
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe mechanism of oxidative addition of Pd(0) to Si–H bonds: electronic effects, reaction mechanism, and hydrosilylation†
Michael R.
Hurst
 ,
Lev N.
Zakharov
and
Amanda K.
Cook
*
,
Lev N.
Zakharov
and
Amanda K.
Cook
*
Department of Chemistry and Biochemistry, University of Oregon, Eugene, OR 97403, USA. E-mail: akcook@uoregon.edu
First published on 14th September 2021
Abstract
The oxidative addition of Pd to Si–H bonds is a crucial step in a variety of catalytic applications, and many aspects of this reaction are poorly understood. One important yet underexplored aspect is the electronic effect of silane substituents on reactivity. Herein we describe a systematic investigation of the formation of silyl palladium hydride complexes as a function of silane identity, focusing on electronic influence of the silanes. Using [(μ-dcpe)Pd]2 (dcpe = dicyclohexyl(phosphino)ethane) and tertiary silanes, data show that equilibrium strongly favours products formed from electron-deficient silanes, and is fully dynamic with respect to both temperature and product distribution. A notable kinetic isotope effect (KIE) of 1.21 is observed with H/DSiPhMe2 at 233 K, and the reaction is shown to be 0.5th order in [(μ-dcpe)Pd]2 and 1st order in silane. Formed complexes exhibit temperature-dependent intramolecular H/Si ligand exchange on the NMR timescale, allowing determination of the energetic barrier to reversible oxidative addition. Taken together, these results give unique insight into the individual steps of oxidative addition and suggest the initial formation of a σ-complex intermediate to be rate-limiting. The insight gained from these mechanistic studies was applied to hydrosilylation of alkynes, which shows parallel trends in the effect of the silanes' substituents. Importantly, this work highlights the relevance of in-depth mechanistic studies of fundamental steps to catalysis.
Introduction
Palladium hydrides are a unique class of organometallic compounds1,2 which are invoked as key intermediates in a variety of catalytic reactions, including the Mizoroki–Heck reaction,3–5 alcohol and alkene oxidation,6–8 nucleocarbonylation reactions, hydrofunctionalization reactions,9 (co)polymerization reactions,10,11 and alkene and alkyne (cyclo)isomerization reactions.12–14 However, despite the fact that these species are often proposed as intermediates, the direct observation or isolation of complexes containing a formal Pd–H bond is uncommon, and fundamental study of their behaviour has largely been precluded.1,2,12Organosilicon compounds are broadly significant, since they are used as monomers to synthesize polysiloxanes (silicones),15,16 and they have also recently gained attention for their use as biocompatible yet non-natural carbon isosteres17 in the synthesis of bioactive molecules.18–20 Transition metal-catalysed hydrosilylation of alkenes and alkynes is the primary method for synthesizing organosilanes,21–25 with most development and mechanistic work focusing primarily on Pt catalysts.22,25,26 On the other hand, Pd catalysts show complementary and modular regioselectivity25,27,28 and are more useful in enantioselective hydrosilylation.29–32
A key step in the mechanism of transition metal-catalysed hydrosilylation is the oxidative addition of the metal to the Si–H bond.21–23,25,26,33–35 While this fundamental step of organometallic chemistry, to form (silyl)M(H) complexes, has been well-studied for Pd's neighbouring elements Rh,36–48 Pt,49–64 and most other transition metals,33–35 it is much less understood for Pd. The oxidative addition of Rh and Pt to Si–H is facile, and complexes have been reported with a variety of ligand scaffolds and silanes. In addition to hydrosilylation (Fig. 1b), the oxidative addition of Pd to Si–H bonds (Fig. 1a) is a vital step of carbene insertion reactions (Fig. 1c),65–67 C–Si cross-coupling reactions (Fig. 1d),68,69 and hydrodehalogenation reactions (Fig. 1e),70–75 amongst others.76–81A key limitation that unifies all of these reactions is the dependence on silane identity; most reactions are heavily dependent on the number of substituents (primary vs. secondary silanes, etc.) on silicon, and most protocols do not investigate the electronic and/or steric effects of the silane substituents. Notably, the electronic effects of the silane substituents have been experimentally investigated for Rh and Pt,37,44,46,61 but remain unexplored for Pd. Because of the importance of this step in many Pd-catalysed reactions, we sought to determine the mechanism of Si–H oxidative addition to Pd(0) with a focus on elucidating the effect of silane substituents.
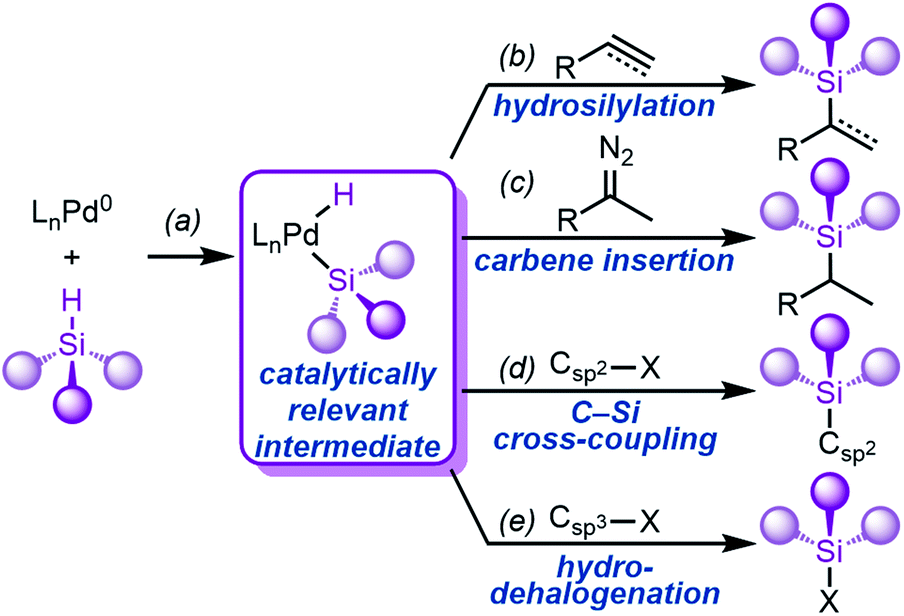 | ||
| Fig. 1 (a) Oxidative addition to form silyl palladium hydride species implicated in (b) hydrosilylation (only one isomer shown for clarity), (c) carbene insertion, (d) C–Si cross-coupling, and (e) hydrodehalogenation reactions. | ||
Despite being implicated as catalytic intermediates for more than half of a century, it was not until the early 2000's that the Fink group reported the isolation of the first (silyl)Pd(H) complex.82,83 Since this seminal work, Ishii84 and Iluc85,86 have both also published related work. Importantly, the effect of silane substituent has not been systematically explored; the silane substituents were restricted to alkyl and phenyl groups (Fig. 2a), yet trends found computationally suggest that inductive electronic effects of silane substituents dictate reactivity.87,88 Furthermore, while some characteristics of the (silyl)Pd(H) compounds were investigated, the overall reaction mechanism remains unsupported with experimental evidence. The Si–H bond is relatively non-polar, and consequently, the mechanism of the reaction likely proceeds through a concerted pathway.33–35 Experimental evidence to support this hypothesis is needed, including the identification of the precise rate-limiting step. Related work with Ni, Rh, and Pt complexes has shown that oxidative addition with these metals proceeds via a concerted oxidative addition mechanism. The formed complexes show similar characteristics; (silyl)PtII(H) complexes exhibit square planar geometry and are proposed to undergo fluxional exchange of the ligands via transient η2-H–SiR3 species.61 This fluxional behaviour is also proposed for related Ni complexes.89–92 A large variety of Rh complexes have been studied for oxidative addition with silanes,33–48 and the mechanism has been elucidated in detail as well.46
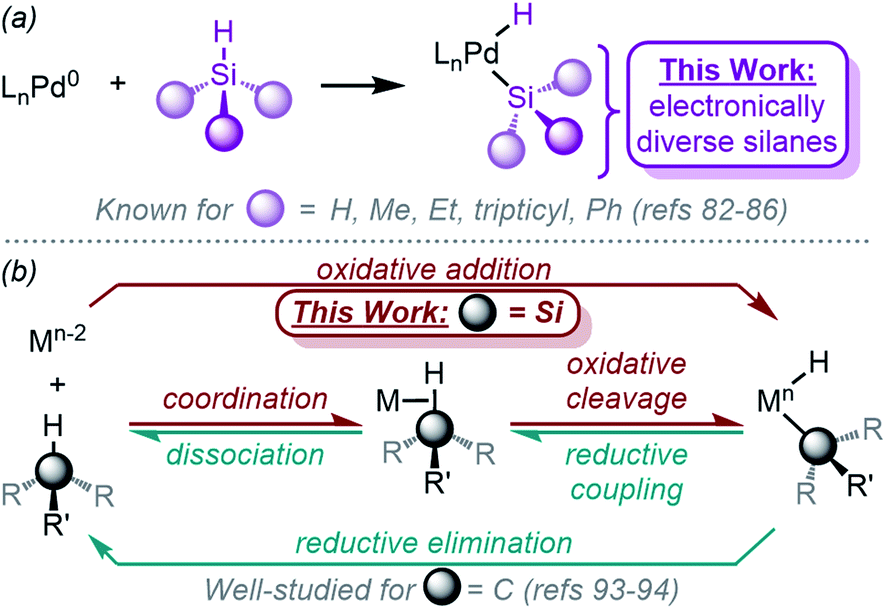 | ||
| Fig. 2 (a) Previously examined silanes for oxidative addition with Pd, and (b) mechanistic relationship between oxidative addition of Si–H and reductive elimination of Si–H and C–H bonds. | ||
Elucidation of the details of the reaction mechanism is also relevant in the context of the oxidative addition of C–H bonds, since Si–H bonds often react analogously; however, Si–H bonds are more basic and therefore more reactive with transition metals. Taking advantage of this reactivity difference, studying Si–H oxidative addition can offer unique insight into the oxidative addition of C–H bonds. One important challenge in studying the mechanism of oxidative addition of non-polar bonds is decoupling the individual steps of the oxidative addition process, which is composed of coordination and oxidative cleavage steps (Fig. 2b). Examining Si–H oxidative addition reactions offers a chance to address that challenge. Ground-breaking work by Jones93 and Parkin94 has investigated the reverse of oxidative addition: reductive elimination of C–H bonds using KIEs and EIEs as tools to decouple the reductive cleavage and dissociation steps.
Lastly, the known (silyl)Pd(H) compounds have yet to be shown to be relevant to catalysis. In this report, we fill in the crucial gaps in knowledge regarding the (i) effect of silane electronics on the thermodynamics and kinetics, (ii) overall reaction mechanism, and (iii) relevance to catalysis (namely hydrosilylation) of silane oxidative addition. Using a variety of mechanistic tools ranging from van't Hoff and Eyring analyses to competition experiments to rate and order studies, we discuss the overall reaction mechanism and the contribution of silane electronics to reactivity. These results are then shown to be relevant in the hydrosilylation of alkynes and provide mechanistic rational behind observed changes in reactivity.
Results and discussion
Initial reactivity studies of [(μ-dcpe)Pd]2 (1) with silanes
We began by investigating the reactivity of a library of tertiary silanes with [(μ-dcpe)Pd]2 (1) by 1H and 31P NMR (Fig. 3). These reactions proceed cleanly to afford a new product, as initially observed by NMR, and only the reactants and the product are observed. For silanes with at least one phenyl ring or alkoxide substituent (3–6), the reactions proceed in good to excellent conversions, but peralkyl-substituted silanes (2) are unreactive. | ||
| Fig. 3 Scope of silane reactivity in oxidative addition with [(μ-dcpe)Pd]2 (1). Percent conversions were determined by NMR integrations. Reaction conditions: 1.00 equiv. of 1, 2.05 equiv. of 2–6, 13.33 mM in C6D6, and 4–6 h. (a) Initial silane scope. (b) Scope of electronically diverse dimethylaryl silanes. (c) Scope of electronically diverse triaryl silanes. | ||
In the 1H NMR spectrum, the peak corresponding to silane Si–H (4–6 ppm) is consumed during the reaction, along with concomitant appearance of a new resonance below 0 ppm (−1.23 to −2.34 ppm), which is consistent with the Pd centre interacting with the silane Si–H bond to form a (silyl)Pd(H), a Pd(η2-H–SiR3) complex, or some structure in between these extremes.33–35,95,96 In addition to the 1H NMR signal, a new band in the IR spectrum (1838 to 1881 cm−1) is observed, consistent with known analogous Ni and Pd complexes.91,92,97 The hydride signal in the 1H NMR spectrum for most products appears as a triplet at room temperature with 2JH–P values of 75–80 Hz, which is the average of the cis and trans coupling constants. Only one signal in the 31P{1H} NMR spectrum is observed (55–60 ppm, corresponding to a downfield shift from 1 at 23 ppm), showing that there is only one phosphorus environment. At low temperatures, the hydride signal in the 1H NMR resolves into a doublet (2JH–P(trans) = ∼160 Hz), the 31P{1H} spectra show two signals between 57–64 ppm, and in the 1H-coupled 31P spectra, the upfield signal is a doublet due to trans coupling to the hydride. Values for 2JH–P(cis) were in most cases too small to measure (<8 Hz). These NMR data are consistent with a Pd–H complex that is undergoing dynamic exchange (vide infra), as has been shown for related complexes.61,82,89 The chemical shift and coupling patterns of the hydride and phosphorus signals align well with related Pd and Pt hydrides stabilized by bidentate phosphine ligands.54,63,82,83 To distinguish where our products lie on the spectrum from σ-complex to (silyl)Pd(H), the NMR spectra of the product from reaction of 1 and 5a (HSiPh3) were analysed in more detail. The hydride peak in the 1H NMR shifts from 5.71 ppm in 5a to −1.74 ppm upon coordination/oxidative addition to Pd and changes from a singlet with 29Si satellites (JH–Si = 198 Hz) to a triplet (2JH–P = 78 Hz) with 29Si satellites (JH–Si = 30 Hz; see ESI, Table S1†). In related Ni,89,91 and Pt,61 and Rh complexes,41,46,98,99 H–Si coupling constants were used to determine whether the complex is the result of oxidative addition to form the (silyl)M(H), or formation of the σ-complex: coupling constants smaller than 20 Hz support the formation of (silyl)M(H) and larger than ∼70 Hz support non-classical interactions.96 The JH–Si values of our complexes are in between these markers yet are closer to values known for (silyl)M(H) complexes of group 10 metals61,82,89,91,92 than for their σ-complexes.62,100–103 Therefore, our data support the formation of the (silyl)Pd(H) complexes, as drawn in Fig. 3.
