
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceClustering of catalytic nanocompartments for enhancing an extracellular non-native cascade reaction†
Viviana
Maffeis‡
ab,
Andrea
Belluati‡
 a,
Ioana
Craciun
a,
Dalin
Wu
a,
Samantha
Novak
a,
Cora-Ann
Schoenenberger
ab and
Cornelia G.
Palivan
*ab
a,
Ioana
Craciun
a,
Dalin
Wu
a,
Samantha
Novak
a,
Cora-Ann
Schoenenberger
ab and
Cornelia G.
Palivan
*ab
aDepartment of Chemistry, University of Basel, Mattenstrasse 24a, BPR 1096, 4058 Basel, Switzerland. E-mail: cornelia.palivan@unibas.ch
bNCCR-Molecular Systems Engineering, BPR 1095, Mattenstrasse 24a, CH-4058 Basel, Switzerland
First published on 31st August 2021
Abstract
Compartmentalization is fundamental in nature, where the spatial segregation of biochemical reactions within and between cells ensures optimal conditions for the regulation of cascade reactions. While the distance between compartments or their interaction are essential parameters supporting the efficiency of bio-reactions, so far they have not been exploited to regulate cascade reactions between bioinspired catalytic nanocompartments. Here, we generate individual catalytic nanocompartments (CNCs) by encapsulating within polymersomes or attaching to their surface enzymes involved in a cascade reaction and then, tether the polymersomes together into clusters. By conjugating complementary DNA strands to the polymersomes' surface, DNA hybridization drove the clusterization process of enzyme-loaded polymersomes and controlled the distance between the respective catalytic nanocompartments. Owing to the close proximity of CNCs within clusters and the overall stability of the cluster architecture, the cascade reaction between spatially segregated enzymes was significantly more efficient than when the catalytic nanocompartments were not linked together by DNA duplexes. Additionally, residual DNA single strands that were not engaged in clustering, allowed for an interaction of the clusters with the cell surface as evidenced by A549 cells, where clusters decorating the surface endowed the cells with a non-native enzymatic cascade. The self-organization into clusters of catalytic nanocompartments confining different enzymes of a cascade reaction allows for a distance control of the reaction spaces which opens new avenues for highly efficient applications in domains such as catalysis or nanomedicine.
Introduction
In nature, compartmentalization is a prerequisite for the spatiotemporal control of signalling pathways and for intra- and intercellular communication. The distance between compartments is critical for intercompartmental interactions,1,2 with distances ranging between 20–50 nm in intracellular and synaptic communication3,4 to above 250 μm in paracrine signalling.5 In addition, there are various reactions or conditions that require bio-compartments such as organelles or cells to directly interact in order to communicate or transfer molecules. Significant efforts have been made to exploit nature's designs and develop compartments in which specific enzymatic reactions take place,6,7 or that can support cascade reactions.8,9 Of particular interest are nanometric polymer vesicles (polymersomes), with their hollow spherical architecture, as they allow insertion of hydrophilic molecules inside their lumen (e.g. enzymes) and/or hydrophobic molecules such as membrane proteins in their membrane.10,11 Polymersomes show improved stability compared to lipid-based vesicles, and allow tuning properties such as membrane thickness, polarity or toxicity based on the chemical versatility of polymers.12–14 Moreover, it is possible to modify their surface with biological molecules that mediate cellular targeting or surface attachment or, more recently, the self-organization of polymersomes into clusters.10,15–21 When loaded with enzymes and made permeable to substrates and products, polymersomes serve as effective catalytic nanocompartments (CNCs) with a broad range of applications, such as organelle models,12,22,23 biosensors,10 detoxifying agents,24 and production or release/activation of prodrugs.10,25 More complex setups rely on enzymatic cascades, where the product of one enzyme becomes the substrate of another, similar to many biochemical pathways in cells.26 As in communicating cellular compartments, the distance between the enzymes has been demonstrated to be a fundamental parameter in cascade reaction efficiency.26,27 However, co-encapsulation of enzymes in the same polymersome was shown to have some limitations, in particular a lack of modularity,28 issues that are avoided in CNCs harbouring only one kind of enzyme at a time. As polymersomes are colloidal systems, the main way to modulate the inter-vesicle distance is to vary their concentration, which affects the cargo concentration as well. Another strategy to keep the distance between polymersomes small and constant is to link them together, for example by DNA,20 resulting in polymersome clusters which, unlike their liposome counterparts,29–32 do not precipitate. Polymersome clusters have been applied to the co-delivery of enzymes and dyes for theranostic applications.15,33 However, their unique potential to increase the efficiency of cascade reactions by mimicking the conditions in which natural organelles or cells are in close contact has not yet been explored.Here we present how different CNCs can be tethered together at a controlled distance and carry out a cascade reaction involving segregated compartments (Fig. 1). We advance from compartments acting in tandem without fixed distance26,34 to a new construct where the distance between compartments is pre-determined. This architecture facilitates the diffusion of molecules between compartments: the products of the first reaction (located in one type of CNC) will reach the second type of compartment where they will become substrates for the in situ reaction, thus supporting the overall cascade reaction. Catalytic nanocompartments are tethered together by DNA hybridization of complementary single-stranded DNA (ssDNA) exposed on the surface of different polymersomes, to promote clusterization.20 As a model cascade, we used glucose oxidase (GOX) and lactoperoxidase (LPO): GOX oxidizes glucose into gluconic acid and H2O2 which is used by LPO to oxidize a variety of substrates. More generally, the oxidase-peroxidase system is an antibacterial cascade found in many animal secretions. Based on the hydrogen peroxide coming from organic substrates, peroxidases can produce bacteriostatic compounds such as hypothiocyanites from thiocyanates, and thus have been suggested for biomedical applications such as oral plaque treatment or to counter to opportunistic infections developed in cystic fibrosis.35–37 Moreover, indications of the GOX–LPO cascade in anticancer and antiviral activities38,39 make it an interesting enzyme pair for possible applications.
 | ||
| Fig. 1 Concepts of a GOX–LPO cascade between two clustered CNCs, tethered via complementary ssDNA, in order to facilitate the diffusion of H2O2 and thus improve the overall reaction efficiency. Similarly, an AMG–GOX–LPO cascade achieves an improved diffusion of the glucose derived from amylose, and the enzyme on the surface allows the access to bulky substrates that would otherwise be out of reach for encapsulated enzymes. | ||
To provide one step further in the cascade complexity, we conjugated amyloglucosidase (AMG), an exoenzyme that hydrolyses amylose into small glucose units,48 to the surface of GOX-nanocompartments. Due to the external location of AMG, thousands of g mol−1 large amylose could be hydrolyzed to glucose that could enter the compartment and serve as substrate for the GOX encapsulated.
The co-localization of enzymes via DNA had been used for several enzymes in bulk,40–42 but never for catalytic compartments. Our approach has the advantage of allowing the enzymes to move freely inside the polymersomes where they are protected from harmful environmental conditions, while inspired by organelles and cells, the compartments are kept at a close but constant distance.26 In addition, the distance between the CNCs within a cluster can be easily controlled by modifying the length of DNA strands which affects the overall performance of the cascade reaction.
More importantly, CNC clusters were not limited to colloidal suspensions: DNA single strands that are not engaged in linking CNCs act as ligands for cell surface receptors and thereby attach the clusters to the surface where they are too big for uptake.15,33 Such decoration endows the cell with a novel compartment, a satellite organelle bound to their plasma membrane with which cells can perform cascade reactions at the surface. With our clusters, cells could break down an otherwise inadequate substrate (amylose) to feed into a totally bio-orthogonal cascade reaction. Both features of our clusters, distance control between consecutive steps of cascade reactions and cell association are essential for developing applications in catalysis or medicine.
