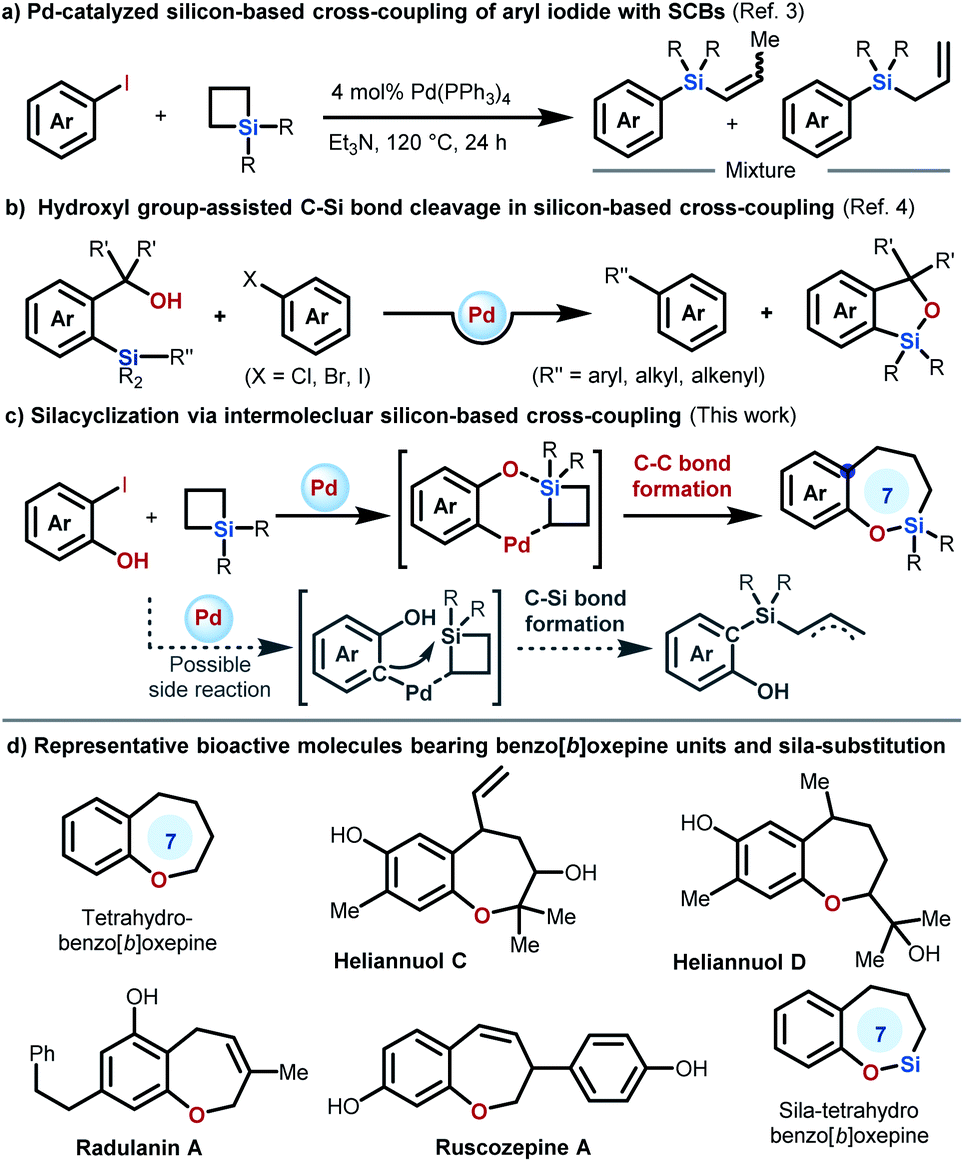 | ||
| Scheme 1 Research background, inspiration, challenge and our study. | ||
Results and discussion
Condition optimization
We first conducted an intermolecular strain-release silicon-based ring expansion between 2-iodophenol 1a and silacyclobutane 2a to try to directly access sila-tetrahydrobenzo[b]oxepine 3aa under the reported conditions for intramolecular strain-release organosilicon-based ring expansion.2 Unfortunately, the reaction did not proceed at all. Then, a range of reaction parameters (e. g., solvents, bases, palladium sources, and ligands) were evaluated (Table S1 in the ESI†), and the reaction occurred smoothly to yield sila-tetrahydrobenzo[b]-oxepine 3aa in 72% isolated yield under the optimized conditions, which were identified to be 5 mol% Pd(MeCN)2Cl2, 20 mol% tBuDavePhos, NEt3/1,2,2,6,6-pentamethylpiperidine (2.0 equiv.; 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) and a mixture of acetonitrile and DMF (10:1) as the solvent at 100 °C for 18 h (Table 1, entry 1). An array of control experiments were subsequently performed to investigate the function of each component (Table 1). The mixture of triethylamine and pempidine in a ratio of 1:4 as the base is essential to ensure the yield of 3aa. Employment of single-component Et3N as the base led to lower yield with incomplete consumption of 2a and 1a (entry 2). On the other hand, pempidine improved the reactivity in the conversion of 2a and 1a but along with more serious unknown side reactions, leading to a lower yield of 3aa (entry 3). Modification of the Et3N/pempidine ratio to 1:1 also afforded inferior results (entry 4). The use of K2CO3 as the base or the absence of a base resulted in termination of this reaction (entries 5 and 6). The reaction also suffered low yields if the MeCN–DMF binary solvent was switched to a single solvent or the MeCN/DMF ratio was modified to 1:1 (entries 7–9). Addition of a small amount of DMF would facilitate opening of the strained four-membered SCB 2avia transient coordination with the silicon atom.10 However, the use of DMF as the solvent would lead to the consumption of the product 3aa during the reaction. Replacement of Pd(MeCN)2Cl2 with [Pd(allyl)Cl]2 or Pd2(dba)3 resulted in a decrease in the yield (entries 10 and 11). The [Pd] catalyst was shown to be essential to initiate this reaction (entry 12). The effect of ligands was also investigated by replacement of tBuDavePhos by DavePhos or RockPhos (entries 13 and 14). Prolonging the reaction time was shown to be ineffective for improving the yield (entry 15). 2-Bromophenol was incapable of cross-coupling with SCB 2a under the optimized conditions (entry 16).
:4) and a mixture of acetonitrile and DMF (10:1) as the solvent at 100 °C for 18 h (Table 1, entry 1). An array of control experiments were subsequently performed to investigate the function of each component (Table 1). The mixture of triethylamine and pempidine in a ratio of 1:4 as the base is essential to ensure the yield of 3aa. Employment of single-component Et3N as the base led to lower yield with incomplete consumption of 2a and 1a (entry 2). On the other hand, pempidine improved the reactivity in the conversion of 2a and 1a but along with more serious unknown side reactions, leading to a lower yield of 3aa (entry 3). Modification of the Et3N/pempidine ratio to 1:1 also afforded inferior results (entry 4). The use of K2CO3 as the base or the absence of a base resulted in termination of this reaction (entries 5 and 6). The reaction also suffered low yields if the MeCN–DMF binary solvent was switched to a single solvent or the MeCN/DMF ratio was modified to 1:1 (entries 7–9). Addition of a small amount of DMF would facilitate opening of the strained four-membered SCB 2avia transient coordination with the silicon atom.10 However, the use of DMF as the solvent would lead to the consumption of the product 3aa during the reaction. Replacement of Pd(MeCN)2Cl2 with [Pd(allyl)Cl]2 or Pd2(dba)3 resulted in a decrease in the yield (entries 10 and 11). The [Pd] catalyst was shown to be essential to initiate this reaction (entry 12). The effect of ligands was also investigated by replacement of tBuDavePhos by DavePhos or RockPhos (entries 13 and 14). Prolonging the reaction time was shown to be ineffective for improving the yield (entry 15). 2-Bromophenol was incapable of cross-coupling with SCB 2a under the optimized conditions (entry 16).
