
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiastereoselective formation of homochiral flexible perylene bisimide cyclophanes and their hybrids with fullerenes†‡
Iris
Solymosi§
 a,
Swathi
Krishna§
b,
Edurne
Nuin
c,
Harald
Maid
a,
Barbara
Scholz
a,
Dirk M.
Guldi
b,
M. Eugenia
Pérez-Ojeda
*a and
Andreas
Hirsch
*a
a,
Swathi
Krishna§
b,
Edurne
Nuin
c,
Harald
Maid
a,
Barbara
Scholz
a,
Dirk M.
Guldi
b,
M. Eugenia
Pérez-Ojeda
*a and
Andreas
Hirsch
*a
aDepartment of Chemistry and Pharmacy, Friedrich-Alexander-University Erlangen-Nuremberg, Nikolaus-Fiebiger-Straße 10, 91058 Erlangen, Germany. E-mail: andreas.hirsch@fau.de; eugenia.perez-ojeda@fau.de
bDepartment of Chemistry and Pharmacy, Friedrich-Alexander-University Erlangen-Nuremberg, Egerlandstraße 3, 91058 Erlangen, Germany. E-mail: dirk.guldi@fau.de
cInstituto de Ciencia Molecular (ICMol), Universidad de Valencia, Catedrático José Beltrán 2, Paterna 46980, Spain
First published on 8th October 2021
Abstract
Cyclophanes of different ring sizes featuring perylene-3,4:9,10-tetracarboxylic acid bisimide (PBI) linked by flexible malonates were designed, synthesized, and investigated with respect to their structural, chemical and photo-physical properties. It is predominantly the number of PBIs and their geometric arrangement, which influence dramatically their properties. For example, two-PBI containing cyclophanes reveal physico-chemical characteristics that are governed by strong co-facial π–π interactions. This is in stark contrast to cyclophanes with either three or four PBIs. Key to co-facial π–π stackings are the flexible malonate linkers, which, in turn, set up the ways and means for diastereoselectivity of the homochiral PBIs at low temperatures, on one hand. In terms of selectivity, diastereomeric (M,M)/(P,P)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :(M,P)/(P,M) pairs with a ratio of approximately 10:1 are discernible in the 1H NMR spectra in C2D2Cl4 and a complete diastereomeric excess is found in CD2Cl2. On the other hand, symmetry-breaking charge transfer as well as charge separation at room temperature are corroborated in steady-state and time-resolved photo-physical investigations. Less favourable are co-facial π–π stackings in the three-PBI containing cyclophanes. For statistical reasons, the diastereoisomers (M,M,M)/(P,P,P) and (M,M,P)/(P,P,M) occur here in a ratio of 1:3. In this case, symmetry-breaking charge transfer as well as charge separation are both slowed down. The work was rounded-off by integrating next to the PBIs, for the first time, hydrophobic or hydrophilic fullerenes into the resulting cyclophanes. Our novel fullerene–PBI cyclophanes reveal unprecedented diastereoselective formation of homochiral (M,M)/(P,P) pairs exceeding the traditional host–guest approach. Hybridization with fullerenes allows us to modulate the resulting solubility, stacking, cavity and chirality, which is of tremendous interest in the field.
:(M,P)/(P,M) pairs with a ratio of approximately 10:1 are discernible in the 1H NMR spectra in C2D2Cl4 and a complete diastereomeric excess is found in CD2Cl2. On the other hand, symmetry-breaking charge transfer as well as charge separation at room temperature are corroborated in steady-state and time-resolved photo-physical investigations. Less favourable are co-facial π–π stackings in the three-PBI containing cyclophanes. For statistical reasons, the diastereoisomers (M,M,M)/(P,P,P) and (M,M,P)/(P,P,M) occur here in a ratio of 1:3. In this case, symmetry-breaking charge transfer as well as charge separation are both slowed down. The work was rounded-off by integrating next to the PBIs, for the first time, hydrophobic or hydrophilic fullerenes into the resulting cyclophanes. Our novel fullerene–PBI cyclophanes reveal unprecedented diastereoselective formation of homochiral (M,M)/(P,P) pairs exceeding the traditional host–guest approach. Hybridization with fullerenes allows us to modulate the resulting solubility, stacking, cavity and chirality, which is of tremendous interest in the field.
Introduction
Following the discovery of [2.2]paracyclophane by Brown and Farthing1 and its targeted synthesis with high dilution by Cram and Steinberg,2 cyclophanes have gained more and more importance in recent decades. For our macrocycles, PBIs were chosen as building blocks owing to their unique physico-chemical as well as thermal properties and, in turn, great potential for applications.3 PBIs are, for example, used as semiconductors in field effect transistors (FETs)4–6 or as components in various solar cells.7,8 In addition, they self-organize based on π–π interactions.9,10 PBI containing cyclophanes have recently received increasing attention. Leading examples from Würthner's group are the demonstration of tunable electronic interactions between PBIs11 or of fluorescent sensing to detect electron-rich and electron-poor guests.12 Within the cyclophanes, the PBIs are held together via either flexible13–15 or rigid linkers.12,16 Notably, the linker conditions the cavity given by the cyclophanes. This has resulted in incorporating small aromatic hydrocarbons,12,17 homochiral molecules,17,18 and fullerenes.19 Despite the fact that flexible and long linkers make the incorporation rather difficult due to PBI self-aggregation, fullerenes have even been encapsulated in a PBI-containing macrocycle with flexible linkers.19 Strong π–π interactions are, however, advantageous for promoting the cyclization through self-organization14,20 Traditionally, cyclization has been favored over oligomerization by either high dilutions13,19,21 or templating effects.12,22 Up to six23 or even nine PBIs24 within a macrocycle have been incorporated and isolated so far.Controlling the size of the cyclophanes and the arrangement of the molecular building blocks therein ultimately governs the properties and, in turn, determines the incorporation of molecular guests. Thus, in the current contribution we have also tackled the template effect to favor a dimeric PBI-arrangement and compared the π–π stacking interactions in two-PBI-containing cyclophanes with those featuring either three or four PBIs. Importantly, PBIs were linked through flexible malonate linkers to enable the functionalization of the cyclic dimer with hydrophobic and hydrophilic fullerenes.25 Incentives to control the cyclophane surroundings were to tune the physico-chemical characteristics and to alter bindings of guests through, for example, hydrophobic effects and other non-covalent interactions. To the best of our knowledge, no PBI-based cyclophanes have ever been functionalized with fullerenes with the objective to investigate such a sophisticated molecular design.
Results and discussion
Synthesis
Scheme 1 shows the synthetic procedure of cyclophanes starting from N,N′-dihydroxypropyl-1,6,7,12-tetrakis-(4-tert-butylphenoxy)-3,4:9,10-perylenetetracarboxydiimide 1, which was prepared in a two-step synthesis according to modified literature procedures.26,27 Cyclization of 128,29 with malonyl dichloride using pyridine30 as a base provides P2, P3, and P4 as purple solids in 8.1%, 3.4%, and 1.3% yields, respectively. 2–4 indicate the associated number of PBIs (P) in the ring.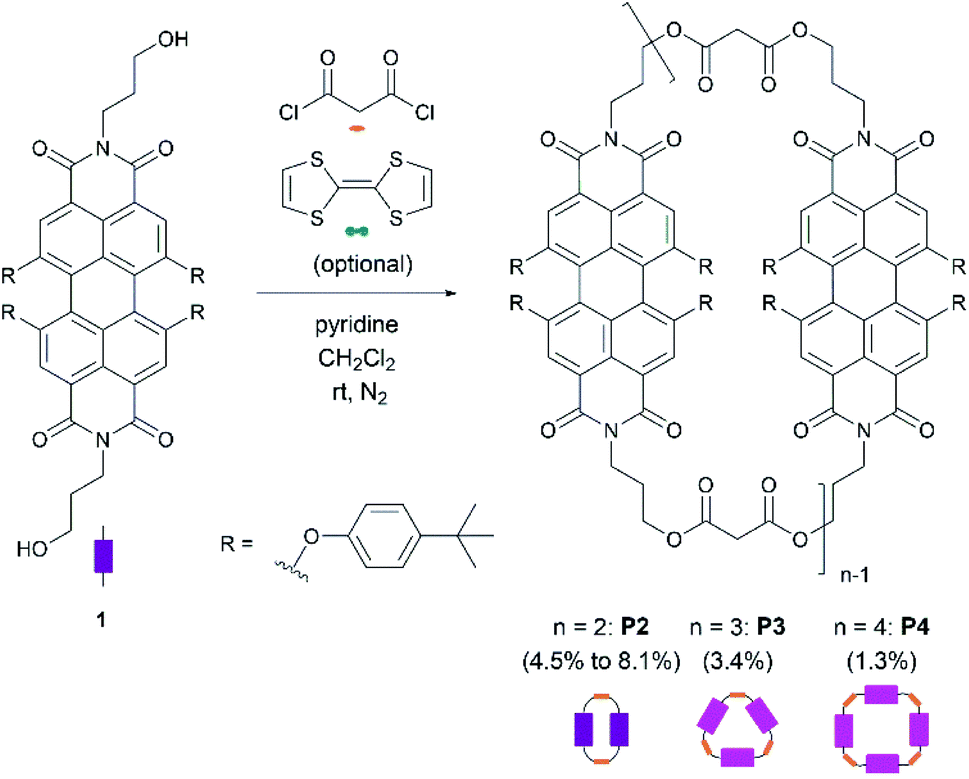 | ||
| Scheme 1 Cyclization reaction for the synthesis of PBI-based macrocycles P2, P3, and P4. | ||
A generalized methodology, which promotes intramolecular ring closure based on the Ziegler–Ruggli principle, takes place under high dilution conditions to avoid polymerization reactions.31 For this purpose, malonyl dichloride was slowly added using an automatic syringe pump over time during the reaction. In addition to P2, larger macrocycles as well as open-chain oligomers and polymers were formed. They were detected in MALDI-TOF experiments (Fig. 1). Due to the difficult isolation and scarce yields, macrocycles larger than P4 were not further investigated.