A comparison of the data shown in Fig. 3 reveals that higher conversions are obtained with increasing number of aryl substituents and alkoxide substituents on the silane. This trend correlates well with the electronegativity of the atoms attached to Si (11 > 10a > 9 > 8a > 7).104 While this trend is revealing, we sought to decouple the kinetic effects from the thermodynamic effects and systematically investigate the electronic influence on each parameter of the reaction.
Electronic effects in oxidative addition of Pd(0) to silanes
To probe our hypothesis that the oxidative addition of silanes to Pd(0) is more facile with electron-poor silanes, a library of electronically diverse triarylsilanes were synthesized and evaluated in the reaction. The targeted silanes span a gradient of electronic character, with substituents in the para position ranging from electron-donating (X = NMe2, OMe, Me) to electron-withdrawing (X = F, CF3). Reactions of these silanes 5 with 1 cleanly yield the expected (dcpe)Pd(H)(SiAr3) complexes 10a–f (Fig. 3c), all in excellent NMR conversions except for the reaction with the electron-rich 4-dimethylaminophenyl-substituted silane (5b → 10b, mass balance is remaining starting material).A translucent, off-white crystal of (dcpe)Pd(H)(Si(4-CF3-Ph)3) (10f) suitable for single crystal diffraction was grown from a reaction mixture of 1 and HSi(4-CF3-Ph)3 (5f) in benzene/hexane (Fig. 4). Product 10f displays nearly ideal square planar geometry around the palladium atom, with the summed total of bond angles equal to 360°. To get a more precise measure of the structure's geometry, the structural parameter τ4 was calculated from crystallographic data.105,106 Analysis of 10f gives τ4 = 0.15, where τ4 = 0 indicates an ideally square planar complex, and τ4 = 1 represents a tetrahedral complex. The τ4 value of 10f can be compared to similar complexes whose characterization includes the Pd–H bond length (allowing calculation of τ4): Ishii's (dcpe)Pd(H)(SiH2Trip) (Trip = 9-triptycyl)84 complex gives τ4 = 0.11 and Iluc's (P–P)Pd(H)(SiHPh2) complex gives τ4 = 0.39 (Table S44†).8610f is therefore comparable to known square planar silyl palladium hydride complexes. Deviation from square planarity is likely due to steric repulsion between the cyclohexyl substituents of dcpe and the aryl groups of the silane.
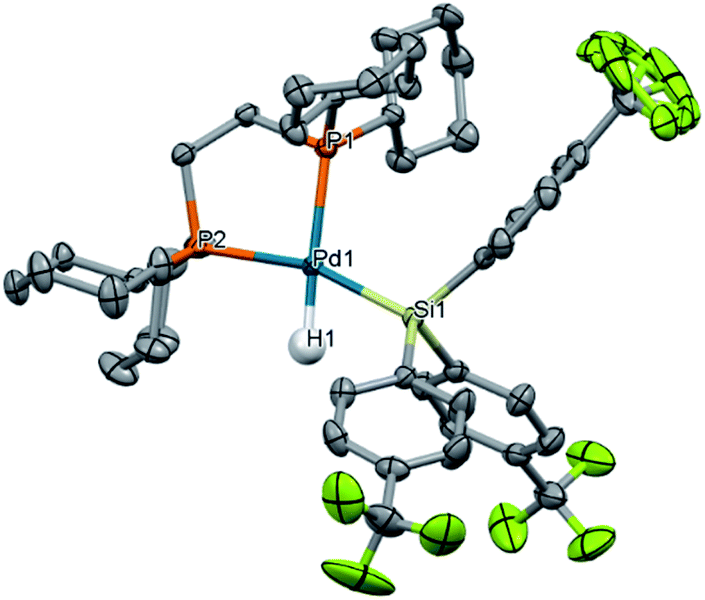 | ||
| Fig. 4 Molecular structure of (dcpe)Pd(H)(Si(4-CF3-Ph)3) (10f). Thermal ellipsoids are drawn at 50% probability level. Hydrogen atoms except for Pd–H are omitted for clarity. Selected bond lengths (Å) and angles (deg): Pd1–H1 1.59(3), Pd1–Si1 2.3413(5), Pd1–P1 2.3422(5), Pd1–P2 2.3584(5), H1–Pd1–Si1 67(1), H1–Pd1–P2 96(1), P1–Pd1–P2 86.46(2), P1–Pd1–Si1 111.01(2). One CF3 group is highly disordered and was refined as being located in three positions with an occupation factor of 0.33 each. | ||
Solid-state geometry also gives information on the strength of each ligand's trans influence. Silyl ligands are known for being among the strongest trans-influencing ligands.107–109 Therefore, we expect that the Pd–P bond trans to the silyl ligand will be longer than the Pd–P bond trans to the H; indeed, this is the case for complex 10f, which has Pd–P bond lengths of 2.3584(5) Å and 2.3422(5) Å, respectively.
Comparing the Pd–P bond lengths in related complexes gives insight into the trans-influencing ability of the 4-CF3-phenyl substituents of the silane. The Pd–P bond length trans to the silyl ligand in complex 10a (ref. 82) and Ishii's complex84 are 2.350(2) Å and 2.3319(14) Å, respectively. With no change in ancillary ligand (dcpe) across all three cases, these results indicate that the –Si(4-CF3–Ph)3 group has a stronger trans influence than the –SiPh3 group, which in turn is stronger than –SiH2Trip.
Returning to our hypothesis on the effect of electronic character of the silane on oxidative addition, no useful distinctions could be made using the data in Fig. 3c, as most triaryl silanes exhibit similar reactivity. We thus turned to less reactive dimethylaryl silanes to further probe the influence of silane electronics on oxidative addition. The reaction of each dimethylaryl silane with 1 yielded the desired products (8a–f) (Fig. 3b), though to lower conversion than with triaryl silanes. The formed (silyl)Pd(H) complexes exhibit similar spectroscopic characteristics as described for their triaryl counterparts. The data in Fig. 3b show a clear trend with respect to the electronic nature of the silane: more electron-poor silanes give higher conversions to the silyl palladium hydrides 8.
Reaction equilibrium
To embark upon mechanistic studies, we first sought to determine if this oxidative addition reaction is reversible or irreversible. 1 and 3a were reacted in an NMR tube at 298 K and allowed to reach equilibrium (Fig. S10†). The reaction was then cooled to 253 K, and the conversion of 8a was measured to be 88%. Warming the reaction to 293 K and then 333 K resulted in concomitant decreases in conversion to 8a (44% and 17%, respectively). Subsequent cooling back to 298 K resulted in a conversion of 41%, which is consistent with the data in Fig. 3. Because the conversion is consistently ∼42% at ambient temperature independent of whether the reaction had previously been heated or cooled, we deemed this reaction to be reversible.We also tested reversibility by taking advantage of the differential reactivity of electron-rich and -poor silanes: if the reaction is indeed reversible, then we expect that a silyl palladium hydride formed from an electron-rich silane should convert to the corresponding product of oxidative addition of an electron-poor silane if one is added. This reaction is expected to occur through reductive elimination of the electron-rich silane, followed by preferential oxidative addition of the electron-poor silane. (dcpe)Pd(H)(Si(4-tolyl)3) (10d, formed in situ from 1 and 5d) was reacted with triphenyl silane (5a), and conversion to an equilibrium mixture of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.3 (10d/10a) was observed (Scheme 1). An analogous experiment with tris(4-fluorophenyl)silane (5e) added to a solution of pre-formed 10a shows an equilibrium ratio of 1:2.8 (10a/10e) (Table S16†). The formation of H2 or bis-silyl by-products was not observed, and mass balance is consistently 100% against an internal standard. These data imply that the formation of silyl palladium hydrides is completely reversible with respect to product distribution.
:3.3 (10d/10a) was observed (Scheme 1). An analogous experiment with tris(4-fluorophenyl)silane (5e) added to a solution of pre-formed 10a shows an equilibrium ratio of 1:2.8 (10a/10e) (Table S16†). The formation of H2 or bis-silyl by-products was not observed, and mass balance is consistently 100% against an internal standard. These data imply that the formation of silyl palladium hydrides is completely reversible with respect to product distribution.
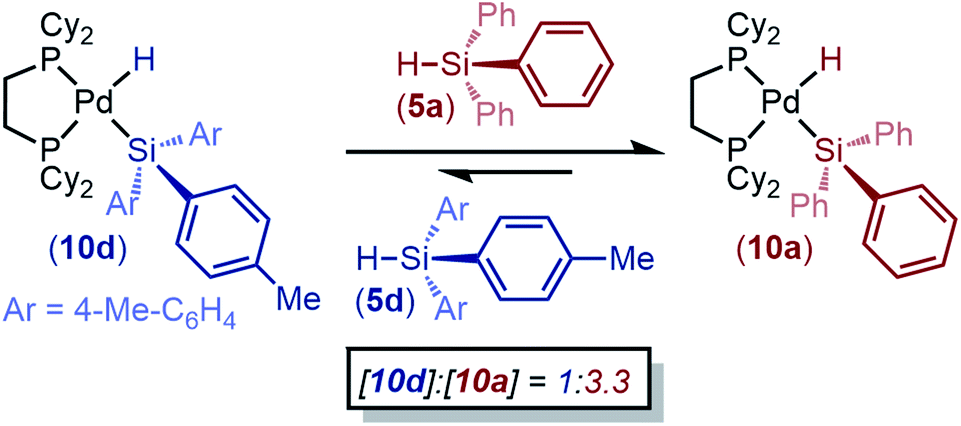 | ||
| Scheme 1 Conversion of 10d into 10avia reversible oxidative addition. Experiment was performed in C7D8 and relative conversions were determined by 31P{1H} NMR integration; Reaction conditions: 1.00 equiv. of 1, 2.05 equiv. each of 5a and 5d, 13.33 mM in C7D8, and 3 h. | ||
To quantify the trend imparted by silane electronic character, we measured the equilibrium constants (Keq) for the reactions of 1 with each silane 3a–f and constructed a Hammett plot. This analysis gave a sensitivity constant, ρeq (eq indicates that this value is derived from equilibrium constants, as opposed to rates of reaction) of 3.5 ± 0.5 (Fig. 5), consistent with the data in Fig. 3b showing that the reaction is more favourable with electron-poor silanes.