|
|
||
|---|---|---|
| Entry | Deviation from standard condition | Yieldb [%] |
| a Reactions were carried out by using [M] precatalyst (5 mol%), ligand (20 mol%), 1a (0.2 mmol), and 2a (0.22 mmol) in solvent for 18 h at 100 °C under an N2 atmosphere. b Isolated yield after purification of the product 3aa by column chromatography. N. P.: No desired products. | ||
| 1 | None | 72 |
| 2 | NEt3 instead of NEt3/pempidine | 54 |
| 3 | Pempidine instead of NEt3/pempidine | 50 |
| 4 | NEt3/pempidine (1:1) |
61 |
| 5 | K2CO3 as the base | Trace |
| 6 | Without base | Trace |
| 7 | MeCN as the solvent | 41 |
| 8 | DMF as the solvent | 33 |
| 9 | MeCN/DMF (1:1) |
44 |
| 10 | [Pd(allyl)Cl]2 instead of Pd(MeCN)2Cl2 | 61 |
| 11 | Pd2(dba)3 instead of Pd(MeCN)2Cl2 | 34 |
| 12 | Without [Pd] catalyst | N. P. |
| 13 | DavePhos instead of tBuDavePhos | Trace |
| 14 | RockPhos instead of tBuDavePhos | 41 |
| 15 | Reaction at 100 °C for 36 h | 71 |
| 16 | 2-Bromophenol instead of 1a | N. P. |
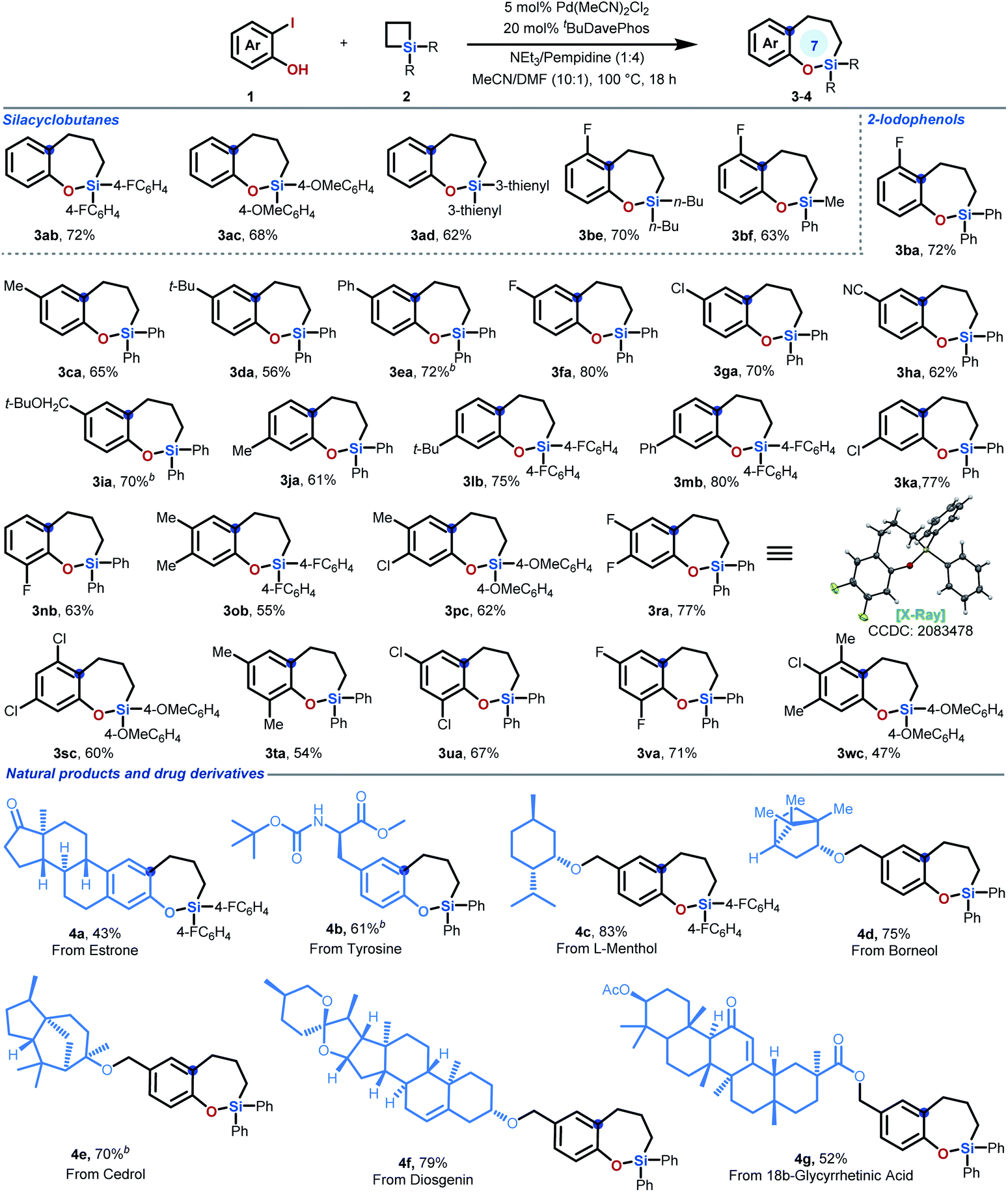
Substrate scope
Then, the scope of the ring expansion reaction between 2-iodophenols 1 and SCBs 2 was explored. As shown in Table 2, a series of silacyclobutanes with different aryl and alkyl substituents on silicon could be employed as substrates, affording the corresponding sila-tetrahydrobenzo[b]oxepines efficiently (3aa–ad & 3be). Additional experiments revealed that the current protocol for sila-tetrahydrobenzo[b]oxepine synthesis was efficient, as a wide array of 2-iodophenols could react well with silacyclobutane 2a. For monosubstituted 2-iodophenols, the starting materials bearing different functional groups in the para-, meta-, and ortho-positions with different electronic characters all proceeded well in the reaction (3ba–nb). In addition to monosubstituted 2-iodophenols, 2-iodophenols bearing two or three substituents at different positions on the aromatic ring are also suitable substrates for this transformation, affording cyclic products 3nb–wc in moderate to good yields. Notably, given the importance of fluorinated aryl systems in drug discovery,9,11 we tried many 2-iodophenols bearing one or two fluoro groups at different positions of the phenol coupling partners for this ring expansion reaction. Fortunately, all of these substrates can react smoothly with moderate to high yields under the optimized conditions (3ba, 3fa, 3nb, 3ra, 3va & 3wc). It is worthwhile to mention that the chloro group on the substrates remained intact (3ga, 3ka, 3pc, 3sc & 3ua), offering opportunities for downstream transformations. The structure of our products was clearly confirmed by X-ray structural analysis of single-crystal 3ra,12 which was generated by recrystallization from a toluene/hexane solution.| a Reaction conditions: Pd(CH3CN)2Cl2 (5 mol%), tBuDavePhos (20 mol%), NEt3 (0.08 mmol), pempidine (0.32 mmol), CH3CN/DMF (1 mL; 10/1), 100 °C, 18 h. Values represent isolated yields. b The reaction was performed in the presence of 2-iodophenol 1 (0.22 mmol), silacyclobutane 2 (0.2 mmol), Pd(MeCN)2Cl2 (2.6 mg, 5 mol%) and tBuDavePhos (13.6 mg, 20 mol%) in DMF (91 μL) and MeCN (0.91 mL) at 100 °C under N2 for 8 h. Then additional Pd(MeCN)2Cl2 (2.6 mg, 5 mol%) was added to the reaction and heated for another 10 h. For more experimental details, see ESI. |
