
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTopological engineering of two-dimensional ionic liquid islands for high structural stability and CO2 adsorption selectivity†
Chenlu
Wang
ab,
Yanlei
Wang
 *ab,
Zhongdong
Gan
a,
Yumiao
Lu
a,
Cheng
Qian
d,
Feng
Huo
a,
Hongyan
He
*abce and
Suojiang
Zhang
*ab
*ab,
Zhongdong
Gan
a,
Yumiao
Lu
a,
Cheng
Qian
d,
Feng
Huo
a,
Hongyan
He
*abce and
Suojiang
Zhang
*ab
aBeijing Key Laboratory of Ionic Liquids Clean Process, State Key Laboratory of Multiphase Complex Systems, CAS Key Laboratory of Green Process and Engineering, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, China. E-mail: ylwang17@ipe.ac.cn; hyhe@ipe.ac.cn; sjzhang@ipe.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cInnovation Academy for Green Manufacture, Chinese Academy of Sciences, Beijing 100190, China
dSchool of Materials Science and Engineering, Ulsan National Institute of Science and Technology, Ulsan 44919, South Korea
eDalian National Laboratory for Clean Energy, Dalian 116023, Liaoning, China
First published on 4th November 2021
Abstract
Ionic liquids (ILs) as green solvents and catalysts are highly attractive in the field of chemistry and chemical engineering. Their interfacial assembly structure and function are still far less well understood. Herein, we use coupling first-principles and molecular dynamics simulations to resolve the structure, properties, and function of ILs deposited on the graphite surface. Four different subunits driven by hydrogen bonds are identified first, and can assemble into close-packed and sparsely arranged annular 2D IL islands (2DIIs). Meanwhile, we found that the formation energy and HOMO–LUMO gap decrease exponentially as the island size increases via simulating a series of 2DIIs with different topological features. However, once the size is beyond the critical value, both the structural stability and electrical structure converge. Furthermore, the island edges are found to be dominant adsorption sites for CO2 and better than other pure metal surfaces, showing an ultrahigh adsorption selectivity (up to 99.7%) for CO2 compared with CH4, CO, or N2. Such quantitative structure–function relations of 2DIIs are meaningful for engineering ILs to efficiently promote their applications, such as the capture and conversion of CO2.
Introduction
Due to the green and environmentally-friendly nature,1,2 ionic liquids (ILs) have been used in many cutting-edge fields, including green chemistry,3–5 electrochemistry,6,7 energy storage,8,9 gas separation,10,11 and catalysis.12,13 In the real application environment, solid-surface supported IL (SSIL) films always exist and show several advantages compared with the bulk ones,14,15 for example, higher stability, less usage of ILs, faster adsorption kinetics, etc. Consequently, SSIL films have attracted broad attention in the academy and industrial community.16,17 The structure, property, and function of SSILs should be the result of the balance between hydrogen bonding, van der Waals (vdW), Coulomb, and interfacial interactions, which are far more complex than those of simple systems such as water, CO, etc. Hence, quantitative understanding of the structure–function relationship of SSIL films has great significance in the rational design and management of IL-based chemistry.Recently, plenty of experiments and simulations have been conducted to clarify the correlation between the structure and performance of SSIL films.18–20Via using the physical vapor deposition (PVD) technique,18 Cremer et al.19 demonstrated that an ultrathin IL film consisting of a closed monolayer structure could be produced, which is stable under ambient conditions and should be the sub-structure of SSILs. Moreover, through scanning tunneling microscopy (STM) and theoretical simulations, previous pioneering studies21–24 identified that ILs could form the so-called ordered checkerboard or disordered glass structure of the monolayer ILs on the metal surface. Considering that the ordered monolayer IL structure should be grown from subunits, Meusel et al.25 studied the subunits within the monolayer ILs on the Au(111) substrate via atomic force microscopy (AFM) and STM, discovering that the striped and hexagonal subunits were composed of two and three cation–anion pairs. Meanwhile, subunits can assemble into different two-dimensional IL islands (2DIIs), for instance, IL islands with branched or quasi-dendritic shapes on the Cu(111) surface.26 Hence, the 2DIIs can indeed be prepared, and their structure depends greatly on the substrate surface, temperature (T), and other external conditions.
Originating from the designable microenvironments and functions, thin IL films or islands have broad application prospects, especially in CO2 capture and fixation. For example, Su et al.27 improved the catalytic activity of ILs in converting CO2 into cyclic carbonates via confining the ILs in the mesoporous silica nanopore, in addition, Xie et al.28,29 and Tang et al.30 substantially strengthened CO2 adsorption capacity and selectivity by just decreasing the thickness of IL films. These excellent performances of SSIL films suggest that the 2DII should be a preferred candidate in IL-based applications.31 However, up to now, beyond qualitative understanding of 2DIIs, the quantitative characterization is still a major challenge, for instance, the detailed subunit structure, electronic properties, and gas adsorption performance. Such limitations restrict the theoretical understanding, rational design, and applications of ILs in the field of highly efficient and low-cost gas separation and capture processes.
In the present work, the image charge augmented quantum mechanics/molecular mechanics (IC-QM/MM) and full-atomistic molecular dynamics (MD) simulations are performed to reveal the structure and properties of 2DIIs on graphite. We aimed at exploring the potential subunits of the ordered 2DIIs, gaining deeper insights into how the hydrogen bond (HB) network drives subunits and how the topological feature determines the thermodynamic stability, electronic properties, and even the gas adsorption mechanism of 2DIIs. Four different subunits are theoretically proposed, and the critical size (NC) for various 2DIIs is demonstrated, where the thermodynamic stability converges once the size is beyond NC. The mechanisms that the subunit and size jointly control the melting behavior and electronic structure of 2DIIs are further revealed, showing the easy-regulating feature of 2DIIs. Finally, the dynamic gas adsorption processes on 2DIIs are conducted, suggesting that the edges of 2DIIs are dominant adsorption sites for CO2 and possess high selectivity for CO2/CO, CO2/CH4, and CO2/N2.
