
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctional and protective hole hopping in metalloenzymes
Harry B.
Gray
 and
Jay R.
Winkler
and
Jay R.
Winkler
Beckman Institute, California Institute of Technology, 1200 E California Boulevard, Pasadena, CA 19925, USA. E-mail: hbgray@caltech.edu; winklerj@caltech.edu
First published on 27th September 2021
Abstract
Electrons can tunnel through proteins in microseconds with a modest release of free energy over distances in the 15 to 20 Å range. To span greater distances, or to move faster, multiple charge transfers (hops) are required. When one of the reactants is a strong oxidant, it is convenient to consider the movement of a positively charged “hole” in a direction opposite to that of the electron. Hole hopping along chains of tryptophan (Trp) and tyrosine (Tyr) residues is a critical function in several metalloenzymes that generate high-potential intermediates by reactions with O2 or H2O2, or by activation with visible light. Examination of the protein structural database revealed that Tyr/Trp chains are common protein structural elements, particularly among enzymes that react with O2 and H2O2. In many cases these chains may serve a protective role in metalloenzymes by deactivating high-potential reactive intermediates formed in uncoupled catalytic turnover.
Introduction
Life on our planet changed dramatically after oxygenic photosynthesis began remaking the atmosphere about 3 billion years ago. Over the next 2.5 billion years, as the atmospheric O2 concentration increased to the current level of 21%,1 the anaerobic respiratory chains of some microorganisms evolved to take advantage of this newly available oxidant.2 The oxidizing power of O2 also stimulated the evolution of new enzymatic transformations involving high-potential intermediates. During this period, living organisms had to learn how to live with the dark side of oxygen – toxic reactive oxygen species (ROS).3The last six of the canonical amino acids introduced into the genetic code are thought to be (in order of appearance) cysteine (Cys), histidine (His), phenylalanine (Phe), methionine (Met), tyrosine (Tyr), and tryptophan (Trp).4,5 The introduction of Cys and His into the genetic code allowed the incorporation of metals, particularly Fe, into protein structures, creating new opportunities for catalysis of redox processes. As the property that distinguishes Met, Tyr, and Trp from all other amino acids (except Cys) is susceptibility to oxidation, it is plausible that their introduction into the genetic code was a response to the oxygenation of our atmosphere.6–8 Tyr and Trp radicals have been found to participate in many enzymatic processes involving oxygen and hydrogen peroxide.9 Of special interest is that we and others have suggested that an additional critical role for Met, Tyr, and Trp is to protect enzymes from damage by serving as endogenous antioxidants.10–15
Single-step electron tunneling through proteins
The understanding of biological electron flow transformed from rudimentary to refined in the second half of the 20th century. In the 1940s, researchers knew that electrons flowed down the aerobic respiratory chain through a series of immobilized (membrane bound) proteins, ultimately reducing molecular oxygen, but the mechanism by which electrons “wander directly from enzyme to enzyme” in this chain remained a mystery.16 At the same time, abiological electron transfer research was stimulated by the availability of radioactive isotopes after the Second World War. Experimental investigations of electron exchange kinetics of transition metal complexes revealed rate constants differing by more than 15 orders of magnitude.17–20 These empirical studies provided the foundation for the development of electron transfer (ET) theory in the 1950s.21 The semiclassical formulation of this theory (eqn (1)) revealed that ET rates depend on three critical reaction parameters: (1) the standard free-energy change (ΔG°); (2) the degree of structural and solvent reorganization that accompanies the electron transfer (λ); and (3) the strength of the electronic interaction between the electron donor and acceptor (HAB).22–25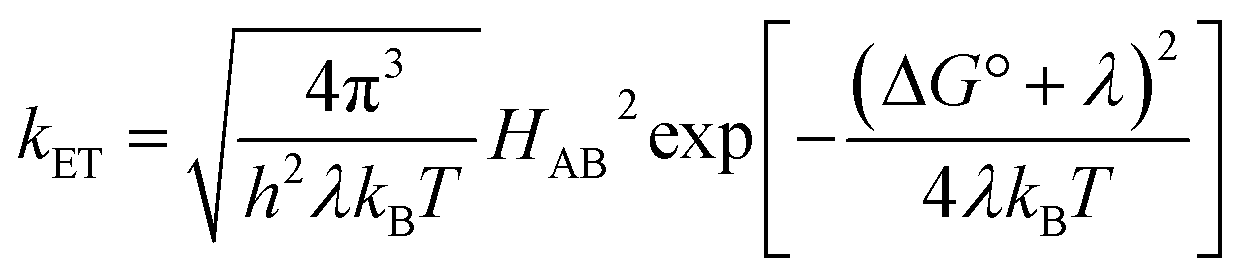 | (1) |
Beginning in the 1970s, rapid progress in structural biology provided keen insights into the arrangements of atoms in metalloproteins.26 Electron transfer protein structures created a new puzzle: redox cofactors are typically buried inside globular polypeptide matrices. How could electrons exchange if the cofactors did not come into contact? In the context of the semiclassical theory, is HAB large enough to support biological ET when redox centers are 15 Å apart? A series of investigations of electron-transfer kinetics in metalloproteins covalently labeled with ruthenium redox reagents demonstrated that superexchange mediated electronic coupling between redox sites could indeed sustain electron tunneling through protein matrices.27–31 In the superexchange model, donor–acceptor coupling is mediated by excess electron or hole states of the intervening medium.30 In proteins, the structure and composition of the polypeptide matrix determines the magnitude of the superexchange coupling between donor and acceptor but, as a rough approximation, HAB decays exponentially with the separation distance and rates decrease by a factor of 6 × 104 for every 10 Å increase in tunneling distance (exponential decay constant β = 1.1 Å−1).27 Electron tunneling timetables (Fig. 1) illustrate that, when driving force (−ΔG°) and reorganization energy are balanced, electrons can tunnel through as much as 25 Å of protein in a millisecond. It is important to remember that tunneling timetables show driving-force optimized times; when the free-energy change does not match the reorganization energy, electron transfer is slower (Fig. 1B).
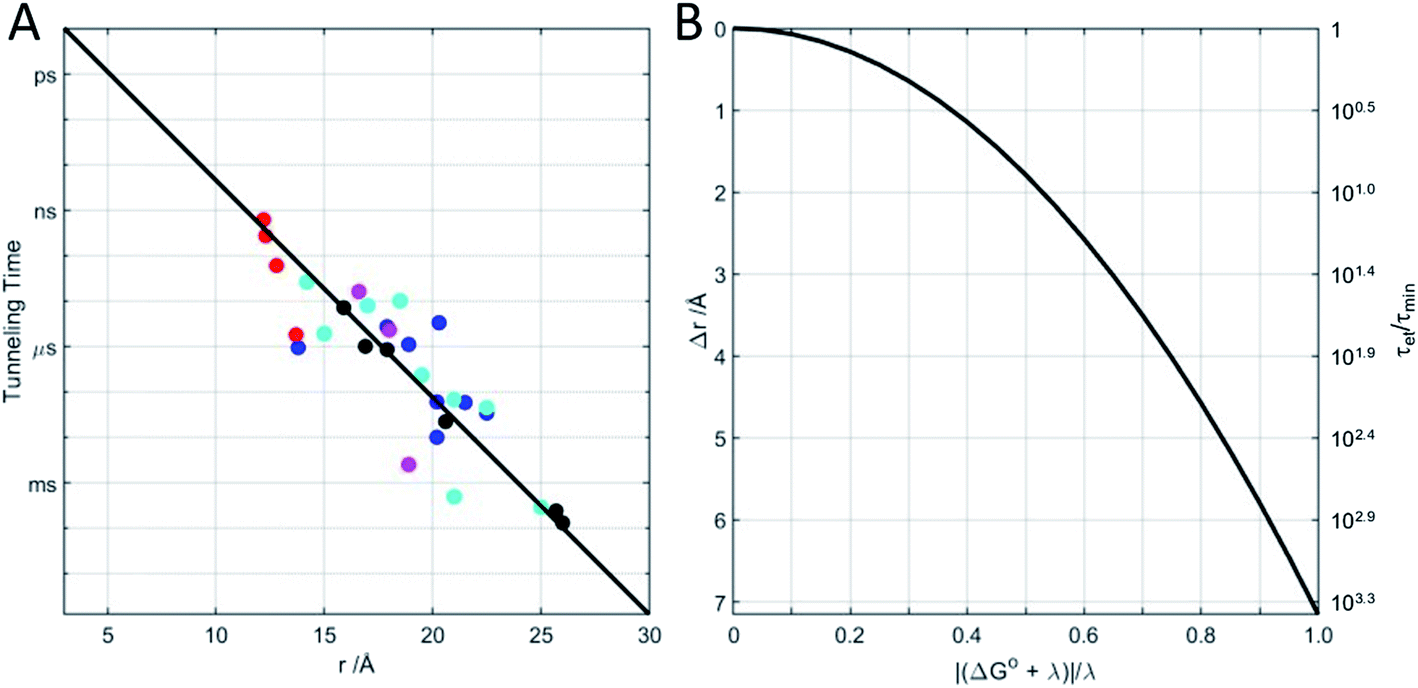 | ||
| Fig. 1 (A) Driving-force optimized (−ΔG° = λ) electron tunneling timetable for Ru-modified proteins: azurin (black); cytochrome c (blue); cytochrome c-b562 (cyan); myoglobin (magenta); high-potential iron protein (red). The solid line shows an exponential dependence on metal center-to-center distance with decay factor β = 1.1 Å−1. (B) When −ΔG° ≠ λ electron tunneling times (τet) increase above their minimum values (τmin). The solid curve illustrates the tunneling-distance change (Δr) that increases the tunneling time by the same factor as the deviation of −ΔG° from λ (calculated with λ = 0.8 eV and β = 1.1 Å−1). Equivalently, Δr is the reduction in tunneling distance necessary to maintain a driving-force-optimized tunneling time. | ||
Multistep electron tunneling (hopping) through proteins
Reorganization energies in biological ET reactions tend to be in the 0.5–1.0 eV range. Consequently, these reactions are rarely driving-force optimized, limiting tunneling distances to 10–15 Å. Moving electrons across large protein complexes or membranes, then, requires multiple tunneling steps. Multistep ET along a continuously decreasing free-energy gradient overcomes the single-step distance limit at the price of somewhat reduced chemical potential in the reaction products. Even in the absence of a ΔG° gradient, and with modestly endergonic intervening steps, multistep tunneling can move charges over several nanometers in under a millisecond. The key to rapid, long range movement of electrons through proteins, then, is the strategic placement of electron or hole waystations between the origin and destination of the transferring charge.32–34The polypeptide matrix plays a critical role in mediating electronic coupling in both single- and multistep ET reactions.35–38 When biological redox processes generate powerful oxidants or reductants, the polypeptide can, in principle, participate directly in electron transfer. Reducing amides or the side chains of aromatic amino acids requires potentials more negative than −2 V vs. NHE. An example of this type of reactivity is found in the quenching of the singlet excited state of tryptophan (Trp) indole groups by electron transfer to the proximal amide.39 On the oxidative side, there are several targets for reaction. The indole group of Trp has a formal potential for radical cation formation of 1.3 V vs. NHE.40,41 Formation of the neutral phenol radical of tyrosine (Tyr) occurs with a formal potential of 1.0 V.41,42 The sulfur amino acids cysteine (Cys) and methionine (Met) have potentials near 1.0 and 1.5 V, respectively.43,44 Oxidations of these residues also involve proton transfer processes that can modulate or limit overall reaction rates.45 Many redox enzymes, particularly those that react with dioxygen or hydrogen peroxide generate high-potential reactive intermediates (formal potentials in the 1.0–1.25 V range) that could transfer holes to these oxidizable amino acids. Indeed, several enzymes are known to utilize hole hopping in their natural catalytic cycles. The question arises, then, how do enzymes whose function requires high-potential intermediates keep oxidizing power at the active site and prevent holes from diffusing (hopping) through the protein until they reach the lowest potential site, often at the protein surface? One strategy would involve designs in which oxidizable amino acids are kept far (>10 Å) from the active site, but many oxygenases, oxidases, and peroxidases seem to have eschewed that option. Instead, we have found that chains of closely spaced Trp and Tyr residues often extend from active sites to protein surfaces, creating a conduit for hole migration.11–15,46–49 We have suggested that these conduits serve as protection for the enzymes under conditions where normal catalysis has gone awry.