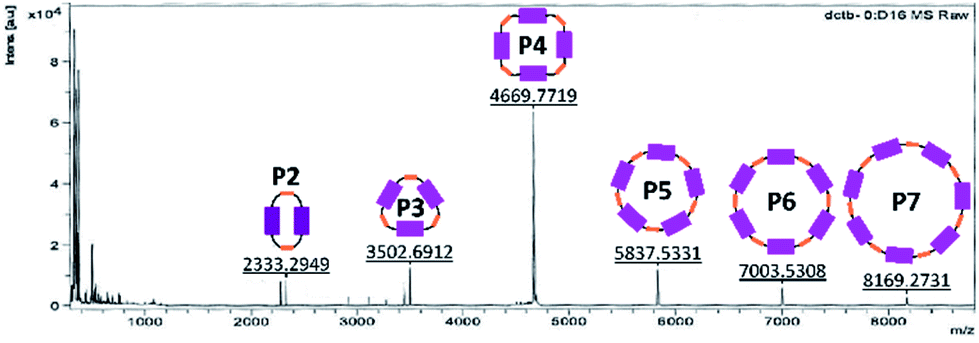 | ||
| Fig. 1 MALDI-TOF spectrum of a fraction already purified by size exclusion and column chromatography in order to identify the bigger cycles (pos. mode, DCTB in CH2Cl2). | ||
To promote the formation of a specific macrocycle size, numerous intramolecular ring closure conditions were tested. To this end, several addition time spans, dilutions, and stoichiometries were investigated – Table 1. First, the addition time of one equivalent of malonyl dichloride was investigated in CH2Cl2 in a range from 1 to 30 h.
| ENa | Ratiob
|
c
|
c
|
t add [h] | t stir [d] |
|
|---|---|---|---|---|---|---|
|
a Number of experiment.
b Ratio of precursor 1:malonyl dichloride:TTF.
c Concentration of precursor molecule 1 in the reaction mixture.
d Concentration of malonyl dichloride in the syringe.
e Addition time span of malonyl dichloride.
f Time of stirring (r.t.).
|
||||||
| A | 1:1:0 |
197 | 2.44 | 3 | 6 | 2.7 |
| B | 1:1:0 |
515 | 20.4 | 3 | 8 | 4.5 |
| C | 1:1:0 |
515 | 20.4 | 3 | 6 | 4.2 |
| D | 1:1:0.5 |
515 | 20.4 | 3 | 7 | 7.2 |
| E | 1:4:0.5 |
515 | 4 × 24.4 | 4 × 3 | 3 | 8.1 |
| F | 1:3:0 |
515 | 48.8 | 3 | 1 | 8.1 |


Based on MALDI-TOF and HPLC experiments, which revealed only traces of P2 even after prolonged malonyl additions, the addition time span was set to 3 h per equivalent (experiments A–E). Second, the dilution conditions were examined. Reaction of 1 with malonyl dichloride at very low concentrations affords P2 in a 2.7% yield (Table 1, experiment A). Higher concentrations of both building blocks resulted in an increased two-PBI cyclophane yield (Table 1, experiments B and C) revealing that dilution optimization of the reaction conditions is crucial in the overall formation of P2. Furthermore, the reproducibility of all of these conditions was proven in experiment C. The macrocycle fraction was purified by size exclusion chromatography (Biobeads S-X1, dry mesh size: 28–74 μm, CH2Cl2) followed by column chromatography (SiO2, using different solvent gradients depending on the macrocycle). Moreover, the separation and characterization of P3 (3.4%) and P4 (1.3%) were successful as well.
Prearranging the building blocks through a template effect32,33 is expected to favor their spatial disposition to afford specific cyclophane geometries and sizes. Specifically, considering the electron-acceptor strength of PBIs, we argued that an electron-donating tetrathiafulvalene34 (TTF) should serve as a template.35 TTF is oxidized in cyclic voltammetry experiments at +0.34 and +0.71 V vs. Ag/AgCl in CH2Cl2,36 while PBI is reduced at −1.08 and −1.23 V vs. Fc/Fc+ in CH2Cl2.37 According to these reduction and oxidation potentials, electron donor–acceptor interactions are likely to promote a pre-arranged, sandwich-like structure, which favors the formation of the two-PBI ring, similar to the template-controlled synthesis of tetracationic macrocycles by Stoddart.38 As such, the cyclization reaction was carried out in the presence of TTF (experiment D) to give P2 in 7.2% yield. This is a 1.6-fold enhancement relative to experiment B. TTF was easily removed from P2 by size-exclusion chromatography using CH2Cl2 as the eluent due to its relatively weak supramolecular interaction.
Non-reacted starting material 1 was recovered under the reaction conditions in experiments A to D as detected by HPLC (vide infra). Thus, in another approach malonyl dichloride was added to the reaction mixture until all of 1 was consumed with or without any templating agent. Here, when TTF is present in the reaction mixture (experiment E), the yield of P2 was raised to 8.1%, which is slightly larger than the 7.2% yield seen in the templated experiment D. An excess of malonyl dichloride improves the yields of P2, but at the expense of higher polymer formation. The latter were successfully separated from the crude mixture by size-exclusion chromatography as part of a more straightforward purification process. When the cyclization reaction was carried out with an excess of malonyl dichloride and without any TTF (experiment F), P2 was furnished in 8.1% as in experiment E. This fact reveals that either the TTF-templated synthesis or the use of an excess of malonyl dichloride results in increased P2 formation. However, the combination of both strategies in the same cyclization reaction does not result in a cooperative yield increase. To better exploit the template effect, other planar electron-rich aromatic hydrocarbons such as anthracene are foreseeable as template molecules given that the binding interaction with PBI cyclophanes has already been demonstrated.12
Reaction control by analytical HPLC
The reaction course as well as the product distribution under each reaction conditions were monitored with analytical high-performance liquid chromatography (HPLC) with a Nucleosil column (EC250/4 Nucleosil 100–5) using CH2Cl2:ethyl acetate:MeOH 1:0:0 → 75:20:5 as the mobile phase. HPLC profiles of experiment A (high dilution), experiment C (low dilution) and isolated P2, P3, and P4 are shown in Fig. S1 (in the ESI‡). Despite the fact that P2, P3, and P4 were detected in the crude mixture, as well as other by-products, their UV-vis spectra could not be compared quantitatively, due to their different spectral intensities as well as their different ε values (the detailed photophysical information is shown below).
When HPLC measurements of experiments A, C, and D were performed, a relatively large amount of unreacted starting material 1 was detected at an elution time of 17 min. Another peak with an elution time of 20 min was assigned to P2open in agreement with MALDI-TOF measurements. Mass spectra as well as the corresponding molecular structure of P2open, are presented in Fig. S1 (in the ESI‡). P2open paves the way for the synthesis of more complex asymmetric hetero-structures when combined with other building blocks.
In addition, HPLC studies of the crude mixture using the TTF-template (experiment D) were carried out. Under these conditions, P2 and TTF were detected separately (Fig. S3 in the ESI‡). This confirms that the interactions between P2 and TTF are weak, which is in agreement with our previous observations during the purification size-exclusion by chromatography. TTF ensures the necessary pre-arrangement of 1 to undergo cyclization, but also enables the target to be separated from the template. Finally, when an excess of malonyl dichloride was used in the cyclization reaction (experiment E, Fig. S4 in the ESI‡), an increased P2 peak intensity in the HPLC profiles went hand-in-hand with decreased peak intensities for 1 and P2open.
Functionalisation with fullerene pentakisadducts