Results and discussion
Hydrogen bonds driving subunits of 2DIIs
Due to the strong correlation originating from special HBs in ILs,32 the gaseous particles will be cation–anion pairs in the PVD process (Fig. 1a), which can be deposited on the solid surface and easily assembled into different island structures, especially when IL pairs are few. As illustrated in Fig. 1b, the HBs in 2DIIs mainly include three kinds, C2–H2⋯F, C4–H4⋯F, and C5–H5⋯F, namely HB2, HB4, and HB5. Owing to the unique directional feature of HBs, the cation–anion pair can construct several subunit, as shown in Fig. 1c, namely PN, where N = 1, 2, 3, and 4, representing the number of cation–anion pairs. For P1, P2, and P4, the differences are mainly the combination styles of HBs and relative orientations within the neighboring cations. However, for P3, the subunit is composed of three pairs of ILs and is an annular structure. The unique structure of subunits can further dominate the permutation structures of 2DIIs and meanwhile form feature edges.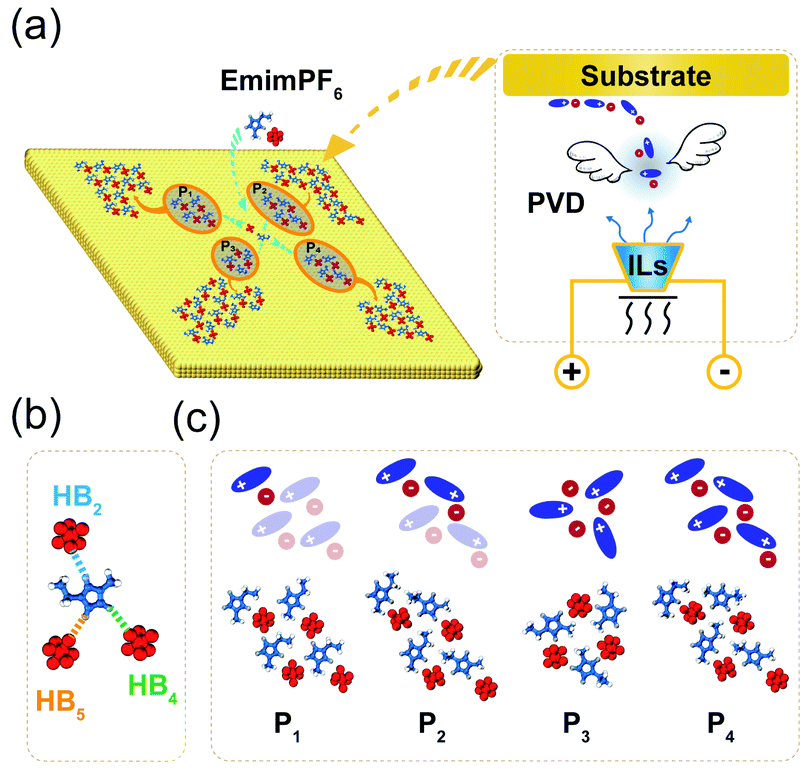 | ||
| Fig. 1 (a) A schematic diagram of 2DIIs on the graphite surface via PVD, where red, blue, yellow, white, and silver colors represent [PF6]−, [Emim]+, C atoms in graphite, H2/4/5 atoms, and H atoms in the alkyl chain, respectively. (b) Three main HBs in the ILs, where HB2,4,5 represents the HB between F and H in different sites of the imidazole ring. (c) The illustrated diagrams and atomic structures of the P1-based 2DII with four subunits, the P2-based 2DII with two subunits, the P3-based 2DII with one subunit, and the P4-based 2DII with one subunit, respectively. | ||
Based on the formation mechanism of various graphene polycrystals grown on the surface of liquid copper reported by Dong et al.,33 taking subunits as an origin, they can grow into different 2DIIs in the form of parallelograms (m,n), where m and n are the number of cation–anion pairs (P1/2/4-based 2DIIs) or subunits (P3-based 2DIIs) along two edges (ESI Movie M1†). The size of 2DIIs, Npairs, is defined as the total number of cation–anion pairs. To represent the structural feature of 2DIIs, the simulated STM images of P1/2/3/4-based 2DIIs with typical combinations of (m,n) are displayed in Fig. 2a and b and S4.† For P1/2/4-based 2DIIs, two different feature edges exist, namely Z4 and Z5, where Z4 comprises HB2 and HB4 while Z5 includes HB2 and HB5. Similarly, edges of P3-based 2DIIs are defined as E1 and E2 (Fig. S5†), where the types of HBs in E1 are almost the same as E2. Moreover, the thermodynamic stability of 2DIIs can be measured by the formation energy (Ef, see ESI Note†). Comparing Ef of 2DIIs with the same size but different combinations of (m,n), it can be found that P1/2/4-based 2DIIs with longer Z5 are more stable than those with longer Z4. For instance, Ef of P1-based 2DIIs with (4,2) and (2,4) is −44.97 and −46.32 kcal mol−1, respectively. However, there is no obvious difference between E1 and E2. The anisotropic nature of P1/2/4-based 2DIIs and the isotropic nature of P3-based 2DIIs coincide well with the styles of HBs in the feature edges, indicating the feasibility of structure and property regulation via controlling the HBs within 2DIIs.
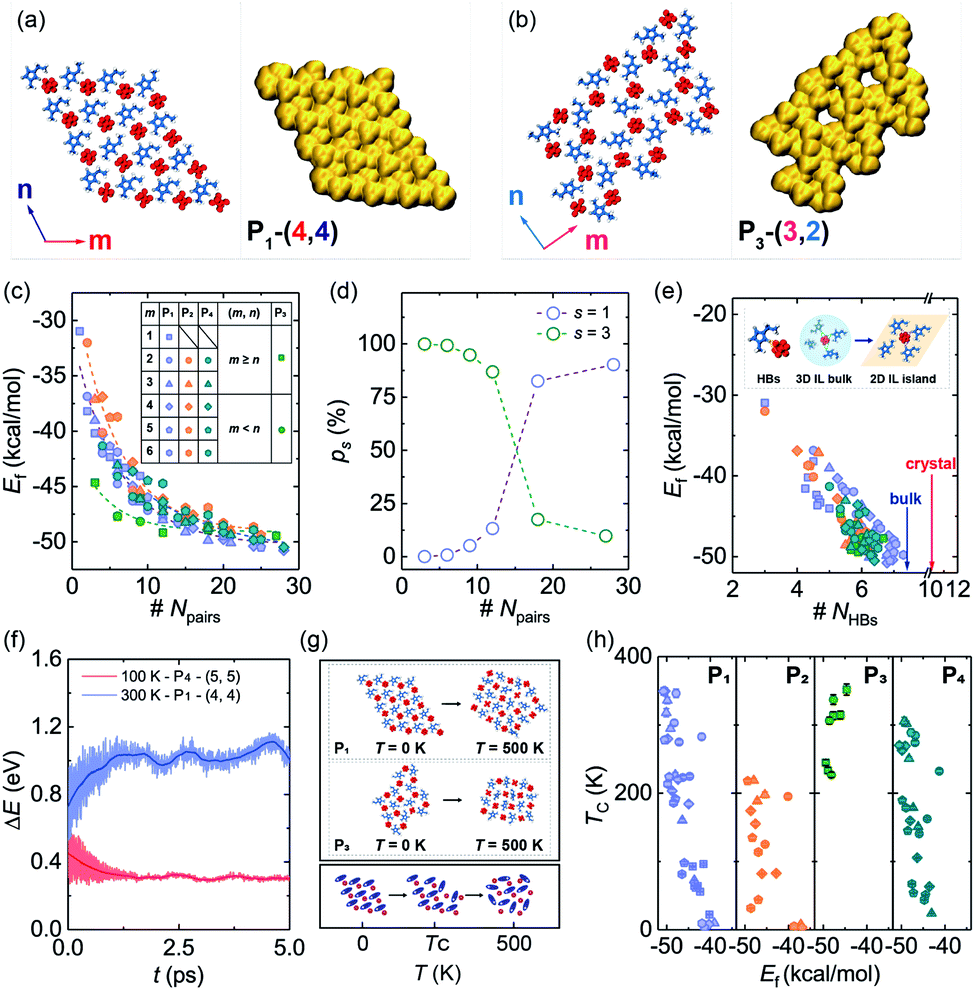 | ||
| Fig. 2 (a and b) Atomic structures and simulated STM images of the P1-based 2DIIs with (4,4) and the P3-based 2DIIs with (3,2), respectively. The bias energy for the generation of all STM images is 20 eV, and the iso-contour value of all STM images is 5.0 × 10−5 Hartree. (c) Ef as a function of Npairs, where the dashed lines represent exponential decay fitting. (d) ps for different 2DIIs changes with Npairs. (e) The correlation between Ef and NHBs, where the inset shows the atomic structures of HBs in the bulk and a 2DII. (f) ΔE of two typical 2DIIs from the AIMD simulations. (g and h) TC in the melting process of various 2DIIs as a function of Ef. | ||
Size-dependent thermodynamic stability and melting process of 2DIIs