Results and discussion
Generation and characterization of catalytic nanocompartments
As building block for the CNCs, we synthesized an amphiphilic diblock copolymer, poly(2-methyl-2-oxazoline)-block-poly(dimethylsiloxane) PMOXA10-PDMS29 according to an established procedure (Fig. S1†).20 Copolymers with PDMS as hydrophobic block and PMOXA as hydrophilic block have been used previously for producing catalytic compartments because generally the membrane of polymersomes is impermeable to small molecules but sufficiently flexible to enable the insertion of membrane proteins.10,11 The same polymer was also functionalized with poly(ethylene glycol)–N3 (PEG4–N3) (Fig. S2†) on its PMOXA block to both provide a clickable moiety on the surface of the self-assembled structures and to ensure maximum miscibility. PEG4–N3 allowed the conjugation of dibenzocyclooctyne (DBCO)–DNA strands, via strain-promoted alkyne–azide cycloaddition (SPAAC).20 Subsequently, 50% (mol%) PMOXA10–PDMS29 were mixed with 50% N3–PEG4–PMOXA10–PDMS29 to have available a high density of accessible azides upon the self-assembly of polymersomes.15,20Self-assembly of polymersomes encapsulating either GOX or LPO was achieved via film rehydration and resulting nanoassemblies were extruded to decrease the size polydispersity. Melittin, a pore-forming peptide derived from bee venom, was added to the polymersomes in order to render membranes permeable to molecular flow to and from the confined enzymes. Melittin has been shown to induce pore formation in membranes formed by triblock PMOXA–PDMS–PMOXA and diblock PMOXA-PDMS without destabilizing the vesicles.21,43 In addition, melittin-induced pores are less prone to hindering to the diffusion of substrates compared to outer membrane porin (OmpF) pores,26,34,45 making it well-suited for the permeabilization of polymersomes. The characterization of the assemblies by static and dynamic light scattering (SLS/DLS) showed that GOX-CNCs had a hydrodynamic radius (Rh) of 119 ± 8 nm and a radius of gyration (Rg) of 110 ± 2 nm, whereas LPO-CNCs were slightly bigger at 170 ± 24 nm and 150 ± 11 nm, respectively. The ratio between Rh and Rg, 0.9 in both cases, confirmed the production of vesicular assemblies. The difference in size between CNCs could be explained by the fact that LPO interacts with the PMOXA block and thus preferentially localizes to the membrane surface. Similarly, such interaction was seen with the related horseradish peroxidase,45 and recently reported for LPO in giant unilamellar vesicles as well.44 The vesicular architecture of LPO- and GOX CNCs was confirmed by TEM micrographs revealing the deformed spherical morphology typical of this kind of polymersome (Fig. 2A, B and S1†).
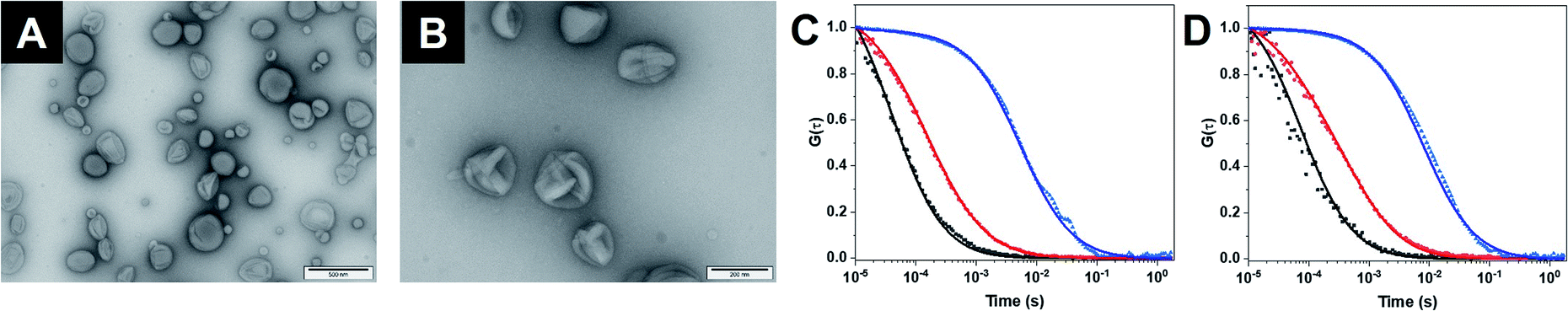 | ||
| Fig. 2 (A) TEM micrograph of GOX-CNC. (B) TEM micrograph of LPO-CNC. (C) FCS autocorrelation curves of free Atto-488 (black), Atto-488–GOX (red) and Atto-488–GOX-loaded CNCs (blue). Dots: raw data. Line: fitted model. (D) FCS autocorrelation curves of free DyLight-633 (black), DyLight-633–LPO (red) and DyLight-633–LPO-loaded CNCs (blue). | ||
The polymersome concentration, determined by nanoparticle tracking analysis (NTA), showed a 30% higher concentration for GOX-CNCs than for the smaller LPO-CNCs. At approximately 200 μg mL−1, the concentration of encapsulated enzyme was determined to be similar for both types of CNCs (Table S1†). To further characterize CNCs, we encapsulated fluorescently labelled GOX (with Atto-488) or LPO (with DyLight 633), and analyzed the diffusion times of CNCs compared to free labelled enzyme and free dye by fluorescence correlation spectroscopy (FCS).19 The corresponding shift of the FCS autocorrelation curves to higher diffusion times indicated that both enzymes were labelled (reduced diffusion time of labelled enzyme compared to free dye) and that polymersomes were associated with labelled enzyme (significant reduction of the diffusion time of CNCs compared to free enzyme) (Fig. 2C, D and Table S2†). Moreover, the brightness intensity of the single species (free enzyme, CNC) allowed us to quantify the average number of dye molecules per enzyme and the enzyme molecules per polymersome.19 The resulting 11 ± 4 GOX/polymersome and 52 ± 32 LPO/polymersome indicated that there was no overcrowding within the CNCs (Table S2†). Calculations of the average space in the cavity of polymersomes occupied by the enzymes revealed the total volume of GOX molecules to be around 1900 times smaller than that of the compartment, and the volume of LPO 330 times smaller.46,47 Furthermore, only 2% of the total enzyme in the samples remained free after purification (Table S2†).
Having characterized the lumen of the CNCs, we moved on to their surface (Fig. S3 and Table S3†). By conjugating a small fluorescent dye (Atto-488 DBCO) via SPAAC to azide groups of vesicles and measuring the fluorescence intensity per polymersome via FCS, we estimated the number of easily accessible azides on the CNC surface to be 104 ± 24 which is in line with published data.15
For linking together the CNCs, we designed two DBCO-coupled, 22 nucleotides long, complementary ssDNA, each comprising a short, 5′ non-complementary thymidine sequence as spacer to improve the DNA hybridization (Table S4†).20 DBCO–ssDNA was then conjugated to the azide-functionalized polymersome membrane via SPAAC. We quantified the number of ssDNA conjugated to each type of CNC by the means of hybridizing complementary ssDNA labelled with a fluorescent dye (Atto-488 for GOX- and Cy5 for LPO-CNCs) and FCS (Fig. S3†). While the surface density of the ssDNA on the polymersomes was quite disperse (ranging from few to above 100 ssDNA per polymersome), the numbers were in the range shown to promote the clustering of CNCs (Table S3†).15
Finally, amyloglucosidase (AMG) was conjugated via an NHS-PEG3-DBCO linker to free azide moieties on preformed, ssDNA–GOX-CNCs. The resulting AMG(GOX)-CNCs hold one type of enzyme (GOX) in their cavity and expose another (AMG) on the outer surface where it is available to substrates too big to enter CNCs through melittin pores. By further clicking Atto-488-DBCO to the remaining azides not occupied by either ssDNA or AMG, we estimated that about 35 ± 4 AMG were present on each GOX-CNC (Table S3†).