The incentive to incorporate fullerenes was to exploit them as bulky molecular scaffolds, which assist in modifying the chemical environment of the cyclophane. For this purpose, flexible malonates were chosen as linkers between the PBIs to open the pathway towards the macrocycle functionalization with fullerenes. Thus, P2 was derivatized with fullerene pentakisadducts bearing different malonate ester groups. Chemical modification of the environment via functionalized fullerenes with hydrophobic or hydrophilic substituents will, in turn, tune the molecular properties and facilitate the study of inter-PBI interactions. We envisioned that the modulation of π–π stacking forces will drive the dynamics of intramolecular PBI self-assembly and chiral interaction. Preparation of P2F2Et and P2F2TEG (Scheme 3) as well as reference P1F2Et (Scheme 2) was carried out by means of the Bingel–Hirsch cyclopropanation of the corresponding malonates with ethyl (Et) or tetraethylene glycol (TEG) substituted fullerene pentakisadducts. These were prepared in a two-step synthesis as described by Pérez-Ojeda et al.25 Under these conditions, P2F2Et, P2F2TEG, and P1F2Et were obtained in 70%, 27%, and 62% yields, respectively.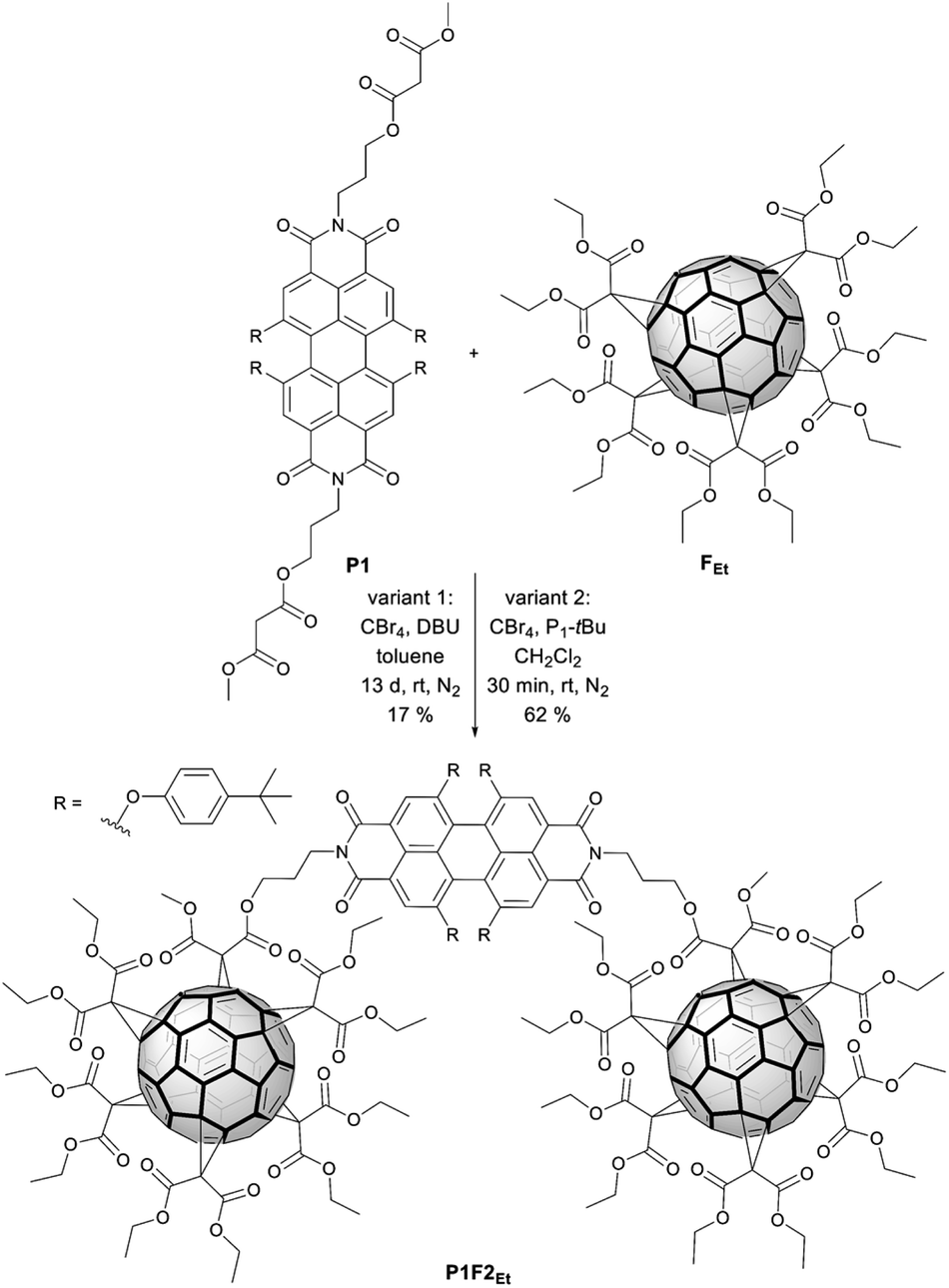 | ||
| Scheme 2 Two different variants for the synthesis of reference P1F2Et. | ||
Synthesis of a model compound
Reference P1F2Et with only one PBI (P1, in the ESI‡) as a bridge between two fullerenes with ethyl substituents (F2Et) was synthesized (Scheme 2). This is similar to two fullerenes bridged to one another via a naphthalene bisimide-containing bis(ethyl malonate) as described by Nishimura et al.39,¶ Typical Bingel–Hirsch conditions were used for the preparation of the reference compound. P1F2Et was obtained in one-step from P1 by a reaction with fullerene pentakisadduct FEt using CBr4 and 1,8-diazabicycloundec-7-ene (DBU) in toluene (Scheme 2, variant 1).40P1F2Et was isolated as a purple solid in 17% yield. To overcome the low yield of P1F2Et, we modified in a second approach our reaction conditions, which are shown in Scheme 2 (variant 2). DBU was replaced by the Schwesinger phosphazene base P1-tBu for the in situ generation of α-bromo-malonate. CH2Cl2 was used as a solvent rather than toluene.41–43 Under these conditions, P1 was completely converted after a few minutes and P1F2Et was afforded in 62% yield.Synthesis of functional hybrids
With the optimized conditions at hand, P2F2Et and P2F2TEG were prepared in 70% and 27% yields, respectively (Scheme 3). It is worth noting that for a complete derivatization of P2 more equivalents of CBr4 and P1-tBu had to be used than in the P1F2Et preparation. Owing to the large steric hindrance of the TEG chains, the reaction time for forming P2F2TEG was significantly longer and only a moderate yield of 27% was isolated in comparison with P2F2Et. Interestingly, in the synthesis of the cyclophanes endowed with two fullerenes, a by-product P2F1TEG with only one fullerene and two bromine substituents was isolated. Its structure was confirmed by the isotopic pattern of a high-resolution mass spectrum (Fig. S37 in the ESI‡) and by 1H-NMR integration. Our methodology allows PBI-cyclophane functionalization not only with fullerene derivatives bearing different substituents to tune their solubility, but also with pristine fullerenes to use the resulting hybrid for optoelectronic applications of high interest and demand.44 Thus, this simple and innovative molecular design represents a groundbreaking strategy towards a new generation of optical materials with tailored properties.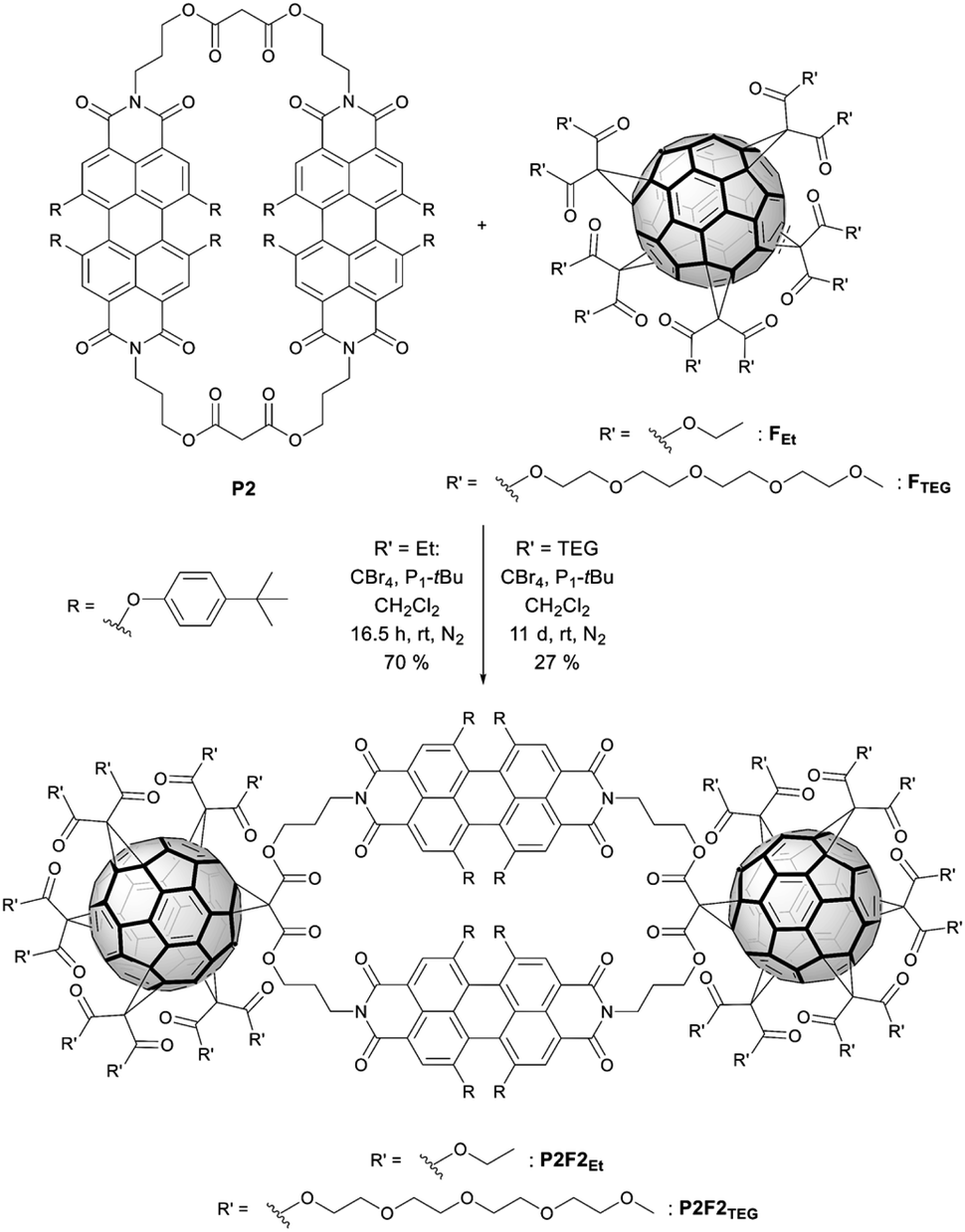 | ||
| Scheme 3 Synthesis of the functional hybrids P2F2Et and P2F2TEG. | ||
The reference as well as the functional hybrids were investigated using spectroscopic methods. The corresponding structures were unambiguously characterized by HRMS, NMR (1H and 13C) as well as UV-vis and fluorescence spectroscopies.
Due to the different amount of PBIs and their arrangements within the cyclophanes, intramolecular π–π stacking interactions prevail in P2. This leads to a spectrum, which differs from P3 and P4. In addition, the presence of fullerenes has a profound influence on the properties in comparison to P2. To examine the structure–property relationship, temperature-dependent NMR, steady-state UV-vis, and fluorescence spectroscopy and transient absorption experiments were carried out.
Temperature-dependent 1H NMR spectroscopy
Concerning NMR spectroscopy, symmetry changes due to dynamic processes and their timescales play an important role and have to be considered. Bay-substituted perylenes are twisted rather than planar due to the steric hindrance of their substituents.45 Therefore, two atropisomers (M/P) of lower symmetry, compared to flat perylene, exist (Scheme 4). For substituents like 4-tert-butylphenoxy it is, however, known that the M ↔ P interconversion (butterfly dynamic) is fast at room temperature.46 For this reason, NMR spectra are expected, which reflect higher symmetry, including symmetry elements of the second kind such as mirror planes. In particular, we awaited 1H-NMR spectra compatible with point groups of D2h for P1, P2, P1F2Et, and P2F2Et, D3h for P3 as well as D4h for P4. | ||
| Scheme 4 Representation of the stereochemistry of the PBI atropisomers M (purple) and P (grey). | ||
The 1H-NMR spectra of P1–P4 in CDCl3 measured at room temperature are presented in Fig. 2. Reference P1 shows a single set of sharp signals indicating fast atropisomerization of the M and P isomers. Between 8.5 and 6.5 ppm, we see a singlet for the PBI-protons (Fig. 2, a) and two multiplets for the AA′BB′ spin system of the para-substituted system of the aromatic bay-substituents (Fig. 2, b and c). The three CH2-groups of the alkyl chain appear as two signals, a triplet like multiplet at 4.2 ppm for the almost isochronous protons of the methylene groups linked to oxygen and nitrogen (Fig. 2, d and e), and a pseudo quintet at 2.0 ppm for the central CH2-group. A singlet was found for the malonate CH2-groups at 3.3 ppm. The spectrum is completed with two singlets for OCH3 and tert-butyl groups at 3.7 and 1.3 ppm, respectively. For P3 and P4, the same set of signals was observed reflecting the high symmetry of D2h for P1, D3h for P3, and D4h for P4. In contrast, the 1H-NMR spectrum of P2 is rather different. All signals are strongly broadened or barely visible (Fig. 2, second row). Additionally, a split of the signals for the OCH2- and the central CH2-groups (Fig. 2, eA, eB gA, gB) is observed. Obviously, atropisomerization is slow at the NMR timescale.
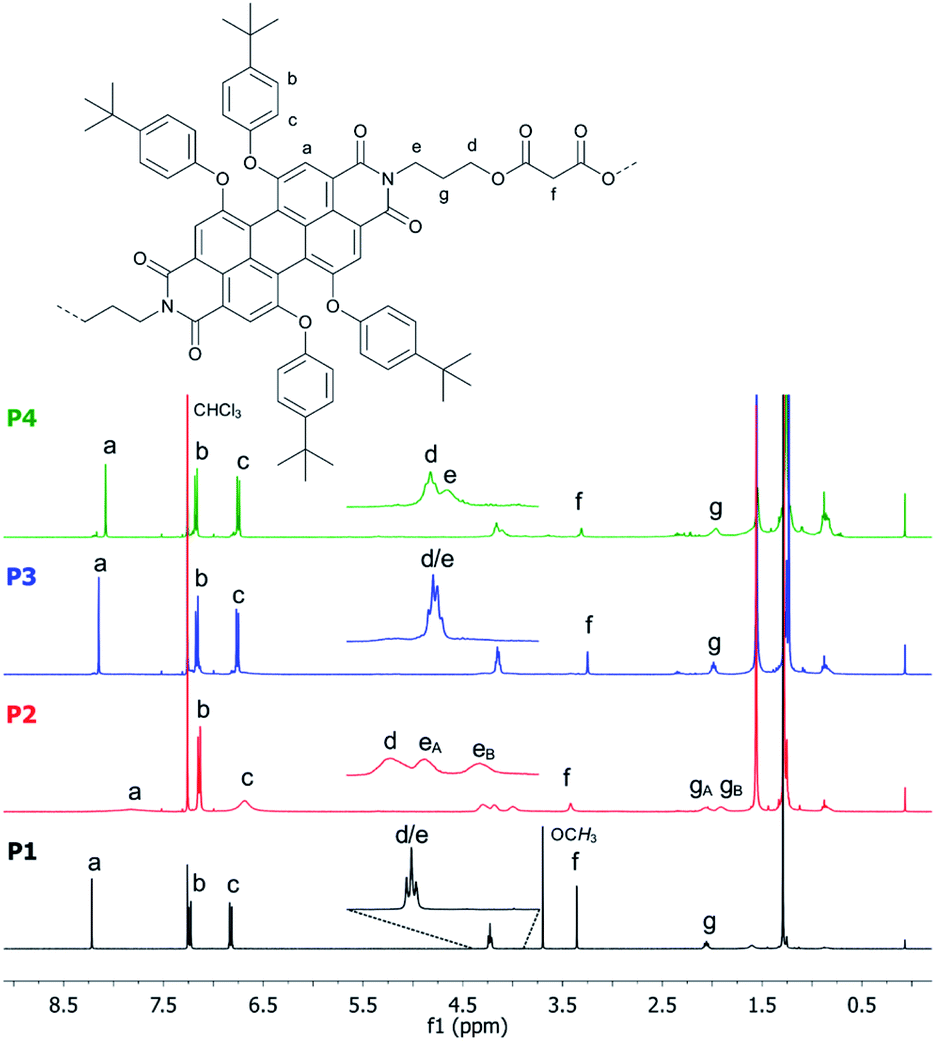 | ||
| Fig. 2 Comparison of the 1H NMR (400 MHz) spectra of P1 (bottom), P2, P3 and P4 (top) recorded in CDCl3 at room temperature. | ||
To further investigate the dynamic behaviour and to understand the stereo-chemical consequences, we conducted a detailed NMR study of P2 and P3 at high and low temperatures. Upon cooling of P2 in [D2]-tetrachloroethane we observed a sharpening of the peaks and a splitting of the signals in the aromatic region, most clearly visible at −15 °C (Fig. 3). Here, the epimerization via a butterfly twist of the perylene is slow on the NMR timescale. In principle, P2 exists as a racemic mixture of two diastereoisomers, homochiral (M,M)-P2/(P,P)-P2 and mesocate (M,P)-P2/(P,M)-P2 (Fig. 4a). In the 1H NMR spectrum, we attribute the main signals to the racemic mixture of homochiral (M,M)-P2/(P,P)-P2 for the following reason. Here, the symmetry is reduced to D2 and, consequently, the protons of the PBIs (Fig. 3, a) as well as the protons of the aromatic bay-substituents (Fig. 3, b and c) form two heterotopic sets of four and eight homotopic protons, respectively.