|---|
|
The broad substrate scope encouraged the evaluation of this newly developed ring expansion method with natural products for potential application in the area of fragment-based drug discovery. The terpene derivative 4-iodoestrone successfully underwent ring expansion to deliver product 4a (43% yield), enabling access to 7-membered silacycles in the parent hormone. Similarly, starting from commercially available 3-iodotyrosine, a seven-membered silacycle moiety can be efficiently introduced to give product 4b in 61% yield. In addition to natural product derivatives, this reaction can also be applied to natural product-tethered 2-iodophenols. 2-Iodophenols with ether-linked L-menthol, borneol, cedrol, diosgenin, and ester-linked 18β-glycyrrhetinic acid smoothly participated in this ring expansion reaction, furnishing the desired products 4c–g in moderate to good yields.
Moreover, we also demonstrated the synthetic usefulness of the sila-tetrahydrobenzo[b]oxepine skeletons obtained by our developed transformation as versatile synthetic intermediates via diverse downstream transformations (Scheme 2). Treatment of 3aa with 40% HF in THF/H2O resulted in Si–O bond cleavage to deliver the compound 5a in 99% yield at room temperature for 1 h. We proved that the Si–O bond of product 3aa can also be preferentially cleaved by treatment with organolithium reagent or methanol through a nucleophilic ring-opening reaction, permitting access to phenolic silane 5b (98% yield) or 5c (83% yield). Additionally, product 3aa can be reduced by LiAlH4 in the presence of 10 mol% HMPA and nOct4NBr to afford noncyclic phenolic hydrosilane 5d (81% yield), which can further participate in hydrosilylation reactions as the synthetic intermediate to increase the complexity of molecules. In addition to ring-opening of the Si–O bond, the aromatic moiety on 3aa can be decorated with a bromo substituent, resulting in 7-bromo-sila-tetrahydrobenzo[b]oxepine 5e (72% yield), which could be readily modified through diverse cross-coupling reactions. For example, Stille coupling of 5e with tributyl(phenyl)stannane proceeds smoothly to afford the product 3ea in 65% yield. Friedel–Crafts acylation of the product 3aa by employment of CH3COCl/AlCl3 also proceeded smoothly to give 7-acetyl-sila-tetrahydro-benzo[b]oxepine 5f in 81% yield. Furthermore, we wondered whether the alkyl chain of our products could also be further functionalized. Taking 5b as a substrate, we first protected the hydroxyl group by treatment with methyl iodide and K2CO3 in DMSO to produce product 5g in 82% yield. Compound 5g could further smoothly undergo tandem oxidation/elimination to yield allylic silane 5h in 87% yield.