The molecular weight48 and dimensions of AMG from Aspergillus niger (75 Å × 45 Å × 40 Å, PDB code 6YQ7, manually measured using Pymol) indicate that the size of AMG is orders of magnitude smaller than that of polymersomes. Accordingly, FCS measurements did not reveal any significant shift in the diffusion times if AMG was conjugated to GOX-CNCs (6000 μs for AMG-conjugated CNCs versus 5486 ± 2510 μs for CNCs without AMG).
Single CNC activity
We tested the enzyme functionality of the single CNCs (without DNA conjugates) based on the oxidative conversion of Amplex Red (AR) to resorufin. LPO-CNCs were directly incubated with AR and H2O2 (Fig. 3A), whereas GOX-CNCs were incubated with glucose, AR and free LPO, ensuring that the H2O2 produced by glucose oxidation can directly diffuse to the peroxidase catalyzing the conversion of AR to resorufin (Fig. 3B). Both types of CNC showed activity only when rendered permeable by melittin (blue curve), confirming that substrates need to have access to the confined enzymes. The small amount of residual non-encapsulated LPO detected in FCS measurements was probably inactivated when testing enzyme activity of LPO-CNCs. The absence of LPO activity outside of LPO-CNCs corroborates the protection of enzymes inside polymersomes, which is in line with previously reported CNCs.26,34,49 AMG(GOX)-CNCs behaved similarly: when supplied externally with amylose, the glucose released by AMG-catalyzed hydrolysis could enter the CNC only if the membrane was permeabilized by melittin (Fig. 3C). We could detect only minimal non-specific oxidation of AR by free LPO (Fig. 3B and C). Notably, the influence of permeabilization via melittin, reflected by the difference between blue (permeabilized) and red (non-permeabilized) curves, was less evident as the system gained complexity: from a large difference when only LPO was involved, to a relatively small difference with a 3-enzyme system.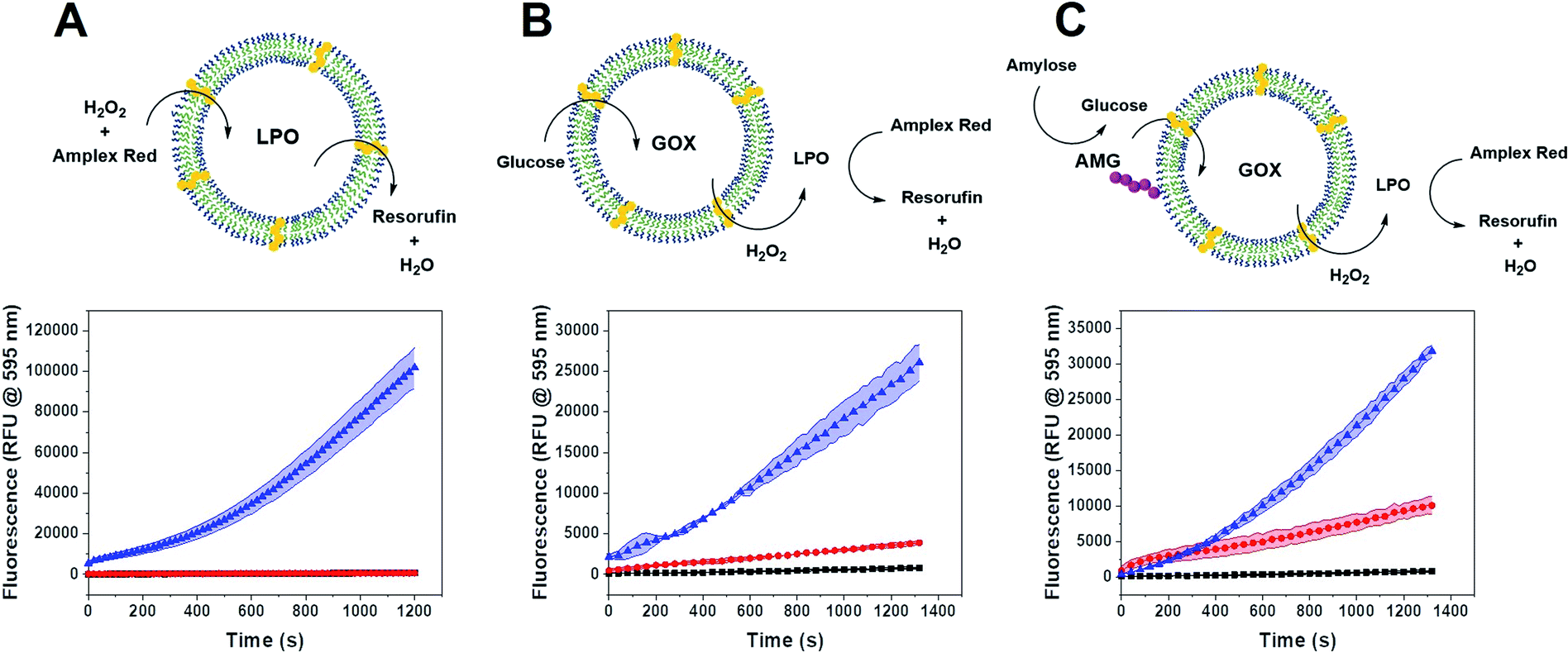 | ||
| Fig. 3 (A) Activity of LPO-CNC with melittin (blue), CNCs without melittin (red) and substrates alone (black, hidden behind red). (B) Activity of GOX-CNC with melittin (blue), CNCs without melittin (red) and substrates alone (black), using LPO as reporter enzyme. (C) Activity of AMG(GOX)-CNC with melittin (blue), without melittin (red) and substrates alone (black). Error bands represent ±SD, n = 3 replicates. | ||
CNC clustering
Having confirmed the enzymatic activity of individual CNCs, we next aimed to establish clusters of CNCs by taking advantage of DNA hybridization. For this purpose, complementary ssDNAs were conjugated to the azide groups on the membranes of the respective polymersomes. After mixing the functionalized CNCs at an equal ratio, we followed the clustering process over time via dynamic light scattering (DLS) (Fig. 4A). The number of ssDNA per polymersome was sufficient to induce clustering. After 14 h, a plateau was reached where LPO–GOX clusters had an average diameter, DH of about 700 nm. AMG(GOX)–LPO-CNCs formed smaller clusters of around 500 nm in diameter, possibly due to AMG-associated repulsion. We then used fluorescence cross-correlation spectroscopy (FCCS) to characterise GOX–LPO cluster formation (Fig. 4B), as both confined enzymes could be fluorescently labelled with different fluorophores. FCCS can be used to detect the association of fluorescent species in Brownian motion when their separate signals are correlated (higher G(τ)).50 We observed an increase in the cross-correlation between the fluorescently-labelled CNCs upon clustering (Fig. 4B, blue curve). The signal was absent if fluorescent CNCs without ssDNA were mixed (Fig. 4B, black curve), confirming that DNA hybridization is key to cluster formation. In addition, the association of fluorescently labelled CNCs into small clusters (3–4 vesicles) was visualized by TEM (Fig. 4C). | ||
| Fig. 4 (A) Time course of cluster growth starting form unclustered CNCs by DLS. Blue: LPO–GOX-CNC clusters; red: AMG(GOX)–LPO-CNC clusters. (B) FCS autocorrelation curves of unclustered GOX–ATTO488 – loaded CNCs (red) and unclustered LPO–DyLight 633 – loaded CNCs (green), and FCCS curves of clustered (blue) and unclustered (black) CNCs. Dots: raw data. Line: fitted model. (C) TEM micrographs of CNC clusters at different magnifications: left pannel, scale bar = 200 nm, right pannel, scale bar = 100 nm. | ||