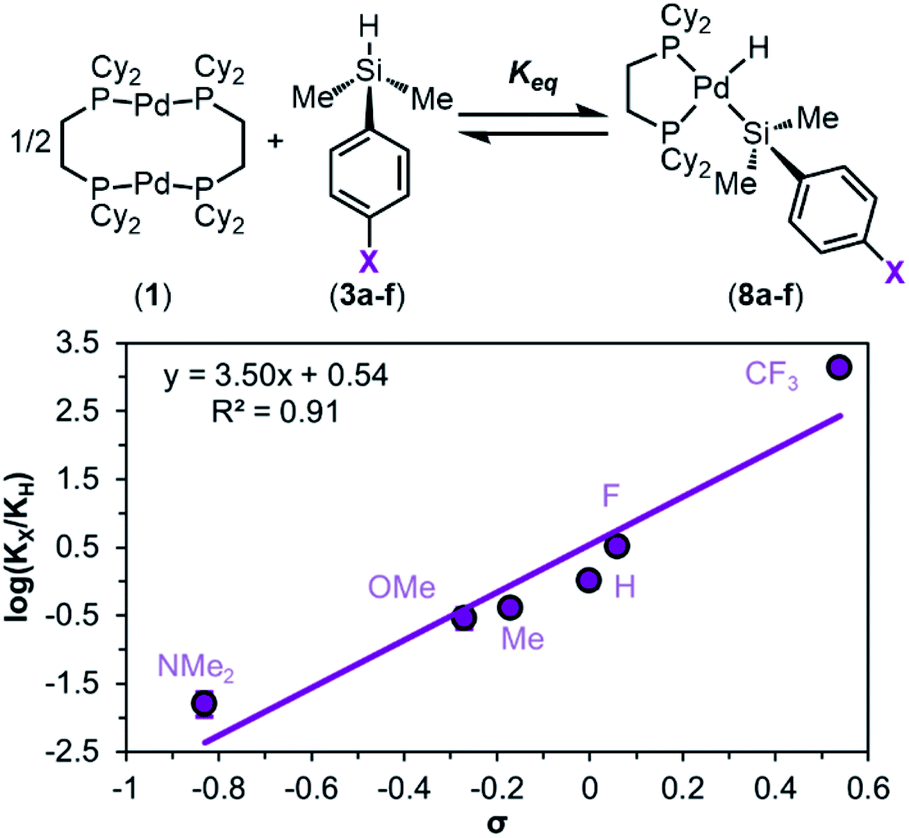 | ||
| Fig. 5 Hammett plot of the equilibrium constants for the reaction between dimethylaryl silanes 3 and 1. Keq values were determined using the NMR integrations of each product. Reaction conditions: 1.00 equiv. of 1, 2.05 equiv. of 3a–f, 13.33 mM in C6D6 at 298 K. | ||
We hypothesized that this observed trend in Keq, and thus ΔGOA (OA, oxidative addition), of the reaction is dictated by the varying bond strengths of the Pd–Si bonds, where stronger Pd–Si bonds are formed with electron-poor silanes. To deconvolute the enthalpic and entropic contributions to ΔGOA, the equilibrium constants as a function of temperature were measured. van't Hoff analysis was carried out (Fig. 6) for the reactions of 1 with 3a (X = H), 3c (X = OMe), and 3f (X = CF3). These particular silanes were chosen to represent a breadth of electronic character. The ΔSOA values are all large and negative, reflecting the loss of entropy in this reaction between three reactants to form two products. ΔHOA is negative for all three substrates, but more importantly, a clear trend is observed: 8f < 8a < 8c (CF3 < H < OMe) in ΔHOA. These data show that the reaction between 1 and silanes 3 is more favourable with electron-poor silanes, which we interpret to mean that there is an increased strength of the Pd–Si bond in products 8.87
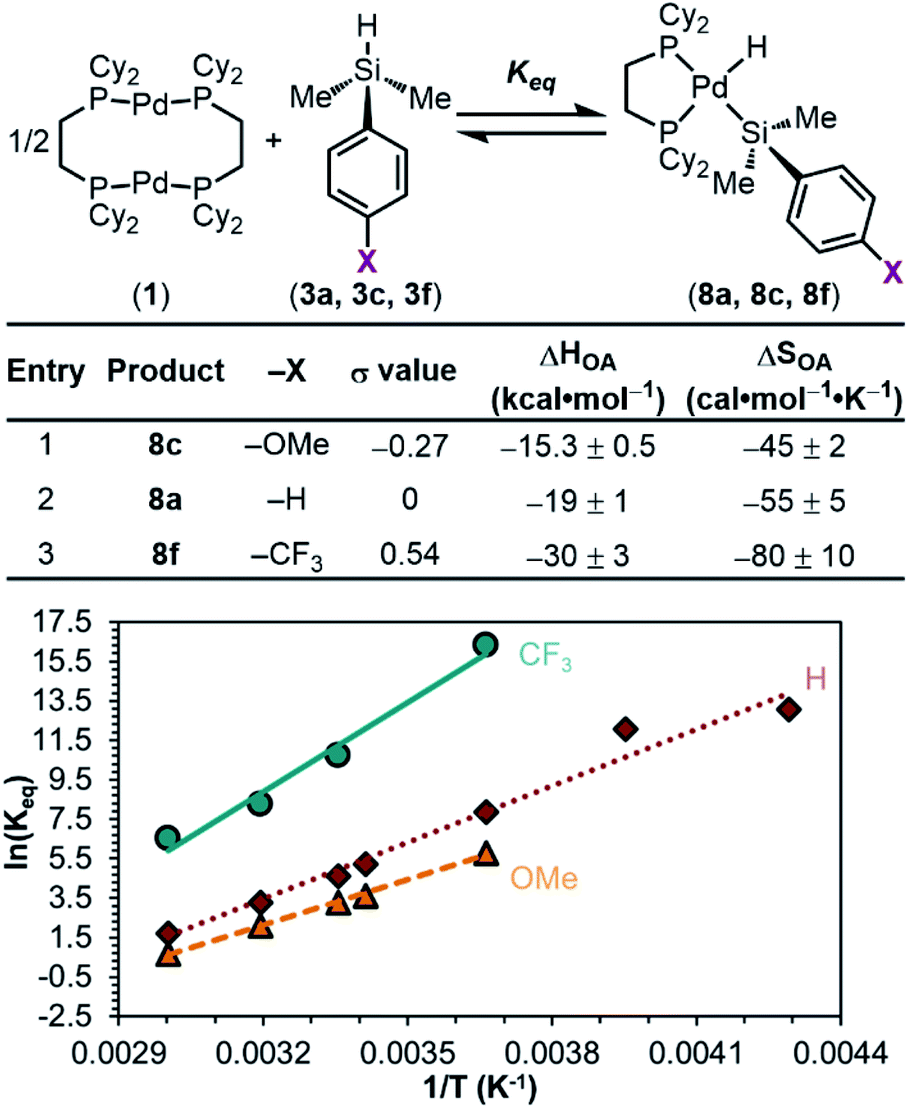 | ||
| Fig. 6 van't Hoff analyses of the reaction equilibrium between dimethylaryl silanes 3 and 1. Reaction conditions: 1.00 equiv. of 1, 2.05 equiv. of 3a, 3c or 3f, 13.33 mM in C7D8, 253–333 K. | ||
To determine if this trend in the electronic influence of silanes is more general, we returned to triaryl silanes 5. Because the equilibrium constants are too large to measure at room temperature under our standard conditions, we took advantage of the reaction's full reversibility in product distribution by performing competition experiments. In these experiments, 2.05 equivalents of each HSiPh3 (5a) and HSi(4-X–Ph)3 (5b–f, X = NMe2, OMe, Me, F, CF3) were added to one equivalent of 1 (Fig. 7).
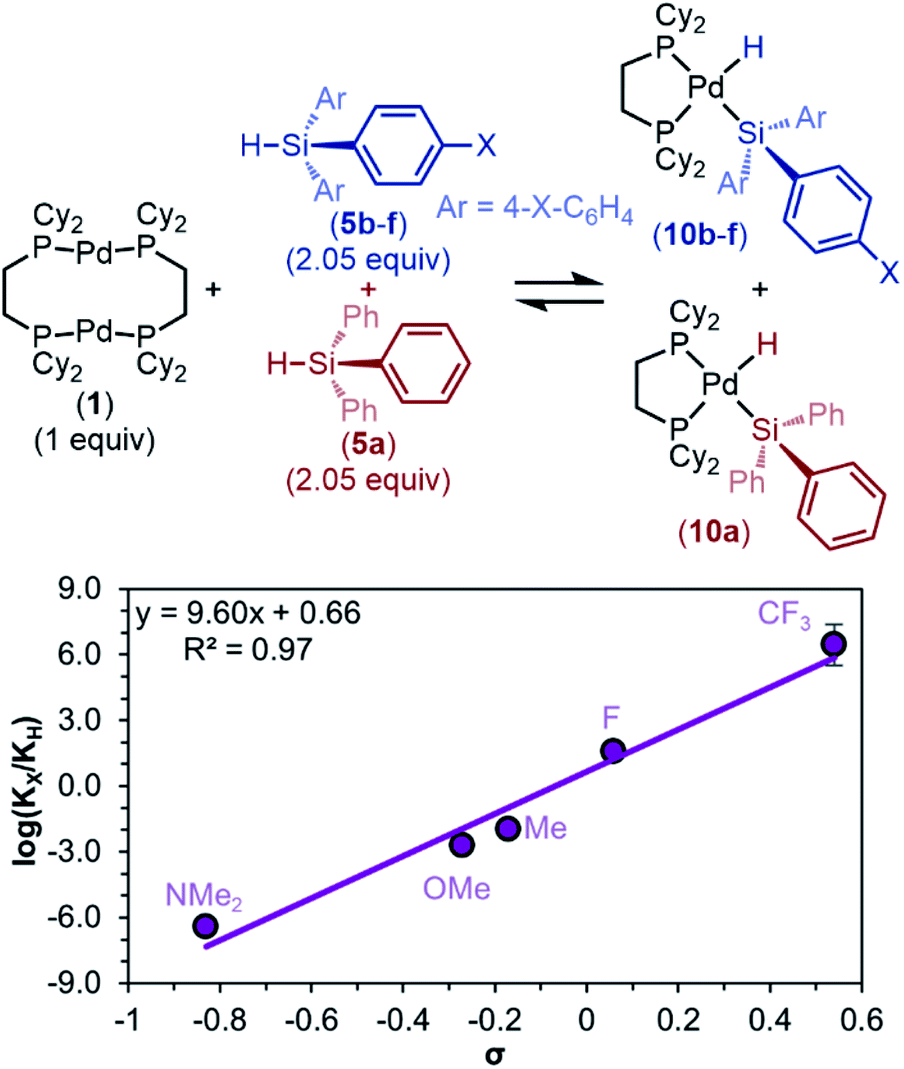 | ||
| Fig. 7 Hammett plot of competition experiments between 5a and electronically diverse silanes 5b–f for oxidative addition with 1. Reaction conditions: 1.00 equiv. of 1, 2.05 equiv. of 5a plus 2.05 equiv. of 5b–f, 13.33 mM in C6D6, 298 K. Hammett plot constructed from Keq values derived from conversions. | ||
As with the non-competitive oxidative addition reactions of dimethylaryl silanes 3a–f (Fig. 5), these data show that oxidative addition occurs preferentially with the more electron-poor silane. For example, when 5a (a comparatively electron-neutral silane) and 5e (an electron-poor silane) compete over the reaction with 1, oxidative addition preferentially occurs with 5e over 5a, forming 10e in larger concentrations than 10a. Because the relative ratios of products are proportional to the ratios of equilibrium constants (see ESI Section 5b† for derivation), a Hammett plot was constructed to quantify the effect of electronic factors on the equilibrium constant of oxidative addition (Fig. 7). For triaryl silanes, Hammett analysis gives ρeq(comp) = 9.6 ± 0.9, showing a more dramatic effect on Keq than monoaryl silanes (non-competitive equilibrium data in Fig. 5, ρeq = 3.5 ± 0.5). However, to allow for a direct comparison of the two systems, we additionally performed the same competition studies with dimethylaryl silanes 3a–f (Table S18 and Fig. S16†). ρeq(comp) was found to be 3.4 ± 0.3 with these dimethylaryl silanes. Notably, these data trend proportionally to the number of changing substituents at silicon.
To conclude studies on the reaction equilibrium, we sought to determine the effect of deuterium substitution in the silane. Reaction of 1 and 3a–d1 proceeded as expected, affording product 8a–d1; the 31P{1H} NMR spectrum at room temperature shows a 1:1:1 triplet (2JD–P = 11 Hz) in 40% conversion. As with 3a, the coupling constant is an average of the cis and trans coupling due to dynamic exchange behaviour (vide infra). The equilibrium constants for the reactions of proteo-silane 3a and deutero-silane 3a–d1 were determined independently (Table S11†). The equilibrium isotope effect (EIE) was determined to be 3.7 ± 0.2 at 298 K. The room temperature IR stretching frequencies for the Si–H and Si–D bonds in 3a and 3a–d1 are 2116 cm−1 and 1539 cm−1, respectively. The EIE measured for this reaction is consistent with the fact that deuterium prefers to form the stronger bond;94,110,111i.e., Keq(D) is lower than Keq(H) because the difference in energy between the Si–D and Pd–D bonds is larger than the difference in energy between the Si–H and Pd–H bonds.
Reaction kinetics
Having examined the ground state, we next turned our attention to understanding the kinetics and reaction mechanism. In probing the mechanism, three possible pathways for oxidative addition were considered: concerted, SN2-like, or radical. A concerted pathway was deemed most reasonable as Si–H bonds are non-polar and have been shown to proceed via concerted pathways in related systems. We ruled out the SN2-like pathway on the basis of relative acidities of Pd–H (pKa ≈ 24)112 and Si–H (pKa ≈ 35)113 bonds. Additionally, no by-products were observed that would result from a radical pathway (Si–Si bond coupling, H2, etc.), so we shifted our attention from that pathway as well.In line with a concerted pathway, the proposed mechanism for oxidative addition is seen in Fig. 8. Step i is dissociation of dimer 1 into monomers 12 in solution.114 The 14-electron complex 12 can then react with silane in step ii to form σ-complex 13, which then undergoes oxidative cleavage of the Si–H bond to form the product in step iii. These steps were taken as the basis for determination of the rate determining step (RDS) of the reaction.
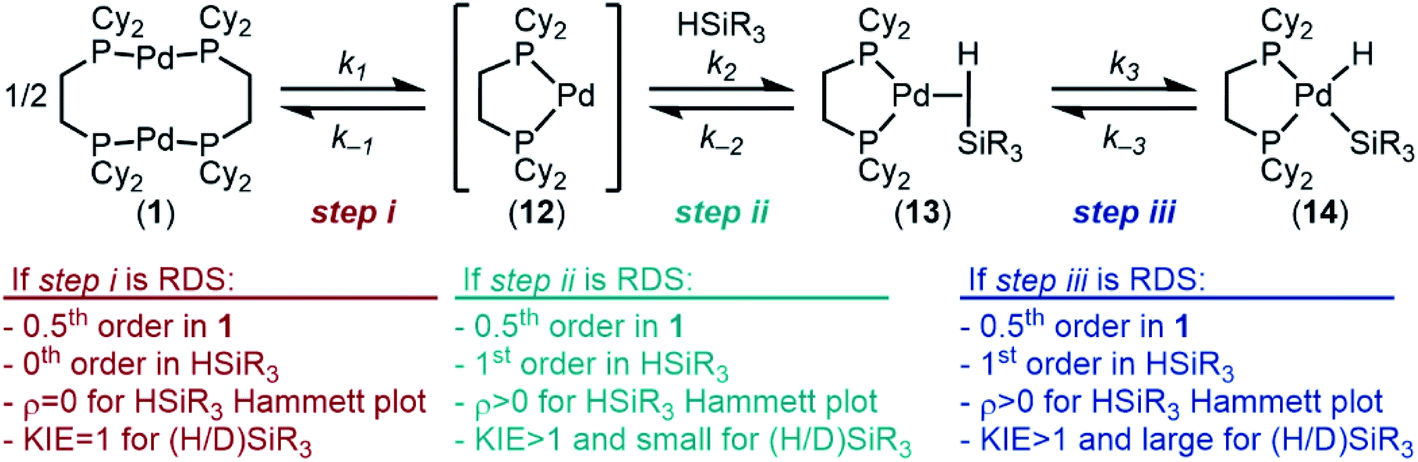 | ||
| Fig. 8 Proposed mechanism for the oxidative addition of silanes to 1. | ||
If step i is rate-determining, then the rate law will show 0.5th order in 1, 0th order in silane, and there will be no effect of electronic or deuterium substitution in the silane on the rate of the reaction (ρ‡ = 0 for a Hammett plot and KIE = 1, respectively (KIE, kinetic isotope effect)). If step ii or step iii is the RDS, then the reaction will be 0.5th order in 1 and 1st order in silane; electron poor-silanes are expected to react faster (ρ‡ > 0 in a Hammett plot); additionally, a normal, primary KIE is predicted. We expect that a small KIE would point toward step ii being the RDS, since the Si–H bond is only weakened in the transition state (12 → 13). A large, primary KIE would point toward step iii being the RDS, since it is during that step that the Si–H bond is cleaved.
Initial attempts to collect kinetic data by monitoring the reaction of 1 with 5a by NMR proved unsuccessful, since the reaction was complete within a minute at room temperature. We thus set out to determine conditions that are amenable to reaction monitoring by NMR; however, even at low reagent concentrations (1.67 mM 1, 3.42 mM 5a) and low temperature (233 K), the formation of 10a was too rapid to obtain reliable kinetic data. In line with our findings on reaction equilibria, we reasoned that Me2PhSiH 3a may also react more slowly in oxidative addition, yet still would have the advantage of allowing for electronic modulation via substitution on the aryl ring. Indeed, the gradual formation of a silyl palladium hydride 8a is observable via31P{1H} and 1H NMR at 233 K.