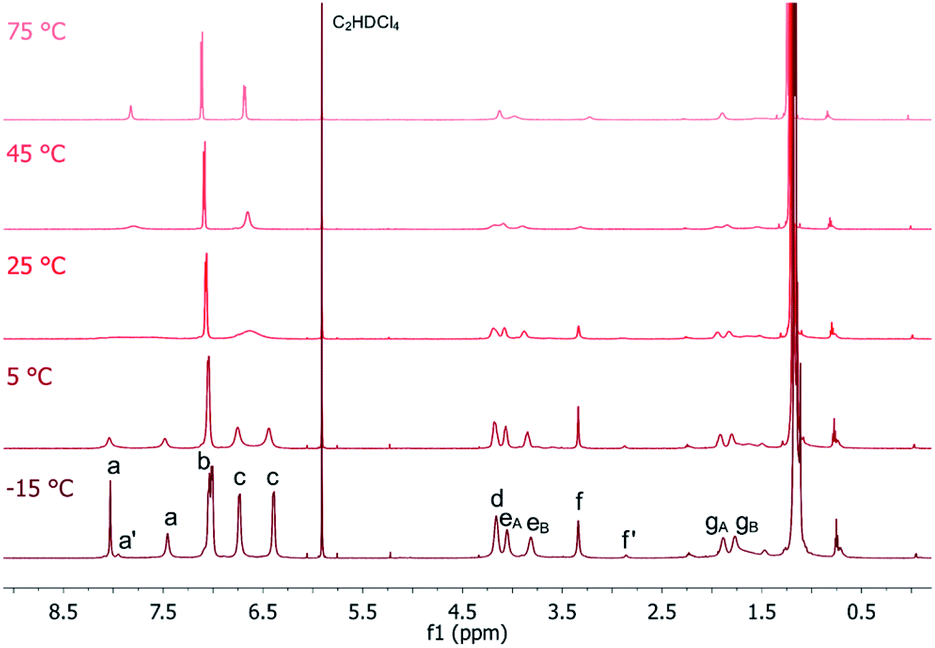 | ||
| Fig. 3 Temperature-dependent 1H NMR (600 MHz) spectra of P2 recorded in C2D2Cl4. | ||
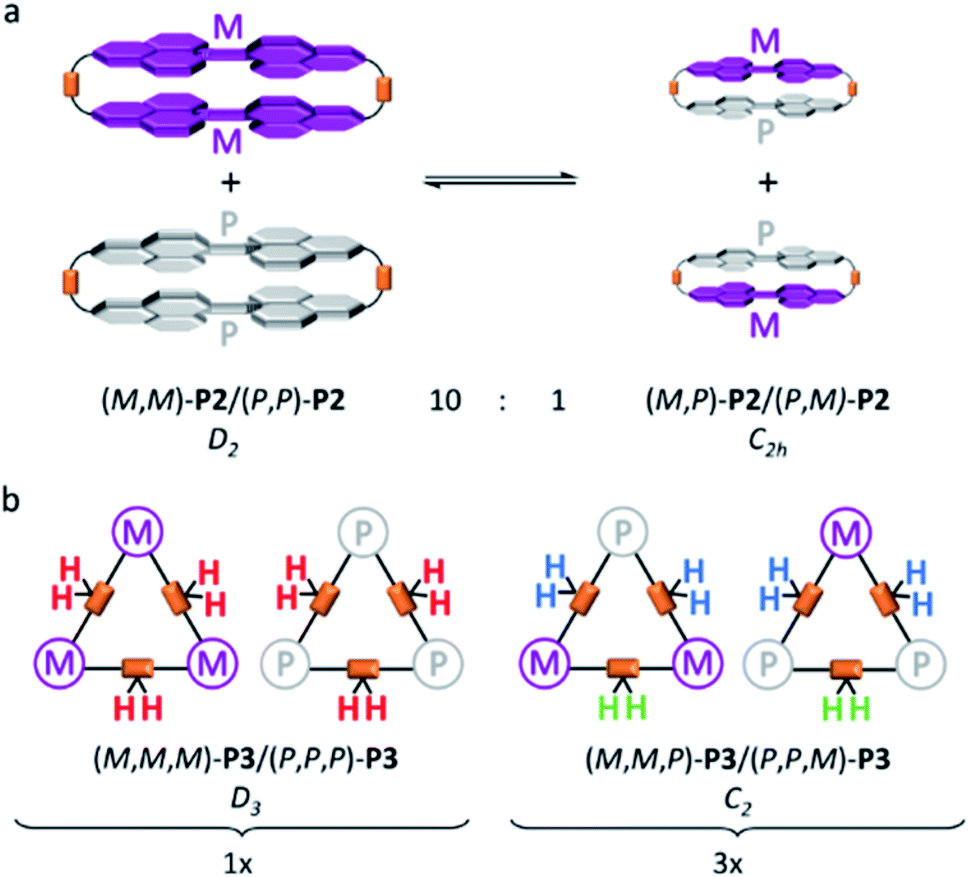 | ||
| Fig. 4 Schematic representation of conformational equilibrium between the homochiral (M,M)-P2/(P,P)-P2 and the achiral mesocate (M,P)-P2/(P,M)-P2 with a ratio of 10:1 in C2D2Cl4 at −15 °C (a) and statistical distribution of (M,M,M)-P3/(P,P,P)-P3 and (M,M,P)-P3/(P,P,M)-P3 of 1:3 (b). | ||
We distinguish between one set of protons, which protrudes into the cage of the cyclophane (endo) and one set which protrudes from it (exo). The protons of the alkyl chains in this environment are diastereotopic and due to the presence of three C2-axes again two sets of four homotopic protons for each CH2-group appear (Fig. 3, eA/B and gA/B). For one methylene-group (Fig. 3, f), no splitting was observed due to isochrony. Interestingly, the malonate protons are homotopic, because they are interchanged by the three C2-axes. Consequently, they appear as one singlet. This would also happen, if diastereotopic protons are isochronous. However, upon close inspection of the 1H-NMR at −15 °C we found additional signals for the PBI- (Fig. 3, a’) and malonate-protons (Fig. 3, f’), which we attribute to the achiral mesocate (M,P)-P2/(P,M)-P2.
Due to the presence of a mirror plane (point group C2h) this diastereomer is achiral. But, no symmetry element interchanges the malonate-protons. These appear as two doublets (2JHH = 17 Hz), best visible at −5 °C, and make our assignment unambiguous (Fig. S5 in the ESI‡). To prove that this signal group belongs to the different diastereoisomers of P2, we performed 1D-EXSY in the rotating frame at 5 °C (Fig. S6 in the ESI‡). Upon irradiation of each signal group exchange peaks appeared, showing the slow interconversion of (M,P)-P2 and its enantiomer into (M,M)-P2 and its enantiomer and vice versa.
The integral ratio between (M,M)-P2/(P,P)-P2 and (M,P)-P2/(P,M)-P2 is 10:1 in C2D2Cl4 at −15 °C (Fig. S7 in the ESI‡). Such an impressive diastereoselectivity for (M,M)-P2/(P,P)-P2 is rationalized on grounds of attractive π–π interactions between the PBIs which are facilitated by the flexible linkers. This is even more pronounced in the case of P2F2Et. No evidence for the presence of (M,P)-P2F2Et/(P,M)-P2F2Et in a variable temperature 1H NMR assays was gathered (Fig. S8 in the ESI‡). Furthermore, the diastereoselectivity is solvent dependent. In CD2Cl2 only (M,M)-P2/(P,P)-P2 is present as shown in Fig. S9 (in the ESI‡). The homochiral self-assembly has been previously observed in solid crystal structures47 and under chiral guest encapsulation conditions.17 However, to the best of our knowledge, this is the first time that it is observed quantitatively in solution without the need for any particular guest requirements.
Upon heating, every set of signals coalesces for P2 and P2F2Et to yield spectra, which are comparable to those of high symmetric molecules of point group D2h. Coalescence temperatures were higher for P2F2Et indicating a higher inversion barrier (Fig. S8 in the ESI‡). To quantify this observation, the activation energy ΔG‡ for the conformational interconversion of the diastereomers was calculated for P2 and P2F2Et in CD2Cl2 and C2D2Cl4 using the coalescence method. The associated equation and the respective coalescence temperatures as well as chemical shifts are listed in the ESI (Table S1‡). In both solvents, the activation energy for P2 (ΔG‡ (CD2Cl2) = 52.8 kJ mol−1 and ΔG‡ (C2D2Cl4) = 57.0 kJ mol−1) is lower than that of P2F2Et (ΔG‡ (CD2Cl2) = 61.3 kJ mol−1 and ΔG‡ (C2D2Cl4) = 64.0 kJ mol−1). P2 is less sterically hindered than P2F2Et and has a high flexibility due to the malonate linker, which favours the interconversion process. Even the values for the sterically much more demanding P2F2Et are lower than the free energy for a comparable PBI cyclophane with rigid linkers (ΔG‡ (C2D2Cl4) = 68.7 kJ mol−1).17
In contrast to P2 and P2F2Et, P3 shows no sharpening at higher temperatures (Fig. S10 in the ESI‡). However, we note an interesting behaviour for P3 at low temperatures. Upon cooling, signals in the 1H-NMR as well as in the 13C-NMR spectra broaden and finally split. To understand the aforementioned thorough symmetry analyses of all isomers together with statistics are necessary. Like P2, P3 exists as a racemic mixture of two diastereoisomers, that is, homochiral (M,M,M)-P3/(P,P,P)-P3 and (M,M,P)-P3/(P,P,M)-P3 (Fig. 4b). Homochiral P3 shows D3 symmetry. In contrast to P2, the PBIs of P3 are free to rotate around the flexible linkers. In other words, a single set is expected for the PBI protons.