Functional hole hopping through metalloenzymes
Ribonucleotide reductase
Class Ia ribonucleotide reductases (RNRs) catalyze the reduction of nucleotides to deoxynucleotides.50–52 The active holo-enzyme from E. coli, with an α2β2 structure, has been the subject of intense study for more than 50 years. The discovery in 1972 of a stable Tyr122˙ radical in the β2 subunit of the enzyme stimulated research to understand its role in catalysis.53–56 Until recently no structure of the intact α2β2 enzyme was available, although structures of both the α2 and β2 subunits had been determined.57,58 A docking model of the holo-enzyme constructed from the two subunits suggested that the distance from Tyr122˙ in β2 to the Cys439 in residue α2 that initiates nucleotide reduction could be as long as 35 Å.57 A recent cryo-EM structure of the holo-enzyme confirmed that estimate (PDB ID 6W4X).59 Decades of biochemical and biophysical study have elucidated a radical transfer pathway composed of: βTyr122-βTrp48-βTyr356-αTyr731-αTyr730-αCys439.57,60–62 The cryo-EM structure indicates that the first three radical transfers involve 8–9 Å hops. The final two transfers span distances of just 3.5 and 2.8 Å. The two E. coli RNR subunits have relatively high proportions of Tyr residues (α2, 5.39%; β2, 4.27%; UniProtKB average, 2.92%), whereas the number of Trp residues is on the low side (α2, 0.66%; β2, 1.87%; UniProtKB average, 1.10%). The long-term stability of the Tyr122˙ radical amid so many alternate residues suggests that it has the lowest formal potential of the Tyr and Trp residues in the enzyme. The high Tyr content, nevertheless, raises the possibility of additional radical transfer pathways. Yet, the fidelity of RNR radical transfer is maintained even though several Tyr residues are near the key pathway residues (Fig. 2). Indeed, the Trp/Tyr branched chains connected to residues in the radical transfer pathway at distances less than 10 Å contain 37 residues (Fig. 3). That holes from Tyr122˙ do not diffuse off-path to nonproductive Tyr residues demonstrates that hole-hopping distance alone does not define a functional radical transfer pathway. Fine tuning of reaction driving forces by local environments and the availability of proton exchange partners are key factors for maintaining the fidelity of the functional radical transfer pathway.62–64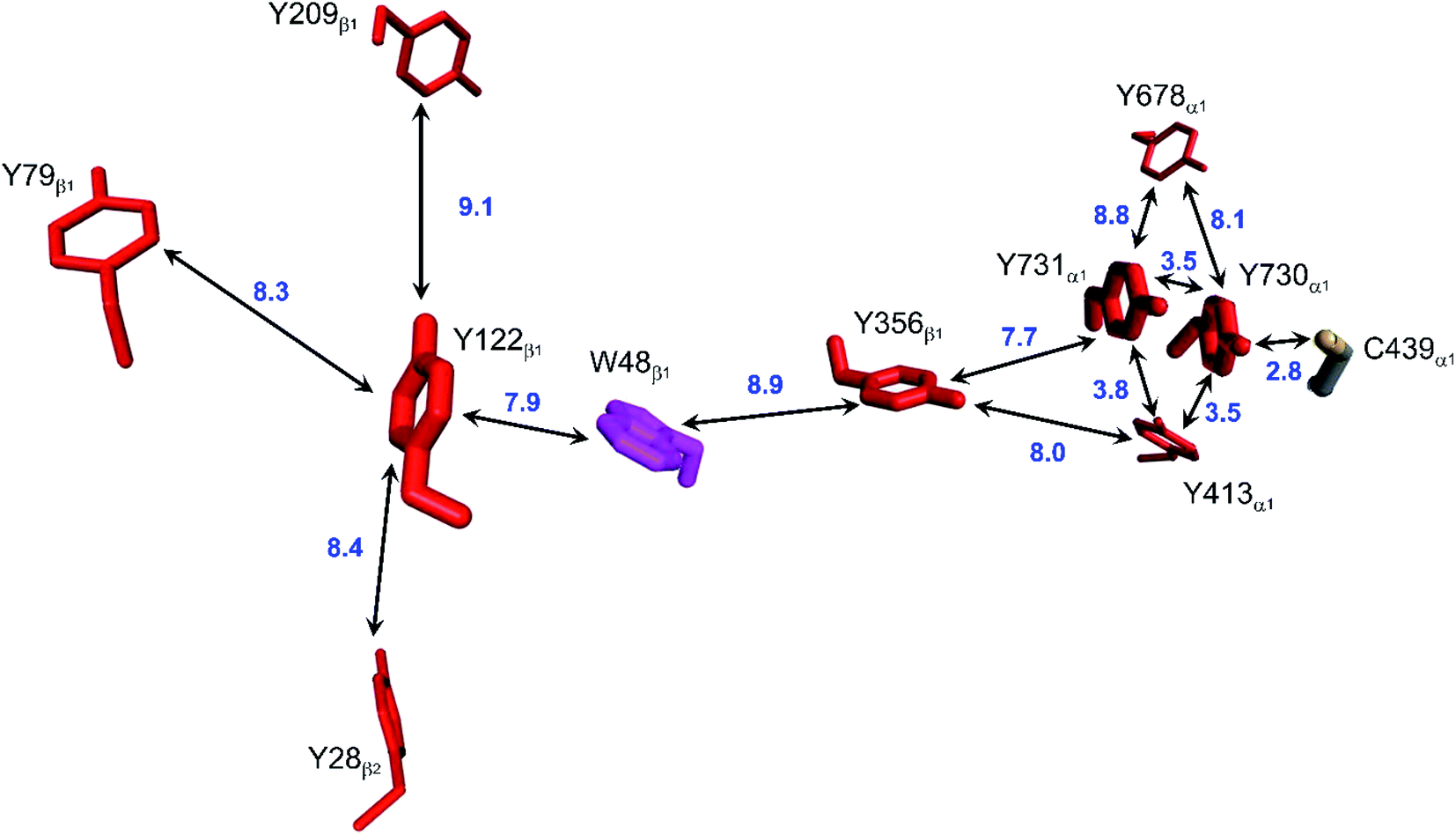 | ||
| Fig. 2 Structural model of the functional radical transfer pathway (Y122-W48-Y356-Y731-Y730-C439) in E. coli RNR (PDB ID 6W4X).59 Surrounding Trp and Tyr residues within 10 Å of pathway residues are shown along with shortest edge–edge distances (Å, blue numbers). | ||
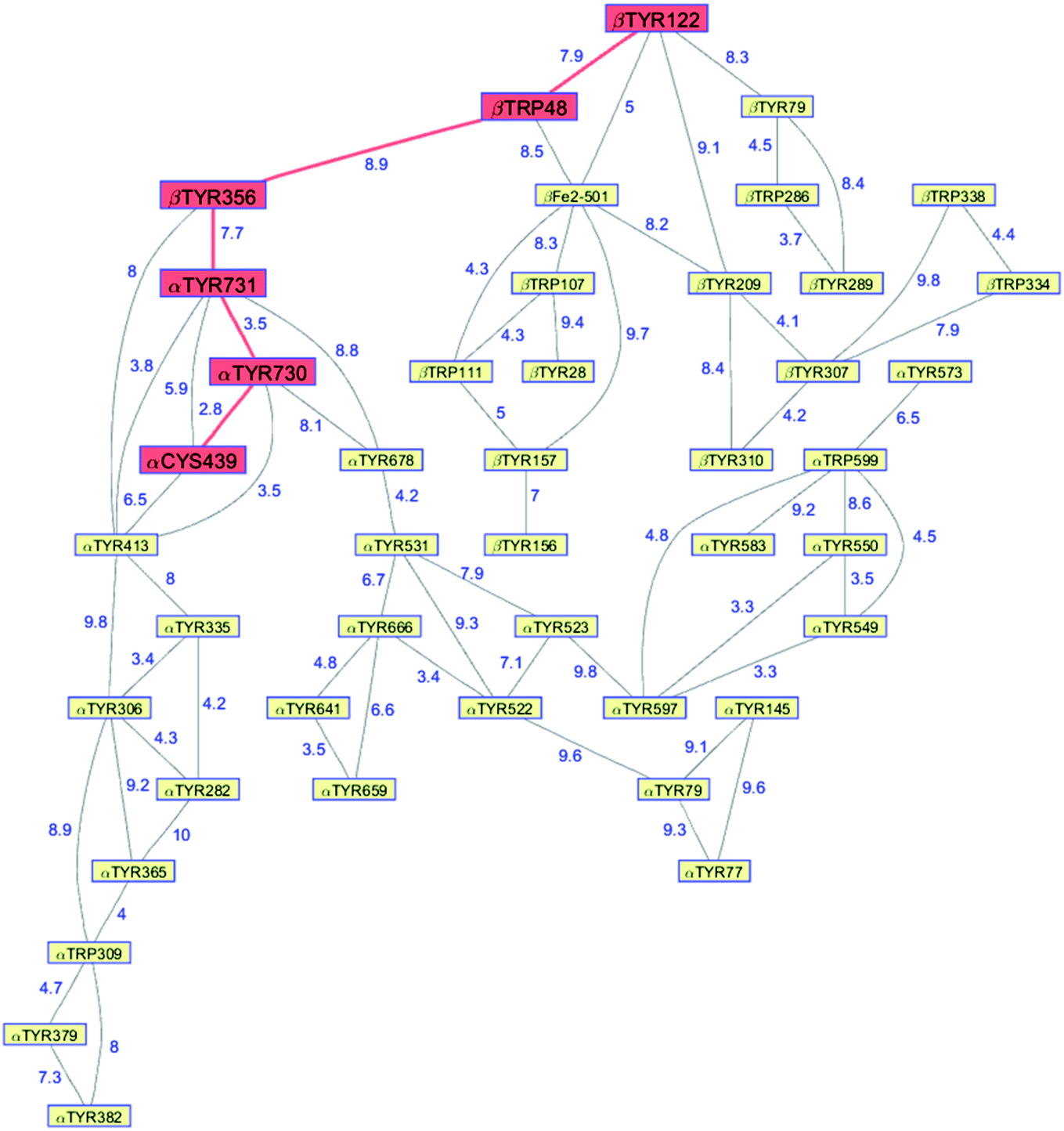 | ||
| Fig. 3 Network of Tyr and Trp residues in branched chains (10 Å maximum contact distance) connected to the E. coli RNR radical transfer pathway residues (βTyr122-βTrp48-βTyr356-αTyr731-αTyr730-αCys439, red). Distances (Å, blue numbers) are from the cryo-EM structure of the holo-enzyme (PDB ID 6W4X).59 Residue βTYR122 was 2,3,5-trifluoro-tyrosine in the structure. | ||
Photolyase and cryptochromes
Whereas the radical transfer pathway in RNR is comprised largely of Tyr residues, those pathways in DNA photolyases and the related cryptochromes primarily involve Trp residues.65 Photoactivation of DNA photolyase involves hole transfer from an electronically excited flavin (*Fla) in a flavin adenine dinucleotide (FAD) cofactor to a nearby Trp residue. Transfer through two more Trp residues brings the hole to the enzyme surface where it is scavenged by solution reductants. Because hole transfer is initiated by photoexcitation, this system is ideally suited to time-resolved laser spectroscopic investigations.66,67 Ultrafast measurements on the E. coli enzyme revealed that hole transfer across the 15 Å from the *Fla to surface-exposed Trp306 is complete in about 0.5 ns. The rate constants for the individual ET steps in the pathway have been determined for on-pathway hopping steps and, through the use of site-directed mutants, for some off-pathway and charge-recombination steps as well (Fig. 4).66,67 Driving-force estimates indicate that the formal potentials for Trp˙+ formation decrease as the distance from *Fla increases. Although the Trp and Tyr content is only slightly greater than the UniProtKB database averages (Trp, 1.49%; Tyr, 3.40%), the branched Trp/Tyr chains (7.5 Å maximum contact distance) connected to the FAD-Trp382-Trp359-Trp306 pathway contain 9 members. The Trp/Tyr network connectivity (PDB ID 1DNP, Fig. 5)68 highlights the potential for off-pathway hole transfers yet the overall quantum efficiency of Trp306˙ formation is estimated to be about 40% (Fig. 4). Detailed kinetics and driving-force data revealed that unfavorable driving forces for off-pathway transfers contribute substantially to the bias toward on-pathway charge flow.66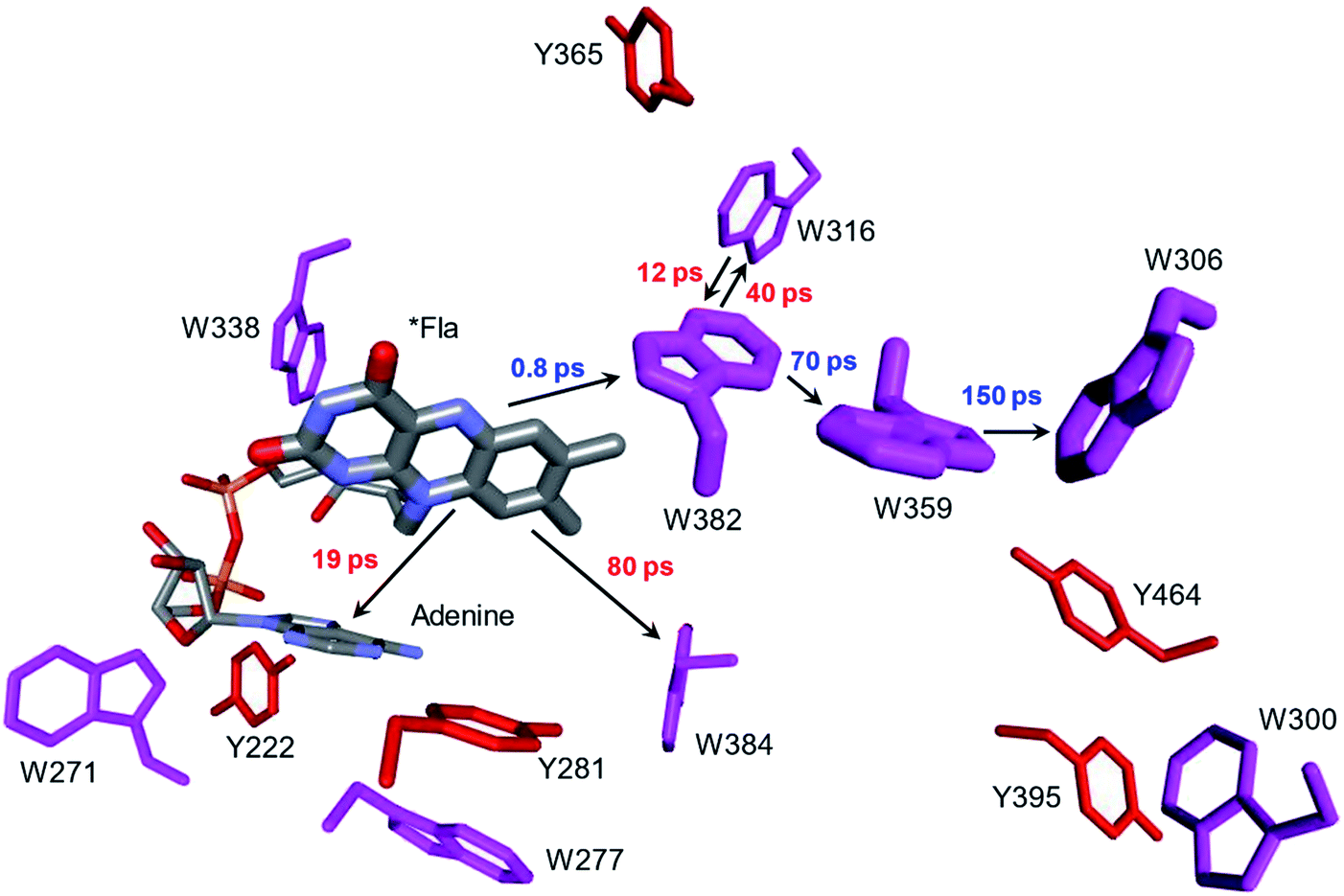 | ||
| Fig. 4 Structural model (PDB ID 1DNP)68 of the functional hole transfer pathway (*Fla-W382-W359-W306) in E. coli DNA photolyase. Surrounding Trp (magenta) and Tyr (red) residues within 7.5 Å of pathway residues are shown along with time constants for on-pathway (blue) and off-pathway (red) hole transfers.66 | ||
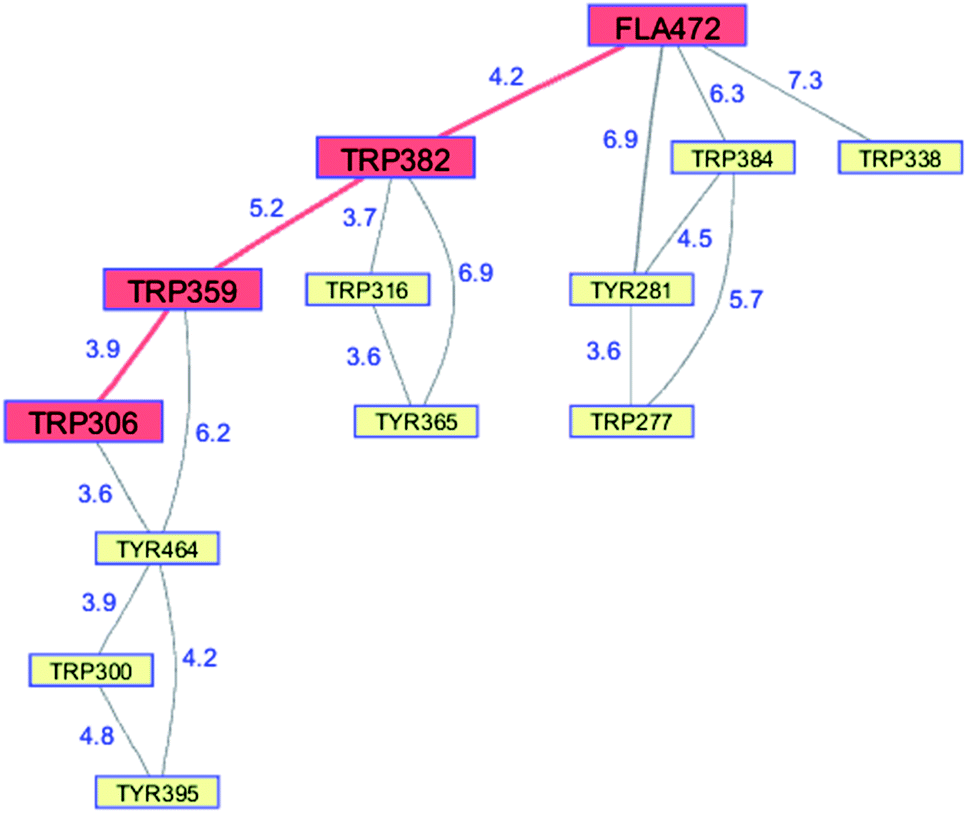 | ||
| Fig. 5 Network of Tyr and Trp residues in branched chains (7.5 Å maximum contact distance) connected to the E. coli DNA photolyase radical transfer pathway residues (FAD472-Trp382-Trp359-Trp306, red). Distances (Å, blue numbers) are from the X-ray crystal structure of the enzyme (PDB ID 1DNP).68 | ||