To further quantify the influence of size on the structural stability, Ef for 2DIIs with different Npairs is summarized in Fig. 2c, authenticating that Ef will decline with growing Npairs for all 2DIIs. When Npairs exceeds the critical value (NC = 18), Ef for various 2DIIs will only fluctuate in a small range, that means as Npairs increases, the thermodynamic stability of 2DIIs will keep increasing until convergence. Interestingly, Ef for P1/2/4-based 2DIIs is almost the same, implying that relative orientations within the neighboring cations only have little effect on the thermodynamic stability. Moreover, the tendencies of Ef–Npairs for P1/2/4-based 2DIIs are fully different from those for P3-based 2DIIs: when the size of 2DIIs is small (Npairs < NC), the Ef of P3-based 2DIIs is the most negative; in contrast, the Ef of P1/2/4-based 2DIIs will be the lowest once the size is beyond NC.Assuming that Npairs is fixed, the Boltzmann factor can be used to describe the distribution of P1/3-based 2DIIs, that is, 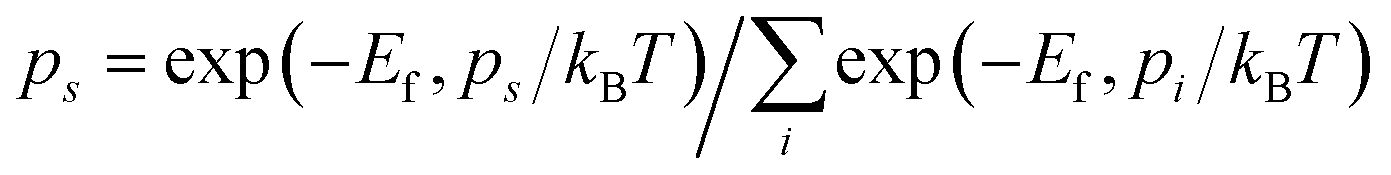 , where s = 1 and 3, i = 1 versus 3, kB is the Boltzmann constant, and T is set as 300 K.34 As displayed in Fig. 2d, there exist two stages in ps–Npairs. For instance, when Npairs = 12, p1 and p3 are respectively 13.18 and 86.82%, while when Npairs = 18, p1 and p3 are 82.51 and 17.49%, respectively. Hence, P3 will be the likeliest subunit if Npairs < 18, which is the opposite when Npairs ≥ 18. The turnover of ps implies that the structural transition from P3 to P1 may occur during the assembly process. Furthermore, the detailed internal arrangements of many large-sized monolayer IL films from experiments20,21,25 are almost like those in P1/2/4-based 2DIIs rather than those in P3-based 2DIIs, reflecting the rationality of the simulated results herein.
, where s = 1 and 3, i = 1 versus 3, kB is the Boltzmann constant, and T is set as 300 K.34 As displayed in Fig. 2d, there exist two stages in ps–Npairs. For instance, when Npairs = 12, p1 and p3 are respectively 13.18 and 86.82%, while when Npairs = 18, p1 and p3 are 82.51 and 17.49%, respectively. Hence, P3 will be the likeliest subunit if Npairs < 18, which is the opposite when Npairs ≥ 18. The turnover of ps implies that the structural transition from P3 to P1 may occur during the assembly process. Furthermore, the detailed internal arrangements of many large-sized monolayer IL films from experiments20,21,25 are almost like those in P1/2/4-based 2DIIs rather than those in P3-based 2DIIs, reflecting the rationality of the simulated results herein.
The HBs are further analyzed, as shown in Fig. S6,† to understand the correlation between the structure and thermodynamic stability. The angle and energy of HBs in 2DIIs are respectively larger and lower than those in the bulk and crystal ILs, showing that graphite strengthens HBs in 2DIIs and further enhances the structural stability of 2DIIs. As displayed in Fig. 2e, Ef almost decreases linearly with the number of HBs per cation–anion pair (NHBs), indicating the dominant role of HBs in constructing 2DIIs. When Npairs > NC, NHBs will catch up with the bulk value, agreeing well with convergent Ef. Furthermore, the 5 ps long ab initio MD (AIMD) simulations of representative 2DIIs at T = 300 and 100 K were performed. The energy difference per cation–anion pair (ΔE, see ESI Note†) with Npairs = 16 and 25 is only 0.28 and −0.15 eV, respectively (Fig. 2f), and the structures at different simulation times are almost unchanged (Fig. S7†). Hence, the AIMD simulations verify the structural stability of the corresponding 2DIIs.
To further confirm the dynamic stability of 2DIIs, the melting processes are elucidated via classical MD simulations. As T increases, 2DIIs first maintain initial structures and then the in-plane transition at the critical temperature (TC) occurs, along with losing the ordered internal arrangements (Fig. 2g). Fig. 2h shows the evolution of TCversus Ef, demonstrating that a negative relation exists in P1/2/4-based 2DIIs while a positive relation exists in P3-based 2DIIs. For instance, Ef of P1-based 2DIIs (Npairs = 28) and P3-based 2DIIs (Npairs = 3) is respectively −50.77 and −44.64 kcal mol−1, and TC for them is 349.14 and 351.73 K (Fig. S8†), respectively. Hence, for some small P3-based 2DIIs and large P1/2/4-based 2DIIs, TC is above room temperature, indicating their excellent dynamic stability. The different relationships between P1/2/4-based and P3-based 2DIIs coincide well with two stages of ps–Npairs (Fig. 2d). Combining Ef–Npairs, Ef–NHBs, TC–Ef, and TC–Npairs, it can be concluded that HB networks can determine the stability of 2DIIs.