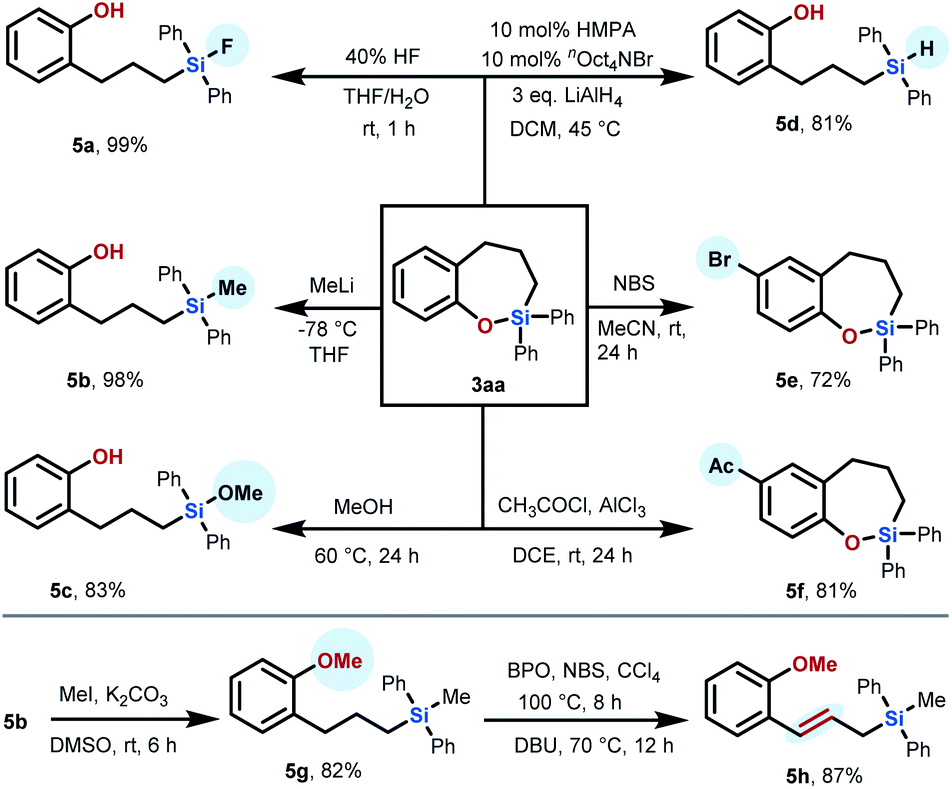 | ||
| Scheme 2 Synthetic applications of sila-benzosuberane 3aa. | ||
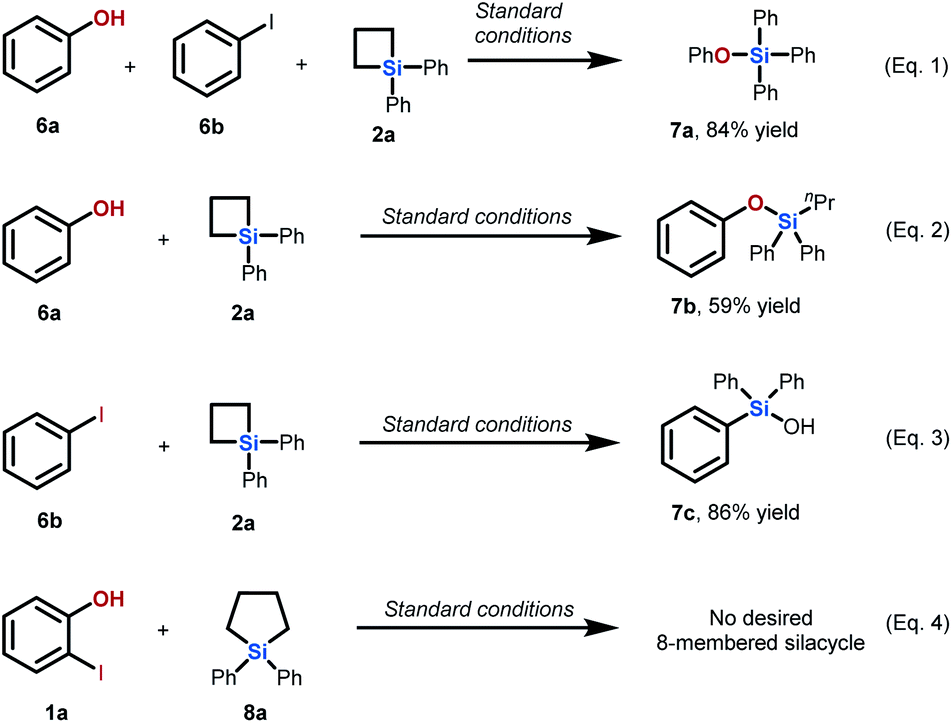
We then conducted several control experiments to support our proposal on the mechanism of this palladium-catalyzed intermolecular ring expansion reaction (Scheme 3). First, we found that no ring expansion occurred when phenol 6a, iodobenzene 6b, and SCB 2a were used as substrates under standard conditions (eqn (1)). This finding indicates that assembly of the iodo group and hydroxy group at adjacent positions of the aromatic ring is necessary to initiate this intermolecular cyclization. The ring opening reaction of SCB 2a occurred in the presence of phenol 6a to deliver product 7b in 59% yield (eqn (2)). This result indicates that phenol as the substrate is capable of attacking the silicon atom on SCB 2a under standard conditions. The reaction of iodobenzene 6b with SCB 2a produced triphenyl silanol 7c by C–Si bond formation (eqn (3)), which is in accordance with results reported in the literature. This proved that the hydroxy group on the aryl iodide played a pivotal role in inhibiting problematic C–Si bond formation and facilitating transmetallation to form C–C bonds in our newly developed reaction. In addition, we subjected 2-iodophenol 1a and 5-membered silacycle 8a to the standard conditions. No desired 8-membered silacycle was observed (eqn (4)). This result indicates that the high ring strain of SCBs is essential in this intermolecular Pd-catalyzed ring expansion reaction.
 | ||
| Scheme 3 Control experiments. | ||
Conclusions
In summary, we have described the first silacyclization strategy involving intermolecular silicon-based cross-coupling reaction enabled by a Pd-catalytic system, which offers a straightforward but strategically distinct approach for preparing sila-benzo[b]oxepines. Moreover, the formed sila-benzo[b]oxepines were shown to be versatile synthetic intermediates for the synthesis of an array of important building blocks. The key to success for this reaction is that silicon atoms have a stronger affinity for oxygen nucleophiles than carbon nucleophiles, and SCBs have inherent ring-strain-release Lewis acidity.Data availability