Peroxidases
Peroxidases catalyze the oxidation of a wide variety of substrates by hydrogen peroxide. The reaction of H2O2 with heme peroxidases produces a potent oxidant known as Compound I (CI),69 a ferryl porphyrin and a protein radical. One-electron reduction of CI usually proceeds at the radical, producing ferryl porphyrin Compound II (CII).69,70 The location of the radical in CI varies with the enzyme. In horseradish peroxidase, the prototypal heme peroxidase, the radical is located on the porphyrin. The radical in CI of Saccharomyces cerevisiae cytochrome c peroxidase (CCP) is one of the first reported examples of a stable protein amino acid radical (Trp191˙).71,72 It is likely that the reaction between Fe3+-CCP and H2O2 first generates a porphyrin radical that subsequently migrates to Trp191˙ by intraprotein hole transfer.73,74The CCP Trp191˙ radical plays a critical functional role in the reaction with its prime substrate, cytochrome c (cyt c). At high ionic strength (310 mM), yeast iso-1 Fe2+-cyt c reduces CCP CI at Trp191˙ with a rate constant of 2.5 × 108 M−1 s−1.75 The rate constant for delivery of the second electron is 5 × 107 M−1 s−1.75 Following decades of study, a consensus has emerged that reduction of both CCP CI and CII proceeds via Trp191˙; that is to say, reduction of CII involves hole hopping from the ferryl center to Trp191 and then to Fe2+-cyt c. Direct (single step) hole transfer from the CCP ferryl to Fe2+-cyt c is at least 100 times slower than the 2-step process. In CCP, diffusion of a hole from its original site on the porphyrin to Trp191 is a functional adaption that leads to enhanced substrate oxidation.
Although CCP peroxidase activity has been exhaustively investigated in vitro, recent in vivo evidence points to an important H2O2 sensing role for the enzyme in yeast.76 Yeast CCP has unusually high Trp (2.38%) and Tyr (4.76%) content (compared to UniProtKB/Swiss-Prot database averages), forming an extensive network (contact distance < 10 Å) connected to the heme (PDB ID 2CYP, Fig. 6).77 Exposure of CCP to excess H2O2 in the absence of Fe2+-cyt c leads to extensive oxidation of Trp and Met residues, as well as Tyr crosslinking to form dityrosine.78,79 Glutathione (GSH) inhibits oxidation at most of these residues, consistent with the presence of protective chains that direct holes away from the heme toward surface residues for scavenging by GSH.79 Respiring yeast produce high levels of mitochondrial H2O2 and holo-CCP recovered from mitochondria after 7 days exhibits extensive oxidation of Trp, Tyr, and Met residues.79 After 7 days of respiration significant levels of extramitochondrial apo-CCP are present with different patterns of amino acid oxidation. The in vivo data demonstrate that H2O2 is produced faster than it can be reduced by Fe2+-cyt c. Under these circumstances, CCP itself becomes a sacrificial antioxidant, delivering reducing equivalents from its own oxidizable amino acids.
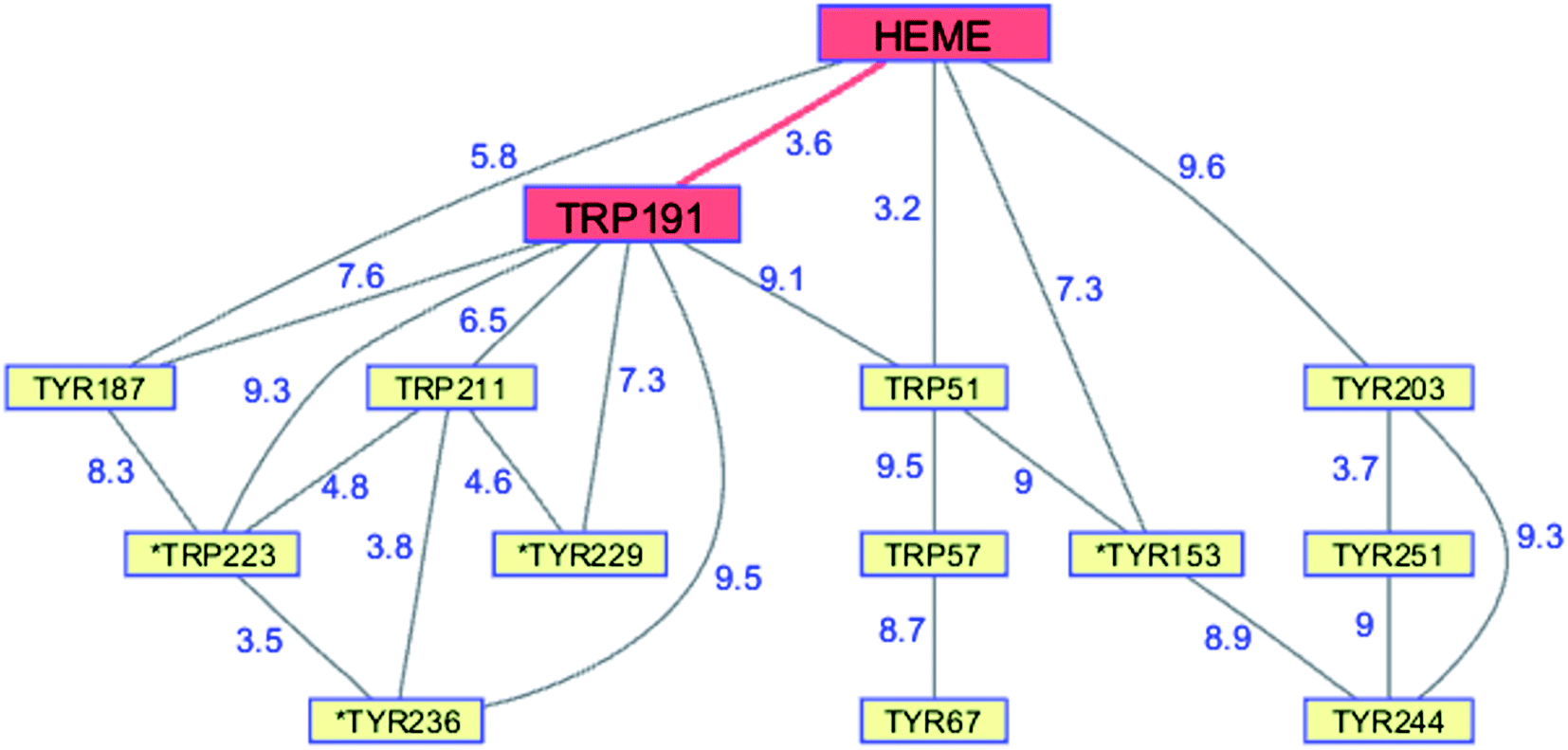 | ||
| Fig. 6 Network of Tyr and Trp residues in branched chains (10 Å maximum contact distance) connected to the yeast CCP hole transfer pathway residues (HEME-Trp191, red). Distances (Å, blue numbers) are from the X-ray crystal structure of the enzyme (PDB ID 2CYP).77 Residues with >20% sidechain solvent exposure are indicated with an asterisk. | ||
Several other peroxidases exploit amino acid radicals for substrate oxidations, typically those enzymes in which the substrate cannot access the heme. The extracellular lignin peroxidase (LiP) from Phanerochaete chrysosporium requires surface exposed Trp171 to oxidize veratryl alcohol (VA), thought to serve as a redox mediator for lignin oxidation.80 The radical in LiP CI is localized on the porphyrin, suggesting that substrate oxidation requires endergonic hole transfer to Trp171. Transfer across the 11.5 Å distance from the heme to Trp171 might be facilitated by hopping through the intervening Met172 residue. In contrast to CCP, P. chrysosporium LiP has relatively few oxidizable residues (Trp, 0.87%; Tyr, 0%; Met, 2.33%). The surface Trp164 in the Pleurotus eryngii ligninolytic peroxidase known as versatile peroxidase (VP) is required for VA oxidation.81,82 EPR and ENDOR measurements confirmed that, unlike P. chrysosporium LiP, the radical in VP CI is located on the surface Trp164 residue.82
The LiP from Trametopsis cervina has one Tyr and two Trp residues. Reaction with H2O2 generates a radical on Tyr181 that forms an adduct with VA.83 Interestingly, CI in the VA-LiP adduct is somewhat less reactive toward VA, but the specific rate of CII reduction by VA in the adduct is 100 times greater than in pristine LiP.
Enzymes known as dye-decolorizing peroxidases (DyPs) also utilize protein radicals for substrate reactions.84 Although the natural substrates are difficult to identify, DyPs can oxidize high potential substrates and are of interest for their role in lignin degradation. Both bacterial and fungal enzymes contain much higher levels of Trp and Tyr in their amino acid sequences than LP and VP. Multiple proteins radicals (both Trp and Tyr) have been detected in these enzymes following treatment with H2O2, and mutagenesis studies indicated their involvement in substrate oxidation.85–87 In the enzyme from Thermomonospora curvata, an initially formed CI porphyrin radical decays with a 0.5 s time constant to a ferryl heme with a radical distributed among a Tyr and two Trp residues.86 It is not clear why DyPs employ multiple Trp and Tyr radicals, while LiP and VP have far fewer options available for radical formation.