Once the clusters were formed, we determined the mean distance between GOX- and LPO–polymersomes, both when clustered and un-clustered, as the distance between surface-conjugated AMG and encapsulated GOX is given. For clustered polymersomes, the distance for productive molecule transfer (i.e. H2O2 from GOX to LPO) depends on the length of ssDNA. Having both paired and unpaired bases, the average DNA length, L in nm, (corresponding to the vesicle-to-vesicle direct surface distance) is based on eqn (1):51
| L = nBP × 0.34 + nS × 0.676 | (1) |
For non-clustered vesicles, we adapted a previously developed equation for lattices of heterogeneous, rigid particles (eqn (2)):53
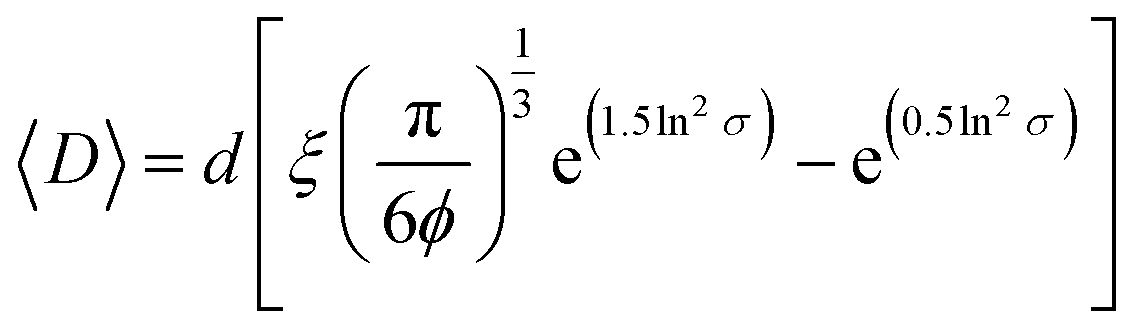 | (2) |
A more in-depth explanation of the geometric meaning of the equations and the rationale behind the further derivation of eqn (3) are presented in Fig. S4 and the related section in ESI.†
 | (3) |
Applying the enzyme concentrations used to test the un-tethered CNC cascade (see Materials and methods), we calculated 〈D〉 = 2.3 ± 0.2 μm, in agreement with the distance calculated for other CNCs working in similar conditions.26 This means that the same type of polymersome could carry the same concentration of enzymes, but then communicate at distances ranging from 10 (when untethered) to less than 0.1 times (when tethered) their average diameter. Interestingly, the longest distance is in the order of magnitude of autocrine signalling, the shortest in that of synaptic signalling.4,5 Thus, these CNCs could be an interesting way to mimic distance effects in cellular communication.
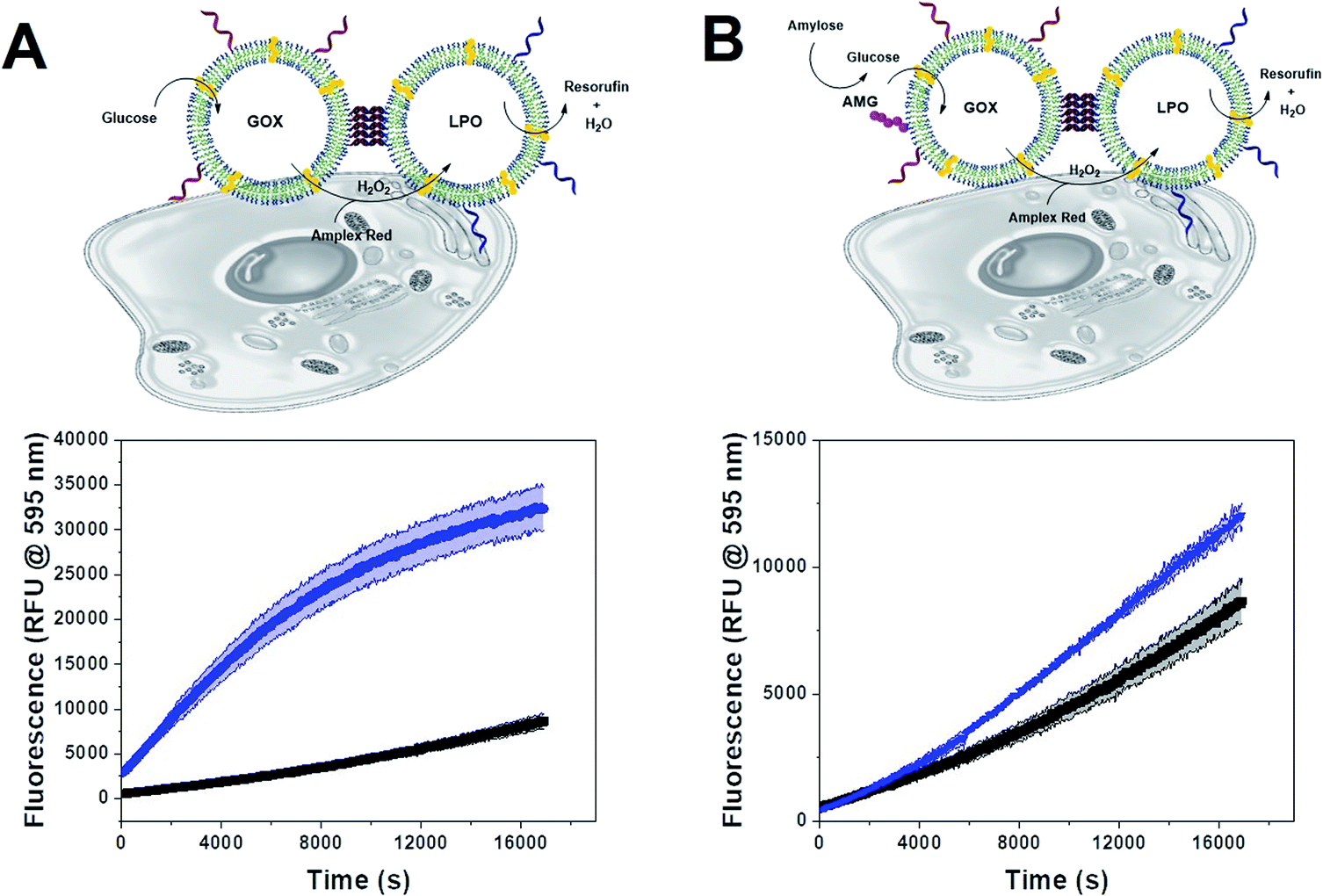
Cascade within clusters
Since polymersome suspensions can be easily nebulized and inhaled,54 airways and lungs could be possible targets for in situ prodrug metabolism or anti-microbial protection. To mimic the conditions of the lungs, we monitored the oxidation of AR catalyzed by clustered and unclustered LPO at 37 °C using glucose concentrations close to those found in the airway surface liquid.55 As the concentration of enzymes was constant, both with and without clusters, only the distance between CNCs of a different kind varied.The inter-cluster distance was calculated with eqn (2), without weights, as all clusters had both types of CNC and thus, no effect would come from the different relative populations of GOX and LPO-CNC. The inter-cluster distance was calculated 2.3 ± 0.1 μm at the same enzyme concentrations used for untethered CNCs. Therefore, CNCs clusters were effectively acting like a “2-enzyme system”, as inter-cluster distances were comparable to un-clustered inter-polymersome distances. The comparison between CNCs inside clusters and when freely moving un-clustered, clearly shows an increase in enzymatic activity. On the contrary, un-clustered CNCs were threefold slower than CNC clusters (Fig. 5A), an effect of controlled co-compartmentalization already observed in giant polymersomes and other cell-mimics, but never in colloidal vesicle networks.44,56 In a previous study we had showed that in the case of un-clustered CNCs, inter-polymersome distances above 1.5 μm significantly hinder the cascade reaction due to the slow diffusion of the “bridging” molecule (the product of the first reaction that is released from the first type of CNC and serves as substrate for the second reaction inside the second type of CNC).26 On the contrary, CNC clustering averted this limitation, resulting in a considerably increased reaction rate at a constant amount of encapsulated enzymes.19
 | ||
| Fig. 5 (A) Concept of a clustered GOX–LPO-CNC cascade and the enzymatic activity of CNC clusters (blue), unclustered CNCs (red) and Amplex Red autoxidation (black). (B) Concept of a clustered AMX(GOX)–LPO-CNC cascade and the enzymatic activity of CNC clusters (blue), unclustered CNCs (red), GOX–LPO-CNC with AMG in solution (magenta) and Amplex Red autoxidation (black). Error bands given as ±SD, n = 3. | ||
We observed the same effect with AMG(GOX)–LPO clusters (Fig. 5B). The effect was less pronounced due to the inherently low catalytic efficiency of AMG which therefore presents an additional bottleneck in the cascade. Interestingly, we found that GOX–LPO clusters in the presence of non-conjugated AMG showed a lower activity than in clusters with conjugated AMG (magenta curve): evidently, they appeared to accelerate the kinetics of the first, slowest reaction. Despite AMG being rate-limiting, cascade efficiency depended on this step and could not be bypassed by additional glucose, as shown by clusters supplied with glucose and amylose at the same time (Fig. S5†).