The arrangement-size binary control mechanism of the electronic structure in 2DIIs
Considering the SSILs widely used in electrochemistry and energy storage, where the electronic properties of 2DIIs can play a dominant role, the highest occupied molecular orbital (HOMO), the lowest unoccupied molecular orbital (LUMO), and energy gap (Egap) are summarized in Fig. 3a, displaying that the overall trend of the HOMO and the LUMO respectively increases and declines as Npairs expands, leading to the reduction of Egap. For example, when Npairs goes up from 1 to 18, Egap of P1,2,4-based 2DIIs will drop by 44.87%, 44.06%, and 46.08%, respectively. However, Egap of P3-based 2DIIs only drops by 12.07%. Meanwhile, Egap of P1/2/3/4-based 2DIIs with Npairs > 24 is 2.74, 2.68, 4.34, and 2.71 eV, respectively, demonstrating that P3-based 2DIIs always possess the largest Egap, no matter how big the 2DII is.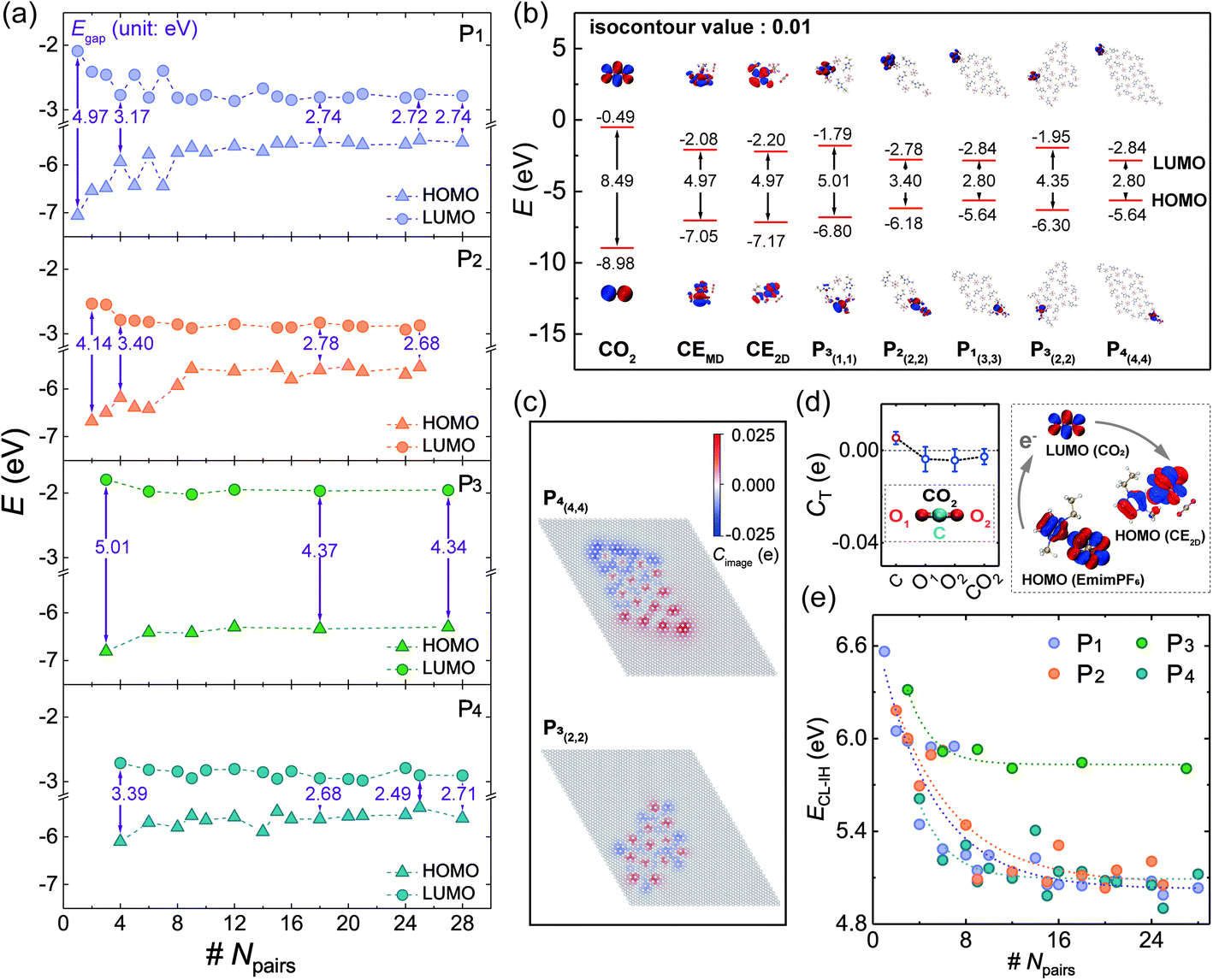 | ||
| Fig. 3 (a) The HOMO, the LUMO, and Egap of 2DIIs change with Npairs. (b) Electron distributions of the HOMO and the LUMO in different IL systems. (c) The image charge distributions of first-layer graphite in P4-based and P3-based 2DIIs. (d) The charge transfer quantity of the CO2 molecule before and after adsorption on the 2DII, where the schematic diagram shows electron transfer from ILs to CO2. (e) The energy gap between HOMOIL and LUMOCO2 changes with Npairs, where the dotted lines represent exponential decay fitting. | ||
Furthermore, the electron distributions of the HOMO and LUMO in 2DIIs are calculated, as shown in Fig. 3b. Interestingly, the electron of the HOMO and LUMO in P1/2/4-based 2DIIs is localized in two different corners, far different from the overlapping distribution of that in P3-based 2DIIs. The localized electron distribution should originate from the anisotropic image charge (Cimage) distribution of graphite. Taking the P4-based 2DII with (4,4) as an example, the C atoms at the upper left corner have negative Cimage while those at the lower right corner have positive Cimage, that means graphite will help decouple the electron in the HOMO and LUMO, especially in the ions at the edges of P1/2/4-based 2DIIs. Thus, for P1/2/4-based 2DIIs, with more ions at the edge comes more obvious decoupling, enhancing the electrical insulation feature of central ions and the electron transferability between edge ions and the substrate, leading to the plunge of Egap–Npairs. However, for P3-based 2DIIs, the Cimage distribution of graphite differs from P1/2/4-based 2DIIs, and the decoupling effect is extremely weak, producing the largest Egap. Thus, the subunit and size can jointly regulate the electronic properties of 2DIIs, the longer edges of P1/2/4-based 2DIIs, and the stronger electron transferability of 2DIIs.