Data for all compounds in this manuscript are available in the ESI,† which includes experimental details, characterization and copies of 1H and 13C NMR spectra. Crystallographic data for compound 3ra has been deposited at the CCDC under CCDC 2083478.Author contributions
Y. Q., L. L., J.-Y. L. and K. L. performed the experiments. D. Z. conceived the concept, directed the project and wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (22071114, 22022103, and 21871146), the National Key Research and Development Program of China (2019YFA0210500), the “Frontiers Science Center for New Organic Matter”, Nankai University (Grant Number 63181206), the Natural Science Foundation of Tianjin (18JCYBJC20400), and the Fundamental Research Funds for the Central Universities and Nankai University.Notes and references
- For some reviews on silicon-based cross-coupling reactions, see: (a) S. E. Denmark and R. F. Sweis, Chem. Pharm. Bull., 2002, 50, 1531 CrossRef CAS PubMed; (b) S. E. Denmark and C. S. Regens, Acc. Chem. Res., 2008, 41, 1486 CrossRef CAS PubMed; (c) Y. Nakao and T. Hiyama, Chem. Soc. Rev., 2011, 40, 4893 RSC; (d) W.-T. T. Chang, R. C. Smith, C. S. Regens, A. D. Bailey, N. S. Werner and S. E. Denmark, Org. React., 2011, 75, 213 CAS; (e) H. F. Sore, W. R. J. D. Galloway and D. R. Spring, Chem. Soc. Rev., 2012, 41, 1845 RSC; (f) S. E. Denmark and A. Ambrosi, Org. Process Res. Dev., 2015, 19, 982 CrossRef CAS PubMed; (g) F. Foubelo, C. Nájera and M. Yus, Chem. Rec., 2016, 16, 2521 CrossRef CAS PubMed; (h) T. Komiyama, Y. Minami and T. Hiyama, ACS Catal., 2017, 7, 631 CrossRef CAS; (i) T. Komiyama, Y. Minami and T. Hiyama, Synlett, 2017, 28, 1873 CrossRef CAS; (j) S. Bähr, W. Xue and M. Oestreich, ACS Catal., 2019, 9, 16 CrossRef; (k) Y. Minami and T. Hiyama, Chem.–Eur. J., 2019, 25, 391 CrossRef CAS PubMed; (l) T. Hiyama, Y. Minami, and A. Mori onTransition-Metal-Catalyzed Cross-Coupling of Organosilicon Compounds in Organosilicon Chemistry, Novel Approaches and Reactions, ed. T. Hiyama and M. Oestreich, Wiley-VCH, Weinheim, 2019, pp. 271–332 Search PubMed.