Cluster localization and functionality on cells
Envisioning possible biological applications, we first tested the effect of unclustered, empty polymersomes up to a concentration of 1 mg mL−1 (Fig. S6A†), unclustered LPO- and GOX-CNCs at 0.2 mg mL−1 (Fig. S6B†), and clustered (AMG)GOX–LPO and GOX–LPO at 0.5 mg mL−1 (Fig. S6C†) on the viability of A549 cells, a lung carcinoma cell line used as a model for lung protection from bacterial infections and ROS therapy.57 Notably, GOX–LPO slightly decreased cell viability compared to (AMG)GOX–LPO which had no effect (Fig. S6C†). This difference may be attributed to the slower H2O2 production associated with AMG (see Fig. 5). Polymersome clusters were previously shown to accumulate at the surface of epithelial cells by the interaction of DNA strands that remain unpaired on the polymersomes after clustering.15,33 We probed the interactions of GOX–LPO clusters where GOX- and LPO-CNCs were labelled with different fluorophores, with A549 cells. Atto488–GOX/DyLight633–LPO-clusters were incubated at 0.2 mg mL−1 with A549 cells for 24 hours and examined by confocal laser scanning microscopy (CLSM) (Fig. 6A–D, S7 and S8†). Confocal Z-stacks revealed a colocalization of fluorescence signals at the surface of the cells. The colocalization analysis of the dyes (Fig. 6C, S7, S8 and Table S5†) yielded a Pearson's coefficient of 0.27 (P = 1) for clusters, and −0.73 for non-clustered vesicles. These results show that clustered CNCs are present on the cell surface where they appear to remain linked,15,33 whereas unlinked CNCs, known to be taken up by cells,19,34 are independently distributed throughout the confocal planes. The relatively low Pearson's and Manders' coefficients (Table S5†) -the closer to 1, the higher the colocalization- are not surprising: if the clusters are well-spaced and big enough, the resolution might be sufficient to resolve the separate polymersomes, at least partially (Fig. 6), as observed in previous studies.15,20,33 The association of clusters with the cell surface allowed us to test whether the cascade reaction could take place ectopically, in the immediate vicinity of the cell surface. Our group has previously described the use of clustered CNCs to localize confined enzyme activity to the cell surface.15,33 However, in these studies, clusters comprised only one type of CNC, i.e. the enzymatic activity was limited and the full potential of clustering was not exploited. In contrast, by positioning clustered GOX–LPO CNCs and AMG(GOX)–LPO at the cell surface, the cells were endowed with an extracellular, two- and three-step cascade reaction. The latter in particular, provided A549 cells with the ability to metabolize amylose (Fig. 7).58 | ||
| Fig. 6 (A) Localization of clustered GOX-CNCs (green) on A549 cells. (B) Same area with localization of LPO-CNCs (red). (C) Merged channels revealing colocalization of clustered GOX- and LPO CNCs (white). (D) Transmission channel. (E) Overlay including the transmission channel. Insets show higher magnifications of the areas boxed in white. The blurred appearance of the brightfield image indicates that the clusters are on the cell surface. Scalebars, 10 μm. | ||
 | ||
| Fig. 7 (A) Activity of GOX–LPO clusters on the surface of cells (blue) and AR non-specific oxidation by cells (black). (B) Activity of AMG(GOX)–LPO clusters on the surface of cells (blue), and AR non-specific oxidation by cells (black). Error bands given as ±SD, n = 3. | ||
Our findings support the potential of clusters as co-delivery systems to epithelial cells, where they modulate the microenvironment at the cell surface, or if internalized, function as artificial organelles. Moreover, this very cascade might be suitable for future on-site bio-catalysis based on starch, as peroxidase bio-transformations have been developed for environmental and industrial applications.59,60
Conclusions
The distance between enzymes is an important factor in the optimization of natural and man-made cascade reactions. Adequate spacing aids in offsetting limiting factors, such as diffusion across membranes, and increases the overall efficiency of the system. Inspired by organelles and intra- and intercellular interactions, we developed clusters of catalytic nanocompartments that sustain efficient cascade reactions. Separate nanocompartments were first independently loaded with enzymes that are able to act in tandem, and then tethered together by complementary DNA strands such as to self-organise into clusters. In addition, a three-step cascade reaction has been successfully established by coupling an upstream enzyme to the surface of GOX-CNCs. Linking compartments by DNA hybridization lends itself to precise tuning of the inter-compartment distance, ranging from distances typical of paracrine signalling for non-adjacent compartments, to those found in some inter-organelle interactions or synaptic signalling for neighbouring compartments. The ability to control the distance between the CNCs supports their use as non-living models for bio-communication. In addition, clusters can modulate the extracellular microenvironment by harnessing cells and improving the efficiency of cascade reactions by segregating reaction steps in physically defined spaces. Besides, such compartment clusters are generated in a modular fashion, such that they can readily be expanded by changing the encapsulated enzymes, by combining more than two types of compartments or by modifying the DNA links. Owing to the combinatorial und functional diversity, clusters of catalytic nanocompartments open new avenues in various domains including bio-catalysis, therapeutics and other biomedical applications.Materials and methods
Materials
DyLight 633 NHS ester and Atto 488-NHS ester were purchased from ThermoFisher Scientific (USA). All other reported compounds were purchased from Sigma-Aldrich (USA) unless stated otherwise.Synthesis of diblock copolymers
The OH-terminated diblock PMOXA10-b-PDMS29 was synthesized according to the already reported procedure.20 Briefly, OH-terminated PMOXA10-b-PDMS29 was dissolved into 5 mL anhydrous chloroform, then succinic anhydride (6.5 mg, 0.066 mmol), 4-dimethylaminopyridine (1.32 mg, 0.011 mmol) and TEA (8.7 mg, 0.088 mmol) were added. After deoxygenating by three vacuum-argon cycles, the mixture was stirred for another 72 h at RT under Ar atmosphere. Finally, 180 mg of a colorless solid product was obtained after the ultrafiltration, yield 90%. 1H NMR (500 MHz, CDCl3) δ 3.65–3.23 (m, 39H), 2.28–2.00 (m, 28H), 1.30 (tt, J = 7.7, 4.5 Hz, 5H), 0.87 (t, J = 6.8 Hz, 4H), 0.56–0.44 (m, 4H), 0.06 (s, 171H) (Fig. S1†). To produce PMOXA10-b-PDMS29–PEG4–N3, the polymer (100 mg) was then first dissolved into anhydrous chloroform, then 11-azido-3,6,9-trioxaundecan-1-amine (11.80 mg, 0.055 mmol), N,N′-dicyclohexylcarbodiimide (15.6 mg, 0.078 mmol) and 4-dimethylaminopyridine (1.2 mg, 0.01 mmol) were added into the above solution. After deoxygenating three times, the mixture was further stirred for another 48 h, at RT. Finally, a colorless solid product was obtained after ultrafiltration. The polymer was characterized again via NMR and GPC. 1H NMR (500 MHz, CDCl3) δ 3.65–3.28 (m, 43H), 2.23–2.03 (m, 29H), 1.59–1.52 (m, 2H), 1.30 (tt, J = 7.5, 3.8 Hz, 5H), 0.87 (t, J = 6.8 Hz, 3H), 0.50 (ddd, J = 15.9, 8.9, 4.5 Hz, 4H), 0.06 (s, 172H) (Fig. S2†).Preparation of GOX- and LPO-CNCs