Catalase-peroxidases
Heme enzymes known as KatGs exhibit both peroxidase and catalase activity. KatGs possess a methionine-tyrosine-tryptophan cross-linked triad that hosts a radical necessary for catalase activity. In the KatG from Mycobacterium tuberculosis (MtKatG), this radical is generated by hole transfer from a porphyrin radical formed in the reaction of the ferric enzyme with H2O2.88 In the absence of peroxidase substrates, MtKatG loses its catalase activity after about 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 turnovers, owing to formation of off-pathway protein radicals, with Trp321 foremost among them. Peroxidase substrates can reduce these off-pathway radicals as well as the resulting CII species to restore catalase activity. MtKatG, like CCP and DyPs, is rich in oxidizable amino acids (Trp, 4.2%; Tyr, 3.7%; Met, 3.2%; Cys, 0.5%).88 In the absence of peroxidase substrates, these residues serve as sacrificial electron donors to extend enzyme survival and catalase activity.88
000 turnovers, owing to formation of off-pathway protein radicals, with Trp321 foremost among them. Peroxidase substrates can reduce these off-pathway radicals as well as the resulting CII species to restore catalase activity. MtKatG, like CCP and DyPs, is rich in oxidizable amino acids (Trp, 4.2%; Tyr, 3.7%; Met, 3.2%; Cys, 0.5%).88 In the absence of peroxidase substrates, these residues serve as sacrificial electron donors to extend enzyme survival and catalase activity.88
Metalloenzyme protection
Cytochrome P450
The cytochromes P450 are members of a superfamily of dioxygen-utilizing enzymes that are found in virtually every living species.89 These heme monooxygenases use CI and CII intermediates to insert an oxygen atom from O2 into C–H bonds of organic substrates. Whereas CI formation in peroxidases involves the direct reaction between H2O2 and the ferric enzyme, CI generation in P450 involves a multistep reductive pathway. Substrate binding to the resting Fe3+ enzyme increases its formal potential, permitting reduction to the Fe2+ level by a reductase.90 Dioxygen binding to the Fe2+-heme produces a ferric-superoxide species (Fe3+–O2−), and delivery of a second electron produces a peroxy adduct (Fe3+–O22−). Heterolytic cleavage of the peroxide generates CI. Reaction of CI with substrate involves H-atom abstraction, generating intermediate CII and a substrate radical, followed by hydroxyl radical rebound to the organic radical forming the hydroxylated product. P450 CII lacks the thermodynamic driving force required to hydroxylate a typical C–H bond, nor is it competent to abstract a substrate H-atom, owing to protonation of the P450 CII ferryl at neutral pH. Consequently, if the radical in P450 CI were to diffuse away from the heme, it would likely be unreactive toward organic substrates.Although P450 CI has yet to be observed directly in the multistep reductive pathway, this intermediate has been detected using peracids in a peroxide shunt reaction. In P450cam from Pseudomonas putida, UV-visible spectroscopic measurements provided a glimpse of CI when the substrate-free ferric enzyme was rapidly mixed with a large excess of m-chloroperbenzoic acid (mCPBA).91,92 The first intermediate formed in the reaction survived just 50 ms, but its UV-visible spectrum closely resembled that of the stable CI species in chloroperoxidase, another Cys-ligated heme enzyme. Freeze-quench EPR measurements on highly purified P450cam samples frozen 2 ms after mixing with mCPBA revealed a spectrum consistent with an S = 1 Fe4+ unit exchange-coupled with an S = 1/2 ligand (porphyrin) radical.93 Longer reaction times (40 ms) produce EPR spectra characteristic of Tyr radicals.94 Measurements on mutant enzymes provided compelling evidence for the involvement of radical formation on Tyr96 and Tyr75. Both residues are within 8 Å of the P450cam heme edge (Tyr 96, 7.4 Å; Tyr75, 7.6 Å; PDB ID 1PHC)95 and have basic groups near the phenol hydroxyl that can accept a proton upon Tyr oxidation (Fig. 7A). The high mCPBA concentrations required to ensure that CI forms more rapidly than it decays also induce rapid heme bleaching that competes with Tyr radical formation.
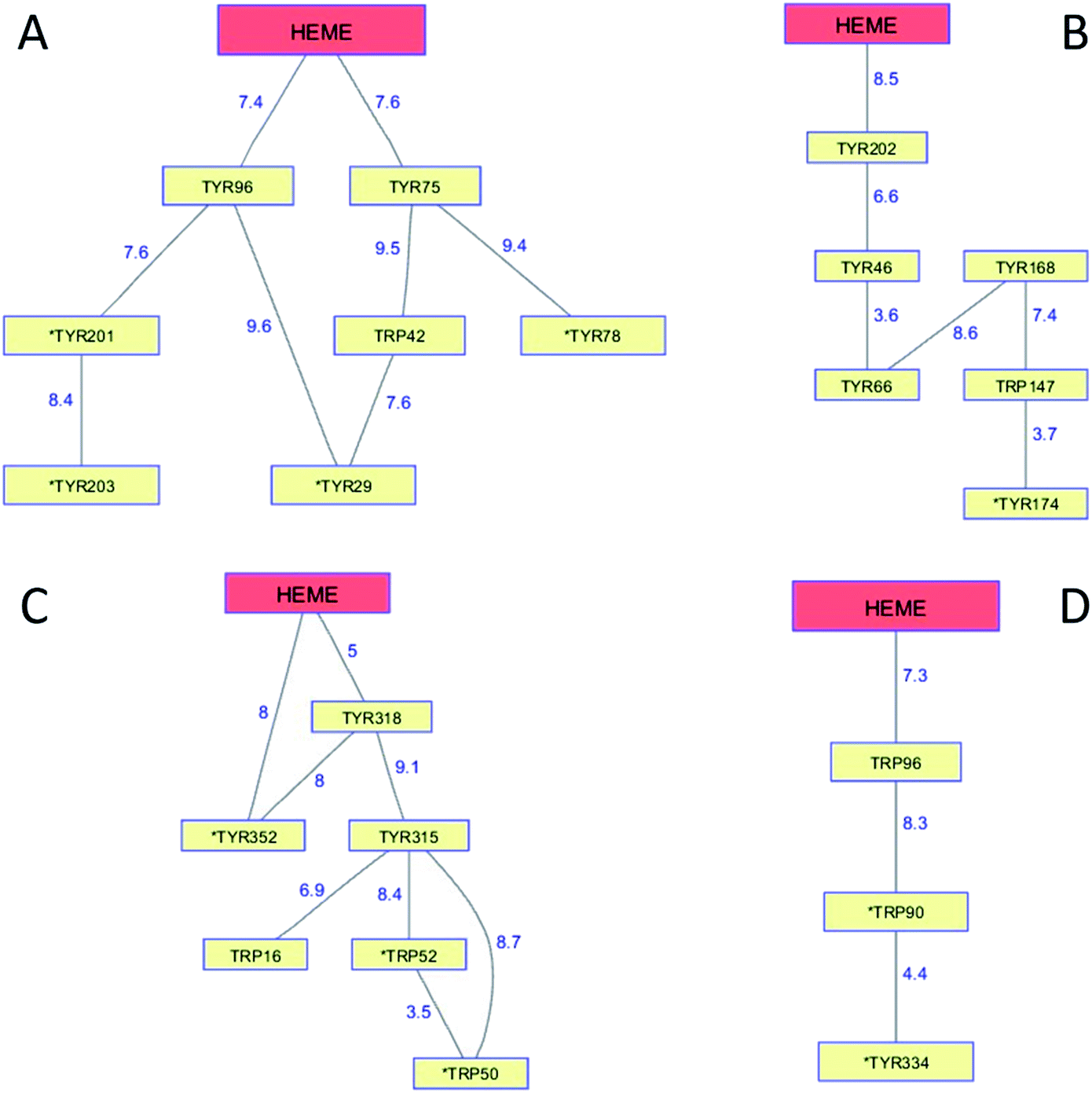 | ||
| Fig. 7 Network of Tyr and Trp residues in branched chains (10 Å maximum contact distance) connected to the hemes in (A) P450cam (PDB ID 1PHC),95 (B) CYP119 (1F4U),98 (C) CYP158 (1S1F),101 and (D) P450BM3 (2IJ2).102 Distances (Å) between residues appear as blue numbers. Solvent-exposed residues are labeled with an asterisk. | ||
Generation of the cytochrome P450 from the thermophilic archaeon Sulfolobus acidocaldarius (CYP119) allowed detailed study of the CI intermediate.96,97 Reaction of highly purified Fe3+-CYP119 with mCPBA produces a CI intermediate that lives several hundred milliseconds at 4 °C, enabling characterization by UV-visible, EPR, and Mössbauer spectroscopies.97 Moreover, the CI yield and stability were great enough to permit kinetics measurements of its hydroxylation reactions. One reason for the greater CYP119 CI stability may be that the closest Tyr residue (Tyr202) is 8.5 Å from the heme edge and no proton-accepting groups are within hydrogen-bonding distance (PDB ID 1F4U) (Fig. 7B).98
Analogous investigations of bacterial P450s from Streptomyces coelicolor (CYP158) and Bacillus megaterium (P450BM3) have shown that the porphyrin radical in CI rapidly diffuses into the protein matrix to form Tyr and Trp radicals.99,100 The Trp/Tyr network connectivity maps for these bacterial P450s (Fig. 7) indicate that P450cam, CYP158, and P450BM3 have short 2-hop paths from the heme to a surface residue (PDB ID 1S1F, 2IJ2).101,102 A hole in the heme of CYP119, however, would have to traverse 4 Tyr and 1 Trp before reaching the modestly surface exposed (18%) Tyr174 residue. CYP119 adopts the typical P450 fold, albeit with a relatively short polypeptide (CYP119, 368 residues; P450cam, 414), but the closest oxidizable residue to the heme is Tyr202 (8.5 Å away).98 Placing oxidizable residues near the heme seems to guarantee CI short-circuiting. We have suggested that these chains provide an antioxidant protection mechanism for the enzyme. If CI does not react with substrate, a nearby Trp or Tyr residue is available to direct holes to the enzyme surface where they can be scavenged by cellular reductants. The CI survival time then depends primarily on the position of the first residue in the chain. Indeed, the longest-lived CI species is in CYP119, the enzyme with the longest link between the heme and a six-residue linear path to a surface Tyr174 residue. The measurements on these bacterial P450s demonstrate that hole hopping from CI along Trp/Tyr chains operates in opposition to productive substrate oxidation. Hole-hopping deactivation of CI is a plausible explanation for high levels of uncoupled turnover observed in many of these heme monooxygenases.