As detailed above (Fig. 8), the rate law distinguishes between step i and steps ii or iii being the RDS; therefore, we first embarked on measuring the orders in the reactants. We utilized the method of initial rates to ensure that the rate of the backward reaction (from 8 to 1 and 3) is negligible. Data was collected on samples of varied concentrations of 1 (1.67–13.3 mM) or 3a (3.42–27.3 mM) while the concentration of the unexamined reagent was kept constant. The observed rate of each reaction (Fig. S17 and S19†) was then plotted against initial reagent concentration to determine the reaction order.
As shown in Fig. 9, the reaction was determined to be 0.5th order (0.57 ± 0.09) in dimer 1. This result is consistent with any of the three steps in the mechanism of Fig. 8 being the RDS, and supports the hypothesis that the dimer 1 dissociates into two [(dcpe)Pd] fragments (12) prior to or during the RDS.
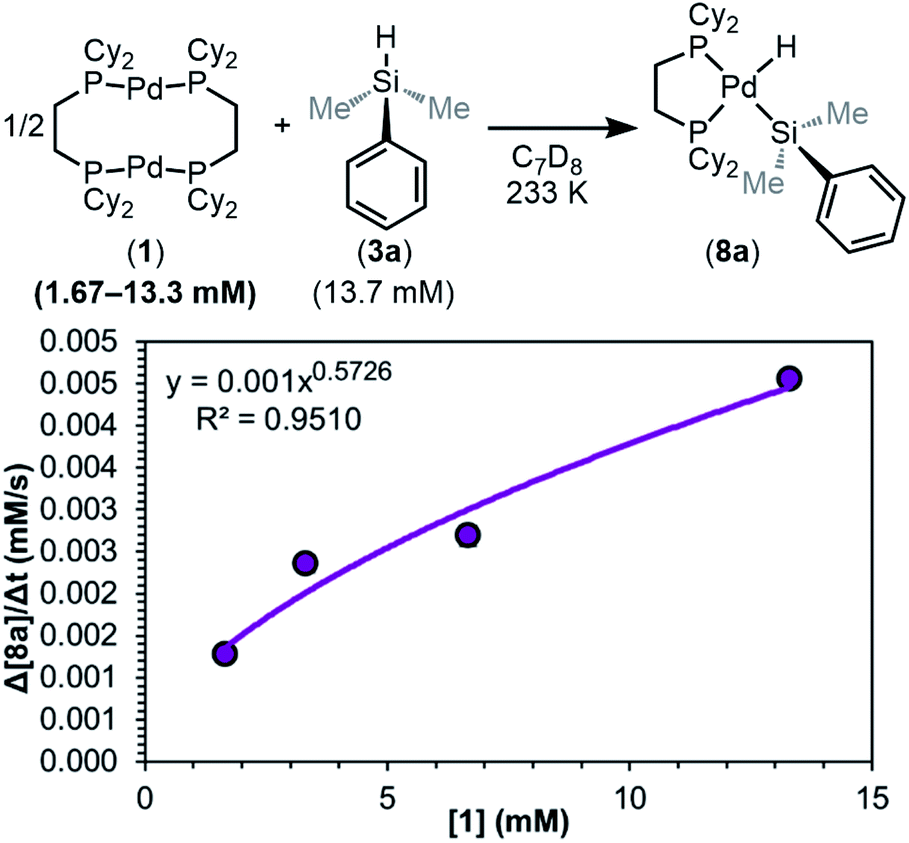 | ||
| Fig. 9 Plot of initial rate versus [1]. Kinetics data was collected in C7D8 as the solvent at 233 K. Product concentration was determined by 31P{1H} NMR integration against an internal standard. | ||
We next determined the order in silane (Fig. 10). A plot of the initial rate versus the concentration of 3a shows that the reaction is 1st order (0.9 ± 0.1) in silane. This suggests the silane is involved in or before the RDS, and thus dimer dissociation (step i) of the mechanism in Fig. 8 can be ruled out as the RDS. With these data, the simplified rate law of the reaction can be written (eqn (1)), showing an overall 1.5th order reaction.
| Rate = kobs[1]0.5[3a]1 | (1) |
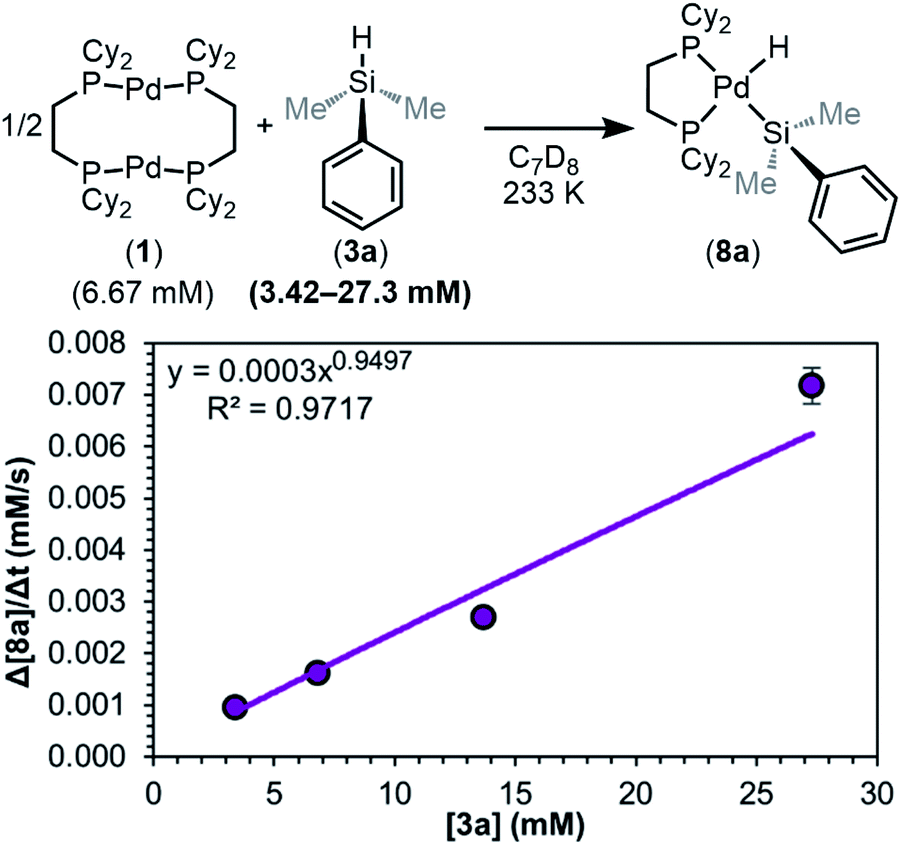 | ||
| Fig. 10 Plot of initial rate versus [3a]. Kinetics data was collected in C7D8 as the solvent at 233 K. Product concentration was determined by 31P{1H} NMR integration against an internal standard. | ||
Having ruled out dimer dissociation as the RDS in the mechanism, we next examined the effect of electronic substitution at silicon on rates of reaction via a Hammett analysis. Rates of reaction were measured using dimethylaryl silanes 3a, 3c, and 3f, and these experiments reveal a marked influence of silane electronics on the initial rates of product formation (Fig. S21–S24†). Analysis of kinetic trials for each silane gives a trend in kobs of X = CF3 > H > OMe, which follows the same trend as in equilibrium studies: strongly electron-withdrawing substituents at silicon favour rapid oxidative addition, while electron-donating substituents hinder the reaction rate.
A Hammett plot was constructed from these data (Fig. 11), which shows ρ‡ = 1.2 ± 0.3 (‡ notation is used to distinguish this sensitivity constant obtained from rates of reaction from the sensitivity constant obtained from reaction equilibrium constants (ρeq)). Comparing the ρ value obtained for the reaction equilibrium (ρeq = 3.5 ± 0.5) with that obtained for rates (ρ‡ = 1.2 ± 0.3) reveals that the observed rate of oxidative addition is approximately three times less sensitive to changes in electronics as the equilibrium constant (Keq). The result of this Hammett analysis provides further evidence that either silane coordination (Fig. 8, step ii) or Si–H oxidative cleavage (Fig. 8, step iii) is the RDS of oxidative addition.
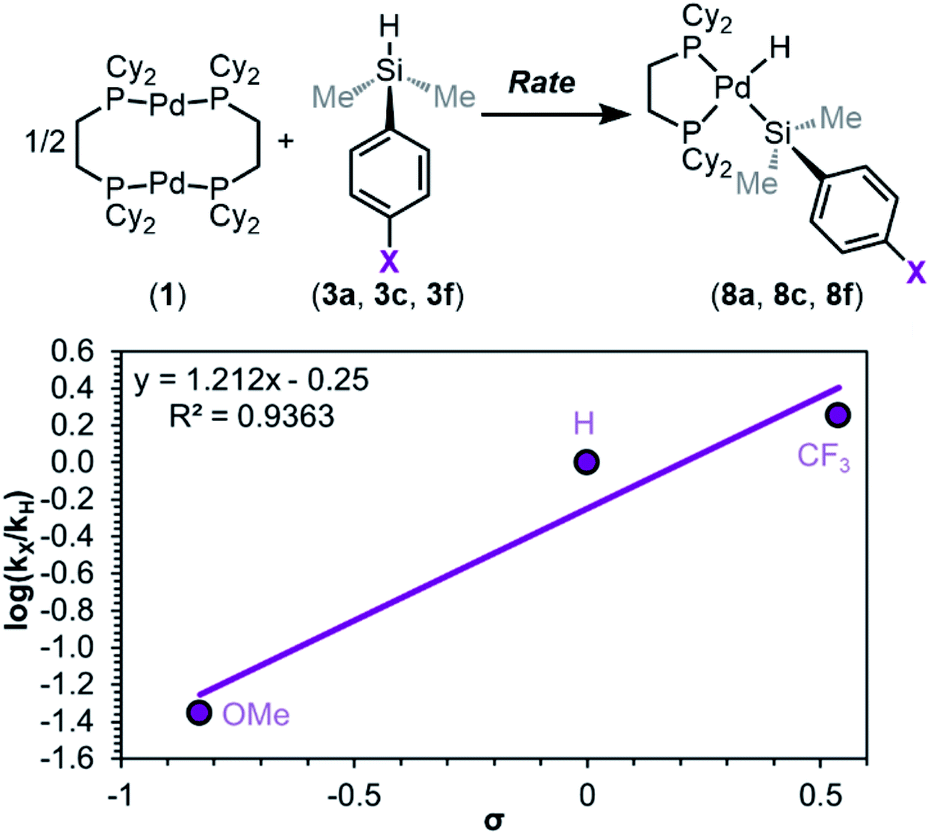 | ||
| Fig. 11 Hammett plot constructed from kinetic data. Kinetic data was collected in C7D8 (13.33 mM) as the solvent at 233 K. Product concentration was determined by 31P{1H} NMR integration against an internal standard; 1.00 equiv. of 1 and 2.05 equiv. of 3a, 3c, or 3f were used. | ||
To help differentiate between these two steps being the RDS, the KIE of the reaction was determined. Recognizing the convoluted nature of KIEs and EIEs,93,94,115,116 we continued using the method of initial rates to discount any equilibria involving reaction of the Si–H bond, and therefore measured rates of reaction are approximately independent of equilibrium effects. The initial rate of reaction for Me2PhSiD (3a–d1) was measured, and it was found to react more slowly than 3a, with a KIE of 1.21 ± 0.04 (Fig. 12). The maximum primary KIE possible for Si–H/Si–D without invoking tunnelling is ∼4 (see ESI, Section 9f† for details). Therefore, a KIE of 1.21 is large enough to be considered a primary isotope effect, but the magnitude is small and challenging to interpret. Either step ii is the RDS, in which a smaller magnitude KIE is rationalized by the Si–H bond not being formally broken in the transition state; or step iii is the RDS, in which a very early transition state (which is expected due to the high energy of the σ-complex) would lead to a lower magnitude KIE. Notably, the KIE reported here is consistent with those of other oxidative addition studies and catalytic reactions involving Si–H and C–H bond cleavage.117
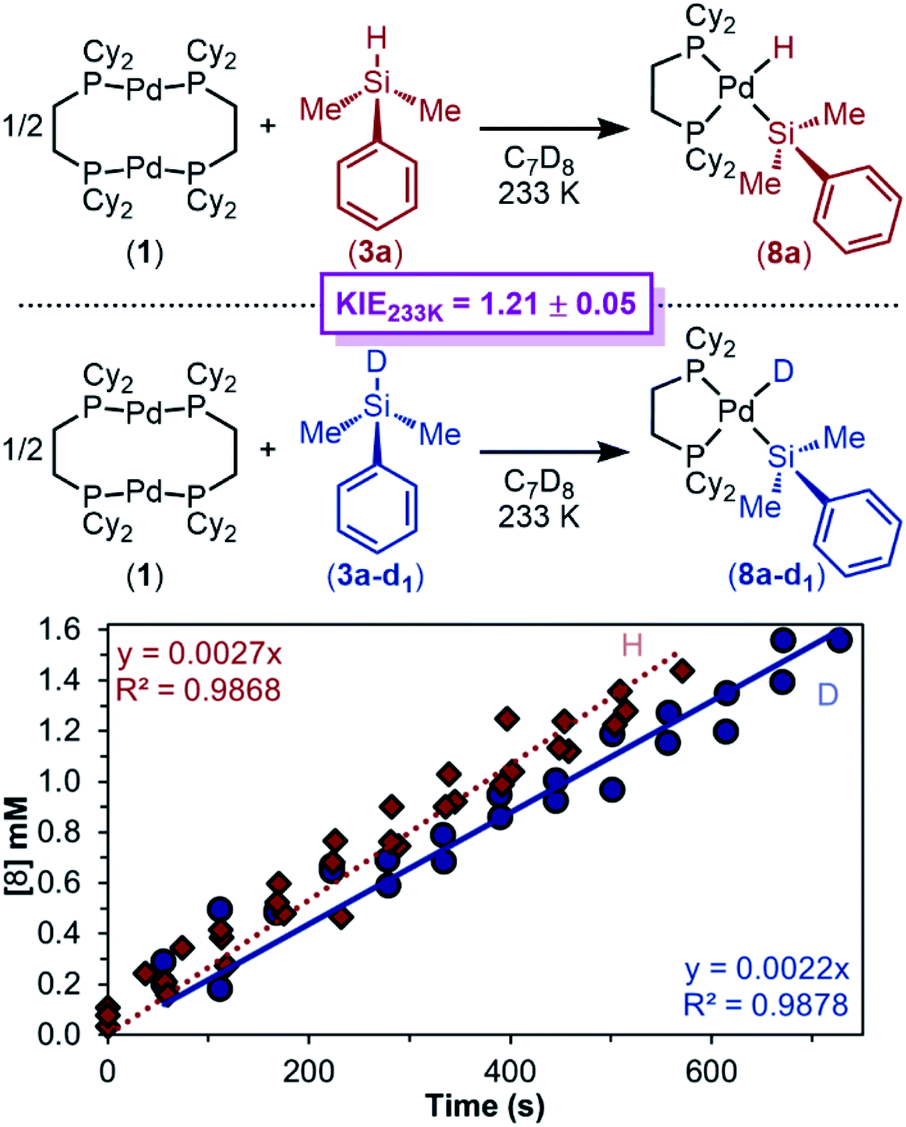 | ||
| Fig. 12 Plot of [8a] or [8a–d1] versus time. Kinetics data was collected in C7D8 (13.33 mM) as the solvent at 233 K. Product concentration was determined by 31P{1H} NMR integration against an internal standard; 1.00 equiv. of 1 and 2.05 equiv. of 3a or 3a–d1 were used. | ||
We next sought to measure the barrier (ΔG‡OA) for the overall reaction by performing an Eyring analysis. The rate of reaction was measured as a function of temperature, and the results are seen in Fig. 13. ΔH‡OA and ΔS‡OA are obtained from the equation of the fit of the data, and as a result, ΔG‡OA at a given temperature can be calculated. From the data in Fig. 13, ΔH‡OA was determined to be 6.0 ± 0.3 kcal mol−1, ΔS‡OA was determined to be −40 ± 20 cal mol−1 K−1, and therefore ΔG‡OA at 233 K was calculated to be 16.3 ± 0.3 kcal mol−1. This large, negative value for ΔS‡OA supports the proposed mechanism in Fig. 8. The transition state of the RDS is highly ordered relative to the ground state, consistent with step ii or iii being the RDS. These kinetic data have been compiled, and the observed rate constants (kobs) have been determined using the rate law in eqn (1) (Table 1).