CNCs were prepared at RT, with 50% (molar ratio) of the azide-functionalized polymer. Films were rehydrated to a final polymer concentration of 10 mg mL−1 with 1 mg of GOX or LPO in PBS (pH 7) and 25 μl of melittin 1 mM (from bee venom). Samples were extruded through an Avanti mini-extruder (Avanti Polar Lipids, USA) with a 200 nm pore diameter polycarbonate membrane for GOX; LPO-CNCs were first extruded through 400 nm and then 200 nm, 11 times each. Non-encapsulated enzyme was removed through size exclusion chromatography (SEC) (Sepharose 4B column; 30 cm length) and recovered for quantification.AMG(GOX)-CNCs
1 mL of amyloglucosidase from Aspergillus niger (AMG) ≥260 U mL−1 aqueous solution, was mixed with 200 μL of dibenzocyclooctyne–PEG4–N-hydroxysuccinimidyl ester (DBCO–PEG4–NHS ester) in DMF and 800 μL of 0.2 M sodium bicarbonate at pH 8–9 was added. The solution was let it stir at room temperature for 16 h. The functionalized protein was purified with Amicon Ultra-0.5 mL–30 kDa cutoff by washing 3 times in PBS for 5 min at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. 200 μM stock solutions of the DBCO-modified DNA strands (Microsynth, Switzerland) were prepared in nuclease-free water. 50 μL of each solution was added to 150 μL of corresponding CNCs, and made to react at 37 °C overnight. The vesicles were thus purified with a 10 cm-Sepharose 2B column, mixed 1:1 (volumetrically) and let to rest at 37 or 4 °C overnight to allow clustering for further experiments and then used with no further purification.
000 rpm. 200 μM stock solutions of the DBCO-modified DNA strands (Microsynth, Switzerland) were prepared in nuclease-free water. 50 μL of each solution was added to 150 μL of corresponding CNCs, and made to react at 37 °C overnight. The vesicles were thus purified with a 10 cm-Sepharose 2B column, mixed 1:1 (volumetrically) and let to rest at 37 or 4 °C overnight to allow clustering for further experiments and then used with no further purification.
Catalytic nanocompartment characterization – static and dynamic light scattering
SLS and DLS experiments were performed on a setup from LS instruments (Switzerland), equipped with a He–Ne 21 mW laser (λ = 632.8 nm) at scattering angles from 30° to 55° at 25 °C. The radius of gyration (Rg) was obtained from the SLS data with a Guinier plot. The intensity versus angle curve of a diluted sample (to suppress multiple scattering) was fit with a linear regression and the slope of the curve m was used to calculate Rg according to the equation | (4) |
The error was calculated on the standard error of the slope.
In the case of SLS, second order cumulant analysis of the data between 30° and 155° was performed to obtain the Rh.
Clustering was followed on a Zetasizer Nano ZSP (Malvern Instruments, UK) at 20 °C, where 50 μL of each DNA-functionalized CNC were added to 200 μL of PBS, measuring the Rh for 14 hours.
Catalytic nanocompartment characterization – transmission electron microscopy (TEM)
CNC suspensions in PBS at 0.25 mg mL−1 were deposited on glow-discharged carbon grids (Quantifoil, Germany) stained with 1.5% uranyl acetate solution and deposited on carbon-coated copper grids. A transmission electron microscope (Philips Morgagni 268D) at 293 K was used.Nanoparticle tracking analysis (NTA)
NTA was used as further analysis of particle size and concentration, on a NanoSight NS300 (Malvern Panalytical Ltd., UK), using a flow cell (100 μL min−1), 1:1000 concentration in freshly filtered PBS, yielding particle Rh and concentration (particle per mL).
Enzyme quantification
Unencapsulated enzyme was recovered from melittin-less samples, and quantified at 280 nm, using a Nanodrop 2000 UV-vis spectrophotometer (ThermoFisher, USA).DNA functionalization and clustering
200 μM stock solutions of the DBCO-modified DNA strands (Microsynth, Switzerland) were prepared in nuclease-free water. 50 μL of each solution was added to 150 μL of corresponding CNCs, and made to react at 37 °C overnight. The vesicles were thus purified with a 10 cm-Sepharose 2B column, mixed 1:1 (volumetrically) and let to rest at 37 or 4 °C overnight to allow clustering for further experiments and then used with no further purification.
Fluorescence (cross) correlation spectroscopy
Stock solutions of GOX (2 mg mL−1) and LPO (2 mg mL−1) were prepared in 0.1 M Na2CO3 buffer. 5 μL of a 1.5 mM Atto-488 NHS ester in DMSO solution was added to 1 mL of the GOX stock solution and 5 μL of 1.5 mM DyLight 633-NHS ester in DMSO was added to 1 mL of LPO solution. Both labelling reactions were mixed overnight at 4 °C. Free dye was removed by spin filtration with Amicon Spin Filters 30 MWCO (Merck, Germany). Upon purification, labelled enzymes were used directly and polymersomes were formed as previously described, with no melittin added. A 488 nm argon laser was used to excite ATTO 488 and a 633 nm HeNe laser was used for DyLight633. The two lasers were passed through MBS488 and MBS488/561/633 filters and the signals were detected in the range of 500–532 nm and 657–690 nm, respectively. The pinholes were adjusted to maximize the count rate using the respective free dye in PBS and the sample volumes were 15 μL. Fluorescent fluctuations over time were recorded for 20 × 5 s. The raw data was processed and analyzed using ZEN software. Autocorrelation curves were fitted by a two-component model, except for dye-only samples. | (5) |
The degree of labelling (DOL) was obtained from the ratio of the counts per molecule (CPM)
 | (6) |
Similarly, the enzymes per vesicle were obtained by
 | (7) |
To quantify DNA, 11T-less strands (thus, 22a and 22b) were used, labeled with Cy5 and Atto-488, respectively. An excess amount (10 μL of a 200 μM stock) was added to vesicles with complementary strands, clustered and then purified via SEC.
 | (8) |
Similar to FCS, dual-colour fluorescence cross-correlation spectroscopy (FCCS) was performed, with the same system, on CNCs containing the labelled enzymes and either non-functionalized, or conjugated to their respective DNA strands, then mixed, incubated at 37 and measured with both lasers simultaneously, in FCCS mode.