Human P450s catalyze critical oxidative transformations and their disruption is associated with a vast array of disease conditions.89,103 A critically important role for these oxygen-utilizing enzymes is in drug metabolism.104 The human CYP3A4 enzyme represents 14–24% of the microsomal P450 pool,104 and is responsible for metabolism of roughly half of all therapeutic drugs.105 This substrate diversity is likely a contributing factor to the poor coupling between O2 consumption and substrate oxidation reported for this enzyme;106 an antioxidant rescue pathway could provide a mechanism to increase enzyme survival. A Trp126 residue is located 7.5 Å from the heme, and 3.6 Å from surface exposed Tyr99 (PDB ID 1TQN)107 in CYP3A4. This pathway is analogous to that found in P450BM3. Kinetics simulations suggest that this hole-escape pathway could deactivate CI within 1 μs if substrate reaction is unsuccessful (Fig. 8).13
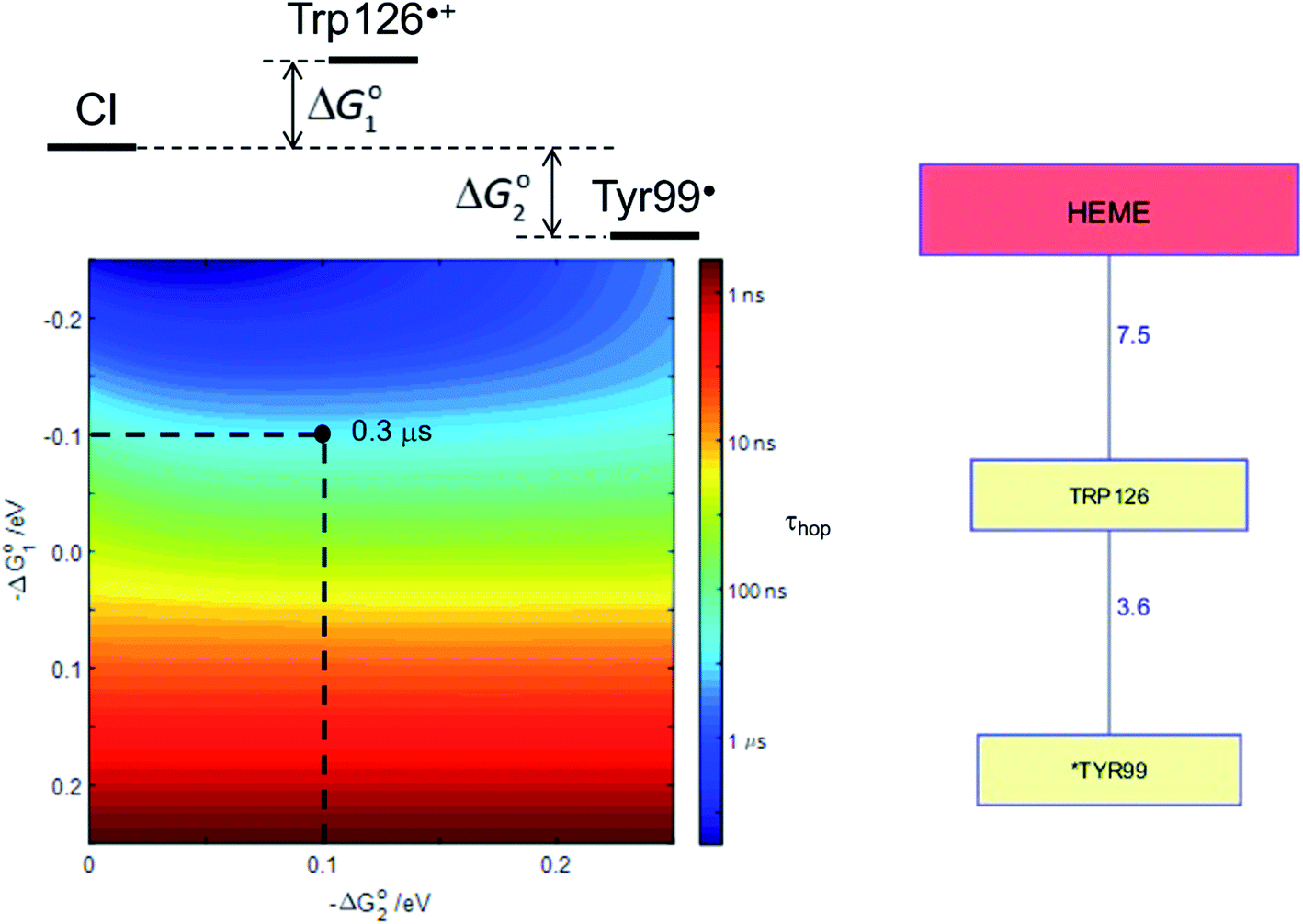 | ||
| Fig. 8 The Tyr/Trp hole migration pathway (10 Å contact distance) from the CYP3A4 (PDB ID 1TQN)107 heme to the enzyme surface is composed of just two residues: Trp126, Tyr99. The colormap illustrates the driving-force dependence of the kinetics modeling for this pathway. If formation of Trp126˙+ is endergonic by 100 meV and subsequent hole transfer to Tyr99 is exergonic by 200 meV, the CI survival time (τhop) is estimated to be 0.3 μs. τhop is given by the integral of the normalized CI decay curve. | ||
Lytic polysaccharide monooxygenases (LPMO)
LPMOs are redox active copper enzymes that oxidatively degrade polysaccharides (cellulose and chitin).108–110 The single type 2 Cu center is coordinated to two His imidazole groups, the N-terminal amino group, and an exogenous ligand, usually H2O, in a configuration known as a histidine brace.109 A Tyr residue in the second coordination sphere of Cu is found in many LPMOs. Early work implicated O2 as the source of oxidizing equivalents for substrate oxidation, leading to the characterization of the enzyme as a monooxygenase.111 Subsequent investigations, however, cast doubt on this interpretation and raised the possibility that H2O2 is the primary oxidant, characterizing the enzyme as a peroxygenase.112 The identity of the “natural” oxidant is still a matter of debate and studies have shown that LPMOs will degrade polysaccharides using either oxidant.113 In the presence of a reductant and O2, but in the absence of polysaccharide substrate, LPMOs will produce H2O2.111,113 Given that the targets of LPMO activity are insoluble polysaccharides, it is plausible that uncoupled reduction of O2 to H2O2 occurs in parallel with coupled substrate oxidation. It remains to be resolved whether the two oxidants produce identical reactive intermediates, as is believed to be the case for P450.In the absence of a polysaccharide substrate, many LPMOs suffer oxidative damage when H2O2 is used as a co-substrate.112,114,115 When exposed to H2O2 in the absence of substrate, the fungal LPMO from Lentis similis (LsAA9) is converted into a catalytically inactive purple variant characterized by an antiferromagnetically coupled Cu2+-Tyr164˙ pair in the active site. Mass spectrometric analysis of the purple enzyme revealed several sites of protein oxidation, including residues identified in a hole-hopping pathway from Tyr164 to a surface Trp5 residue.47,115 It is noteworthy that the presence of polysaccharide substrates inhibits formation of the purple species and suppresses protein oxidation.112,115 As was the case with the four bacterial P450s, a hole-hopping pathway in LsAA9 LPMO is available to direct holes away from the metal active site in opposition to substrate reaction. Particularly in the case of enzymes acting on insoluble refractory substrates, incorporation of a route for hole escape from a powerfully oxidizing active site would prolong enzyme activity.
Multicopper oxidases (MCOs)
MCOs catalyze the oxidation of organic substrates and metal ions by O2. They are constructed from 2 or 3 cupredoxin domains (2dMCO, 3dMCO), one of which retains a type 1 copper. The locus of O2 reduction to H2O is a trinuclear Cu active site (TNC) comprised of one type 2 Cu center and a type 3 Cu dimer inserted into the interface between two cupredoxin domains.116 Closely coupled Tyr/Trp chains are not common structural elements in single-domain cupredoxin ET proteins,12 but they do appear clustered around the TNC in 2dMCOs and 3dMCOs.12,13The homotrimeric 2dMCO from Streptomyces coelicolor (SLAC)117,118 has a Tyr108 residue 4.3 Å (PDB ID 3KW8)119 from the type 2 Cu of the TNC. During catalysis of substrate oxidation, a Tyr108˙ radical ferromagnetically coupled to the TNC Cu2+ center develops.120,121 Replacement of Tyr108 with Phe or Ala reduces the catalytic rate constant (kcat) by a factor of 2–3, but has only a minor impact on second-order rate constants (kcat/KM) for reaction with O2 and substrate (N,N,N′N′-tetramethyl-p-phenylenediamine).121 The accepted mechanism for MCO catalysis involves O2 binding to the fully reduced (4 × Cu+) enzyme, reduction of O2 by 2 electrons to a peroxy intermediate (PI), followed by delivery of two more electrons to produce the native intermediate (NI).122 With just three electrons available in the TNC for O2 reduction, the possibility of hydroxyl radical formation arises if the fourth electron is not delivered rapidly from the type 1 Cu center. The Tyr108 residue may serve as an obligatory intermediate formed in the reduction of PI to NI, or as a safety valve to deliver electrons when they are not available from the type 1 Cu.
Ceruloplasmin (Cp) is a mammalian ferroxidase enzyme that is structurally similar to SLAC, but comprised of a single polypeptide chain folded into six cupredoxin domains containing three type 1 Cu centers and a single TNC (PDB ID 2J5W).123 Early Cp work indicated that an intermediate with an absorption maximum at 420 nm formed following oxidation of the reduced enzyme by O2.124 More recent investigations have revealed UV-visible absorption signatures of a Tyr˙ radical following O2 oxidation of the 3- and 4-electron reduced enzyme.125 Cp Tyr107 was suggested to be the likely candidate for radical formation; this residue 4.6 Å from the type 2 Cu is in a position analogous to that of Tyr108 in SLAC. A hole escape pathway from Tyr107 to Tyr108 was identified in Cp. Substrate (Fe2+) is limiting for Cp in vivo, and the dissolved O2 concentration is high so partially reduced enzyme will be the norm. It is likely that Tyr107 radical formation protects the enzyme from damage under these low-substrate conditions.
Flavordiiron proteins (FDPs)
The flavodiiron proteins are a group of enzymes that catalyze the reduction of O2 to H2O and NO to N2O. The FDPs, found in many obligate and facultative anaerobic microorganisms, have modular multidomain structures with a minimal requirement of a catalytic diiron center in a metallo-β-lactamase domain linked to a C-terminal flavodoxin domain containing a flavin mononucleotide (FMN).126 These enzymes were originally believed to be O2 reductases that protect anaerobes from oxygen toxicity,127 although subsequent work demonstrated that the E. coli FDP is an NO reductase.128 Two of the four electrons required for O2 reduction are delivered by two Fe2+ centers in the active site; the remaining two are delivered by the reduced flavin positioned about 6 Å away. The formal potentials for reduction of the diiron core tend to be 50–200 mV more positive than those of the flavin,126 admitting the possibility of forming a partially reduced enzyme. Electrons originating in NAD(P)H move through the FMN domain into the diiron domain, making it likely that O2 would react with a partially reduced enzyme. In a partially reduced FDP, nearby Trp and Tyr residues are available to provide the additional reducing equivalents required for H2O production. The structures of five FDPs reveal at least two Trp/Tyr chains in each protein extending from the diiron center to the enzyme surface.126 The closest Trp or Tyr residues in these chains are 4–6 Å from the diiron core. One of these chains is conserved among the five enzymes, extending from the diiron site to the surface near the FMN cofactor.126,129 The FDP enzyme from Methanothermobacter marburgensis has an extensive network of interconnected Trp and Tyr residues (PDB ID 2OHH, Fig. 9).130 The Trp/Tyr chains provide the means to avoid ROS formation and protect against oxygen toxicity in the FDPs of anaerobic microorganisms.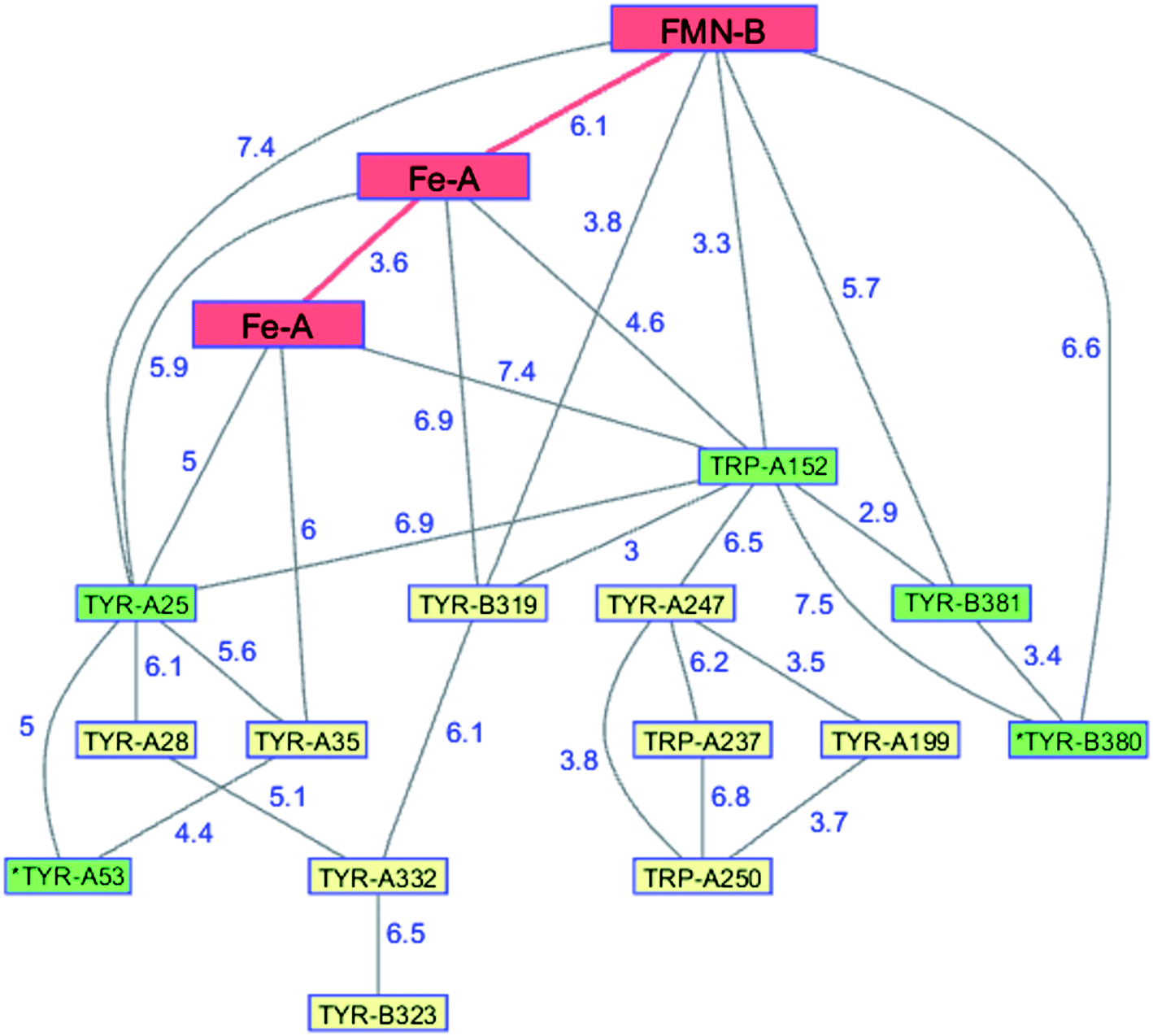 | ||
| Fig. 9 Network of Tyr and Trp residues in branched chains (7.5 Å maximum contact distance) connected to the FMN-Fe2 ET pathway residues (red) in the FDP from Methanothermobacter marburgensis. Distances (Å, blue numbers) are from the X-ray crystal structure of the enzyme (PDB ID 2OHH).130 Previously identified chains are highlighted in green.126 Solvent-exposed residues are labeled with an asterisk. | ||
Biological implications
Many enzymes risk damage or deactivation as a consequence of the reactions they catalyze. Enzymes that catalyze higher risk reactions are likely to be characterized by fewer catalytic cycles performed before failure (i.e., total turnover number, TTN). TTN values range from 1 (for suicide “enzymes”131) to >107.132 No accepted scale of risk factors for enzyme reactivity has been established, although an early study suggested that reactions with O2 or H2O2 increased the risk of enzyme deactivation.133 A more recent attempt to classify enzyme reaction risks suggested that radical mechanisms and those involving highly reactive intermediates increased the likelihood of enzyme deactivation.132 Consistent with this expectation, analysis of catalytic cycles until replacement (CCR, the in vivo analogue of TTN) among 97 Lactococcus lactis and 182 yeast enzymes revealed that CCR correlates inversely with the risk of the reaction catalyzed. A case in point is the radical enzyme RNR, which exhibited among the lowest CCR values (L. lactis RNR, CCR = 1732, rank = 12; yeast RNR, CCR = 53, rank = 1).132 Significant energetic costs are associated with low CCR. A proteomic analysis of L. lactis revealed that protein turnover is a major ATP sink in the cell.134 It is reasonable, then, that increasing enzyme CCR values would be advantageous for the organism.The wealth of structures in the protein data bank provides convincing evidence for aromatic–aromatic, cation–π, and anion–π interactions in folded polypeptides.135–137 But Trp and Tyr do more than simply stabilize protein folds. Indeed, in this perspective we have highlighted several examples of functional and protective redox activities for these two aromatic amino acids. Our analysis of hopping pathways in metalloenzymes is based on structural considerations and knowledge of the factors that regulate electron-transfer rates in proteins. The question that remains is whether these pathways have any biological significance. If enzyme misfunction is rare, then evidence for protective pathways is unlikely to appear in studies of enzyme kinetics, since the protective reaction is a minor contribution to the overall flux. That said, a protective hole-hopping pathway in an enzyme should serve to increase the TTN for an enzyme. Comparisons of TTN values in wild type and mutant enzymes could provide insights into protective capabilities of hole-hopping pathways. We are currently pursuing investigations of this type with various cytochromes P450 and multicopper oxidases. Evaluating the biological significance of protective hole-hopping chains will require in vivo determinations of CCR values for wild type and mutant enzymes. No oxidases, oxygenases, or peroxidases were included among the L. lactis and yeast enzymes in the recent report of in vivo enzyme survival data, but these clearly would be interesting targets of opportunity.132
Author contributions
Harry B. Gray: writing – review & editing, Jay R. Winkler: writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the NIH under Award R01DK019038. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Additional support was provided by the Arnold and Mabel Beckman Foundation.References
- T. W. Lyons, C. T. Reinhard and N. J. Planavsky, The rise of oxygen in Earth's early ocean and atmosphere, Nature, 2014, 506(7488), 307–315, DOI:10.1038/nature13068.