- Y. Qin, J.-L. Han, C.-W. Ju and D. Zhao, Angew. Chem., Int. Ed., 2020, 59, 8481 CrossRef CAS PubMed.
- Y. Tanaka, A. Nishigaki, Y. Kimura and M. Yamashita, Appl. Organomet. Chem., 2001, 15, 667 CrossRef CAS.
- (a) Y. Nakao, H. Imanaka, A. K. Sahoo, A. Yada and T. Hiyama, J. Am. Chem. Soc., 2005, 127, 6952 CrossRef CAS PubMed; (b) Y. Nakao, M. Takeda, T. Matsumoto and T. Hiyama, Angew. Chem., Int. Ed., 2010, 49, 4447 CrossRef CAS PubMed; (c) A. Tsubouchi, D. Muramatsu and T. Takeda, Angew. Chem., Int. Ed., 2013, 52, 12719 CrossRef CAS PubMed.
- Prof. Denmark’s group has proved that alkoxide nucleophiles are capable of promoting silicon-based cross-coupling via coordination with the silicon atom in an intermolecular manner. For some references, see: (a) S. E. Denmark and R. F. Sweis, J. Am. Chem. Soc., 2001, 123, 6439 CrossRef CAS PubMed; (b) S. E. Denmark, J. D. Baird and C. S. Regens, J. Org. Chem., 2008, 73, 1440 CrossRef CAS PubMed; (c) S. E. Denmark and J. M. Kallemeyn, J. Org. Chem., 2005, 70, 2839 CrossRef CAS PubMed.
- For reviews on SCBs, see: (a) L. J. Li, Y. B. Zhang, L. Gao and Z. L. Song, Tetrahedron Lett., 2015, 56, 1466 CrossRef CAS; (b) Q. C. Mu, J. Chen, C. G. Xia and L. W. Xu, Coord. Chem. Rev., 2018, 374, 93 CrossRef CAS . For some recent progress, see:; (c) K. Hirano, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2007, 129, 6094 CrossRef CAS PubMed; (d) R. Shintani, K. Moriya and T. Hayashi, J. Am. Chem. Soc., 2011, 133, 16440 CrossRef CAS PubMed; (e) N. Ishida, W. Ikemoto and M. Murakami, J. Am. Chem. Soc., 2014, 136, 5912 CrossRef CAS PubMed; (f) Q. W. Zhang, K. An, L. C. Liu, S. X. Guo, C. R. Jiang, H. F. Guo and W. He, Angew. Chem., Int. Ed., 2016, 55, 6319 CrossRef CAS PubMed; (g) S. Okumura, F. Sun, N. Ishida and M. Murakami, J. Am. Chem. Soc., 2017, 139, 12414 CrossRef CAS PubMed; (h) Q.-W. Zhang, K. An, L.-C. Liu, Q. Zhang, H. Guo and W. He, Angew. Chem., Int. Ed., 2017, 56, 1125 CrossRef CAS PubMed; (i) W. T. Zhao, F. Gao and D. Zhao, Angew. Chem., Int. Ed., 2018, 57, 6329 CrossRef CAS PubMed; (j) H. Chen, Y. Chen, X. Tang, S. Liu, R. Wang, T. Hu, L. Gao and Z. Song, Angew. Chem., Int. Ed., 2019, 58, 4695 CrossRef CAS PubMed; (k) X.-B. Wang, Z.-J. Zheng, J.-L. Xie, X.-W. Gu, Q.-C. Mu, G.-W. Yin, F. Ye, Z. Xu and L.-W. Xu, Angew. Chem., Int. Ed., 2020, 59, 790 CrossRef CAS PubMed; (l) L. Zhang, K. An, Y. Wang, Y.-D. Wu, X. Zhang, Z.-X. Yu and W. He, J. Am. Chem. Soc., 2021, 143, 3571 CrossRef CAS PubMed; (m) M.-H. Zhu, X.-W. Zhang, M. Usman, H. Cong and W.-B. Liu, ACS Catal., 2021, 11, 5703 CrossRef CAS.