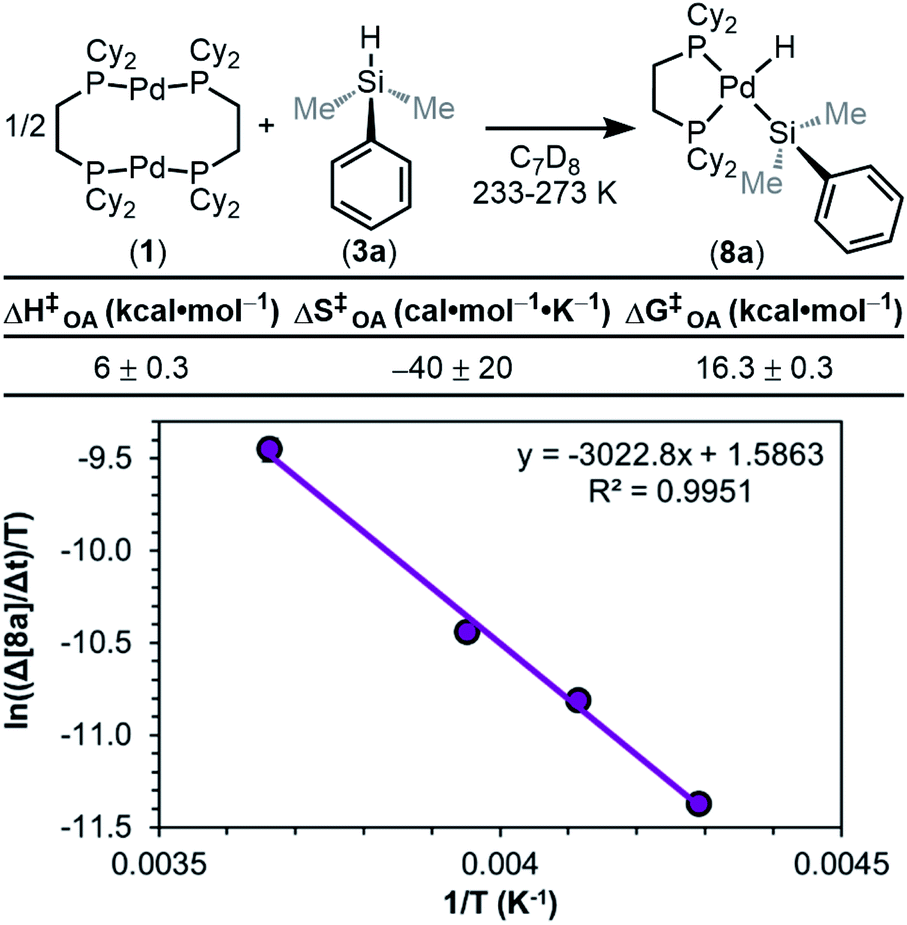 | ||
| Fig. 13 Eyring plot constructed from variable temperature kinetics data in C7D8 (13.33 mM). Product concentration was determined by 31P{1H} NMR integration against an internal standard; 1.00 equiv. of 1 and 2.05 equiv. of 3a was used. ΔG‡OA determined at 233 K. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Pd complex | –H/D | –X | Temperature | k obs (mM0.5 s−1) |
| a k obs for the formation of 8a at 233 K was determined from eight trials, and the value reported is the average (standard deviation is 1 × 10−5). | |||||
| 1 | 8a | –H | –H | 233 K | 9 × 10−5 |
| 2 | 8a | –H | –H | 243 K | 1.4 × 10−4 |
| 3 | 8a | –H | –H | 253 K | 2.1 × 10−4 |
| 4 | 8a | –H | –H | 273 K | 5.9 × 10−4 |
| 5 | 8a-d1 | –D | –H | 233 K | 3.2 × 10−4 |
| 6 | 8c | –H | –OMe | 233 K | 3.3 × 10−6 |
| 7 | 8f | –H | –CF3 | 233 K | 1.4 × 10−4 |
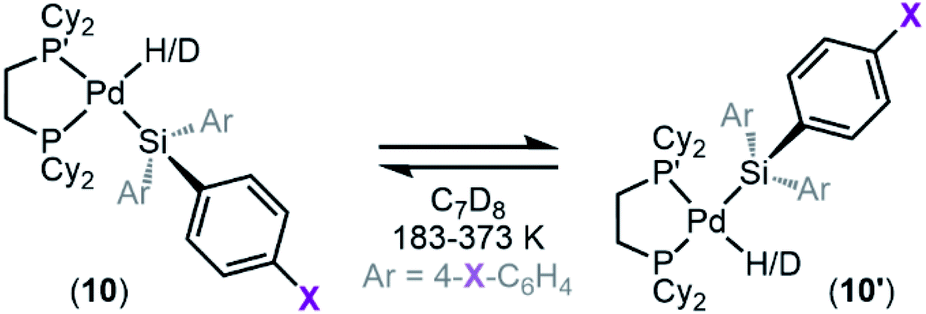
Dynamic exchange behaviour
One interesting feature of these silyl metal hydride complexes is their fluxional behaviour in solution.33–35,118,119 We recognized that investigating this dynamic exchange (DE) behaviour would provide insight into the overall reaction mechanism of oxidative addition because it offers a unique chance to probe the oxidative cleavage and reductive coupling steps (k3 and k−3, respectively, in Fig. 8) independent of the overall oxidative addition process. Accordingly, solution 1H and 31P{1H} NMR spectra of product 8a were recorded at multiple temperatures (Fig. 14). At very low temperatures, the 31P{1H} NMR spectrum shows two singlets (δ 58.4 and 57.4 ppm at 183 K) which undergo coalescence at 213 K, eventually forming a sharp singlet (δ 56.4 ppm at 273 K (Fig. 14a). Similar behaviour is seen for the Pd–H peak in the 1H NMR spectra at low temperatures; a doublet of doublets (δ −1.39 ppm, 2JH–P(trans) = 192.6 Hz, 2JH–P(cis) = 7.9 Hz at 183 K) proceeds through coalescence at 213 K to eventually form an apparent triplet, centring at −1.71 ppm, 2JH–P = 75.4 Hz at 273 K) (Fig. 14b). Line broadening analysis120,121 allows for the calculation of the barriers for this dynamic exchange (DE) process. ΔH‡DE, ΔS‡DE, and ΔG‡DE at 233 K were determined to be 7.7 ± 0.4 kcal mol−1, −10.7 ± 0.5 cal mol−1 K, and 10.2 ± 0.4 kcal mol−1, respectively.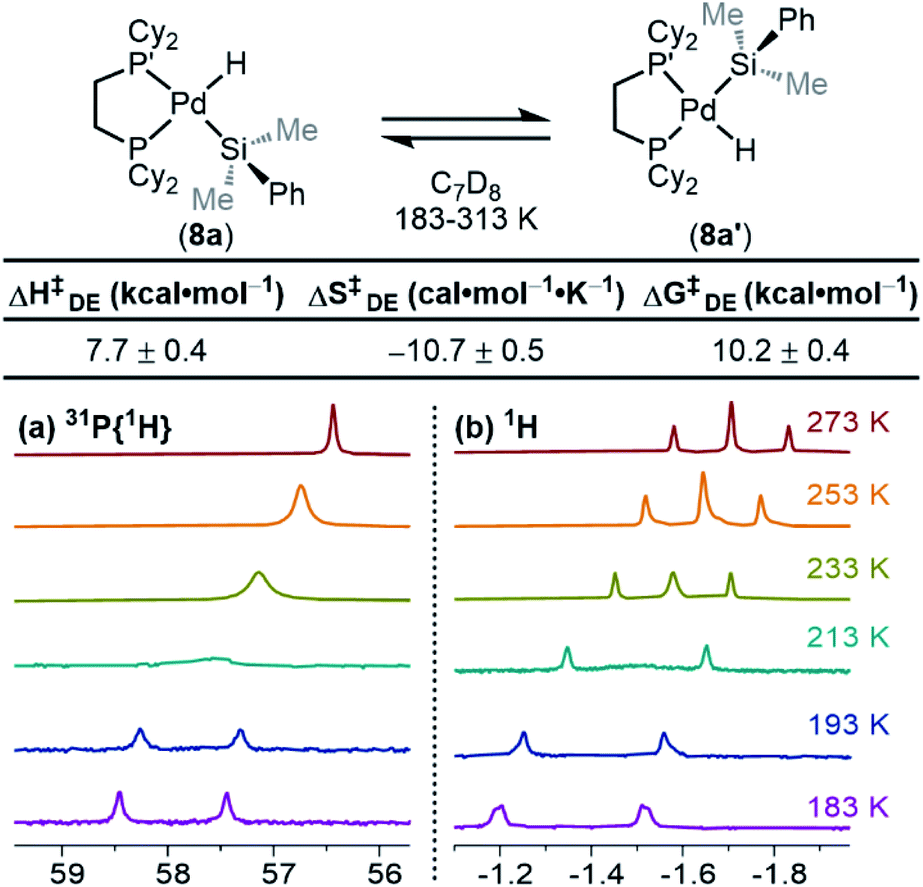 | ||
| Fig. 14 (a) 31P{1H} NMR spectra and (b) 1H NMR spectra of the Pd–H region of 8a/8a′ as a function of temperature with the measured barriers for dynamic exchange (DE). Spectra collected in C7D8 (13.33 mM). ΔG‡DE determined at 233 K. | ||
In analogy to reported complexes and their dynamic exchange behaviour,61,82–84 we propose a mechanism in Scheme 2 that accounts for these NMR spectra. 8a and 8a′ are proposed to interconvert via the σ-complexes 13a and 13a′, which themselves interconvert by rotation around the Pd–(η2-H–SiMe2Ph) bond (k4 and k−4). Conversion of 8a into 13a and 8a′ into 13a′ are reductive coupling steps (k−3), the microscopic reverse of oxidative cleavage (k3).
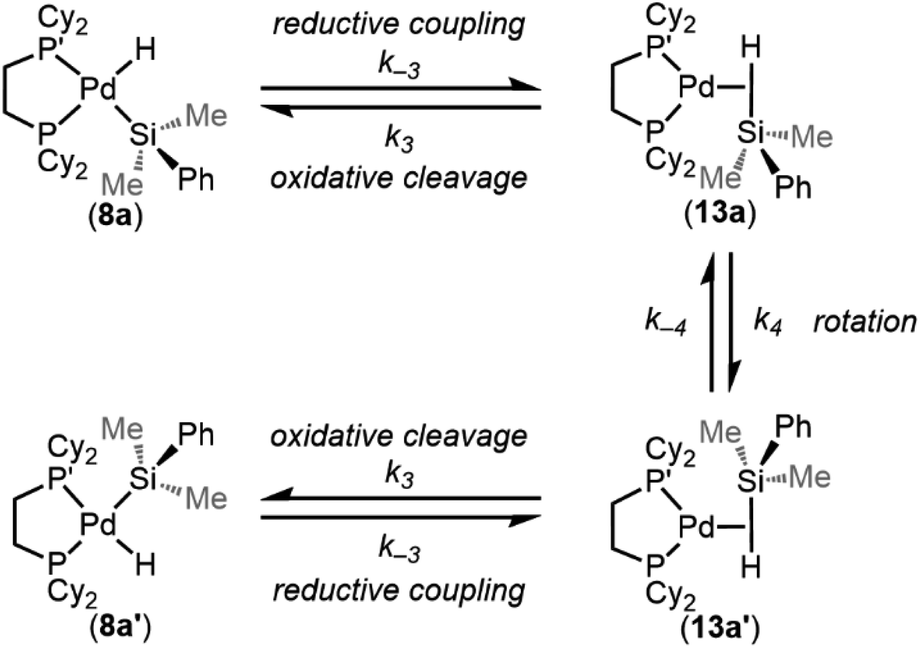 | ||
| Scheme 2 Proposed mechanism for the intramolecular dynamic exchange of –H and –SiMe2Ph environments. | ||
At elevated temperatures, exchange is rapid on the NMR timescale, and the Pd–H signal in the 1H NMR appears as a triplet due to coupling to both P nuclei, and the coupling constant is an average of the trans and the cis2JH–P values. This interpretation is supported by the 31P{1H} NMR data, which shows a singlet at elevated temperatures. Upon cooling, the Pd–H signal (1H NMR) undergoes coalescence and gradually resolves into a doublet of doublets at sufficiently low temperatures. The splitting pattern is accounted for by the inequivalency of the P nuclei; at low temperature in the 31P{1H} NMR, two distinct phosphorous environments are observed, as exchange is sufficiently slowed and both P nuclei environments (P and P′ in Scheme 2) are discrete. The tunability of this process from slow-exchange to fast-exchange over an accessible temperature range indicates a relatively small energy barrier for conversion between species, and is consistent with the measured barrier of 10.2 ± 0.4 kcal mol−1 at 233 K.