The protons of the three CH2 alkyl groups are diastereotopic, but the malonate protons are not (Fig. 4b, red protons). As they are in line with the C2-axes they are all interchanged by action of the C3- and the three C2-axes. Consequently, one singlet for this set of homotopic protons is expected. (M,M,P)-P3/(P,P,M)-P3 has only one C2-axis and is of point group C2. Therefore, two singlets in a 2:1 ratio for the PBI protons are expected. The malonate protons are a special case. In two out of the three CH2-groups the protons are diastereotopic (Fig. 4b, blue protons). But, the third is again in line with the C2-axis (Fig. 4b, green protons) and, therefore, homotopic. Thus, we expected two doublets from the AB-spin system (Fig. 4b, blue protons) and one singlet (Fig. 4b, green protons) in a 2:1 ratio. Fig. 5 shows the 1H-NMR spectrum of P3 at −20 °C with the described splitting for all protons. For the 13C-NMR spectra see Fig. S11 (in the ESI‡).
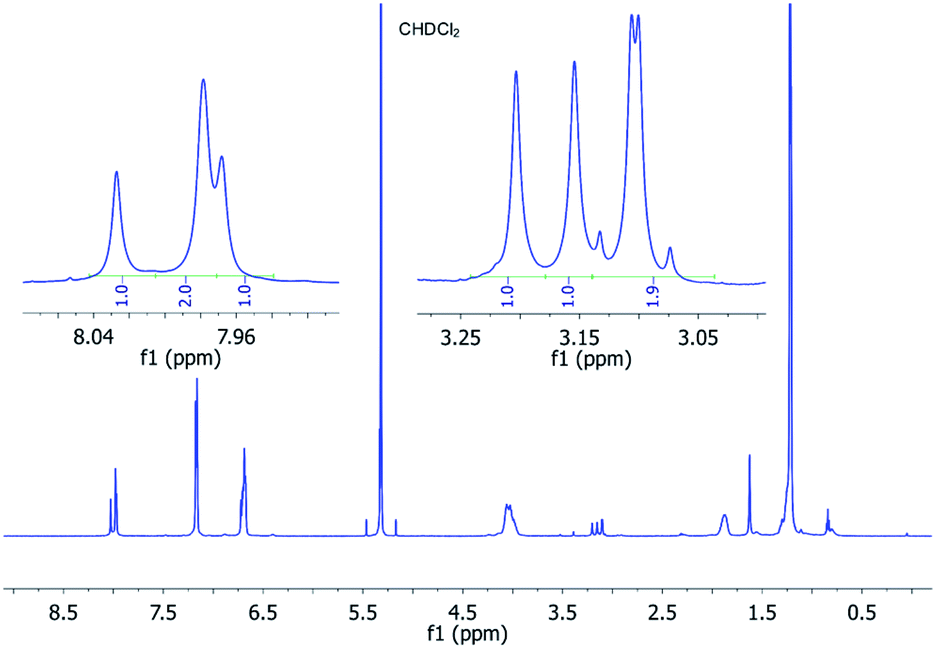 | ||
| Fig. 5
1H (600 MHz) NMR spectrum of P3 dissolved in CD2Cl2 recorded at −20 °C showing the typical ratio of 1:1:2 for the perylene and malonate protons. | ||
The splitting and the multiplicity of the signals are both in perfect agreement with our symmetry considerations. The unexpected integral ratio of 1:1:2 needs, however, explanation. Considering only symmetry, a mixture without any preference for one diastereoisomer would yield a proton spectrum with an integral ratio of 3:2:1 for each signal group. But, when taking statistics into account (M,M,P)-P3/(P,P,M)-P3 would be three times more populated than (M,M,M)-P3/(P,P,P)-P3. Thus, an integral ratio of 1:1:2 would result for each signal group, which is in sound agreement with the experiments. For P3, in contrast to P2, no preference for one of the diastereoisomers is observed. This can be rationalized from the fact that in the three-PBI cyclophane a conformation with two PBIs in close contact with each other is less favored. In the C2-symmetrical P3, two branches of the alkyl chain and two of the PBIs cannot be interchanged. For the third branch, this is possible by action of the C2-axis, which is why an additional splitting with a total ratio of 1:1:1:1 is expected. Interestingly, this does not apply to the malonate CH2-groups. They are located in the middle between the two branches and the ratio, thus, remains at 2:1:1. This additional splitting in the third branch was detected for one of the PBI carbon atoms and for the OCH2-group at even lower temperatures of −38 °C (Fig. S12 in the ESI‡). To prove that all signal groups belong to the different diastereoisomers of P3, we performed 1D-EXSY for the malonate resonances in the rotating frame at −20 °C. Exchange peaks appeared when each signal group was irradiated. This is exemplified for the selective excitation at 3.24 ppm in Fig. S13 (in the ESI‡). It displays the slow interconversion of (M,M,M)-P3 and its enantiomer into (M,M,P)-P3 and its enantiomer and vice versa.
Steady-state absorption and fluorescence spectroscopy
Steady-state absorption and fluorescence spectra of P1, P2, P3, and P4 measured in toluene are shown in Fig. 6. P1 reveals the characteristic S0–S1 PBI features in the 500 to 600 nm range, that is, a set of absorption maxima at 534 and 575 nm, corresponding to the 0–*1 and 0–*0 vibrational transitions. In the 450 nm region, the weaker absorptions are attributed to the S0–S2 transitions. In P2, the 0–*1 vibrational transition is more pronounced than in P1, with a 0.99-to-1 ratio of the corresponding 0–*1 and 0–*0 intensities, respectively. At this point we infer H-type excitonic coupling due to the co-facial stacking of the two PBIs.15,48 According to exciton theory, transitions to the lower-energy exciton-split LUMO, which corresponds to the 0–*0 transition, are symmetrically forbidden in face-to-face stacked PBIs. Instead, transitions to the higher-energy exciton-split LUMOs, which relate to the 0–*1 transition, take the complete oscillator strength.16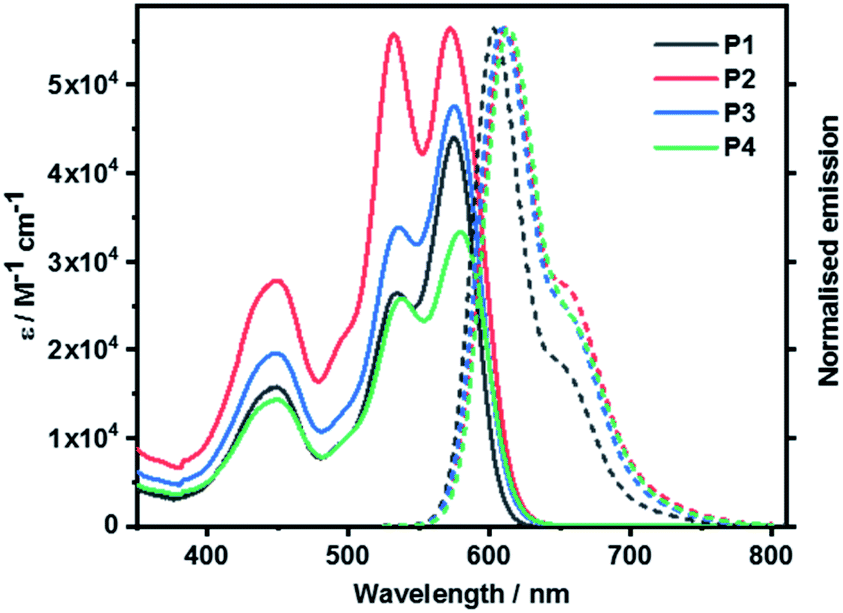 | ||
| Fig. 6 Absorption and normalized fluorescence spectra of P1 as well as cyclophanes P2, P3, and P4 recorded in toluene at room temperature. | ||
PBI stacking in P2 deviates, however, from the ideal scenario. It is the presence of the bulky phenoxy substituents at the bay positions that evokes non-planar PBIs. Considering that the 0–*0 transitions are discernible in P2 a subtle interplay between excitonic and vibronic couplings relieves the symmetry restrictions.49 Intermolecular aggregation of P2 was ruled out by means of corroborating that the absorption features were found to be concentration independent (Fig. S39 in ESI‡). This is in agreement with the fact that the sterically demanding tetra-(4-tert-butylphenoxy) substituents cause significant structural distortions and, in turn, suppress the tendency towards aggregation.48
When turning to P3 and P4, both 0–*1 and 0–*0 transitions are seen with, however, different relative intensities. The intensity ratio follows P2 > P4 > P3. This trend suggests that the flexible linkers of P3 and P4 permit loose co-facial PBI stackings. Solvent viscosity-dependent bathochromic shifts of the absorptions were observed when measured in tetrahydrofuran (THF), benzonitrile (PhCN), and 1,1,2,2-tetrachloroethane (TCE) next to toluene (Fig. S38 in the ESI‡).
Fig. 7 surveys the absorption and fluorescence spectra of P1F2Et and P2F2Et in toluene, and P2F2TEG in toluene as well as 5% THF/water mixture. All P1-related absorption features were retained in P1F2Et. No significant perturbations nor any additional transitions were seen. What was, however, observed was the absorbance increase at wavelengths below 400 nm. This arises from the presence of the fullerenes. Co-facial π–π stacking is present in P2F2Et and P2F2TEG, as evidenced by 1.16-to-1 and 1.12-to-1 ratios of the corresponding 0–*1 and 0–*0 intensities, respectively. P2F2TEG dissolved in a 5% THF/water mixture showed absorptions that are bathochromically shifted by ∼7 nm compared to toluene solutions, which is a trend that has been reported for PBIs in an aqueous environment.50,51 Here, hydrophobic interactions favor interchromophore stacking and result in an even higher intensity ratio of 1.31-to-1. The higher ratios seen in P2F2Et and P2F2TEG corroborate the stronger co-facial π–π stackings as seen, for example, in the NMR investigations. All maxima, molar extinction coefficients, and relative intensities of the 0–*1 and 0–*0 transitions in different solvents are listed in Table S2 (in the ESI‡).