The SSIL films have been widely used in the process of gas capture and conversion, where the interaction between gas and 2DIIs plays a dominant role. Taking CO2 as an example, the charge transfer quantity (CT) between a CO2 molecule and the typical 2DII is displayed in Fig. 3d, by statistical averaging from 9 independent adsorption sites. This shows that the CT of CO2 is negative, demonstrating that CO2 will get electrons from the 2DII in the typical chemical process.35,36 This means electrons may transfer from the HOMO of 2DIIs (HOMOIL) to the LUMO of CO2 (LUMOCO2). The difficulty of the electronic transfer process can be quantitatively described by the energy difference: ECL-IH = LUMOCO2 − HOMOIL, as shown in Fig. 3e. Excitingly, ECL-IH will decline exponentially as Npairs increases for various 2DIIs, especially for P1/2/4-based 2DIIs, representing that larger 2DIIs can transfer electrons to CO2 more quickly and be effective in the capture and fixation process of CO2.
Edge selective adsorption of 2DIIs in the CO2 capture process
During the practical reactive application of gas capture and conversion, the gas adsorption process, especially for CO2, is meaningful for the rational design of the corresponding catalyst. To evaluate the gas adsorption stability, we defined four different adsorption sites of 2DIIs (vertex site – Rvertex, edge sites – RZ4/RZ5/RE1/RE2, above site – Rabove, and inside site – Rin), as shown in Fig. 4a and S9.† The adsorption energy (Egas, see ESI Note†) of different sites is also calculated, as shown in Fig. 4b. For instance, Egas of CO2 at Rvertex, RZ4, RZ5, and Rabove in the P4-based 2DII with (4,4) is −9.88, −10.24, −12.08, and −5.99 kcal mol−1, respectively, reflecting that the edge site (RZ5) provides the strongest adsorption ability (ESI Movie M2 and M3†). The lower Egas should originate from the anisotropic Cimage distribution of graphite in Fig. 3c, providing the additional adsorption force to the target gas molecules. However, for the P3-based 2DII, Egas of CO2 at Rin, RE1, and RE2 is close, agreeing well with the isotropic Cimage distribution. In addition, since the position of the CO2 molecule at Rabove is so far away from the graphite substrate, this leads to weaker substrate contribution, further resulting in a higher Egas compared with that of other sites. Besides, Egas of CO, CH4, and N2 at various sites of the P4-based 2DII also shows the same order as that of CO2.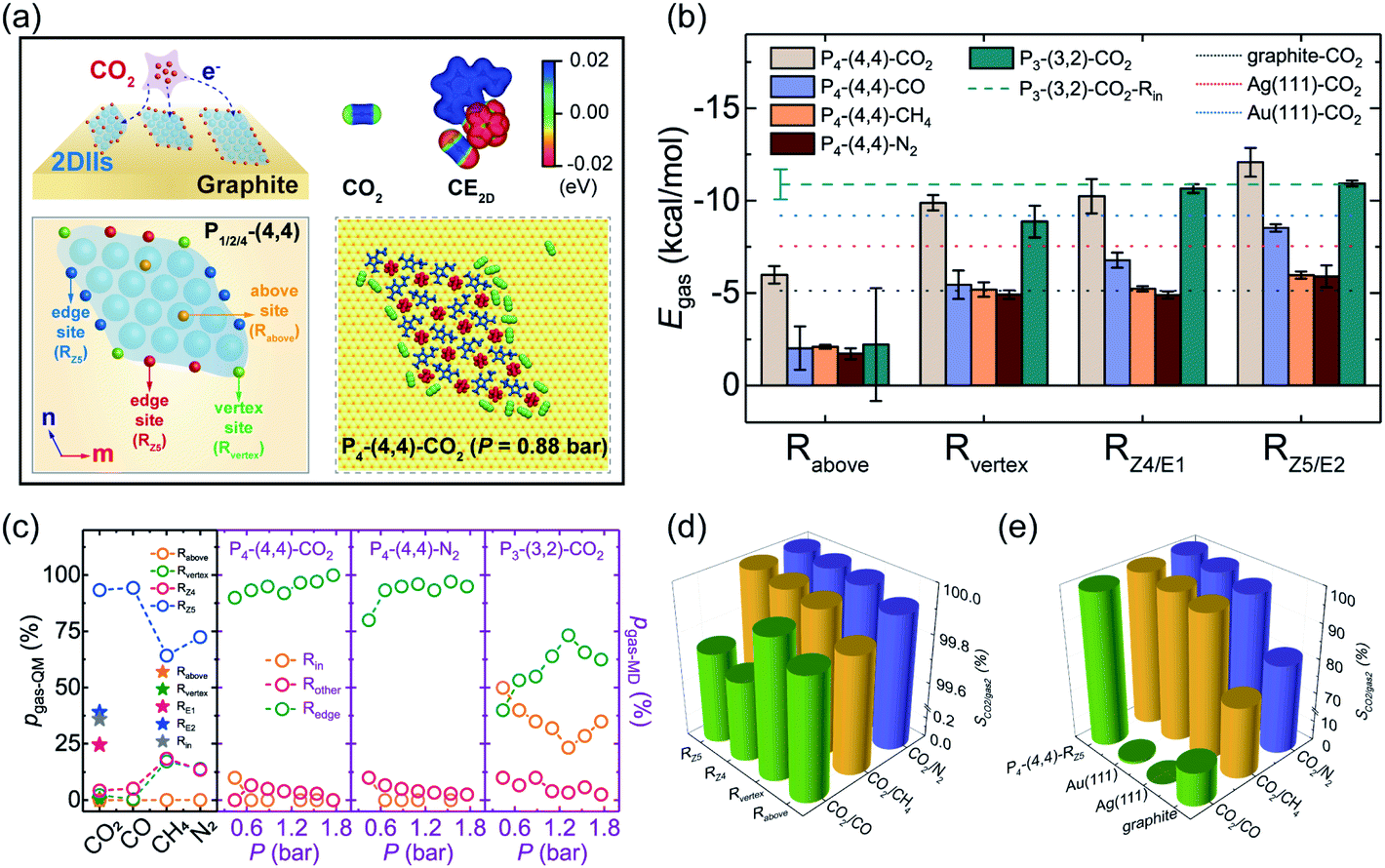 | ||
| Fig. 4 (a) Schematic diagrams of CO2 molecules adsorbed on different sites of 2DIIs, electrostatic potential (ESP) mapped to the van der Waals surface (electron-density isosurface = 0.01 e Bohr−3) of a CO2 molecule before (left) and after (right) adsorption on the P1-based 2DII with (1,1), and the illustration in the dotted box shows the atomic structures of CO2 molecules adsorbed on the 2DII at P = 0.88 bar. (b) Egas for different gases adsorbed on 2DIIs and pure solid surfaces. (c) The pgas-QM and pgas-MD for gases adsorbed on different sites. (d and e) The CO2 adsorption selectivity to other gases on the 2DII and pure solid surfaces. | ||
To quantitatively explore the dynamic stability of gases adsorbed on 2DIIs, MD simulations of the adsorption process were further performed at T = 300 K (Fig. 4a and S10†). As the partial pressure of gases (P) ranges from 0.44 to 1.76 bar, almost all gas molecules adsorb on the edges of the P4-based 2DII, while some adsorb on the sites inside the P3-based 2DII (ESI Movie M4†). Excitedly, as shown in Fig. 4c, the Boltzmann factor of Egas (pgas-QM, see ESI Note†) is well consistent with the proportion of gases adsorbed on different sites (pgas-MD, see ESI Note†), verifying that the edges of 2DIIs are dominant adsorption sites for different gases.