Important for the overall oxidative addition reaction (formation of 8a from 1 and 3a), the RDS of this dynamic exchange process is either the barrier for reductive coupling/oxidative cleavage (k−3/k3) or the barrier for rotation (k4/k−4). Therefore, the oxidative cleavage step (k3) must be equal to or less than 10.2 ± 0.4 kcal mol−1 at 233 K.
Taking advantage of the line broadening observed for additional silyl palladium hydrides, we sought to investigate the effect of silane substitution on the barriers for DE. We focused on triaryl silanes 5 because not all products with dimethylaryl silanes 3 reached the slow exchange regime at temperatures accessible in toluene-d8. The activation parameters (Table 2) show that there is no significant effect of electronic or isotopic substitution on the rate of dynamic exchange for triaryl silanes since the measured barriers are mostly within error of one another. ΔH‡ varies from 10–14 kcal mol−1, showing a trend of more electron-poor silanes having higher enthalpic barriers than electron-rich silanes, and ΔS‡ is close to zero. In this dynamic exchange process, ΔH‡ and ΔS‡ are opposing, and ΔG‡ for products 10a, 10c, and 10f are within error of one another, resulting in a net null electronic effect. Furthermore, the barriers for this dynamic exchange process for 10a–d1 and 10a are also within error, implying a KIE of approximately 1 (within error). Measuring the rates of dynamic exchange at varying concentrations of 10f gave similar values; these data along with small values of ΔS‡ support our hypothesis that the process is intramolecular. These results point toward rotation (k4, Scheme 2) being the RDS of dynamic exchange since, if reductive coupling/oxidative cleavage were the RDS, a measurable electronic effect and KIE would be expected. Notably, this conclusion has also been drawn for silyl-Rh complexes,61 and values are similar to known dynamic exchange processes of silyl Ni, Pd, Pt, and Rh complexes.46,61,82,92
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Pd complex | –H/D | –X | ΔH‡DE | ΔS‡DE | ΔG‡DE |
| 1 | 10a | –H | –H | 12 + 1 | 3.5 + 0.3 | 11 + 1 |
| 2 | 10a-d1 | –D | –H | 10.0 + 0.4 | −2.46 + 0.09 | 10.6 + 0.4 |
| 3 | 10c | –H | –OMe | 10.1 + 0.4 | −2.80 + 0.09 | 10.7 ± 0.4 |
| 4 | 10f | –H | –CF3 | 13.7 + 0.5 | 5.5 ± 0.2 | 12.5 ± 0.5 |
Constructing a reaction coordinate diagram
Taking our findings from equilibrium and kinetic studies, we have constructed a complete mechanistic picture for the oxidative addition of silanes to 1. With the bulk of the data obtained using dimethylphenyl silane 3a, we focus on those results in the discussion below (Fig. 15), but we expect the general trends to be analogous for the other silanes studied in this work.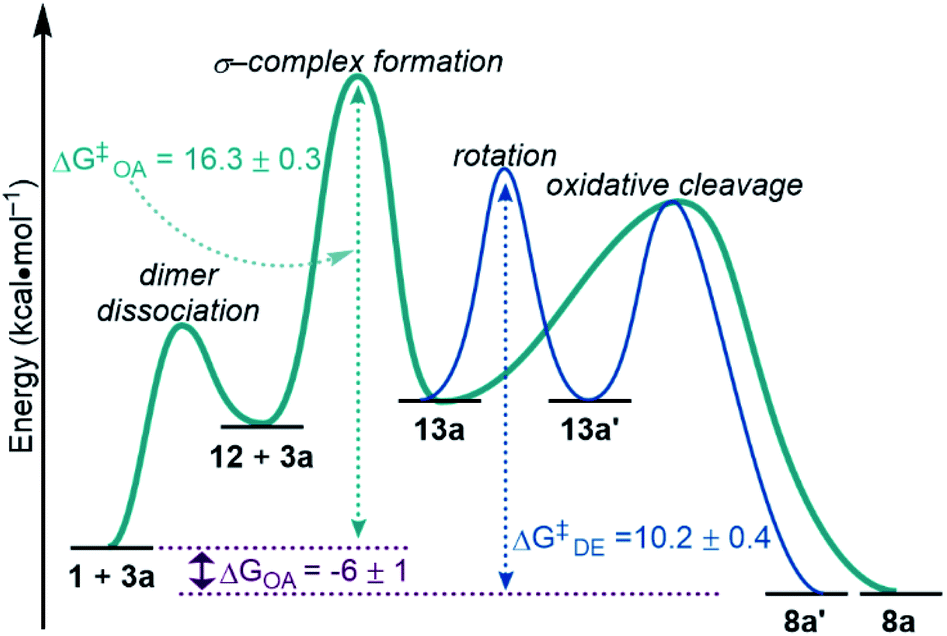 | ||
| Fig. 15 Reaction coordinate diagram with experimentally determined free energy values at 233 K. OA, oxidative addition. DE, dynamic exchange. | ||
ΔGOA was determined in the course of studying the reaction equilibrium. Keq for the reaction between 3a and 1 at 233 K was determined to be 125, corresponding to ΔGOA = −6 ± 1 kcal mol−1 (Fig. 15, purple). Therefore, the silyl palladium hydride 8a is lower in energy by 6 kcal mol−1 than the starting materials 1 and 3a. This energy difference is larger for electron-poor silanes than electron-rich silanes, as shown via Hammett plots (Fig. 5, 7 and S16†).
The reaction was found to be 1st order in silane 3a (Fig. 10) and 0.5th order in palladium dimer 1 (Fig. 9). Because there is a dependence on the silane on the rate of reaction, dimer dissociation (1 → 12) is ruled out as the RDS. Therefore, either formation of the σ-complex 13a or oxidative cleavage of the Si–H bond must be the RDS, since both would account for the observed orders in reactants. Hammett analysis of the dependence of the rate of reaction on the electronic influence of the silane (Fig. 11) also does not differentiate between σ-complex formation and oxidative cleavage being the RDS, since the barriers for both steps are expected to be lowered with electron-poor silanes, in line with the observed trends. Based on the small, but primary KIE of 1.21 ± 0.04 (Fig. 12), we hypothesized that formation of the σ-complex 13a is the RDS, since cleavage of the Si–H bond is expected to result in a larger KIE value. However, this is not definitive evidence.
The reaction barrier for the full oxidative addition process (1 + 3a → 8a) was determined to be 16.3 ± 0.3 kcal mol−1 by an Eyring analysis (Fig. 13). By linewidth analysis of the 31P{1H} NMR spectra of product 8a, we determined that the barrier for the dynamic exchange process between 8a and 8a′via13a/13a′ is 10.2 ± 0.4 kcal mol−1 (Fig. 14). Because no significant KIE or electronic effect was observed for the dynamic exchange process, we concluded that the RDS of dynamic exchange is likely rotation. Therefore, the maximum barrier of reductive coupling (the microscopic reverse of oxidative cleavage, or 8a → 13a) is equal to or lower than 10 kcal mol−1. If oxidative cleavage (13a → 8a) was the RDS, then the maximum overall reaction barrier for oxidative addition would be the sum of ΔGOA (−6 ± 1 kcal mol−1) and ΔG‡DE (10.2 ± 0.4 kcal mol−1). This sum (4 kcal mol−1) is much less than the measured free energy barrier for oxidative addition (16.3 ± 0.3 kcal mol−1). Therefore, formation of the σ-complex (step ii in Fig. 8) is deduced to be the RDS. As a consequence of step ii being the RDS, equilibria involving the silane and reaction of the Si–D/H bond are not included in the rate law, and the KIE can be considered separately from the EIE. Because of the importance in oxidative addition reactions in catalysis, we expect these findings to affect the design of silanes and catalysts for many catalytic reactions.
Application to catalysis
Having fully elucidated the mechanism of the oxidative addition of silanes to palladium(0), we next demonstrate the relevance of this work to catalysis. Hydrosilylation was our first target because of its importance and industrial relevance. Subjecting complex 1 to HSiPh3 (5a) and phenylacetylene (15) in toluene resulted in 66% yield of hydrosilylation products. Two regioisomers were observed: the branched product (α-16a) and the linear E-alkene (E-β-16a) in a 1:8 ratio. These results are consistent with related Pd(0)-catalysed hydrosilylation reactions.122,123 Notably, the effect of silane electronics on alkyne hydrosilylation has not been investigated in the literature. When compared to the neutral silane 5a, the rate of the reaction is faster with the electron-poor silane 5f and slower with the electron-rich silane 5d (Fig. 16). Reaction with 5f gives products α-16f and E-β-16f in 98% yield in a 1:34 ratio, and reaction with 5d gives products α-16d and E-β-16d in 40% yield in a 1:6 ratio. These results directly parallel the data obtained from oxidative addition and highlight the importance of studying this fundamental reaction in organometallic chemistry. Furthermore, using the isolated complex 10a as a catalyst in 5 mol% leads to a reaction profile nearly identical to that generated from 1/5a mixtures (see ESI Section 11†), further supporting the relevance of this oxidative addition to catalysis.
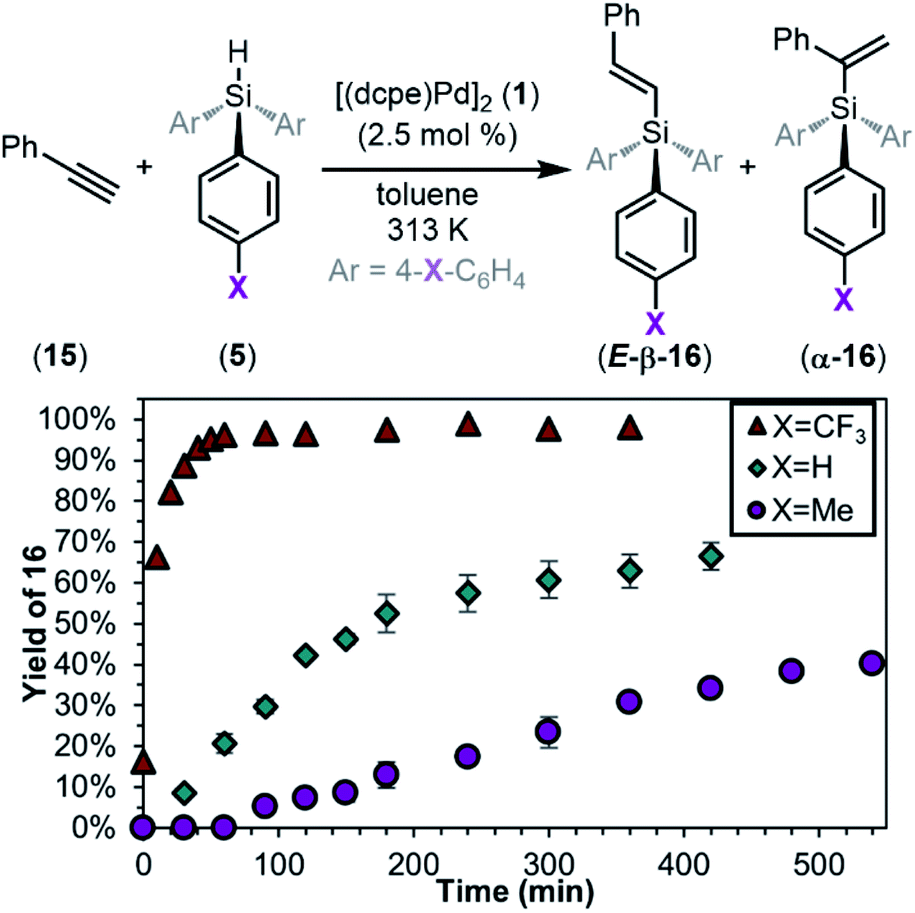 | ||
| Fig. 16
1-Catalysed hydrosilylation of phenylacetylene and silanes 5. Reaction conditions: 15 (1 equiv.), 5 (1 equiv.), 1 (2.5 mol%), 0.04 M in toluene, 313 K. Yields are the sum of the yields of both products (E-β-16 + α-16) determined by GCMS using a calibration curve against 1,3,5-trimethoxybenzene. Selectivities of E-β-16 + α-16 are as follows: E-β-16f/α-16f = 34:1 (X = CF3); E-β-16a/α-16a = 8:1 (X = H); E-β-16d/α-16d = 6:1 (X = Me). | ||
Conclusions
In this work, we have elucidated the mechanism of oxidative addition of Pd(0) to Si–H bonds and shown the relevance of this reaction in catalytic hydrosilylation. We evaluated the impact of silane electronic influence in the oxidative addition of [(μ-dcpe)Pd]2 to form well-defined, mononuclear silyl palladium hydrides. A library of electronically varied silanes have been synthesized and the corresponding silyl palladium hydrides characterized, including via single crystal diffraction of 10f. Extensive equilibrium and competition studies point to a pronounced effect of silane electronics in the oxidative addition reaction, quantified via Hammett analysis to show the reaction with triaryl silanes 5 to be approximately three times more sensitive to electronics than with dimethylaryl silanes 3. Formation of (silyl)Pt(H) and (silyl)Rh(H) complexes via silane oxidative addition have shown a similar trend of being more favourable with electron-withdrawing substituents at silicon.44,49,61 van't Hoff analysis suggests that this trend arises from the corresponding Pd–Si bond strength in products 10 and 8. Our conclusion that Pd–Si bond strength is related to the identity of the silicon substituents is supported by computational work of related Pd complexes.87 The formed silyl palladium hydrides exist in a dynamic equilibrium with 1 that is fully reversible with regard to both temperature and product distribution, which is similar to behaviour of (silyl)Pt(H) complexes.50 Kinetic studies further support the favourability of electron-poor silanes and show that the rate of reaction (kobs) is less sensitive to electronics than the equilibrium constant (Keq).Further, this system is well-suited to answer long-standing questions in the oxidative addition of palladium to hydrosilanes via mechanism elucidation. The reaction is 0.5th order in starting dimer 1 and 1st order in silanes. The reaction barrier was determined to be 16.3 kcal mol−1 using Eyring analysis. This energy barrier far exceeds that of oxidative cleavage of the Si–H bond, (determined to be equal to or less than 10 kcal mol−1). The upper limit of the barrier for oxidative cleavage was determined by studying the dynamic behaviour of the (silyl)Pd(H) complexes on the NMR timescale. Analogous behaviour has been observed with (silyl)Pt(H) complexes, which was shown to be an intramolecular process involving reductive coupling of the hydride and silyl ligands.59,61,124,125 The barriers for dynamic exchange of these Pt complexes are approximately the same as the barriers reported here for (silyl)Pd(H) complexes.61 In conjunction with a small, primary KIE, we conclude that the RDS of oxidative addition is the formation of the σ-complex, not Si–H bond cleavage.