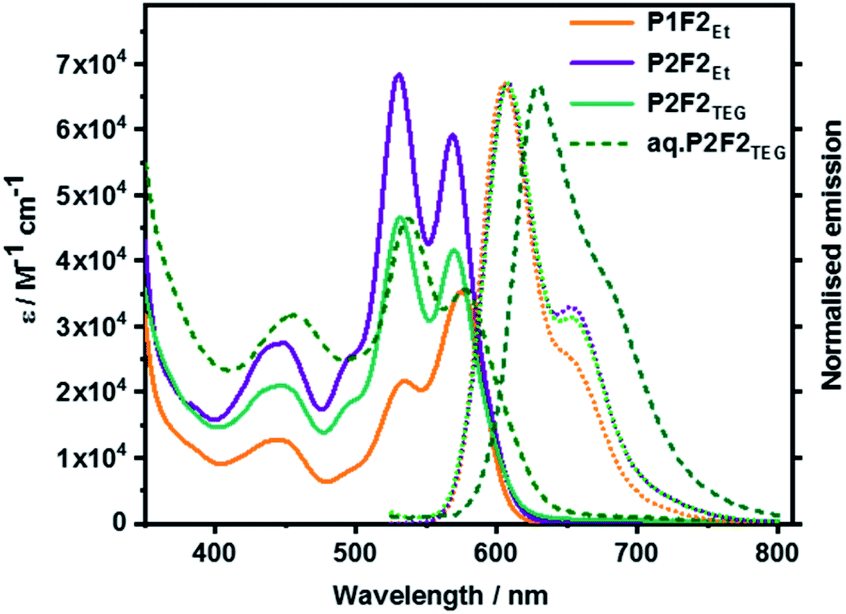 | ||
| Fig. 7 Absorption and normalized fluorescence spectra of P1F2Et, P2F2Et, and P2F2TEG recorded in toluene as well as P2F2TEG recorded in 5% THF/water mixture (dashed line) at room temperature. | ||
Overall, all fluorescence maxima are Stokes-shifted by 30–35 nm. For example, P1 shows a vibrationally resolved fluorescence spectrum with a maximum at 603 nm and a shoulder at 655 nm. P2, P3, and P4 exhibit similar fluorescence features with slightly red-shifted maxima, that is, from 610 to 615 nm. No evidence for any excimer formation was, however, noted. The fluorescence quantum yields decrease from as high as 90% for P1 to less than 26% for the cyclophanes (Table S3 in the ESI‡). Such a trend substantiates a π–π stacking-induced quenching of the PBI fluorescence. A remarkable de-intensification of the quantum yields is also observed when the solvent is changed from non-polar toluene to highly polar benzonitrile. Implicit is the involvement of symmetry-breaking charge transfer and/or symmetry-breaking charge separation, which opens up non-radiative deactivation pathways.52 The emission spectral features of the model compound P1F2Et and cyclophane-fullerene hybrids P2F2Et and P2F2TEG in organic solvents exhibited no significant shifts compared to their pure PBI counterparts.
However, the fluorescence quantum yield is reduced to 40–50% in P1F2Et in different organic solvents, suggesting excited state interactions of PBI with the attached fullerene pentakisadducts. P2F2Et and P2F2TEG also exhibit solvent-dependent quenching much like what is seen for P2. Aqueous solutions of P2F2TEG showed a broad and red-shifted fluorescence with a maximum at 630 nm, a shoulder at 685 nm, and a quantum yield of <1%.
Temperature-dependent absorption and fluorescence spectroscopic studies were performed with P2 and P3 in 1,1,2,2-tetrachloroethane and are gathered in Fig. S43 in the ESI.‡ Both P2 and P3 exhibit hypochromic as well as hypsochromic shifts in the absorption spectra upon heating. We rationalize this by the enhanced flipping of the naphthalenes of the PBI core at higher temperatures.53 However, unlike P3, the ratio of 0–*1 to 0–*0 intensities of P2 shows a subtle decrease towards higher temperature. This indicates weakening of the PBI π–π stackings. P2 reveals a two-fold increase upon heating, whereas no significant changes are seen for P3. In other words, P3 is less rigid than P2 at any given temperature. This finding is in sound agreement with the low-temperature NMR results.
Transient absorption spectroscopy
To understand the solvent-sensitive excited state dynamics, femtosecond (fs) and nanosecond (ns) transient absorption (TA) studies were performed in toluene, THF and PhCN, by photoexciting at 550 nm, with an OD of around 0.4 (Fig. S45–S51 in the ESI‡). In-depth analysis of the TA spectra was performed using the GloTarAn program.54 Kinetic models with four or five species were employed to run the global sequential analysis. The resulting evolution associated spectra (EAS) and the relative population of the excited states are shown in the ESI.‡ To elucidate the deactivation pathway of P1, a kinetic model based on four species was employed (Fig. S52 and S53 in the ESI‡). To this end, P1 shows in toluene the characteristic spectral signatures with ground state bleaching at 452 and 595 nm, singlet excited state absorptions at 700, 955, and 1038 nm, and stimulated emission at 652 nm. The first species, which is a vibrationally hot singlet excited state, transforms to a vibrationally relaxed excited state in 8.5 ps as the second species. It undergoes further relaxation to a third species in 350.6 ps. This third species is the fluorescent singlet excited state, which decays to the ground state with a lifetime of 6.0 ns. In line with a fluorescence quantum yield of 90% in toluene, P1 decays primarily by fluorescence. Still, the remaining 10% is subject to intersystem crossing to afford the triplet excited state. From the ns-TA spectra, we conclude a lifetime of more than 350 μs. Regardless of the solvent polarity, similar kinetics were obtained for P1.In contrast, P2 exhibits distinct, solvent polarity-dependent spectral evolutions (Fig. S46 in the ESI‡). The EAS reconstructed from the global fits of fs-TA studies for P2 in toluene and benzonitrile are shown in Fig. 8a and d.
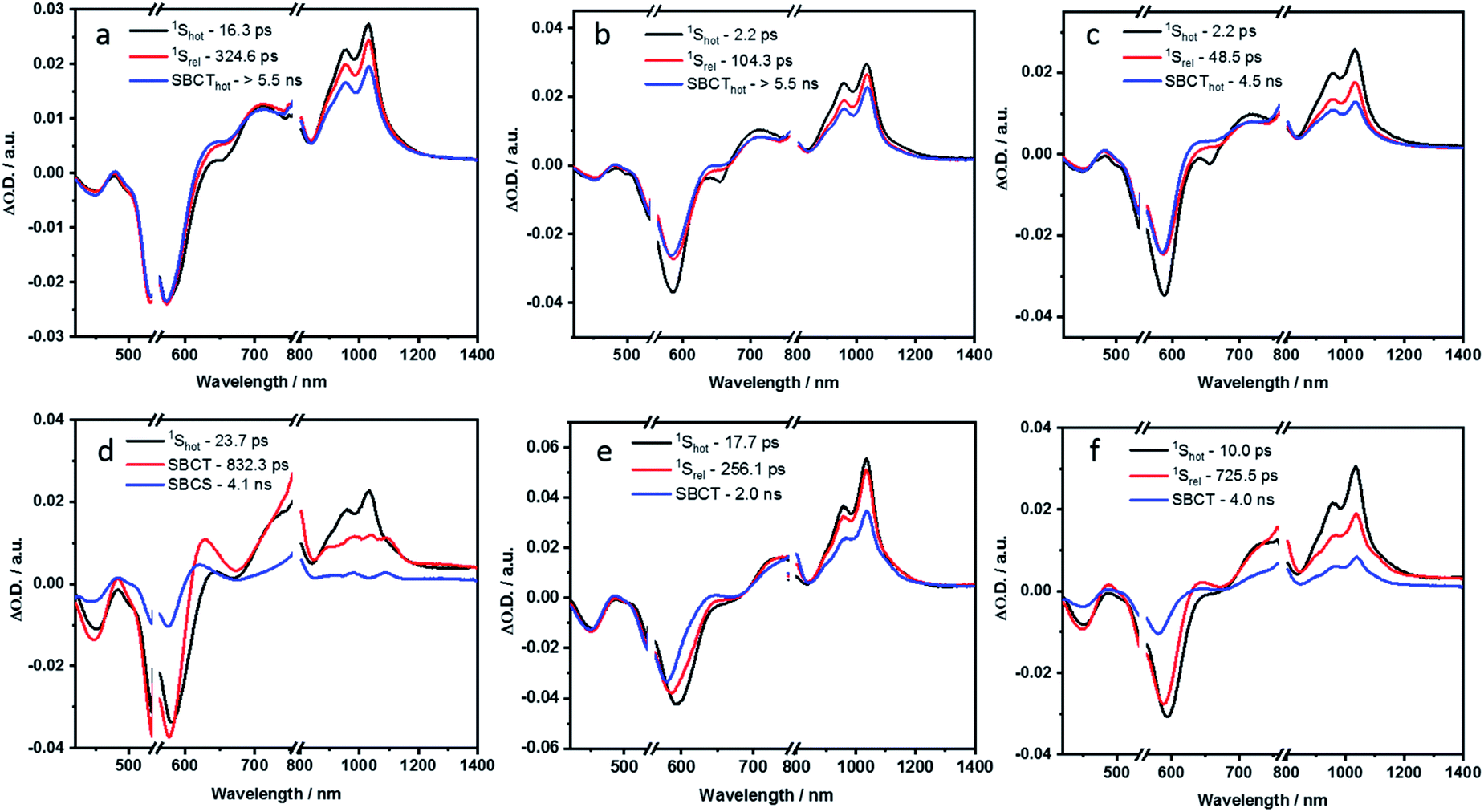 | ||
| Fig. 8 Evolution associated spectra reconstructed from the sequential global analysis of fs-TA spectra of P2, P3 and P4 in toluene (a, b and c respectively) and PhCN (d, e and f respectively). | ||
In the non-polar toluene, the initial vibrationally hot singlet excited state, undergoes vibrational relaxation to form the second species in 16.3 ps and then proceeds to a new long-lived intermediate third species in 324.6 ps. By means of comparing the spectral features with the spectroelectrochemical absorptions of the PBI radical cation (630, 790, 975, and 1083 nm) and radical anion (685, 790, 975, and 1083 nm) (Fig. S44 in the ESI‡), the third species corresponds to the PBIδ+–PBIδ− symmetry-breaking charge transfer (SBCT) state. Its decay dynamics were probed using ns-TA measurements and the corresponding EAS spectra are shown in Fig. 9. Considering that the fourth species also features the SBCT fingerprints, we assign the third and fourth species to a hot SBCT and a relaxed SBCT, respectively. The PBI triplet excited state is the fifth species and is generated with a time constant of 14.7 ns. A weak positive absorption in the 500 nm region is the characteristic of the triplet excited state and it lives for 137 μs.