- Z. Lu and J. A. Imlay, When anaerobes encounter oxygen: mechanisms of oxygen toxicity, tolerance and defence, Nat. Rev. Microbiol., 2021 DOI:10.1038/s41579-021-00583-y.
- J. S. Valentine, D. L. Wertz, T. J. Lyons, L.-L. Liou, J. J. Goto and E. B. Gralla, The dark side of dioxygen biochemistry, Curr. Opin. Chem. Biol., 1998, 2(2), 253–262, DOI:10.1016/S1367-5931(98)80067-7.
- E. N. Trifonov, Consensus temporal order of amino acids and evolution of the triplet code, Gene, 2000, 261(1), 139–151, DOI:10.1016/S0378-1119(00)00476-5.
- E. N. Trifonov, The origin of the genetic code and of the earliest oligopeptides, Res. Microbiol., 2009, 160(7), 481–486, DOI:10.1016/j.resmic.2009.05.004.
- A. Bender, P. Hajieva and B. Moosmann, Adaptive antioxidant methionine accumulation in respiratory chain complexes explains the use of a deviant genetic code in mitochondria, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(43), 16496–16501, DOI:10.1073/pnas.0802779105.
- M. Granold, P. Hajieva, M. I. Toşa, F.-D. Irimie and B. Moosmann, Modern diversification of the amino acid repertoire driven by oxygen, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(1), 41–46, DOI:10.1073/pnas.1717100115.
- B. Moosmann, Redox Biochemistry of the Genetic Code, Trends Biochem. Sci., 2021, 46(2), 83–86, DOI:10.1016/j.tibs.2020.10.008.
- R. J. P. Williams, The necessary and the desirable production of radicals in biology, Philos. Trans.: Biol. Sci. Philos. Trans. R. Soc. London, Ser. B, 1985, 311(1152), 593–603, DOI:10.1098/rstb.1985.0166.
- R. L. Levine, L. Mosoni, B. S. Berlett and E. R. Stadtman, Methionine residues as endogenous antioxidants in proteins, Proc. Natl. Acad. Sci. U. S. A., 1996, 93(26), 15036–15040, DOI:10.1073/pnas.93.26.15036.
- H. B. Gray and J. R. Winkler, Hole hopping through tyrosine/tryptophan chains protects proteins from oxidative damage, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(35), 10920–10925, DOI:10.1073/pnas.1512704112.
- H. B. Gray and J. R. Winkler, The Rise of Radicals in Bioinorganic Chemistry, Isr. J. Chem., 2016, 56(9–10), 640–648, DOI:10.1002/ijch.201600069.
- H. B. Gray and J. R. Winkler, Living with Oxygen, Acc. Chem. Res., 2018, 51(8), 1850–1857, DOI:10.1021/acs.accounts.8b00245.
- J. R. Winkler and H. B. Gray, Could tyrosine and tryptophan serve multiple roles in biological redox processes?, Philos. Trans. R. Soc., A, 2015, 373, 2037, DOI:10.1098/rsta.2014.0178.
- J. R. Winkler and H. B. Gray, Electron flow through biological molecules: does hole hopping protect proteins from oxidative damage?, Q. Rev. Biophys., 2015, 48(4), 411–420, DOI:10.1017/s0033583515000062.
- A. Szent-Györgyi, Towards a New Biochemistry?, Science, 1941, 93(2426), 609–611, DOI:10.1126/science.93.2426.609.
- J. Silverman and R. W. Dodson, The Exchange Reaction between the 2 Oxidation States of Iron in Acid Solution, J. Phys. Chem., 1952, 56(7), 846–852, DOI:10.1021/j150499a007.
- A. Hammershoi, D. Geselowitz and H. Taube, Redetermination of the hexaaminecobalt(III/II) electron-self-exchange rate, Inorg. Chem., 1984, 23(7), 979–982, DOI:10.1021/ic00175a036.
- E. Eichler and A. C. Wahl, Electron-exchange Reactions between Large Complex Cations1, J. Am. Chem. Soc., 1958, 80(16), 4145–4149, DOI:10.1021/ja01549a008.
- A. Anderson and N. A. Bonner, The Exchange Reaction between Chromous and Chromic Ions in Perchloric Acid Solution, J. Am. Chem. Soc., 1954, 76(14), 3826–3830, DOI:10.1021/ja01643a070.
- R. A. Marcus, On the Theory of Oxidation-Reduction Reactions Involving Electron Transfer. I, J. Chem. Phys., 1956, 24(5), 966–978, DOI:10.1063/1.1742723.
- R. A. Marcus and N. Sutin, Electron Transfers in Chemistry and Biology, Biochim. Biophys. Acta, 1985, 811, 265–322, DOI:10.1016/0304-4173(85)90014-X.
- V. G. Levich and R. R. Dogonadze, Theory of Non-Radiation Electron Transitions from Ion to Ion in Solutions, Dokl. Akad. Nauk SSSR, 1959, 124(1), 123–126 CAS.
- G. I. Likhtenshtein, Solar Energy Conversion: Chemical Aspects – Electron Transfer Theories, Wiley, Germany, ch. 1, 2012 Search PubMed.
- A. M. Kuznetsov and J. Ulstrup, Electron Transfer in Chemistry and Biology: An Introduction to the Theory, John Wiley & Sons, Inc., Hoboken, NJ, 1998, p. 350 Search PubMed.
- H. M. Berman, The Protein Data Bank: a historical perspective, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64(1), 88–95, DOI:10.1107/S0108767307035623.
- H. B. Gray and J. R. Winkler, Electron Tunneling through Proteins, Q. Rev. Biophys., 2003, 36(3), 341–372, DOI:10.1017/S0033583503003913.
- H. B. Gray and J. R. Winkler, Electron flow through metalloproteins, Biochim. Biophys. Acta, Bioenerg., 2010, 1797(8), 1563–1572, DOI:10.1016/j.bbabio.2010.05.001.
- J. R. Winkler and H. B. Gray, Electron Flow through Metalloproteins, Chem. Rev., 2014, 114(7), 3369–3380, DOI:10.1021/cr4004715.
- J. R. Winkler and H. B. Gray, Long-Range Electron Tunneling, J. Am. Chem. Soc., 2014, 136(8), 2930–2939, DOI:10.1021/ja500215j.
- H. B. Gray and J. R. Winkler, Electron flow through proteins, Chem. Phys. Lett., 2009, 483, 1–9 CrossRef CAS PubMed.
- C. Shih, A. K. Museth, M. Abrahamsson, A. M. Blanco-Rodriguez, A. J. Di Bilio, J. Sudhamsu, B. R. Crane, K. L. Ronayne, M. Towrie, A. Vlcek, J. H. Richards, J. R. Winkler and H. B. Gray, Tryptophan-accelerated electron flow through proteins, Science, 2008, 320(5884), 1760–1762, DOI:10.1126/science.1158241.
- K. Takematsu, H. R. Williamson, P. Nikolovski, J. T. Kaiser, Y. Sheng, P. Pospíšil, M. Towrie, J. Heyda, D. Hollas, S. Záliš, H. B. Gray, A. Vlček and J. R. Winkler, Two Tryptophans Are Better Than One in Accelerating Electron Flow through a Protein, ACS Cent. Sci., 2019, 5(1), 192–200, DOI:10.1021/acscentsci.8b00882.
- J. J. Warren, M. E. Ener, A. Vlček, J. R. Winkler and H. B. Gray, Electron hopping through proteins, Coord. Chem. Rev., 2012, 256(21–22), 2478–2487, DOI:10.1016/j.ccr.2012.03.032.
- D. N. Beratan, J. N. Betts and J. N. Onuchic, Protein Electron Transfer Rates Set by the Bridging Secondary and Tertiary Structure, Science, 1991, 252(5010), 1285–1288, DOI:10.1126/science.1656523.
- D. N. Beratan, J. N. Onuchic, J. R. Winkler and H. B. Gray, Electron-Tunneling Pathways in Proteins, Science, 1992, 258(5089), 1740–1741, DOI:10.1126/science.1334572.
- K. Kumar, I. V. Kurnikov, D. N. Beratan, D. H. Waldeck and M. B. Zimmt, Use of Modern Electron Transfer Theories to Determine the Electronic Coupling Matrix Elements in Intramolecular Systems, J. Phys. Chem. A, 1998, 102(28), 5529–5541, DOI:10.1021/jp980113t.
- A. A. Stuchebrukhov, Long-distance electron tunneling in proteins, Theor. Chem. Acc., 2003, 110(5), 291–306, DOI:10.1134/S1054660X09170186.
- P. R. Callis and J. R. Tusell, MD + QM Correlations with Tryptophan Fluorescence Spectral Shifts and Lifetimes, in Fluorescence Spectroscopy and Microscopy: Methods and Protocols, ed. Engelborghs Y. and Visser A. J. W. G., 2014, pp. 171–213 Search PubMed.
- S. D. Glover, R. Tyburski, L. Liang, C. Tommos and L. Hammarström, Pourbaix Diagram, Proton-Coupled Electron Transfer, and Decay Kinetics of a Protein Tryptophan Radical: Comparing the Redox Properties of W32˙ and Y32˙ Generated Inside the Structurally Characterized α3W and α3Y Proteins, J. Am. Chem. Soc., 2018, 140(1), 185–192, DOI:10.1021/jacs.7b08032.
- S. V. Jovanic, A. Harriman and M. G. Simic, Electron-Transfer Reactions of Tryptophan and Tyrosine Derivatives, J. Phys. Chem., 1986, 90(9), 1935–1939 CrossRef.
- B. W. Berry, M. C. Martínez-Rivera and C. Tommos, Reversible voltammograms and a Pourbaix diagram for a protein tyrosine radical, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(25), 9739–9743, DOI:10.1073/pnas.1112057109.
- C. J. Gagliardi, C. F. Murphy, R. A. Binstead, H. H. Thorp and T. J. Meyer, Concerted Electron–Proton Transfer (EPT) in the Oxidation of Cysteine, J. Phys. Chem. C, 2015, 119(13), 7028–7038, DOI:10.1021/acs.jpcc.5b00368.
- V. C. Diculescu and T. A. Enache, Voltammetric and mass spectrometry investigation of methionine oxidation, J. Electroanal. Chem., 2019, 834, 124–129, DOI:10.1016/j.jelechem.2018.12.058.
- A. Nilsen-Moe, C. R. Reinhardt, S. D. Glover, L. Liang, S. Hammes-Schiffer, L. Hammarström and C. Tommos, Proton-Coupled Electron Transfer from Tyrosine in the Interior of a de novo Protein: Mechanisms and Primary Proton Acceptor, J. Am. Chem. Soc., 2020, 142(26), 11550–11559, DOI:10.1021/jacs.0c04655.
- N. F. Polizzi, A. Migliore, M. J. Therien and D. N. Beratan, Defusing redox bombs?, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(35), 10821–10822, DOI:10.1073/pnas.1513520112.
- R. D. Teo, R. Wang, E. R. Smithwick, A. Migliore, M. J. Therien and D. N. Beratan, Mapping hole hopping escape routes in proteins, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(32), 15811–15816, DOI:10.1073/pnas.1906394116.
- R. N. Tazhigulov, J. R. Gayvert, M. Wei and K. B. Bravaya, eMap: A Web Application for Identifying and Visualizing Electron or Hole Hopping Pathways in Proteins, J. Phys. Chem. B, 2019, 123(32), 6946–6951, DOI:10.1021/acs.jpcb.9b04816.
- L. Zanetti-Polzi, I. Daidone and S. Corni, Evidence of a Thermodynamic Ramp for Hole Hopping to Protect a Redox Enzyme from Oxidative Damage, J. Phys. Chem. Lett., 2019, 10(7), 1450–1456, DOI:10.1021/acs.jpclett.9b00403.
- B. M. Sjöberg, Ribonucleotide Reductases - A Group of Enzymes with Different Metallosites and a Similar Mechanism, Struct. Bonding, 1997, 88, 139–173, DOI:10.1007/3-540-62870-3_5.
- J. A. Cotruvo Jr and J. Stubbe, Class I Ribonucleotide Reductases: Metallocofactor Assembly and Repair In Vitro and In Vivo, Annu. Rev. Biochem., 2011, 80(1), 733–767, DOI:10.1146/annurev-biochem-061408-095817.