Lastly, we showed the relevance of our findings to catalysis, namely hydrosilylation. Electronic variation of the silane in catalysis directly parallels that of oxidative addition, which further supports the relevance and importance of in-depth mechanistic studies to catalysis.
Data availability
All data collected for this article, including experimental procedures, compound characterization, conversion values and graphical data, and NMR and IR spectra are available in the ESI.†Author contributions
M. R. H. and A. K. C. are responsible for conceptualization, investigation, and manuscript writing. M. R. H. carried out all experimentation except crystallography. L. N. Z. performed all crystallography experimentation, data collection, and analysis. A. K. C. is responsible for supervision and project administration. All authors edited and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Energy and Sustainable Materials Cluster and the Office of the Vice President of Research at the University of Oregon. The authors would like to acknowledge contributions from Dr Nanette Jarenwattanon (NMR experiment preparation and analysis).Notes and references
- V. V. Grushin, Hydrido Complexes of Palladium, Chem. Rev., 1996, 96, 2011–2034 CrossRef CAS.
- K. K. M. Hii, in Handbook of Organopalladium Chemistry for Organic Synthesis, ed. N. Ei-ichi, 2003, pp. 81–90 Search PubMed.
- R. F. Heck, Palladium-catalyzed reactions of organic halides with olefins, Acc. Chem. Res., 1979, 12, 146–151 CrossRef CAS.
- I. P. Beletskaya and A. V. Cheprakov, The Heck Reaction as a Sharpening Stone of Palladium Catalysis, Chem. Rev., 2000, 100, 3009–3066 CrossRef CAS PubMed.
- I. D. Hills and G. C. Fu, Elucidating Reactivity Differences in Palladium-Catalyzed Coupling Processes: The Chemistry of Palladium Hydrides, J. Am. Chem. Soc., 2004, 126, 13178–13179 CrossRef CAS PubMed.
- M. S. Sigman and D. R. Jensen, Ligand-Modulated Palladium-Catalyzed Aerobic Alcohol Oxidations, Acc. Chem. Res., 2006, 39, 221–229 CrossRef CAS PubMed.
- D. Wang, A. B. Weinstein, P. B. White and S. S. Stahl, Ligand-Promoted Palladium-Catalyzed Aerobic Oxidation Reactions, Chem. Rev., 2018, 118, 2636–2679 CrossRef CAS PubMed.
- S. E. Mann, L. Benhamou and T. D. Sheppard, Palladium(II)-Catalysed Oxidation of Alkenes, Synthesis, 2015, 47, 3079–3117 CrossRef CAS.
- J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science, 2010 Search PubMed.
- G. Cavinato and L. Toniolo, Carbonylation of Ethene Catalysed by Pd(II)-Phosphine Complexes, Molecules, 2014, 19, 15116–15161 CrossRef PubMed.
- Z. Chen and M. Brookhart, Exploring Ethylene/Polar Vinyl Monomer Copolymerizations Using Ni and Pd α-Diimine Catalysts, Acc. Chem. Res., 2018, 51, 1831–1839 CrossRef CAS.
- E. Larionov, H. Li and C. Mazet, Well-defined transition metal hydrides in catalytic isomerizations, Chem. Commun., 2014, 50, 9816–9826 RSC.
- J. J. Molloy, T. Morack and R. Gilmour, Positional and Geometrical Isomerisation of Alkenes: The Pinnacle of Atom Economy, Angew. Chem., Int. Ed., 2019, 58, 13654–13664 CrossRef CAS.
- B. M. Trost, Palladium-catalyzed cycloisomerizations of enynes and related reactions, Acc. Chem. Res., 1990, 23, 34–42 CrossRef CAS.
- H.-H. Moretto, M. Schulze and G. Wagner, in Ullmann's Encyclopedia of Industrial Chemistry, 2012 Search PubMed.
- R. Y. Lukin, A. M. Kuchkaev, A. V. Sukhov, G. E. Bekmukhamedov and D. G. Yakhvarov, Platinum-Catalyzed Hydrosilylation in Polymer Chemistry, Polymers, 2020, 12, 2174 CrossRef CAS.
- S. B. J. Kan, R. D. Lewis, K. Chen and F. H. Arnold, Directed evolution of cytochrome c for carbon–silicon bond formation: Bringing silicon to life, Science, 2016, 354, 1048–1051 CrossRef CAS PubMed.
- A. K. Franz and S. O. Wilson, Organosilicon Molecules with Medicinal Applications, J. Med. Chem., 2013, 56, 388–405 CrossRef CAS PubMed.
- E. Rémond, C. Martin, J. Martinez and F. Cavelier, Silicon-Containing Amino Acids: Synthetic Aspects, Conformational Studies, and Applications to Bioactive Peptides, Chem. Rev., 2016, 116, 11654–11684 CrossRef PubMed.
- G. A. Showell and J. S. Mills, Chemistry challenges in lead optimization: silicon isosteres in drug discovery, Drug Discovery Today, 2003, 8, 551–556 CrossRef CAS.
- B. Marciniec, Hydrosilylation, Springer, 2009 Search PubMed.
- B. Marciniec, Catalysis by transition metal complexes of alkene silylation–recent progress and mechanistic implications, Coord. Chem. Rev., 2005, 249, 2374–2390 CrossRef CAS.
- D. Troegel and J. Stohrer, Recent advances and actual challenges in late transition metal catalyzed hydrosilylation of olefins from an industrial point of view, Coord. Chem. Rev., 2011, 255, 1440–1459 CrossRef CAS.
- Y. Nakajima and S. Shimada, Hydrosilylation reaction of olefins: recent advances and perspectives, RSC Adv., 2015, 5, 20603–20616 RSC.
- A. K. Roy, A Review of Recent Progress in Catalyzed Homogeneous Hydrosilation (Hydrosilylation), Adv. Organomet. Chem., 2007, 55, 1–59 CrossRef.
- T. K. Meister, K. Riener, P. Gigler, J. Stohrer, W. A. Herrmann and F. E. Kühn, Platinum Catalysis Revisited—Unraveling Principles of Catalytic Olefin Hydrosilylation, ACS Catal., 2016, 6, 1274–1284 CrossRef CAS.
- K. A. Horn, Regio- and Stereochemical Aspects of the Palladium-Catalyzed Reactions of Silanes, Chem. Rev., 1995, 95, 1317–1350 CrossRef CAS.
- M. Zaranek and P. Pawluc, Markovnikov Hydrosilylation of Alkenes: How an Oddity Becomes the Goal, ACS Catal., 2018, 8, 9865–9876 CrossRef CAS.
- T. Hayashi, Chiral Monodentate Phosphine Ligand MOP for Transition-Metal-Catalyzed Asymmetric Reactions, Acc. Chem. Res., 2000, 33, 354–362 CrossRef CAS.
- S. E. Gibson and M. Rudd, The Role of Secondary Interactions in the Asymmetric Palladium-Catalysed Hydrosilylation of Olefins with Monophosphane Ligands, Adv. Synth. Catal., 2007, 349, 781–795 CrossRef CAS.
- J. W. Han and T. Hayashi, Palladium-catalyzed asymmetric hydrosilylation of styrenes with trichlorosilane, Tetrahedron: Asymmetry, 2014, 25, 479–484 CrossRef CAS.
- L. F. Tietze, H. Ila and H. P. Bell, Enantioselective Palladium-Catalyzed Transformations, Chem. Rev., 2004, 104, 3453–3516 CrossRef CAS PubMed.
- J. Y. Corey and J. Braddock-Wilking, Reactions of Hydrosilanes with Transition-Metal Complexes: Formation of Stable Transition-Metal Silyl Compounds, Chem. Rev., 1999, 99, 175–292 CrossRef CAS PubMed.
- J. Y. Corey, Reactions of Hydrosilanes with Transition Metal Complexes and Characterization of the Products, Chem. Rev., 2011, 111, 863–1071 CrossRef CAS PubMed.
- J. Y. Corey, Reactions of Hydrosilanes with Transition Metal Complexes, Chem. Rev., 2016, 116, 11291–11435 CrossRef CAS PubMed.
- F. de Charentenay, J. A. Osborn and G. Wilkinson, Interaction of silanes with tris(triphenylphosphine)chlororhodium(I) and other rhodium complexes; hydrosilation of hex-1-ene by use of trichlorosilane, J. Turk. Chem. Soc., Sect. A, 1968, 787–790 RSC.
- R. N. Haszeldine, R. V. Parish and D. J. Parry, Organosilicon chemistry. Part V. Rhodium(III)–silyl complexes and the hydrosilation of hex-1-ene, J. Chem. Soc. A, 1969, 683–690 RSC.
- M. J. Fernandez, P. M. Bailey, P. O. Bentz, J. S. Ricci, T. F. Koetzle and P. M. Maitlis, Synthesis, x-ray, and low-temperature neutron-diffraction study of a rhodium(V) complex: dihydridobis(triethylsilyl)(pentamethylcyclopentadienyl)rhodium, J. Am. Chem. Soc., 1984, 106, 5458–5463 CrossRef CAS.
- M. Brockmann, H. t. Dieck and J. Klaus, Rhodium(I)-mono- und -diazadienkomplexe, synthese, spektroskopische charakterisierung, oxidative additionsreaktionen und einsatz in der homogenen katalyse zur hydrosilylierung, J. Organomet. Chem., 1986, 301, 209–226 CrossRef CAS.
- J. Ruiz, P. O. Bentz, B. E. Mann, C. M. Spencer, B. F. Taylor and P. M. Maitlis, The formation and characterisation of (η2-ethene)hydrido(η5-pentamethylcyclopentadienyl)(trisubstituted-silyl)rhodium complexes; intermediates in catalytic dehydrogenative silylation reactions, J. Chem. Soc., Dalton Trans., 1987, 2709–2713 RSC.
- S. B. Duckett, D. M. Haddleton, S. A. Jackson, R. N. Perutz, M. Poliakoff and R. K. Upmacis, Photochemical oxidative addition reactions of (.eta.5-cyclopentadienyl)bis(ethene)rhodium with dihydrogen and trialkylsilanes: formation and isolation of rhodium(III) and rhodium(V) hydrides, Organometallics, 1988, 7, 1526–1532 CrossRef CAS.
- D. P. Drolet and A. J. Lees, Solution photochemistry of (.eta.5-C5R5)Rh(CO)2 (R = H, Me) complexes: pathways for photosubstitution and C-H/Si-H bond activation reactions, J. Am. Chem. Soc., 1992, 114, 4186–4194 CrossRef CAS.
- M. Aizenberg and D. Milstein, Catalytic Activation of Carbon-Fluorine Bonds by a Soluble Transition Metal Complex, Science, 1994, 265, 359–361 CrossRef CAS PubMed.
- K. Osakada, S. Sarai, T.-a. Koizumi and T. Yamamoto, Structure and Chemical Properties of Chlorohydrido(diarylsilyl)rhodium(III) Complexes, mer-RhCl(H)(SiHAr2)(PMe3)3. Thermally Induced Chloro Transfer from Rhodium to Silicon in the Complexes and Silane Exchange, Organometallics, 1997, 16, 3973–3980 CrossRef CAS.
- M. Okazaki, S. Ohshitanai, H. Tobita and H. Ogino, Synthesis, structure, and reactivity of {(2-phosphinoethyl)silyl}rhodium(i) complexes Rh[(κ2Si,P)-Me2Si(CH2)2PPh2](PMe3)n (n = 2, 3), J. Chem. Soc., Dalton Trans., 2002, 2061–2068 RSC.
- K. A. M. Ampt, S. B. Duckett and R. N. Perutz, Photochemical reactions of (η5-cyclopentadienyl)bis(t-butylacrylate) rhodium with silanes: Dynamics of isomer interconversion via Rh(η2-silane) species, Dalton Trans., 2004, 3331–3337 RSC.
- E. Calimano and T. D. Tilley, Synthesis and reactivity of rhodium and iridium alkene, alkyl and silyl complexes supported by a phenyl-substituted PNP pincer ligand, Dalton Trans., 2010, 39, 9250–9263 RSC.
- M. A. Esteruelas, M. Oliván and A. Vélez, POP-Pincer Silyl Complexes of Group 9: Rhodium versus Iridium, Inorg. Chem., 2013, 52, 12108–12119 CrossRef CAS.
- H. C. Clark and M. J. Hampden-Smith, Ligand Interactioins in crowded molecules, Coord. Chem. Rev., 1987, 79, 229–255 CrossRef CAS.
- C. Eaborn, B. Ratcliff and A. Pidcock, The preparation of hydrido(silyl)bis(triphenylphosphine)platinum(II) complexes from bis(triphenylphosphine)platinum-ethylene and silicon hydrides, J. Organomet. Chem., 1974, 65, 181–186 CrossRef CAS.
- E. A. V. Ebsworth, V. M. Marganian, F. J. S. Reed and R. O. Gould, Platinum hydrides containing silyl or germyl ligands. Crystal structure of trans-hydridosilylbis(tricyclohexylphosphine)platinum(II), J. Chem. Soc., Dalton Trans., 1978, 1167–1170 RSC.