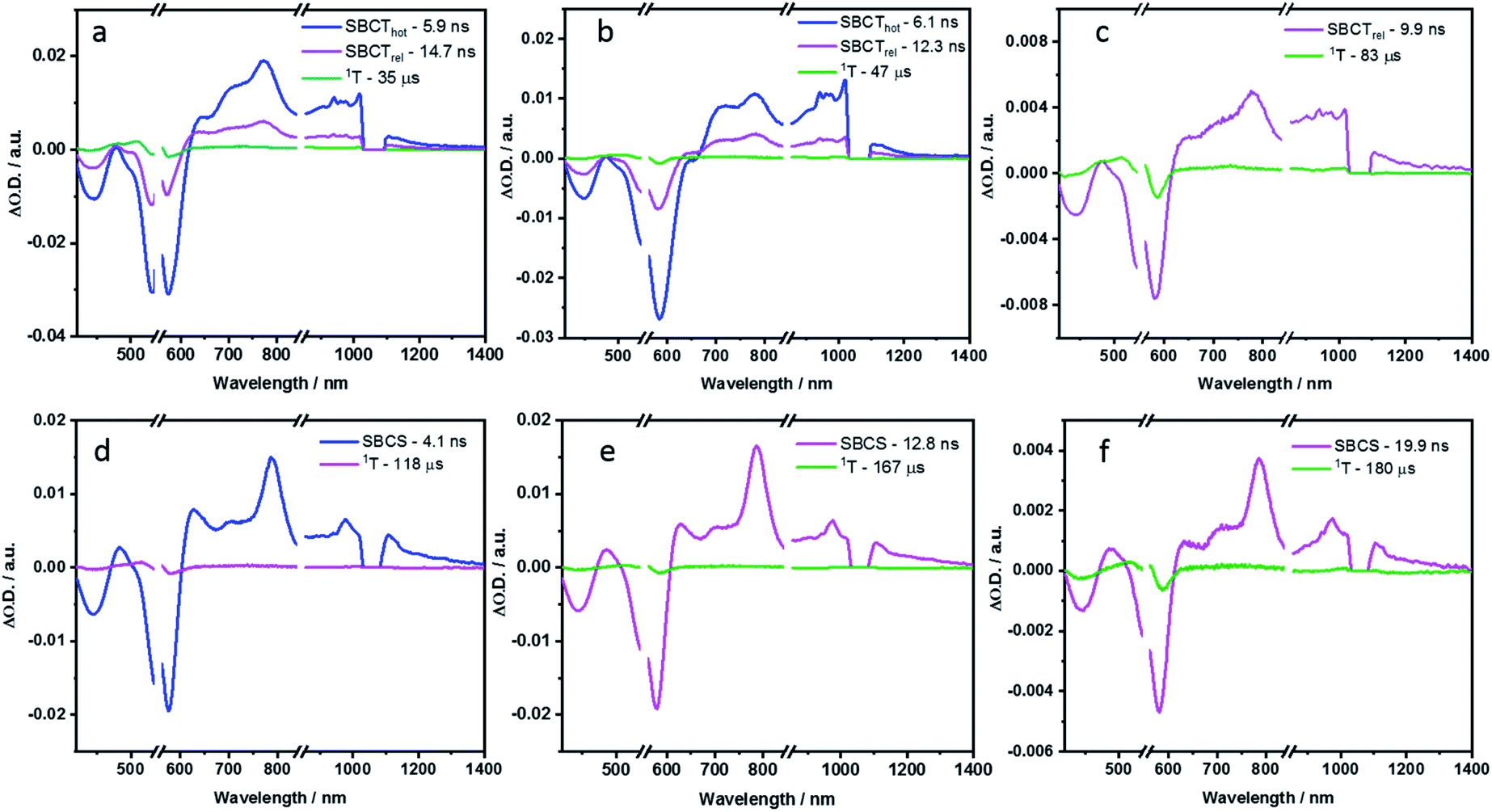 | ||
| Fig. 9 Evolution associated spectra reconstructed from the sequential global analysis of ns-TA spectra of P2, P3 and P4 in toluene (a, b and c respectively) and PhCN (d, e and f respectively). | ||
In polar PhCN, a different excited state relaxation pathway was concluded. Formation of the vibrationally hot excited state is followed by deactivation over a time of 23.7 ps to afford the SBCT state as first and second species, respectively. It lives for 832.3 ps and is characterized by broadening of the excited state absorption in the nIR region and an evolving maximum at 630 nm due to the formation of the PBI radical cation. The SBCT state then transitions to a symmetry-breaking charge-separated (SBCS) state with the respective markers at 630, 685, 790, 975, and 1085 nm. This is the third species. SBCS is unfavorable in non-polar toluene due to the positive free energy of charge separation.55,56 Finally, it is with a time delay of 4.1 ns that the SBCS state populates the PBI triplet excited state as the fourth and final species. 118 μs is the lifetime, by which the triplet excited state decays back to the ground state. The decay kinetics observed for P2 in THF, which has intermediate polarity, was very similar to that obtained in PhCN (Fig. S54 and S55 in the ESI‡).
In toluene, the excited state deactivation mechanism of P3 and P4 is similar to that gathered for P2 in toluene. Excitation populates the first species, which is the singlet excited state but a high vibrational level of it, followed by the formation of a vibrationally relaxed state in 2.2 ps as the second species. It undergoes further relaxation within 104.3 ps for P3 or 48.5 ps for P4 to form the third species or the hot SBCT state with a dipolar character. The corresponding lifetimes are 6.1 and 4.5 ns in P3 and P4, respectively. From the ns-TA spectra, it is evident that the hot SBCT state relaxes to the fourth species, which has a lifetime of 12.3 ns in P3 and 9.9 ns in P4. Finally, this relaxed SBCT state decays to form the triplet excited state, which then repopulates the ground state in <100 μs.
In PhCN, photoexcitation populates the vibrationally hot singlet excited state of P3, which relaxes initially in 17.7 ps, and, which subsequently deactivates in 256.1 ps. This decay is concomitant with a remarkable blue-shift in the ground state bleaching. Within a time span of 2.0 ns, the SBCT state populates the SBCS state, featuring the fingerprint absorption bands of the PBI radical cation and radical anion. The SBCS state populates in 12.8 ns the triplet manifold, which exhibits a lifetime of 167 μs.
Unlike P3, the evolution associated spectrum of P4 in PhCN resembles that of P2 and requires the use of an additional species to fit the TA spectrum. In general, the difference is ascribed to the excited state geometry of P4. For P4, we hypothesize two pairs of π–π stacked PBIs. So, its behavior is expected to be similar to that of P2. For P4 in PhCN, the vibrationally hot singlet excited state undergoes relaxation in 10 ps. By this, the second species, which has a slight dipolar character, is formed. It subsequently populates the SBCT, SBCS, and triplet excited states in 725.5 ps, 4.0 ns, and 19.9 ns, respectively. Finally, the triplet excited state decays back to the ground state with a lifetime of 180 μs. The deactivation mechanism in THF was comparable for P3 and P4 to those in PhCN, as shown in Fig. S56–S59 (in the ESI‡). Based on the results of TA spectral analyses, a simplified energy scheme for the excited state deactivation pathway of the cyclophanes in polar and non-polar solvents is summarized in Fig. 10. The estimated lifetime values are summarized in Tables S4 and S5 in the ESI.‡
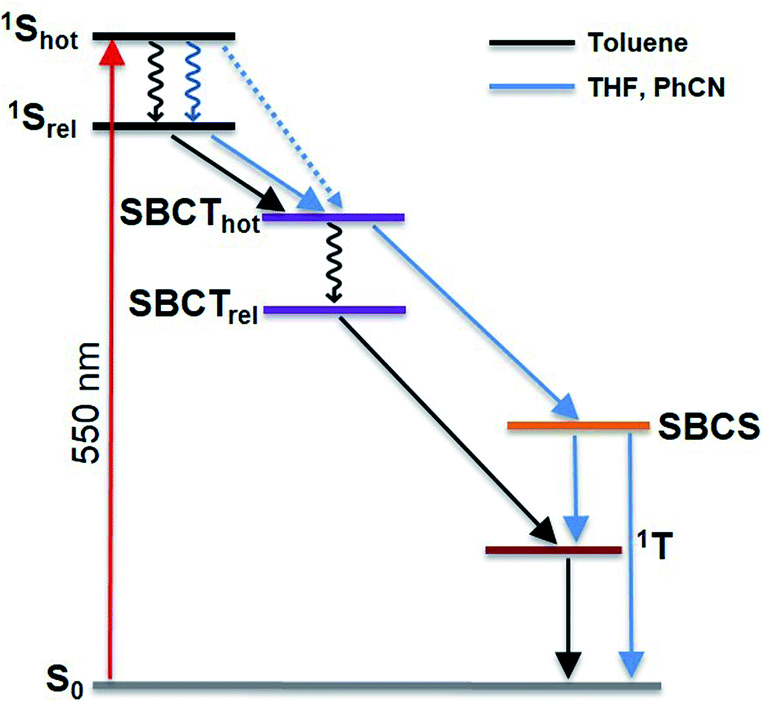 | ||
| Fig. 10 Simplified energy diagram illustrating the excited state deactivation mechanism of the cyclophanes in polar and non-polar solvents. The black arrows represent the decay pathway in toluene. The blue arrows indicate the deviation observed in polar solvents THF and PhCN, which favour the formation of a stabilized SB-CS state. | ||
For all cyclophanes, the population of the PBI triplet excited state from either SBCT in toluene or SBCS in benzonitrile was quantified by measuring the singlet oxygen phosphorescence at 1270 nm (Fig. S66 in ESI‡). Using C60 in air-equilibrated toluene as the reference,57,58 the singlet oxygen quantum yields (ΦΔ) were determined. They are summarized in Table S6.‡ In toluene, particularly high ΦΔ values of 67% and 76% for P2 and P4 and a comparably low ΦΔ value of 37% for P3 correlate well with the fluorescence quantum yields. However, in benzonitrile, charge recombination from SBCS to afford the triplet excited state is reduced for P2 and P4 as indicated by ΦΔ values of 53% and 44% for P2 and P4, respectively. The differences relative to toluene are due to a competing charge-recombination pathway, by which the ground state is directly recovered. Only in P3, it is slightly increased to 43%.
Finally, we analyzed P1F2Et, P2F2Et and P2F2TEG in polar and non-polar solvents. In particular, 550 nm excitation initiated the same excited state decay mechanism as observed for P1 and P2, respectively. As such, we used the kinetic models, which proved to fit the data for P1 and P2 in different solvents quite well with four or five species. The fs-TA, ns-TA, and evolution associated spectra together with the relative population of the corresponding species are shown in Fig. S49–S51 and S60–S65 (in the ESI‡). Lifetimes of the species are summarised in Tables S4 and S5.‡ By virtue of 550 nm photoexcitation, which is selective for PBI, no fullerene-centered transients were detected. In contrast, when photoexciting the fullerenes at around 370 nm a resonance energy transfer by means of dipole–dipole interactions was concluded from the 3D fluorescence heat maps (Fig. S67 and S68 in the ESI‡).
Conclusions
PBI-based cyclophanes P2, P3, and P4 with different sizes and various arrangements were synthesized and the relationship between the structure and properties was thoroughly investigated. By varying the reaction conditions and using TTF in an electron donor–acceptor templated synthesis, the yield of the two-PBI P2 was improved from 2.7% to 8.1%, which is already a reasonably high yield in cyclophane synthesis. The changes in the NMR spectra indicate a different chemical environment in terms of π–π stacking PBIs in the cyclophanes. Remarkably, P2 exhibits the diastereoselective formation of homochiral atropisomers (M,M)-P2/(P,P)-P2 at low temperatures by virtue of the flexible linkers. Owing to the strong intramolecular π–π interactions between co-facially arranged PBIs, P2 shows H-type excitonic coupling. Looser is the co-facial stacking of the PBIs within three-PBI and four-PBI cyclophanes. Therefore, at low temperatures, both diastereomeric pairs (M/M/M)-P3/(P,P,P)-P3 as well as (M/M/P)-P3/(P,P,M)-P3 are detected in the 1H-NMR spectra of P3. Such a dynamic stacking is expected to provide an accessible cavity for hosting various molecular guests.From a fluorescence quantum yield decrease in the order P1 ≫ P3 > P4 > P2 π–π stacking-induced quenching between the PBIs in their excited states was concluded. Moreover, the quantum yields are solvent-dependent. This suggests symmetry-breaking charge-transfer/charge-separation interactions. Indeed, time-resolved pump-probe experiments helped to confirm the presence of both charge-transfer and charge-separation. With a temperature increase, P2 is subject to a weakening of the π–π stacking PBIs. Of great value are the flexible malonate linkers in P2 as they enabled the functionalization with hydrophobic as well as hydrophilic fullerene pentakisadducts. In this context, we synthesized for the first time a covalently linked PBI-cyclophane-fullerene hybrid. The solvent dependent coalescence temperatures as well as the activation energies for the conformational interconversion are higher for P2F2Et than for P2, which is due to a sterically demanding fullerene. The enhanced co-facial π–π stacking present in the cyclophane-fullerene adducts P2F2Et and P2F2TEG leads to a complete diastereoselectivity for the case of P2F2Et. This illustrates that the hybridization strategy with fullerenes allows us to modulate the cyclophanes with respect to solubility, stacking, cavity size, photophysics and sterical arrangement. Given the current undivided attention in dye-based cages for selective purification, sensing, catalysis or chiral recognition, our findings are of utmost interest.