Furthermore, compared with many pure solids, Egas of different gases on edge sites of 2DIIs is even stronger (Fig. 4b and S11†). Taking CO2 as an example, Egas of RZ5 in the P4-based 2DII with (4,4), Au(111), Ag(111), and graphite is −12.08, −9.20, −7.54, and −5.13 kcal mol−1, respectively. Considering that Egas of CO2 is always lower than that of CO, CH4, and N2 when they adsorb on the P4-based 2DII, we further calculated the CO2 adsorption selectivity to other gases (SCO2/gas2, see ESI Note†) on different solid surfaces. Fantastically, SCO2/gas2 for the P4-based 2DII is bigger than 99.70% (Fig. 4d and e), indicating that all adsorption sites of 2DIIs show extremely high CO2 selectivity.
Meanwhile, SCO2/CH4, SCO2/CO, and SCO2/N2 of the P4-based 2DII can be up to 1.31, 1.52, and 1.23 times higher than those of the pure graphite surface. Moreover, SCO2/CH4, SCO2/CO, and SCO2/N2 for Au (111) are 98.59, 1.26, and 98.91%, respectively, lower than those for 2DIIs. The high CO2 adsorption selectivity suggests that the 2DII can be viewed as a promising adsorbent or reaction promoter in the process of carbon capture and utilization, which agrees well with recent experiments on the IL-stabilized single-atom catalysts.37 Considering that some 2DIIs can maintain stable structures above room temperature, it should be feasible to achieve higher gas adsorption and selectivity by extending the edges of 2DIIs or modulating the ionic microenvironment around the catalyst.
Additional discussion
Physically, the selective edge adsorption for different gases should originate from the unique Cimage distribution of the graphite supporting P1/2/4-based 2DIIs, where the structure, thermodynamic stability, electronic properties, and gas adsorption can also be further enhanced by selecting species of ILs and solid substrates. Moreover, the length of edges can measure the potential effectiveness of 2DIIs adsorbing the gas reactants. The longer side length of the 2DIIs is more conducive to playing the function of 2DIIs in the application of gas adsorption and selectivity. However, the bigger 2DII will lead to the large usage of ILs, which will be a challenge, especially considering the high synthesis cost of ILs.Excitingly, the optimal size of a P1/2/4-based 2DII can be determined from the size-dependent Ef and Egap (Fig. 2c and 4a). A too-small P1/2/4-based 2DII (Npairs < NC) will be thermodynamically unstable and possess a large Egap. Meanwhile, a too-large P1/2/4-based 2DII (Npairs > NC) will have almost the same thermodynamic stability and Egap as the 2DII of NC. Hence, the suitable size of P1/2/4-based 2DIIs should exist in practical applications, for instance, highly efficient and low-cost CO2 capture and conversion. Hence, multiple P1/2/4-based 2DIIs with an optimal size rather than a single large 2DII can provide the best performance, including high electrical conductivity and efficient CO2 adsorption and selectivity, and minimize the usage of ILs to the greatest extent, which is valuable to overcome the economic barrier of the high-price of ILs in real applications.
Conclusions
In brief, this study set out to better understand the structure, thermodynamic stability, electronic properties, and gas adsorption performance of 2DIIs via combined IC-QM/MM calculations and classical MD simulations. We proposed four kinds of subunits, which can assemble into various 2DIIs supported by graphite. Both the formation energy and HOMO–LUMO energy gap decrease as the size of the 2DIIs grow, and they converge once the size is beyond the critical value; some 2DIIs can also maintain their stable structures even above room temperature. Meanwhile, the HOMO–LUMO gap of the sparsely arranged (Table S2†) annular 2DII (P3) is far larger than that of close-packed ones (P1/2/4). Furthermore, the edges of 2DIIs are dominant adsorption sites for CO2, CO, CH4, and N2, and also show high CO2/CH4, CO2/CO, and CO2/N2 selectivity, which is up to 1.31, 1.52, and 1.23 times higher than that of the pure graphite surface. The adsorption stability and selectivity are even better than those of some pure metal surfaces. The quantitative structure–function relationship of 2DIIs proves that it is feasible to achieve a higher electronic or gas adsorption performance just by adopting an optimal size of 2DIIs, which is meaningful for the rational design and management of IL-based green chemical engineering applications.Computational details
Atomic structures
The IL considered herein is 1-ethyl-3-methylimidazolium hexafluoro-phosphate, namely EmimPF6, widely used as the electrolyte in supercapacitors and other applications.38–41 Considering the applications of the EmimPF6–graphite interface in the field of batteries, electrochemical reactions, and other chemical engineering processes,42,43 graphite was adopted as the solid surface. A three-layer slab models the graphite substrates, and a periodic boundary condition (PBC) was applied in all three dimensions, as shown in Fig. S1.† To properly accommodate the adsorbates on the surface, avoiding spurious interactions with the periodic images, at least 13 × 13 replicas of the unit cell in graphite are used in the lateral dimensions. To decouple the periodic images in the dimension perpendicular to the surface, a vacuum of at least ∼1.7 nm is added along the z-direction.An IC-QM/MM approach
The optimization of the hybrid IL-graphite system was performed using the CP2K/QUICKSTEP program.44,45 In order to expand the number of atoms in the hybrid IL-graphite system as much as possible, we employed the IC method46 within a QM/MM47,48 framework, where the QM part is treated at the Kohn–Sham (KS) density functional theory (DFT) level within a mixed Gaussian and plane wave49 (GPW) formalism. The accuracy of the IC-QM/MM approach is evaluated by comparing the structural and energetic properties to the full DFT results, as shown in Fig. S2.† “Full DFT” in this context means that graphite and the adsorbate are both treated at the DFT level. For QM calculations, double-ζ valence plus polarization (DZVP) basis sets of the Molopt-type50 are used to represent the valence electrons. The interactions between valence and core electrons are described by norm-conserving Goedecker, Teter, and Hutter pseudopotentials.51–53 The Becke–Lee–Yang–Parr (BLYP) functional54,55 models the exchange and correlation potential. The van der Waals interactions are accounted for by employing the DFT-D3 dispersion correction. The energy cutoff for the auxiliary plane wave expansion of the density is set to 400 Ry. Moreover, The MM-based interactions between EmimPF6 and graphite are modeled with the parameters from the MD simulations.MD simulations