- B. L. Greene, G. Kang, C. Cui, M. Bennati, D. G. Nocera, C. L. Drennan and J. Stubbe, Ribonucleotide Reductases: Structure, Chemistry, and Metabolism Suggest New Therapeutic Targets, Annu. Rev. Biochem., 2020, 89(1), 45–75, DOI:10.1146/annurev-biochem-013118-111843.
- A. Ehrenberg and P. Reichard, Electron Spin Resonance of the Iron-containing Protein B2 from Ribonucleotide Reductase, J. Biol. Chem., 1972, 247(11), 3485–3488, DOI:10.1016/S0021-9258(19)45166-1.
- A. Larsson and B. M. Sjöberg, Identification of the Stable Free-Radical Tyrosine Residue in Ribonucleotide Reductase, EMBO J., 1986, 5(8), 2037–2040, DOI:10.1016/0014-5793(85)80962-5.
- B. M. Sjöberg, P. Reichard, A. Gräslund and A. Ehrenberg, The tyrosine free radical in ribonucleotide reductase from Escherichia coli, J. Biol. Chem., 1978, 253(19), 6863–6865, DOI:10.1016/S0021-9258(17)37999-1.
- B.-M. Sjöberg, P. Reichard, A. Gräslund and A. Ehrenberg, Nature of the free radical in ribonucleotide reductase from Escherichia coli, J. Biol. Chem., 1977, 252(2), 536–541, DOI:10.1016/S0021-9258(17)32750-3.
- U. Uhlin and H. Eklund, Structure of ribonucleotide reductase protein R1, Nature, 1994, 370(6490), 533–539, DOI:10.1038/370533a0.
- P. Nordlund, B.-M. Sjöberg and H. Eklund, Three-dimensional structure of the free radical protein of ribonucleotide reductase, Nature, 1990, 345(6276), 593–598, DOI:10.1038/345593a0.
- G. Kang, A. T. Taguchi, J. Stubbe and C. L. Drennan, Structure of a trapped radical transfer pathway within a ribonucleotide reductase holocomplex, Science, 2020, 368(6489), 424–427, DOI:10.1126/science.aba6794.
- B. Worsdorfer, D. A. Conner, K. Yokoyama, J. Livada, M. Seyedsayamdost, W. Jiang, A. Silakov, J. Stubbe, J. M. Bollinger and C. Krebs, Function of the Diiron Cluster of Escherichia coli Class Ia Ribonucleotide Reductase in Proton-Coupled Electron Transfer, J. Am. Chem. Soc., 2013, 135(23), 8585–8593, DOI:10.1021/ja401342s.
- P. G. Holder, A. A. Pizano, B. L. Anderson, J. Stubbe and D. G. Nocera, Deciphering Radical Transport in the Large Subunit of Class I Ribonucleotide Reductase, J. Am. Chem. Soc., 2012, 134(2), 1172–1180, DOI:10.1021/ja209016j.
- E. C. Minnihan, D. G. Nocera and J. Stubbe, Reversible, Long-Range Radical Transfer in E-coli Class la Ribonucleotide Reductase, Acc. Chem. Res., 2013, 46(11), 2524–2535, DOI:10.1021/ar4000407.
- F. Hecker, J. Stubbe and M. Bennati, Detection of Water Molecules on the Radical Transfer Pathway of Ribonucleotide Reductase by 17O Electron–Nuclear Double Resonance Spectroscopy, J. Am. Chem. Soc., 2021, 143(19), 7237–7241, DOI:10.1021/jacs.1c01359.
- C. Cui, B. L. Greene, G. Kang, C. L. Drennan, J. Stubbe and D. G. Nocera, Gated Proton Release during Radical Transfer at the Subunit Interface of Ribonucleotide Reductase, J. Am. Chem. Soc., 2021, 143(1), 176–183, DOI:10.1021/jacs.0c07879.
- J. Wang, X. Du, W. Pan, X. Wang and W. Wu, Photoactivation of the cryptochrome/photolyase superfamily, J. Photochem. Photobiol., C, 2015, 22, 84–102, DOI:10.1016/j.jphotochemrev.2014.12.001.
- Z. Liu, C. Tan, X. Guo, J. Li, L. Wang, A. Sancar and D. Zhong, Determining complete electron flow in the cofactor photoreduction of oxidized photolyase, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(32), 12966–12971, DOI:10.1073/pnas.1311073110.
- C. Aubert, M. H. Vos, P. Mathis, A. P. M. Eker and K. Brettel, Intraprotein Radical Transfer during Photoactivation of DNA Photolyase, Nature, 2000, 405(6786), 586–590, DOI:10.1038/35014644.
- H. W. Park, S. T. Kim, A. Sancar and J. Deisenhofer, Crystal Structure of DNA Photolyase from Escherichia coli, Science, 1995, 268(5219), 1866–1872, DOI:10.1126/science.7604260.
- P. C. E. Moody and E. L. Raven, The Nature and Reactivity of Ferryl Heme in Compounds I and II, Acc. Chem. Res., 2018, 51(2), 427–435, DOI:10.1021/acs.accounts.7b00463.
- A. Gumiero, C. L. Metcalfe, A. R. Pearson, E. L. Raven and P. C. E. Moody, Nature of the Ferryl Heme in Compounds I and II, J. Biol. Chem., 2011, 286(2), 1260–1268, DOI:10.1074/jbc.M110.183483.
- T. Yonetani, H. Schleyer and A. Ehrenberg, Studies on Cytochrome c Peroxidase : VII. Electron Paramagnetic Resonance Absorptions of the Enzyme and Complex ES in Dissolved and Crystalline Forms, J. Biol. Chem., 1966, 241(13), 3240–3243, DOI:10.1016/S0021-9258(18)96523-3.
- M. Sivaraja, D. B. Goodin, M. Smith and B. M. Hoffman, Identification by ENDOR of TRP191 as the Free-Radical Site in Cytochrome c Peroxidase Compound ES, Science, 1989, 245(4919), 738–740, DOI:10.1126/science.2549632.
- J. E. Erman and L. B. Vitello, Yeast cytochrome c peroxidase: mechanistic studies via protein engineering, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2002, 1597(2), 193–220, DOI:10.1016/S0167-4838(02)00317-5.
- J. E. Erman, L. B. Vitello, J. M. Mauro and J. Kraut, Detection of an oxyferryl porphyrin π-cation-radical intermediate in the reaction between hydrogen peroxide and a mutant yeast cytochrome c peroxidase. Evidence for tryptophan-191 involvement in the radical site of compound I, Biochemistry, 1989, 28(20), 7992–7995, DOI:10.1021/bi00446a004.
- M. A. Miller, R.-Q. Liu, S. Hahm, L. Geren, S. Hibdon, J. Kraut, B. Durham and F. Millett, Interaction Domain for the Reaction of Cytochrome c with the Radical and the Oxyferryl Heme in Cytochrome c Peroxidase Compound I, Biochemistry, 1994, 33(29), 8686–8693, DOI:10.1021/bi00195a009.
- M. Kathiresan, D. Martins and A. M. English, Respiration triggers heme transfer from cytochrome c peroxidase to catalase in yeast mitochondria, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(49), 17468–17473, DOI:10.1073/pnas.1409692111.
- B. C. Finzel, T. L. Poulos and J. Kraut, Crystal Structure of Yeast Cytochrome c Peroxidase Refined at 1.7-Å Resolution, J. Biol. Chem., 1984, 259(21), 13027–13036, DOI:10.1016/S0021-9258(18)90651-4.
- M. Kathiresan and A. M. English, LC-MS/MS suggests that hole hopping in cytochrome c peroxidase protects its heme from oxidative modification by excess H2O2, Chem. Sci., 2017, 8(2), 1152–1162, 10.1039/c6sc03125k.
- M. Kathiresan and A. M. English, LC-MS/MS Proteoform Profiling Exposes Cytochrome c Peroxidase Self-Oxidation in Mitochondria and Functionally Important Hole Hopping from Its Heme, J. Am. Chem. Soc., 2018, 140(38), 12033–12039, DOI:10.1021/jacs.8b05966.
- W. A. Doyle, W. Blodig, N. C. Veitch, K. Piontek and A. T. Smith, Two Substrate Interaction Sites in Lignin Peroxidase Revealed by Site-Directed Mutagenesis, Biochemistry, 1998, 37(43), 15097–15105, DOI:10.1021/bi981633h.
- F. J. Ruiz-Dueñas, M. Morales, E. García, Y. Miki, M. J. Martínez and A. T. Martínez, Substrate oxidation sites in versatile peroxidase and other basidiomycete peroxidases, J. Exp. Bot., 2008, 60(2), 441–452, DOI:10.1093/jxb/ern261.
- R. Pogni, M. C. Baratto, C. Teutloff, S. Giansanti, F. J. Ruiz-Dueñas, T. Choinowski, K. Piontek, A. T. Martínez, F. Lendzian and R. Basosi, A Tryptophan Neutral Radical in the Oxidized State of Versatile Peroxidase from Pleurotus eryngii: a Combined Multifrequency EPR and Density Functional Theory Study, J. Biol. Chem., 2006, 281(14), 9517–9526, DOI:10.1074/jbc.M510424200.
- Y. Miki, R. Pogni, S. Acebes, F. Lucas, E. Fernández-Fueyo, M. C. Baratto, M. I. Fernández, V. de los Ríos, F. J. Ruiz-Dueñas, J. Francisco, A. Sinicropi, R. Basosi, K. E. Hammel, V. Guallar and A. T. Martínez, Formation of a tyrosine adduct involved in lignin degradation by Trametopsis cervina lignin peroxidase: a novel peroxidase activation mechanism, Biochem. J., 2013, 452(3), 575–584, DOI:10.1042/bj20130251.
- Y. Sugano and T. Yoshida, DyP-Type Peroxidases: Recent Advances and Perspectives, Int. J. Mol. Sci., 2021, 22(11), 5556, DOI:10.3390/ijms22115556.
- M. C. Baratto, A. Sinicropi, D. Linde, V. Sáez-Jiménez, L. Sorace, F. J. Ruiz-Duenas, A. T. Martinez, R. Basosi and R. Pogni, Redox-Active Sites in Auricularia auricula-judae Dye-Decolorizing Peroxidase and Several Directed Variants: A Multifrequency EPR Study, J. Phys. Chem. B, 2015, 119(43), 13583–13592, DOI:10.1021/acs.jpcb.5b02961.
- R. Shrestha, X. Chen, K. X. Ramyar, Z. Hayati, E. A. Carlson, S. H. Bossmann, L. Song, B. V. Geisbrecht and P. Li, Identification of Surface-Exposed Protein Radicals and A Substrate Oxidation Site in A-Class Dye-Decolorizing Peroxidase from Thermomonospora curvata, ACS Catal., 2016, 6(12), 8036–8047, DOI:10.1021/acscatal.6b01952.
- K. Nys, P. G. Furtmüller, C. Obinger, S. Van Doorslaer and V. Pfanzagl, On the Track of Long-Range Electron Transfer in B-Type Dye-Decolorizing Peroxidases: Identification of a Tyrosyl Radical by Computational Prediction and Electron Paramagnetic Resonance Spectroscopy, Biochemistry, 2021, 60(15), 1226–1241, DOI:10.1021/acs.biochem.1c00129.
- O. J. Njuma, I. Davis, E. N. Ndontsa, J. R. Krewall, A. Liu and D. C. Goodwin, Mutual synergy between catalase and peroxidase activities of the bifunctional enzyme KatG is facilitated by electron hole-hopping within the enzyme, J. Biol. Chem., 2017, 292(45), 18408–18421, DOI:10.1074/jbc.M117.791202.
- D. W. Nebert, K. Wikvall and W. L. Miller, Human cytochromes P450 in health and disease, Philos. Trans. R. Soc., B, 2013, 368(1612) DOI:10.1098/rstb.2012.0431.
- A. Das, Y. V. Grinkova and S. G. Sligar, Redox Potential Control by Drug Binding to Cytochrome P450 3A4, J. Am. Chem. Soc., 2007, 129(45), 13778–13779, DOI:10.1021/ja074864x.
- T. Egawa, H. Shimada and Y. Ishimura, Evidence for Compound I Formation in the Reaction of Cytochrome-P450cam with m-Chloroperbenzoic Acid, Biochem. Biophys. Res. Commun., 1994, 201(3), 1464–1469, DOI:10.1006/bbrc.1994.1868.
- T. Spolitak, J. H. Dawson and D. P. Ballou, Reaction of ferric cytochrome P450cam with peracids - Kinetic characterization of intermediates on the reaction pathway, J. Biol. Chem., 2005, 280(21), 20300–20309, DOI:10.1074/jbc.M501761200.
- T. H. Yosca and M. T. Green, Preparation of Compound I in P450cam: The Prototypical P450, Isr. J. Chem., 2016, 56(9–10), 834–840, DOI:10.1002/ijch.201600013.
- V. Schünemann, F. Lendzian, C. Jung, J. Contzen, A. L. Barra, S. G. Sligar and A. X. Trautwein, Tyrosine radical formation in the reaction of wild type and mutant cytochrome P450cam with peroxy acids - A multifrequency EPR study of intermediates on the millisecond time scale, J. Biol. Chem., 2004, 279(12), 10919–10930, DOI:10.1074/jbc.M307884200.
- T. L. Poulos, B. C. Finzel and A. J. Howard, Crystal structure of substrate-free Pseudomonas putida cytochrome P-450, Biochemistry, 1986, 25(18), 5314–5322, DOI:10.1021/bi00366a049.