- (a) F. A. Macias, J. M. G. Molinillo, R. M. Varela, A. Torres and F. R. Fronczek, J. Org. Chem., 1994, 59, 8261 CrossRef CAS; (b) J. B. P. A. Wijnberg, A. van Veldhuizen, H. J. Swarts, J. C. Frankland and J. A. Field, Tetrahedron Lett., 1999, 40, 5767 CrossRef CAS; (c) M. Shiraishi, Y. Aramaki, M. Seto, H. Imoto, Y. Nishikawa, N. Kanzaki, M. Okamoto, H. S. Awada, O. N. Ishimura, M. B. Aba and M. Fujino, J. Med. Chem., 2000, 43, 2049 CrossRef CAS PubMed; (d) D. G. Lloyd, R. B. Hughes, D. M. Zisterer, D. C. Williams, C. Fattorusso, B. Catalanotti, G. Campiani and M. J. Meegan, J. Med. Chem., 2004, 47, 5612 CrossRef CAS PubMed; (e) V. K. Tandon, K. A. Singh, A. K. Awasthi, J. M. Khanna, B. Lal and N. Anand, Bioorg. Med. Chem. Lett., 2004, 14, 2867 CrossRef CAS PubMed; (f) S. Kim, Y.-W. Chin, B.-N. Su, S. Riswan, L. B. S. Kardono, J. J. Afriastini, H. Chai, N. R. Farnsworth, G. A. Cordell, S. M. Swanson and A. D. Kinghorn, J. Nat. Prod., 2006, 69, 1769 CrossRef CAS PubMed; (g) P. K. Singh and O. Silakari, ChemMedChem, 2018, 13, 1071 CrossRef CAS PubMed.
- (a) W. Bains and R. Tacke, Curr. Opin. Drug Discovery Dev., 2003, 6, 526 CAS; (b) G. A. Showell and J. S. Mills, Drug Discovery Today, 2003, 8, 551 CrossRef CAS PubMed; (c) P. Englebienne, A. V. Hoonacker and C. V. Herst, Drug Des. Rev., 2005, 2, 467 CAS; (d) A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388 CrossRef CAS PubMed; (e) R. Ramesh and D. S. Reddy, J. Med. Chem., 2018, 61, 3779 CrossRef CAS PubMed; (f) S. J. Barraza and S. E. Denmark, J. Am. Chem. Soc., 2018, 140, 6668 CrossRef CAS PubMed.
- (a) G. Bertrand, P. Mazerolles and J. Ancelle, Tetrahedron, 1981, 37, 2459 CrossRef CAS; (b) C. P. Lenges and M. Brookhart, J. Am. Chem. Soc., 1997, 119, 3165 CrossRef CAS; (c) M. Parasram, V. O. Iaroshenko and V. Gevorgyan, J. Am. Chem. Soc., 2014, 136, 17926 CrossRef CAS PubMed.
- N. Ishida, S. Okumura, T. Kawasaki and M. Murakami, Angew. Chem., Int. Ed., 2018, 57, 11399 CrossRef CAS PubMed.
- (a) P. Jeschke, Chem BioChem., 2004, 5, 570 CrossRef CAS PubMed; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (c) J. Wang, M. Sánchez-Roselló, J. L. Aceño, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed; (d) E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832 CrossRef CAS PubMed.
- CCDC 2083478 (3ra) contains the supplementary crystallographic data.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 2083478. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04180k |
| This journal is © The Royal Society of Chemistry 2021 |