The parameters of the bond, angle, dihedral, van der Waals interactions, and electrostatic interactions of EmimPF6 were described by the all-atom optimized potentials for the liquid simulations (OPLS-AA) force field,56–58 which has been used to successfully obtain the structures and liquid properties of ILs.59,60 Previously, Tang et al. found that the non-polarizable force field could prophesy a rational interfacial structure at the IL-graphite interface via the AIMD simulation.61,62 Therefore, the non-polarizable OPLS-AA force field employed here commendably described the structure and properties of 2DIIs on graphite. The interactions between EmimPF6 and graphite can be divided into electrostatic and van der Waals interactions. The former one, the long-range coulombic interaction, was computed using the particle–particle–particle–mesh (PPPM) algorithm with an accuracy of 0.0001.63 The latter one was represented by using the 12-6 Lennard-Jones (LJ) potential, which was truncated at 1.2 nm. The Lorentz–Berthelot mixing rules were used to calculate the van der Waals interactions between different atomic species. The SHAKE algorithm64 was applied to the C–H bonds to reduce high-frequency vibrations. All MD simulations in this work were carried out using a large-scale atomic/molecular massively parallel simulator (LAMMPS).65 The time step for integrating the Newtonian equations of motion was set as 2.0 fs, which was demonstrated to ensure energy conservation. The in-plane (x–y) size of the graphite surface was set as 3.20 × 3.20 nm2 at least, and the height of the PBC box along the z-direction is 20 nm. The atoms of the graphite substrate were fixed during the MD simulations to maintain the planar structure. P1, P2, P3, and P4 were chosen from the equilibrium configuration of monolayer EmimPF6 in Fig. S1.† Then, P1/2/3/4-based 2DIIs with different Npairs were placed about 0.3 nm above the graphite substrate, which was relaxed in the NVT ensemble at T = 0.01 K, controlled by using the Berendsen thermostat.66 After a 10 ns long MD simulation, the initial structure of the hybrid IL–graphite system can be obtained.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
H. H., Y. W., and S. Z. designed this research; C. W. performed the simulations; Z. G., Y. L., C. Q., and F. H. discussed the results; C. W., Y. W., H. H., and S. Z. contributed to preparing the final manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was funded by the National Natural Science Foundation of China (21922813, 22078322, 22178344, and 21890762), the Youth Innovation Promotion Association of CAS (2021046), the Fund of State Key Laboratory of Multiphase Complex Systems (MPCS-2021-A-10), DNL Cooperation Fund, CAS (DNL180202), and the Innovation Academy for Green Manufacture, Chinese Academy of Sciences (IAGM2020C16).References
- K. Dong, X. Liu, H. Dong, X. Zhang and S. Zhang, Chem. Rev., 2017, 117, 6636–6695 CrossRef CAS PubMed.
- R. Rogers and K. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed.
- J. Avila, L. Lepre, C. Santini, M. Tiano, S. Denis-Quanquin, K. Chung Szeto, A. Padua and M. Costa Gomes, Angew. Chem., Int. Ed., 2021, 60, 12876–12882 CrossRef CAS PubMed.
- B. Li, C. Wang, Y. Zhang and Y. Wang, Green Energy Environ., 2021, 6, 253–260 CrossRef.
- Z. Wu, W. Ding, Y. Zhang, Y. Wang and H. He, Acta Phys.-Chim. Sin., 2021, 37, 2002021 Search PubMed.
- M. Hope, K. Griffith, B. Cui, F. Gao, S. Dutton, S. Parkin and C. Grey, J. Am. Chem. Soc., 2018, 140, 16685–16696 CrossRef CAS PubMed.
- J. Feng, H. Gao, L. Zheng, Z. Chen, S. Zeng, C. Jiang, H. Dong, L. Liu, S. Zhang and X. Zhang, Nat. Commun., 2020, 11, 4341 CrossRef PubMed.
- H. Sun, P. Liang, G. Zhu, W. Hung, Y. Li, H. Tai, C. Huang, J. Li, Y. Meng, M. Angell, C. Wang and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 27847–27853 CrossRef CAS PubMed.
- S. Bi, H. Banda, M. Chen, L. Niu, M. Chen, T. Wu, J. Wang, R. Wang, J. Feng, T. Chen, M. Dincă, A. Kornyshev and G. Feng, Nat. Mater., 2020, 19, 552–558 CrossRef CAS PubMed.
- H. Dou, B. Jiang, M. Xu, Z. Zhang, G. Wen, F. Peng, A. Yu, Z. Bai, Y. Sun, L. Zhang, Z. Jiang and Z. Chen, Angew. Chem., Int. Ed., 2019, 58, 13969–13975 CrossRef CAS PubMed.
- S. Budhathoki, J. Shah and E. Maginn, Ind. Eng. Chem. Res., 2017, 56, 6775–6784 CrossRef CAS.
- M. Babucci, C. Fang, A. Hoffman, S. Bare, B. Gates and A. Uzun, ACS Catal., 2017, 7, 6969–6972 CrossRef CAS.
- W. Sun, M. Wang, Y. Zhang, W. Ding, F. Huo, L. Wei and H. He, Green Energy Environ., 2020, 5, 183–194 CrossRef.
- B. Xin and J. Hao, Chem. Soc. Rev., 2014, 43, 7171–7187 RSC.
- B. Polesso, F. Bernard, H. Ferrari, E. Duarte, F. Vecchia and S. Einloft, Heliyon, 2019, 5, e02183 CrossRef PubMed.
- L. Offner-Marko, A. Bordet, G. Moos, S. Tricard, S. Rengshausen, B. Chaudret, K. Luska and W. Leitner, Angew. Chem., Int. Ed., 2018, 57, 12721–12726 CrossRef CAS PubMed.
- S. El Sayed, A. Bordet, C. Weidenthaler, W. Hetaba, K. Luska and W. Leitner, ACS Catal., 2020, 10, 2124–2130 CrossRef CAS.
- J. Armstrong, C. Hurst, R. Jones, P. Licence, K. Lovelock, C. Satterley and I. Villar-Garcia, Phys. Chem. Chem. Phys., 2007, 9, 982 RSC.
- T. Cremer, M. Killian, J. Gottfried, N. Paape, P. Wasserscheid, F. Maier and H. Steinrück, ChemPhysChem, 2008, 9, 2185–2190 CrossRef CAS PubMed.
- B. Uhl, F. Buchner, D. Alwast, N. Wagner and R. Behm, Beilstein J. Nanotechnol., 2013, 4, 903–918 CrossRef CAS PubMed.