Enzymatic assays
Cell viability (proliferation) assay
For cell viability assessment, a CellTiter 96 Aqueous One Solution Cell Proliferation Assay (MTS, Promega) was used according to manufacturer instructions. A549 cells were seeded (5000 cells per well in 100 μL of cell culture medium) in a 96-well plate and incubated for 24 h (n = 4 per sample). After 24 h, empty polymersomes, CNCs, CNCs with ssDNA, and CNC clusters were added to the cells at the concentrations indicated to reach a final volume of 200 μL per well. After 24 h incubation, 20 μL of MTS reagent was added to each well. After 3 h, absorbance was measured at 490 nm using a Spectramax id3 plate reader. Background absorbance from control wells containing all assay components without cells was subtracted from each well, and data were normalized to control cells containing all components and PBS instead of CNCs. One-way ANOVA followed by Tukey's post hoc test was performed to determine whether cell viability was significantly affected by the treatment.Live cell imaging of A549 cells
Freshly trypsinized A549 human carcinoma cells were seeded at a density of 6 × 104 cells per well in an 8-well glass bottom ibidi plate. After 24 h, the cell supernatant was removed and replaced with fluorescently-labelled CNCs, CNC clusters at a final concentration of 0.2 mg mL−1, or PBS as control.After carefully removing unbound CNCs/clusters, cells were imaged by confocal laser scanning microscopy (CLSM) on a LSM 880 confocal laser microscope with a 40×, 1.2 water immersion C-Apochromat objective lens, using Atto-488 laser and DyLight-633 light path parameters.
Colocalization analysis
CLSM micrographs were analysed with ImageJ Coloc2 plugin, using both Pearson's (PCC) and Manders' threshold coefficient for the red to the green channel (tM1).For PCC,
 | (9) |
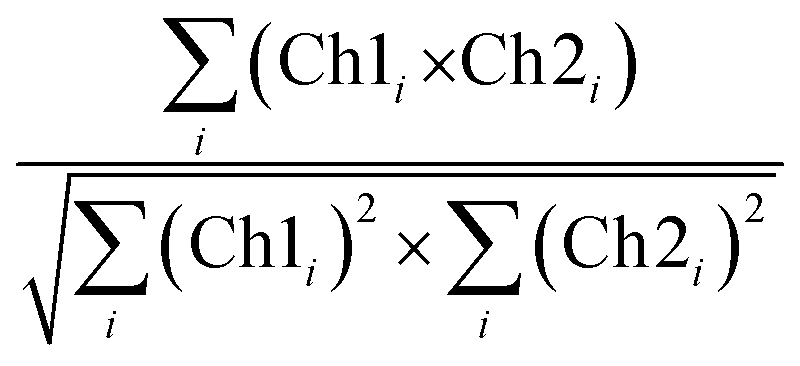 | (10) |
PCC quantifies the degree to which two channels follow a simple linear relationship of intensity. Values can range from −1 (an inverse or “anti-colocalization” relationship), to 0 (a random cloud of no relationship), or +1 (a perfect linear slope); tM1 is similar to PCC but ranges from 0 to +1. It does not incorporate a relationship to mean intensity (as with Pearson's), so it is mostly sensitive to only the overlap alone above the threshold.61 Costes' p-value was calculated by the plugin to determine whether the result was statistically significant.
Extracellular cluster functionality
A549 cells were seeded in a 96-well plate at a density of 5000 cells per well in 100 μL cell culture medium, and incubated for 24 h at 37 °C. Then, cells were dosed for 24 h with 50 μL of GOX–LPO and AMG(GOX)–LPO clusters. AR (1 μM, final concentration in 200 μL final volume) and AR/amylose (1 μM/50 μg mL−1) was added to each well containing GOX–LPO and AMG(GOX)–LPO clusters, respectively. The enzyme kinetics of clusters on the cell surface was determined by monitoring the formation of resorufin at 37 °C for 4 hours.Data availability
All experimental procedures, characterization data supporting this article have been described in the manuscript and its ESI.† Raw data shown in this paper are available on Zenodo from the corresponding author upon reasonable request.Author contributions
The manuscript was largely written by VM, AB, and CAS. VM and AB equally performed most of the experimental work, DW synthesized the polymers used in this study, IC and CAS carried out cell work, SN contributed with preliminary physical and enzymatic characterization. CGP conceived the core idea, supervised the project and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank the Swiss National Science Foundation, the University of Basel and the National Centre of Competence in Research – Molecular Systems Engineering for the funding. They also thank Gabriele Persy (Department of Chemistry, University of Basel) for providing the TEM images, Dr Ludovit Zweifel (Biozentrum, University of Basel) for his advice on FCCS data interpretation, and Dr Roman Jakob (Biozentrum, University of Basel) for determining AMG dimensions.References
- J. Krishnan, L. Lu and A. Alam Nazki, J. R. Soc., Interface, 2020, 17, 20200251 CrossRef CAS.
- K. Francis and B. O. Palsson, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 12258–12262 CrossRef CAS PubMed.
- G. Achleitner, B. Gaigg, A. Krasser, E. Kainersdorfer, S. D. Kohlwein, A. Perktold, G. Zellnig and G. Daum, Eur. J. Biochem., 1999, 264, 545–553 CrossRef CAS PubMed.
- A. Barberis, E. M. Petrini and J. W. Mozrzymas, Front. Cell. Neurosci., 2011, 5, 6 Search PubMed.
- S. Y. Shvartsman, H. S. Wiley, W. M. Deen and D. A. Lauffenburger, Biophys. J., 2001, 81, 1854–1867 CrossRef CAS.
- T. W. Giessen and P. A. Silver, J. Mol. Biol., 2016, 428, 916–927 CrossRef CAS.
- N. Ichihashi and T. Yomo, Curr. Opin. Chem. Biol., 2014, 22, 12–17 CrossRef CAS.
- S. Tsitkov and H. Hess, ACS Catal., 2019, 9, 2432–2439 CrossRef CAS.
- R. J. R. W. Peters, M. Marguet, S. Marais, M. W. Fraaije, J. C. M. van Hest and S. Lecommandoux, Angew. Chem., Int. Ed., 2014, 53, 146–150 CrossRef CAS.
- A. Belluati, I. Craciun, C. E. Meyer, S. Rigo and C. G. Palivan, Curr. Opin. Biotechnol., 2019, 60, 53–62 CrossRef CAS PubMed.
- M. Garni, S. Thamboo, C.-A. Schoenenberger and C. G. Palivan, Biochim. Biophys. Acta, 2017, 1859, 619–638 CrossRef CAS.
- M. Marguet, C. Bonduelle and S. Lecommandoux, Chem. Soc. Rev., 2013, 42, 512–529 RSC.
- C. G. Palivan, R. Goers, A. Najer, X. Y. Zhang, A. Car and W. Meier, Chem. Soc. Rev., 2016, 45, 377–411 RSC.
- F. Itel, M. Chami, A. Najer, S. Lörcher, D. Wu, I. A. Dinu and W. Meier, Macromolecules, 2014, 47, 7588–7596 CrossRef CAS.
- J. Liu, I. Craciun, A. Belluati, D. Wu, S. Sieber, T. Einfalt, D. Witzigmann, M. Chami, J. Huwyler and C. G. Palivan, Nanoscale, 2020, 12, 9786–9799 RSC.
- S. Egli, H. Schlaad, N. Bruns and W. Meier, Polymers, 2011, 3, 252–280 CrossRef CAS.
- J. K. Patra, G. Das, L. F. Fraceto, E. V. R. Campos, M. d. P. Rodriguez-Torres, L. S. Acosta-Torres, L. A. Diaz-Torres, R. Grillo, M. K. Swamy, S. Sharma, S. Habtemariam and H.-S. Shin, J. Nanobiotechnol., 2018, 16, 71 CrossRef.
- C. Zelmer, L. P. Zweifel, L. E. Kapinos, I. Craciun, Z. P. Güven, C. G. Palivan and R. Y. H. Lim, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 2770 CrossRef CAS PubMed.
- P. Tanner, O. Onaca, V. Balasubramanian, W. Meier and C. G. Palivan, Chemistry, 2011, 17, 4552–4560 CrossRef CAS.
- J. Liu, V. Postupalenko, S. Lörcher, D. Wu, M. Chami, W. Meier and C. G. Palivan, Nano Lett., 2016, 16, 7128–7136 CrossRef CAS.