Data availability
All experimental data associated to the article is given in the ESI.† Data have not been deposit in any repository.Author contributions
I. Solymosi: investigation (synthesis and characterization); writing-original draft. S. Krishna: investigation (photophysics), writing-original draft. E. Nuin: conceptualization, investigation (synthesis), writing – review & editing. H. Maid: investigation (NMR characterization), writing – original draft. B. Scholz: investigation (precursor synthesis). D. M. Guldi: conceptualization, supervision, writing – review & editing, funding acquisition. M. E. Pérez-Ojeda: conceptualization, investigation (synthesis), supervision, writing – review & editing, funding acquisition. A. Hirsch: supervision, writing – review & editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (DFG) which supported this work through the SFB 953 projects A1, A9 and B10 as well as the emerging talents initiative (ETI) and Marie Sklodowska-Curie IF European Actions 747734 HysolFullGraph to M. E. Pérez-Ojeda.Notes and references
- C. J. Brown and A. C. Farthing, Nature, 1949, 164, 915–916 CrossRef CAS.
- D. J. Cram and H. Steinberg, J. Am. Chem. Soc., 1951, 73, 5691–5704 CrossRef CAS.
- F. Würthner, Chem. Commun., 2004, 14, 1564–1579 RSC.
- D. H. Harris, S. Brixi, B. S. Gelfand, B. H. Lessard and G. C. Welch, J. Mater. Chem. C, 2020, 8, 9811–9815 RSC.
- J. D. Yuen, V. A. Pozdin, A. T. Young, B. L. Turner, I. D. Giles, J. Naciri, S. A. Trammell, P. T. Charles, D. A. Stenger and M. A. Daniele, Dyes Pigm., 2020, 174, 108014 CrossRef CAS.
- R. K. Gupta, A. Dey, A. Singh, P. K. Iyer and A. A. Sudhakar, ACS Appl. Electron. Mater., 2019, 1, 1378–1386 CrossRef CAS.
- W. Yan, Z. He, J. Jiang, D. Lu, Y. Gong, W. Yang, R. Xia, W. Huang and H. Xin, J. Mater. Chem. C, 2020, 8, 14773–14781 RSC.
- R. Xin, C. Zeng, D. Meng, Z. Ren, W. Jiang, Z. Wang and S. Yan, ACS Omega, 2020, 5, 18449–18457 CrossRef CAS PubMed.
- J. Huang, H. Ren, R. Zhang, L. Wu, Y. Zhai, Q. Meng, J. Wang, Z. Su, R. Zhang, S. Dai, S. Z. D. Cheng and M. Huang, ACS Nano, 2020, 14, 8266–8275 CrossRef CAS PubMed.
- M. Hecht, P. Leowanawat, T. Gerlach, V. Stepanenko, M. Stolte, M. Lehmann and F. Würthner, Angew. Chem., Int. Ed., 2020, 59, 17084–17090 CrossRef CAS PubMed.
- P. Spenst and F. Würthner, J. Photochem. Photobiol., C, 2017, 31, 114–138 CrossRef CAS.
- P. Spenst and F. Würthner, Angew. Chem., Int. Ed., 2015, 54, 10165–10168 CrossRef CAS PubMed.
- H. Langhals and R. Ismael, Eur. J. Org. Chem., 1998, 9, 1915–1917 CrossRef.
- W. Wang, L. Wang, B. J. Palmer, G. J. Exarhos and A. D. Q. Li, J. Am. Chem. Soc., 2006, 128, 11150–11159 CrossRef CAS PubMed.
- F. Schlosser, M. Moos, C. Lambert and F. Würthner, Adv. Mater., 2013, 25, 410–414 CrossRef CAS PubMed.
- J. Feng, Y. Zhang, C. Zhao, R. Li, W. Xu, X. Li and J. Jiang, Chem.–Eur. J., 2008, 14, 7000–7010 CrossRef CAS PubMed.
- M. Sapotta, P. Spenst, C. R. Saha-Möller and F. Würthner, Org. Chem. Front., 2019, 6, 892–899 RSC.
- M. Weh, J. Rühe, B. Herbert, A.-M. Krause and F. Würthner, Angew. Chem., Int. Ed., 2021, 60, 15323–15327 CrossRef CAS PubMed.
- T. A. Barendt, W. K. Myers, S. P. Cornes, M. A. Lebedeva, K. Porfyrakis, I. Marques, V. Félix and P. D. Beer, J. Am. Chem. Soc., 2020, 142, 349–364 CrossRef CAS PubMed.
- W. Wang, A. D. Shaller and A. D. Q. Li, J. Am. Chem. Soc., 2008, 130, 8271–8279 CrossRef CAS PubMed.
- P. Spenst, A. Sieblist and F. Würthner, Chem.–Eur. J., 2017, 23, 1667–1675 CrossRef CAS PubMed.
- J. Rühe, D. Bialas, P. Spenst, A.-M. Krause and F. Würthner, Org. Mater., 2020, 2, 149–158 CrossRef.
- F. Schlosser, J. Sung, P. Kim, D. Kim and F. Würthner, Chem. Sci., 2012, 3, 2778–2785 RSC.
- P. Spenst, R. M. Young, B. T. Phelan, M. Keller, J. Dostál, T. Brixner, M. R. Wasielewski and F. Würthner, J. Am. Chem. Soc., 2017, 139, 2014–2021 CrossRef CAS PubMed.
- M. E. Pérez-Ojeda, I. Wabra, C. Böttcher and A. Hirsch, Chem.–Eur. J., 2018, 24, 14088–14100 CrossRef PubMed.
- Y. Sun and Z. Li, Polym. Chem., 2017, 8, 4422–4427 RSC.
- E. Nuin, V. Lloret, K. Amsharov, F. Hauke, G. Abellán and A. Hirsch, Chem.–Eur. J., 2018, 24, 4671–4679 CrossRef CAS PubMed.
- J.-J. Shim, C.-W. Lee and M.-S. Gong, Synth. Met., 2001, 124, 435–441 CrossRef CAS.
- C.-W. Lee, S.-W. Joo, J. Ko, J.-S. Kim, S.-S. Lee and M.-S. Gong, Synth. Met., 2002, 126, 97–104 CrossRef CAS.
- N. Chronakis, T. Brandmüller, C. Kovacs, U. Reuther, W. Donaubauer, F. Hampel, F. Fischer, F. Diederich and A. Hirsch, Eur. J. Org. Chem., 2006, 10, 2296–2308 CrossRef.
- K. Ziegler, H. Eberle and H. Ohlinger, Liebigs Ann. Chem., 1933, 504, 94–130 CrossRef CAS.
- R. Hoss and F. Vögtle, Angew. Chem., Int. Ed. Engl., 1994, 33, 375–384 CrossRef.
- J. C. Barnes, M. Juríček, N. A. Vermeulen, E. J. Dale and J. F. Stoddart, J. Org. Chem., 2013, 78, 11962–11969 CrossRef CAS PubMed.
- D. Canevet, M. Sallé, G. Zhang, D. Zhang and D. Zhu, Chem. Commun., 2009, 17, 2245–2269 RSC.
- H. Y. Au-Yeung, P. Pengo, G. D. Pantoş, S. Otto and J. K. M. Sanders, Chem. Commun., 2009, 4, 419–421 RSC.
- M. R. Bryce, G. J. Marshallsay and A. J. Moore, J. Org. Chem., 1992, 57, 4859–4862 CrossRef CAS.
- F. Würthner and A. Sautter, Chem. Commun., 2000, 6, 445–446 RSC.
- F. Diederich and P. J. Stang, Templated Organic Synthesis, John Wiley & Sons, 2000 Search PubMed.
- Y. Nakamura, S. Minami, K. Iizuka and J. Nishimura, Angew. Chem., Int. Ed., 2003, 42, 3158–3162 CrossRef CAS PubMed.
- X. Camps and A. Hirsch, J. Chem. Soc., Perkin Trans. 1, 1997, 11, 1595–1596 RSC.
- K. L. Maxouti and A. Hirsch, Eur. J. Org. Chem., 2018, 2018, 2579–2586 CrossRef CAS.
- F. Beuerle and A. Hirsch, Chem.–Eur. J., 2009, 15, 7434–7446 CrossRef CAS PubMed.
- P. Witte, F. Hörmann and A. Hirsch, Chem.–Eur. J., 2009, 15, 7423–7433 CrossRef CAS PubMed.
- D. M. Guldi, B. M. Illescas, C. M. Atienza, M. Wielopolski and N. Martín, Chem. Soc. Rev., 2009, 38, 1587–1597 RSC.
- F. Würthner, Pure Appl. Chem., 2006, 78, 2341–2349 Search PubMed.
- J. Schönamsgruber and A. Hirsch, Eur. J. Org. Chem., 2015, 2015, 2167–2174 CrossRef.
- P. U. Osswald, PhD thesis, Julius-Maximilians-Universität Würzburg, Germany, 2007.
- W. Kim, A. Nowak-Król, Y. Hong, F. Schlosser, F. Würthner and D. Kim, J. Phys. Chem. Lett., 2019, 10, 1919–1927 CrossRef CAS PubMed.
- R. L. Fulton and M. Gouterman, J. Chem. Phys., 1964, 41, 2280–2286 CrossRef CAS.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- M. Sapotta, A. Hofmann, D. Bialas and F. Würthner, Angew. Chem., Int. Ed., 2019, 58, 3516–3520 CrossRef CAS PubMed.
- J. M. Giaimo, A. V. Gusev and M. R. Wasielewski, J. Am. Chem. Soc., 2002, 124, 8530–8531 CrossRef CAS PubMed.
- C. Kaufmann, D. Bialas, M. Stolte and F. Würthner, J. Am. Chem. Soc., 2018, 140, 9986–9995 CrossRef CAS PubMed.
- J. J. Snellenburg, S. P. Laptenok, R. Seger, K. M. Mullen and I. H. van Stokkum, J. Stat. Softw., 2012, 49, 1–22 Search PubMed.
- P. Spenst, R. M. Young, M. R. Wasielewski and F. Würthner, Chem. Sci., 2016, 7, 5428–5434 RSC.
- J. Kong, W. Zhang, G. Li, D. Huo, Y. Guo, X. Niu, Y. Wan, B. Tang and A. Xia, J. Phys. Chem. Lett., 2020, 11, 10329–10339 CrossRef CAS PubMed.
- L. Biczok, H. Linschitz and R. I. Walter, Chem. Phys. Lett., 1992, 195, 339–346 CrossRef CAS.
- S. Foley, M. N. Berberan-Santos, A. Fedorov, D. J. McGarvey, C. Santos and B. Gigante, J. Phys. Chem. A, 1999, 103, 8173–8178 CrossRef CAS.
Footnotes |
| † In memory of Prof. Dr Klaus Hafner. |
| ‡ Electronic supplementary information (ESI) available: Experimental details including HPLC, NMR, mass, UV-vis, fluorescence, spectroelectrochemistry, transient absorption spectra and fluorescence emission–excitation matrix. See DOI: 10.1039/d1sc04242d |
| § These authors contributed equally. |
| ¶ This dumbbell molecule was functionalized with unsubstituted [60]fullerene and cannot be used as a photophysical model for our systems given the different optical properties. |
| This journal is © The Royal Society of Chemistry 2021 |