- T. Cremer, M. Stark, A. Deyko, H. Steinrück and F. Maier, Langmuir, 2011, 27, 3662–3671 CrossRef CAS PubMed.
- C. Wang, C. Qian, Z. Li, N. Wei, N. Zhang, Y. Wang and H. He, Ind. Eng. Chem. Res., 2020, 59, 8028–8036 CrossRef CAS.
- S. Zhang, Y. Lu, C. Peng, H. Liu and D. Jiang, J. Phys. Chem. C, 2019, 123, 618–624 CrossRef CAS.
- B. Uhl, H. Huang, D. Alwast, F. Buchner and R. Jürgen-Behm, Phys. Chem. Chem. Phys., 2015, 17, 23816–23832 RSC.
- M. Meusel, M. Lexow, A. Gezmis, S. Schötz, M. Wagner, A. Bayer, F. Maier and H. Steinrück, ACS Nano, 2020, 14, 9000–9010 CrossRef CAS PubMed.
- B. Uhl, F. Buchner, S. Gabler, M. Bozorgchenani and R. Jürgen-Behm, Chem. Commun., 2014, 50, 8601–8604 RSC.
- Q. Su, Y. Qi, X. Yao, W. Cheng, L. Dong, S. Chen and S. Zhang, Green Chem., 2018, 20, 3232–3241 RSC.
- W. Xie, X. Ji, X. Feng and X. Lu, AIChE J., 2015, 61, 4437–4444 CrossRef CAS.
- W. Xie, X. Ji, X. Feng and X. Lu, Ind. Eng. Chem. Res., 2016, 55, 366–372 CrossRef CAS.
- Z. Tang, L. Lu, Z. Dai, W. Xie, L. Shi and X. Lu, Langmuir, 2017, 33, 11658–11669 CrossRef CAS PubMed.
- S. Zhang, Y. Wang, H. He, F. Huo, Y. Lu, X. Zhang and K. Dong, Green Energy Environ., 2017, 2, 329–330 CrossRef.
- K. Dong, S. Zhang and J. Wang, Chem. Commun., 2016, 52, 6744–6764 RSC.
- J. Dong, D. Geng, F. Liu and F. Ding, Angew. Chem., Int. Ed., 2019, 131, 7805–7809 CrossRef.
- X. Zhou, H. Shu, Q. Li, P. Liang, D. Cao and X. Chen, J. Mater. Chem. C, 2020, 8, 4432–4440 RSC.
- M. Asadi, K. Kim, C. Liu, A. Addepalli, P. Abbasi, P. Yasaei, P. Phillips, A. Behranginia, J. Cerrato, R. Haasch, P. Zapol, B. Kumar, R. Klie, J. Abiade, L. Curtiss and A. Salehi-Khojin, Science, 2016, 353, 467–470 CrossRef CAS PubMed.
- Y. Oh and X. Hu, Chem. Commun., 2015, 51, 13698–13701 RSC.
- S. Ding, Y. Guo, M. Hülsey, B. Zhang, H. Asakura, L. Liu, Y. Han, M. Gao, J. Hasegawa, B. Qiao, T. Zhang and N. Yan, Chem, 2019, 5, 3207–3219 CAS.
- F. Huo, Z. Liu and W. Wang, J. Phys. Chem. B, 2013, 117, 11780–11792 CrossRef CAS PubMed.
- X. Liu, D. Wu, H. Wang and Q. Wang, Adv. Mater., 2014, 26, 4370–4375 CrossRef CAS PubMed.
- T. Zhao, J. Liang, Y. Zhang, Y. Wu and X. Hu, Chem. Commun., 2018, 54, 8964–8967 RSC.
- B. Moghadam, M. Razmkhah, M. Mosavian and F. Moosavi, Phys. Chem. Chem. Phys., 2016, 18, 33053–33067 RSC.
- X. Shi, W. Zhang, J. Wang, W. Zheng, K. Huang, H. Zhang, S. Feng and H. Chen, Adv. Energy Mater., 2016, 6, 1601378 CrossRef.
- Q. Dou, M. Sha, H. Fu and G. Wu, J. Phys.: Condens. Matter, 2011, 23, 175001 CrossRef CAS PubMed.
- J. Hutter, M. Iannuzzi, F. Schiffmann and J. VandeVondele, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 15–25 CAS.
- J. Vandevondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- D. Golze, M. Iannuzzi, M. Nguyen, D. Passerone and J. Hutter, J. Chem. Theory Comput., 2013, 9, 5086–5097 CrossRef CAS PubMed.
- T. Laino, F. Mohamed, A. Laio and M. Parrinello, J. Chem. Theory Comput., 2005, 1, 1176–1184 CrossRef CAS PubMed.
- T. Laino, F. Mohamed, A. Laio and M. Parrinello, J. Chem. Theory Comput., 2006, 2, 1370–1378 CrossRef CAS PubMed.
- B. Lippert and J. Parrinello, Mol. Phys., 1997, 92, 477–488 CrossRef.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef CAS PubMed.
- C. Hartwigsen, S. Goedecker and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 3641–3662 CrossRef CAS.
- M. Krack, Theor. Chem. Acc., 2005, 114, 145–152 Search PubMed.
- A. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- J. Canongia Lopes, J. Deschamps and A. Pádua, J. Phys. Chem. B, 2004, 108, 2038–2047 CrossRef.
- J. Canongia Lopes, J. Deschamps and A. Pádua, J. Phys. Chem. B, 2004, 108, 11250 CrossRef.
- J. Canongia Lopes and A. Pádua, J. Phys. Chem. B, 2004, 108, 16893–16898 CrossRef.
- C. Qian, B. Ding, Z. Wu, W. Ding, F. Huo, H. He, N. Wei, Y. Wang and X. Zhang, Ind. Eng. Chem. Res., 2019, 58, 20109–20115 CrossRef CAS.
- Y. Wang, C. Wang, Y. Zhang, F. Huo, H. He and S. Zhang, Small, 2019, 15, 1804508 CrossRef PubMed.
- F. Tang, T. Ohto, T. Hasegawa, M. Bonn and Y. Nagata, Phys. Chem. Chem. Phys., 2017, 19, 2850–2856 RSC.
- S. Kim, S. Zhou, Y. Hu, M. Acik, Y. Chabal, C. Berger, W. De Heer, A. Bongiorno and E. Riedo, Nat. Mater., 2012, 11, 544–549 CrossRef CAS PubMed.
- R. Hockney and J. Eastwood, Computer simulation using particles, Taylor & Francis, Inc., 1988 Search PubMed.
- J. Ryckaert, G. Ciccotti and H. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- H. Berendsen, J. Postma, W. Van Gunsteren, A. Dinola and J. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc05431g |
| This journal is © The Royal Society of Chemistry 2021 |