- D. Wu, S. Rigo, S. Di Leone, A. Belluati, E. C. Constable, C. E. Housecroft and C. G. Palivan, Nanoscale, 2020, 12, 1551–1562 RSC.
- L. Schoonen and J. C. M. van Hest, Adv. Mater., 2016, 28, 1109–1128 CrossRef CAS.
- A. Belluati, S. Thamboo, A. Najer, V. Maffeis, C. Planta, I. Craciun, C. G. Palivan and W. Meier, Adv. Funct. Mater., 2020, 30, 2002949 CrossRef CAS.
- L. M. P. E. van Oppen, L. K. E. A. Abdelmohsen, S. E. van Emst-de Vries, P. L. W. Welzen, D. A. Wilson, J. A. M. Smeitink, W. J. H. Koopman, R. Brock, P. H. G. M. Willems, D. S. Williams and J. C. M. van Hest, ACS Cent. Sci., 2018, 4, 917–928 CrossRef CAS.
- T. Nishimura and K. Akiyoshi, Adv. Sci., 2018, 5, 1800801 CrossRef PubMed.
- A. Belluati, I. Craciun, J. Liu and C. G. Palivan, Biomacromolecules, 2018, 19(10), 4023–4033 CrossRef CAS.
- A. Perktold, B. Zechmann, G. Daum and G. Zellnig, FEMS Yeast Res., 2007, 7, 629–638 CrossRef CAS PubMed.
- P. Tanner, V. Balasubramanian and C. G. Palivan, Nano Lett., 2013, 13, 2875–2883 CrossRef CAS.
- U. Jakobsen, A. C. Simonsen and S. Vogel, J. Am. Chem. Soc., 2008, 130, 10462–10463 CrossRef CAS PubMed.
- N. Dave and J. Liu, ACS Nano, 2011, 5, 1304–1312 CrossRef CAS PubMed.
- P. A. Beales and T. K. Vanderlick, J. Phys. Chem. A, 2007, 111, 12372–12380 CrossRef CAS PubMed.
- A. Czogalla, H. G. Franquelim and P. Schwille, Biophys. J., 2016, 110, 1698–1707 CrossRef CAS PubMed.
- C. E. Meyer, J. Liu, I. Craciun, D. Wu, H. Wang, M. Xie, M. Fussenegger and C. G. Palivan, Small, 2020, 16, e1906492 CrossRef.
- A. Belluati, I. Craciun and C. G. Palivan, ACS Nano, 2020, 14, 12101–12112 CrossRef CAS PubMed.
- P. Kelly, B. W. Woonton and G. W. Smithers, in Functional and Speciality Beverage Technology, ed. P. Paquin, Woodhead Publishing, 2009, DOI:10.1533/9781845695569.2.170, pp. 170–231.
- M. Magacz, K. Kędziora, J. Sapa and W. Krzyściak, Int. J. Mol. Sci., 2019, 20, 1443 CrossRef CAS PubMed.
- Ł. Minarowski, D. Sands, A. Minarowska, A. Karwowska, A. Sulewska, M. Gacko and E. Chyczewska, Folia Histochem. Cytobiol., 2008, 46, 245–246 Search PubMed.
- M. Stanislawski, V. Rousseau, M. Goavec and H. Ito, Cancer Res., 1989, 49, 5497–5504 CAS.
- U. Patel, A. Gingerich, L. Widman, D. Sarr, R. A. Tripp and B. Rada, PLoS One, 2018, 13, e0199167 CrossRef.
- K. Vogele, J. List, F. C. Simmel and T. Pirzer, Langmuir, 2018, 34, 14780–14786 CrossRef CAS PubMed.
- C. Schmidt-Dannert and F. Lopez-Gallego, Microb. Biotechnol., 2016, 9, 601–609 CrossRef.
- J. Fu, Z. Wang, X. H. Liang, S. W. Oh, E. St. Iago-McRae and T. Zhang, Top. Curr. Chem., 2020, 378, 38 CrossRef CAS.
- A. Belluati, V. Mikhalevich, S. Yorulmaz Avsar, D. Daubian, I. Craciun, M. Chami, W. P. Meier and C. G. Palivan, Biomacromolecules, 2019, 21(2), 701–715 CrossRef.
- E. C. Dos Santos, A. Belluati, D. Necula, D. Scherrer, C. E. Meyer, R. P. Wehr, E. Lortscher, C. G. Palivan and W. Meier, Adv. Mater., 2020, 32, e2004804 CrossRef.
- C. Edlinger, T. Einfalt, M. Spulber, A. Car, W. Meier and C. G. Palivan, Nano Lett., 2017, 17, 5790–5798 CrossRef CAS PubMed.
- R. Shenoy and C. N. Bowman, Biomaterials, 2012, 33, 6909–6914 CrossRef CAS PubMed.
- M. J. Burne, T. M. Osicka and W. D. Comper, Kidney Int., 1999, 55, 261–270 CrossRef CAS.
- N. Ramasesh, K. R. Sreekantiah and V. S. Murthy, Starke, 1982, 34, 346–351 CrossRef CAS.
- M. Spulber, P. Baumann, S. S. Saxer, U. Pieles, W. Meier and N. Bruns, Biomacromolecules, 2014, 15, 1469–1475 CrossRef CAS.
- K. Bacia, S. A. Kim and P. Schwille, Nat. Methods, 2006, 3, 83–89 CrossRef CAS PubMed.
- Q. Chi, G. Wang and J. Jiang, Phys. A (Amsterdam, Neth.), 2013, 392, 1072–1079 CrossRef CAS.
- V. Hertlein, H. Flores-Romero, K. K. Das, S. Fischer, M. Heunemann, M. Calleja-Felipe, S. Knafo, K. Hipp, K. Harter, J. C. Fitzgerald and A. J. García-Sáez, Life Sci. Alliance, 2020, 3, e201900600 CrossRef.
- Z. H. Liu, Y. Li and K. W. Kowk, Polymer, 2001, 42, 2701–2706 CrossRef CAS.
- M. Rudokas, M. Najlah, M. A. Alhnan and A. Elhissi, Medical Principles And Practice: International Journal of The Kuwait University, Health Science Centre, 2016, 25, suppl. 2, pp. 60–72.
- E. H. Baker and D. L. Baines, Chest, 2018, 153, 507–514 CrossRef.
- R. Booth, Y. Qiao, M. Li and S. Mann, Angew. Chem., Int. Ed., 2019, 58, 9120–9124 CrossRef CAS PubMed.
- B. Perillo, M. Di Donato, A. Pezone, E. Di Zazzo, P. Giovannelli, G. Galasso, G. Castoria and A. Migliaccio, Exp. Mol. Med., 2020, 52, 192–203 CrossRef CAS.
- B. I. Brown and D. H. Brown, Biochim. Biophys. Acta, 1965, 110, 124–133 CrossRef CAS.
- J. Gau, P. G. Furtmüller, C. Obinger, M. Prévost, P. Van Antwerpen, J. Arnhold and J. Flemmig, Free Radical Biol. Med., 2016, 97, 307–319 CrossRef CAS PubMed.
- C. Torres-Duarte and R. Vazquez-Duhalt, in Biocatalysis Based on Heme Peroxidases: Peroxidases as Potential Industrial Biocatalysts, ed. E. Torres and M. Ayala, Springer, Berlin, Heidelberg, 2010, DOI:10.1007/978-3-642-12627-7_8, pp. 179–206.
- K. W. Dunn, M. M. Kamocka and J. H. McDonald, Am. J. Physiol.: Cell Physiol., 2011, 300, C723–C742 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Showing the polymer characterization, the complete physical characterization of vesicles, a discussion on the calculation of the inter-vesicle distance, and cluster–cell interaction, including colocalization analysis. See DOI: 10.1039/d1sc04267j |
| ‡ V. M. and A. B. contributed equally to this study. |
| This journal is © The Royal Society of Chemistry 2021 |