- D. G. Kellner, S.-C. Hung, K. E. Weiss and S. G. Sligar, Kinetic Characterization of Compound I Formation in the Thermostable Cytochrome P450 CYP119*, J. Biol. Chem., 2002, 277(12), 9641–9644, DOI:10.1074/jbc.C100745200.
- J. Rittle and M. T. Green, Cytochrome P450 Compound I: Capture, Characterization, and C-H Bond Activation Kinetics, Science, 2010, 330(6006), 933–937, DOI:10.1126/science.1193478.
- J. K. Yano, L. S. Koo, D. J. Schuller, H. Li, P. R. Ortiz de Montellano and T. L. Poulos, Crystal Structure of a Thermophilic Cytochrome P450 from the Archaeon Sulfolobus solfataricus, J. Biol. Chem., 2000, 275(40), 31086–31092, DOI:10.1074/jbc.M004281200.
- T. H. Yosca, J. Rittle, C. M. Krest, E. L. Onderko, A. Silakov, J. C. Calixto, R. K. Behan and M. T. Green, Iron(IV)hydroxide pKa and the Role of Thiolate Ligation in C–H Bond Activation by Cytochrome P450, Science, 2013, 342(6160), 825–829, DOI:10.1126/science.1244373.
- C. Jung, V. Schünemann and F. Lendzian, Freeze-quenched iron-oxo intermediates in cytochromes P450, Biochem. Biophys. Res. Commun., 2005, 338(1), 355–364, DOI:10.1016/j.bbrc.2005.08.166.
- B. Zhao, F. P. Guengerich, A. Bellamine, D. C. Lamb, M. Izumikawa, L. Lei, L. M. Podust, M. Sundaramoorthy, J. A. Kalaitzis, L. M. Reddy, S. L. Kelly, B. S. Moore, D. Stec, M. Voehler, J. R. Falck, T. Shimada and M. R. Waterman, Binding of Two Flaviolin Substrate Molecules, Oxidative Coupling, and Crystal Structure of Streptomyces coelicolor A3(2) Cytochrome P450 158A2*, J. Biol. Chem., 2005, 280(12), 11599–11607, DOI:10.1074/jbc.M410933200.
- H. M. Girvan, H. E. Seward, H. S. Toogood, M. R. Cheesman, D. Leys and A. W. Munro, Structural and spectroscopic characterization of P450BM3 mutants with unprecedented P450 heme iron ligand sets - New heme ligation states influence conformational equilibria in P450BM3, J. Biol. Chem., 2007, 282(1), 564–572, DOI:10.1074/jbc.M607949200.
- E. Stavropoulou, G. G. Pircalabioru and E. Bezirtzoglou, The Role of Cytochromes P450 in Infection, Front. Immunol., 2018, 9(89) DOI:10.3389/fimmu.2018.00089.
- U. M. Zanger and M. Schwab, Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation, Pharmacol. Ther., 2013, 138(1), 103–141, DOI:10.1016/j.pharmthera.2012.12.007.
- L. C. Wienkers and T. G. Heath, Predicting in vivo drug interactions from in vitro drug discovery data, Nat. Rev. Drug Discovery, 2005, 4, 825, DOI:10.1038/nrd1851.
- Y. V. Grinkova, I. G. Denisov, M. A. McLean and S. G. Sligar, Oxidase uncoupling in heme monooxygenases: Human cytochrome P450 CYP3A4 in Nanodiscs, Biochem. Biophys. Res. Commun., 2013, 430(4), 1223–1227, DOI:10.1016/j.bbrc.2012.12.072.
- J. K. Yano, M. R. Wester, G. A. Schoch, K. J. Griffin, C. D. Stout and E. F. Johnson, The Structure of Human Microsomal Cytochrome P450 3A4 Determined by X-ray Crystallography to 2.05-Å Resolution, J. Biol. Chem., 2004, 279(37), 38091–38094, DOI:10.1074/jbc.C400293200.
- G. Vaaje-Kolstad, B. Westereng, S. J. Horn, Z. Liu, H. Zhai, M. Sørlie and V. G. H. Eijsink, An Oxidative Enzyme Boosting the Enzymatic Conversion of Recalcitrant Polysaccharides, Science, 2010, 330(6001), 219–222, DOI:10.1126/science.1192231.
- R. J. Quinlan, M. D. Sweeney, L. Lo Leggio, H. Otten, J.-C. N. Poulsen, K. S. Johansen, K. B. R. M. Krogh, C. I. Jørgensen, M. Tovborg, A. Anthonsen, T. Tryfona, C. P. Walter, P. Dupree, F. Xu, G. J. Davies and P. H. Walton, Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(37), 15079–15084, DOI:10.1073/pnas.1105776108.
- W. T. Beeson, V. V. Vu, E. A. Span, C. M. Phillips and M. A. Marletta, Cellulose Degradation by Polysaccharide Monooxygenases, Annu. Rev. Biochem., 2015, 84(1), 923–946, DOI:10.1146/annurev-biochem-060614-034439.
- C. H. Kjaergaard, M. F. Qayyum, S. D. Wong, F. Xu, G. R. Hemsworth, D. J. Walton, N. A. Young, G. J. Davies, P. H. Walton, K. S. Johansen, K. O. Hodgson, B. Hedman and E. I. Solomon, Spectroscopic and computational insight into the activation of O2 by the mononuclear Cu center in polysaccharide monooxygenases, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(24), 8797–8802, DOI:10.1073/pnas.1408115111.
- B. Bissaro, A. K. Rohr, G. Muller, P. Chylenski, M. Skaugen, Z. Forsberg, S. J. Horn, G. Vaaje-Kolstad and V. G. H. Eijsink, Oxidative cleavage of polysaccharides by monocopper enzymes depends on H2O2, Nat. Chem. Biol., 2017, 13(10), 1123–1128, DOI:10.1038/nchembio.2470.
- J. A. Hangasky, A. T. Iavarone and M. A. Marletta, Reactivity of O2 versus H2O2 with polysaccharide monooxygenases, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(19), 4915–4920, DOI:10.1073/pnas.1801153115.
- S. Kuusk, B. Bissaro, P. Kuusk, Z. Forsberg, V. G. H. Eijsink, M. Sørlie and P. Väljamäe, Kinetics of H2O2-driven degradation of chitin by a bacterial lytic polysaccharide monooxygenase, J. Biol. Chem., 2018, 293(2), 523–531, DOI:10.1074/jbc.M117.817593.
- A. Paradisi, E. M. Johnston, M. Tovborg, C. R. Nicoll, L. Ciano, A. Dowle, J. McMaster, Y. Hancock, G. J. Davies and P. H. Walton, Formation of a Copper(II)–Tyrosyl Complex at the Active Site of Lytic Polysaccharide Monooxygenases Following Oxidation by H2O2, J. Am. Chem. Soc., 2019, 141(46), 18585–18599, DOI:10.1021/jacs.9b09833.
- S. M. Jones and E. I. Solomon, Electron transfer and reaction mechanism of laccases, Cell. Mol. Life Sci., 2015, 72(5), 869–883, DOI:10.1007/s00018-014-1826-6.
- M. C. Machczynski, E. Vijgenboom, B. Samyn and G. W. Canters, Characterization of SLAC: A small laccase from Streptomyces coelicolor with unprecedented activity, Prot. Sci., 2004, 13(9), 2388–2397, DOI:10.1110/ps.04759104.
- J. A. R. Worrall, M. C. Machczynski, B. J. F. Keijser, G. di Rocco, S. Ceola, M. Ubbink, E. Vijgenboom and G. W. Canters, Spectroscopic Characterization of a High-Potential Lipo-Cupredoxin Found in Streptomyces coelicolor, J. Am. Chem. Soc., 2006, 128(45), 14579–14589, DOI:10.1021/ja064112n.
- T. Skálová, J. Dohnálek, L. H. Østergaard, P. R. Østergaard, P. Kolenko, J. Dušková and J. Hašek, Crystallization and preliminary X-ray diffraction analysis of the small laccase from Streptomyces coelicolor, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2007, 63(12), 1077–1079, DOI:10.1107/S1744309107060721.
- A. W. J. W. Tepper, S. Milikisyants, S. Sottini, E. Vijgenboom, E. J. J. Groenen and G. W. Canters, Identification of a Radical Intermediate in the Enzymatic Reduction of Oxygen by a Small Laccase, J. Am. Chem. Soc., 2009, 131(33), 11680–11682, DOI:10.1021/ja900751c.
- A. Gupta, I. Nederlof, S. Sottini, A. Tepper, E. J. J. Groenen, E. A. J. Thomassen and G. W. Canters, Involvement of Tyr108 in the Enzyme Mechanism of the Small Laccase from Streptomyces coelicolor, J. Am. Chem. Soc., 2012, 134(44), 18213–18216, DOI:10.1021/ja3088604.
- S.-K. Lee, S. D. George, W. E. Antholine, B. Hedman, K. O. Hodgson and E. I. Solomon, Nature of the Intermediate Formed in the Reduction of O2 to H2O at the Trinuclear Copper Cluster Active Site in Native Laccase, J. Am. Chem. Soc., 2002, 124(21), 6180–6193, DOI:10.1021/ja0114052.
- I. Bento, C. Peixoto, V. N. Zaitsev and P. F. Lindley, Ceruloplasmin revisited: structural and functional roles of various metal cation-binding sites, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2007, 63(2), 240–248, DOI:10.1107/S090744490604947X.
- T. Manabe, N. Manabe, H. Hatano and K. Hiromi, New Intermediate in Reoxidation of Reduced Human Ceruloplasmin, FEBS Lett., 1972, 23(2), 268, DOI:10.1016/0014-5793(72)80358-2.
- S. Tian, S. M. Jones and E. I. Solomon, Role of a Tyrosine Radical in Human Ceruloplasmin Catalysis, ACS Cent. Sci., 2020, 6, 1835–1843, DOI:10.1021/acscentsci.0c00953.
- M. C. Martins, C. V. Romão, F. Folgosa, P. T. Borges, C. Frazão and M. Teixeira, How superoxide reductases and flavodiiron proteins combat oxidative stress in anaerobes, Free Radical Biol. Med., 2019, 140, 36–60, DOI:10.1016/j.freeradbiomed.2019.01.051.
- L. Chen, M. Y. Liu, J. Legall, P. Fareleira, H. Santos and A. V. Xavier, Rubredoxin Oxidase, a New Flavo-Hemo-Protein, Is the Site of Oxygen Reduction to Water by the “Strict Anaerobe” Desulfovibrio gigas, Biochem. Biophys. Res. Commun., 1993, 193(1), 100–105, DOI:10.1006/bbrc.1993.1595.
- A. M. Gardner, R. A. Helmick and P. R. Gardner, Flavorubredoxin, an Inducible Catalyst for Nitric Oxide Reduction and Detoxification in Escherichia coli *, J. Biol. Chem., 2002, 277(10), 8172–8177, DOI:10.1074/jbc.M110471200.
- C. V. Romão, J. B. Vicente, P. T. Borges, B. L. Victor, P. Lamosa, E. Silva, L. Pereira, T. M. Bandeiras, C. M. Soares, M. A. Carrondo, D. Turner, M. Teixeira and C. Frazão, Structure of Escherichia coli Flavodiiron Nitric Oxide Reductase, J. Mol. Biol., 2016, 428(23), 4686–4707, DOI:10.1016/j.jmb.2016.10.008.
- H. Seedorf, C. H. Hagemeier, S. Shima, R. K. Thauer, E. Warkentin and U. Ermler, Structure of coenzyme F420H2 oxidase (FprA), a di-iron flavoprotein from methanogenic Archaea catalyzing the reduction of O2 to H2O, FEBS J., 2007, 274(6), 1588–1599, DOI:10.1111/j.1742-4658.2007.05706.x.
- A. Chatterjee, N. D. Abeydeera, S. Bale, P.-J. Pai, P. C. Dorrestein, D. H. Russell, S. E. Ealick and T. P. Begley, Saccharomyces cerevisiae THI4p is a suicide thiamine thiazole synthase, Nature, 2011, 478(7370), 542–546, DOI:10.1038/nature10503.
- A. D. Hanson, D. R. McCarty, C. S. Henry, X. Xian, J. Joshi, J. A. Patterson, J. D. García-García, S. D. Fleischmann, N. D. Tivendale and A. H. Millar, The number of catalytic cycles in an enzyme's lifetime and why it matters to metabolic engineering, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(13), e2023348118, DOI:10.1073/pnas.2023348118.
- M. R. Gray, Substrate inactivation of enzymes in vitro and in vivo, Biotechnol. Adv., 1989, 7(4), 527–575, DOI:10.1016/0734-9750(89)90722-2.
- P.-J. Lahtvee, A. Seiman, L. Arike, K. Adamberg and R. Vilu, Protein turnover forms one of the highest maintenance costs in Lactococcus lactis, Microbiology, 2014, 160(7), 1501–1512, DOI:10.1099/mic.0.078089-0.
- M. H. Al Mughram, C. Catalano, J. P. Bowry, M. K. Safo, J. N. Scarsdale and G. E. Kellogg, 3D Interaction Homology: Hydropathic Analyses of the “π–Cation” and “π–π” Interaction Motifs in Phenylalanine, Tyrosine, and Tryptophan Residues, J. Chem. Inf. Model., 2021, 61(6), 2937–2956, DOI:10.1021/acs.jcim.1c00235.
- X. Lucas, A. Bauzá, A. Frontera and D. Quiñonero, A thorough anion–π interaction study in biomolecules: on the importance of cooperativity effects, Chem. Sci., 2016, 7(2), 1038–1050, 10.1039/C5SC01386K.
- S. K. Burley and G. A. Petsko, Aromatic-Aromatic Interaction - A Mechanism of Protein Structure Stabilization, Science, 1985, 229(4708), 23–28, DOI:10.1126/science.3892686.
| This journal is © The Royal Society of Chemistry 2021 |