- R. S. Paonessa, A. L. Prignano and W. C. Trogler, Photochemical generation of bis(phosphine)palladium and bis(phosphine)platinum equivalents, Organometallics, 1985, 4, 647–657 CrossRef CAS.
- D. L. Packett, A. Syed and W. C. Trogler, Associative reactions of dihydridobis(trimethylphosphine)platinum(II). Molecular structures of (diphenylacetylene)bis(trimethylphosphine)platinum and hydridotris(trimethylphosphine)platinum(II) tetraphenylborate, Organometallics, 1988, 7, 159–166 CrossRef CAS.
- D. F. Mullica, E. L. Sappenfield and M. J. Hampden-Smith, Synthesis and X-ray structural investigation of cis-hydrido(triphenylsilyl)-1,4-butanediyl-bis-(dicyclohexylphosphine)platinum(II), Polyhedron, 1991, 10, 867–872 CrossRef CAS.
- L. D. Boardman, (.eta.5-Cyclopentadienyl)trialkylplatinum photohydrosilylation catalysts. Mechanism of active catalyst formation and preparation of a novel bis(silyl)platinum hydride, Organometallics, 1992, 11, 4194–4201 CrossRef CAS.
- S. D. Grumbine, T. D. Tilley, F. P. Arnold and A. L. Rheingold, A Fischer-type silylene complex of platinum: [trans-(Cy3P)2(H)Pt:Si(SEt)2]BPh4, J. Am. Chem. Soc., 1993, 115, 7884–7885 CrossRef CAS.
- L. A. Latif, C. Eaborn, A. P. Pidcock and N. S. Weng, Square planar platinum(II) complexes. Crystal structures of cis-bis(triphenylphosphine)hydro (triphenylstannyl)platinum(II) and cis-bis(triphenylphosphine)hydro(triphenylsilyl)platinum(II), J. Organomet. Chem., 1994, 474, 217–221 CrossRef.
- T.-a. Koizumi, K. Osakada and T. Yamamoto, Intermolecular Transfer of Triarylsilane from RhCl(H)(SiAr3)[P(i-Pr)3]2 to a Platinum(0) Complex, Giving cis-PtH(SiAr3)(PEt3)2 (Ar = C6H5, C6H4F-p, C6H4Cl-p), Organometallics, 1997, 16, 6014–6016 CrossRef CAS.
- J. D. Feldman, G. P. Mitchell, J.-O. Nolte and T. D. Tilley, Isolation and Characterization of Neutral Platinum Silylene Complexes of the Type (R3P)2PtSiMes2 (Mes = 2,4,6-Trimethylphenyl), J. Am. Chem. Soc., 1998, 120, 11184–11185 CrossRef CAS.
- R. S. Simons, L. M. Sanow, K. J. Galat, C. A. Tessier and W. J. Youngs, Synthesis and Structural Characterization of the Mono(silyl)platinum(II) Complex cis-(2,6-Mes2C6H3(H)2Si)Pt(H)(PPr3)2, Organometallics, 2000, 19, 3994–3996 CrossRef CAS.
- D. Chan, S. B. Duckett, S. L. Heath, I. G. Khazal, R. N. Perutz, S. Sabo-Etienne and P. L. Timmins, Platinum Bis(tricyclohexylphosphine) Silyl Hydride Complexes, Organometallics, 2004, 23, 5744–5756 CrossRef CAS.
- J. Takaya and N. Iwasawa, Reaction of bis(o-phosphinophenyl)silane with M(PPh3)4 (M = Ni, Pd, Pt): synthesis and structural analysis of [small eta]2-(Si-H) metal(0) and pentacoordinate silyl metal(ii) hydride complexes of the Ni triad bearing a PSiP-pincer ligand, Dalton Trans., 2011, 40, 8814–8821 RSC.
- N. Nakata, N. Kato, N. Sekizawa and A. Ishii, Si–H Bond Activation of a Primary Silane with a Pt(0) Complex: Synthesis and Structures of Mononuclear (Hydrido)(dihydrosilyl) Platinum(II) Complexes, Inorganics, 2017, 5, 72 CrossRef.
- A. Naka, T. Mihara, H. Kobayashi and M. Ishikawa, Platinum-Catalyzed Reactions of 2,3-Bis(diisopropylsilyl)thiophene with Alkynes, ACS Omega, 2017, 2, 8517–8525 CrossRef CAS.
- H. Keipour, V. Carreras and T. Ollevier, Recent progress in the catalytic carbene insertion reactions into the silicon–hydrogen bond, Org. Biomol. Chem., 2017, 15, 5441–5456 RSC.
- B. D. Bergstrom, L. A. Nickerson, J. T. Shaw and L. W. Souza, Transition Metal Catalyzed Insertion Reactions with Donor/Donor Carbenes, Angew. Chem., Int. Ed. Engl., 2021, 60, 6864–6878 CrossRef CAS.
- Z. Liu, J. Huo, T. Fu, H. Tan, F. Ye, M. L. Hossain and J. Wang, Palladium(0)-catalyzed C(sp3)–Si bond formation via formal carbene insertion into a Si–H bond, Chem. Commun., 2018, 54, 11419–11422 RSC.
- Z. Xu, W.-S. Huang, J. Zhang and L.-W. Xu, Recent Advances in Transition-Metal-Catalyzed Silylations of Arenes with Hydrosilanes: C–X Bond Cleavage or C–H Bond Activation- Synchronized with Si–H Bond Activation, Synthesis, 2015, 47, 3645–3668 CrossRef CAS.
- Z. Xu and L.-W. Xu, Silylations of Arenes with Hydrosilanes: From Transition-Metal-Catalyzed C-X Bond Cleavage to Environmentally Benign Transition-Metal-Free C-H Bond Activation, ChemSusChem, 2015, 8, 2176–2179 CrossRef CAS PubMed.
- F. Alonso, I. P. Beletskaya and M. Yus, Metal-Mediated Reductive Hydrodehalogenation of Organic Halides, Chem. Rev., 2002, 102, 4009–4092 CrossRef CAS PubMed.
- S. Bähr, W. Xue and M. Oestreich, C(sp3)–Si Cross-Coupling, ACS Catal., 2019, 9, 16–24 CrossRef.
- G. M. Noonan, B. R. Hayter, A. D. Campbell, T. W. Gorman, B. E. Partridge and G. M. Lamont, Expanding the scope of silane-mediated hydrodehalogenation reactions, Tetrahedron Lett., 2013, 54, 4518–4521 CrossRef CAS.
- A. Iwata, Y. Toyoshima, T. Hayashida, T. Ochi, A. Kunai and J. Ohshita, PdCl2 and NiCl2-catalyzed hydrogen–halogen exchange for the convenient preparation of bromo- and iodosilanes and germanes, J. Organomet. Chem., 2003, 667, 90–95 CrossRef CAS.
- R. Boukherroub, C. Chatgilialoglu and G. Manuel, PdCl2-Catalyzed Reduction of Organic Halides by Triethylsilane, Organometallics, 1996, 15, 1508–1510 CrossRef CAS.
- A. Kunai, T. Sakurai, E. Toyoda, M. Ishikawa and Y. Yamamoto, Versatile Method for the Synthesis of Iodosilanes, Organometallics, 1994, 13, 3233–3236 CrossRef CAS.
- X.-F. Bai, T. Song, W.-H. Deng, Y.-L. Wei, L. Li, C.-G. Xia and L.-W. Xu, (EtO)3SiH-Promoted Palladium-Catalyzed Isomerization of Olefins: Convenient Synthesis of Internal Alkenes from Terminal Alkenes, Synlett, 2014, 25, 417–422 CAS.
- M. Mirza-Aghayan, R. Boukherroub, M. Bolourtchian, M. Hoseini and K. Tabar-Hydar, A novel and efficient method for double bond isomerization, J. Organomet. Chem., 2003, 678, 1–4 CrossRef CAS.
- M. Mirza-Aghayan, R. Boukherroub and M. Bolourtchian, A mild and efficient palladium–triethylsilane system for reduction of olefins and carbon–carbon double bond isomerization, Appl. Organomet. Chem., 2006, 20, 214–219 CrossRef CAS.
- X.-F. Bai, L.-W. Xu, L.-S. Zheng, J.-X. Jiang, G.-Q. Lai and J.-Y. Shang, Aromatic-Amide-Derived Olefins as a Springboard: Isomerization-Initiated Palladium-Catalyzed Hydrogenation of Olefins and Reductive Decarbonylation of Acyl Chlorides with Hydrosilane, Chem.–Eur. J., 2012, 18, 8174–8179 CrossRef CAS PubMed.
- M. Mirza-Aghayan, R. Boukherroub and M. Rahimifard, Efficient method for the reduction of carbonyl compounds by triethylsilane catalyzed by PdCl2, J. Organomet. Chem., 2008, 693, 3567–3570 CrossRef CAS.
- T. Matsumura, T. Niwa and M. Nakada, Pd-catalyzed reductive cleavage of alkyl aryl sulfides with triethylsilane that is accelerated by trialkylsilyl chloride, Tetrahedron Lett., 2012, 53, 4313–4316 CrossRef CAS.
- R. C. Boyle, J. T. Mague and M. J. Fink, The First Stable Mononuclear Silyl Palladium Hydrides, J. Am. Chem. Soc., 2003, 125, 3228–3229 CrossRef CAS.
- R. C. Boyle, D. Pool, H. Jacobsen and M. J. Fink, Dynamic Processes in Silyl Palladium Complexes: Evidence for Intermediate Si−H and Si−Si σ-Complexes, J. Am. Chem. Soc., 2006, 128, 9054–9055 CrossRef CAS.
- N. Nakata, S. Fukazawa, N. Kato and A. Ishii, Palladium(II) Hydrido Complexes Having a Primary Silyl or Germyl Ligand: Synthesis, Crystal Structures, and Dynamic Behavior, Organometallics, 2011, 30, 4490–4493 CrossRef CAS.
- B. J. Barrett and V. M. Iluc, An Adaptable Chelating Diphosphine Ligand for the Stabilization of Palladium and Platinum Carbenes, Organometallics, 2017, 36, 730–741 CrossRef CAS.
- B. J. Barrett and V. M. Iluc, Metal-ligand cooperation between palladium and a diphosphine ligand with an olefinic backbone, Inorg. Chim. Acta, 2017, 460, 35–42 CrossRef CAS.
- H. Jacobsen and M. J. Fink, Tuning the Palladium−Silicon Bond: Bond Analysis of Bisphosphine Silyl Palladium Hydrides, Organometallics, 2006, 25, 1945–1952 CrossRef CAS.
- S. Sakaki, M. Ogawa and M. Kinoshita, A Theoretical Study on the Oxidative Addition of an Si-X Bond (X = H or Si) to M(PH3)2 (M = Pd or Pt). A Comparison of the Reactivity between Pt(PH3)2 and Pd(PH3)2, J. Phys. Chem., 1995, 99, 9933–9939 CrossRef CAS.
- V. M. Iluc and G. L. Hillhouse, Snapshots of the oxidative-addition process of silanes to nickel(0), Tetrahedron, 2006, 62, 7577–7582 CrossRef CAS.
- E. E. Smith, G. Du, P. E. Fanwick and M. M. Abu-Omar, Dehydrocoupling of Organosilanes with a Dinuclear Nickel Hydride Catalyst and Isolation of a Nickel Silyl Complex, Organometallics, 2010, 29, 6527–6533 CrossRef CAS.
- T. Zell, T. Schaub, K. Radacki and U. Radius, Si-H Activation of hydrosilanes leading to hydrido silyl and bis(silyl) nickel complexes, Dalton Trans., 2011, 40, 1852–1854 RSC.
- D. Schmidt, T. Zell, T. Schaub and U. Radius, Si-H bond activation at {(NHC)2Ni0} leading to hydrido silyl and bis(silyl) complexes: a versatile tool for catalytic Si-H/D exchange, acceptorless dehydrogenative coupling of hydrosilanes, and hydrogenation of disilanes to hydrosilanes, Dalton Trans., 2014, 43, 10816–10827 RSC.
- W. D. Jones, Isotope Effects in C−H Bond Activation Reactions by Transition Metals, Acc. Chem. Res., 2003, 36, 140–146 CrossRef CAS PubMed.
- G. Parkin, Temperature-Dependent Transitions Between Normal and Inverse Isotope Effects Pertaining to the Interaction of H−H and C−H Bonds with Transition Metal Centers, Acc. Chem. Res., 2009, 42, 315–325 CrossRef CAS.
- U. Schubert, in Adv. Organomet. Chem., ed. F. G. A. Stone and R. West, Academic Press, 1990, vol. 30, pp. 151–187 Search PubMed.
- G. I. Nikonov, in Adv. Organomet. Chem., ed. R. West, A. F. Hill and F. G. A. Stone, Academic Press, 2005, vol. 53, pp. 217–309 Search PubMed.
- R. Beck and S. A. Johnson, Structural Similarities in Dinuclear, Tetranuclear, and Pentanuclear Nickel Silyl and Silylene Complexes Obtained via Si–H and Si–C Activation, Organometallics, 2012, 31, 3599–3609 CrossRef CAS.
- S. B. Duckett and R. N. Perutz, Thermal and photochemical reactions of rhodium(trialkylsilyl)hydride complexes: NMR and bonding of poly(silyl)(hydride) complexes, J. Chem. Soc., Chem. Commun., 1991, 28–31 RSC.
- S. B. Duckett and R. N. Perutz, Mechanism of homogeneous hydrosilation of alkenes by (.eta.5-cyclopentadienyl)rhodium, Organometallics, 1992, 11, 90–98 CrossRef CAS.
- S. Wu, X. Li, Z. Xiong, W. Xu, Y. Lu and H. Sun, Synthesis and Reactivity of Silyl Iron, Cobalt, and Nickel Complexes Bearing a [PSiP]-Pincer Ligand via Si–H Bond Activation, Organometallics, 2013, 32, 3227–3237 CrossRef CAS.
- J. Takaya and N. Iwasawa, Bis(o-phosphinophenyl)silane as a Scaffold for Dynamic Behavior of H−Si and C−Si Bonds with Palladium(0), Organometallics, 2009, 28, 6636–6638 CrossRef CAS.
- S. J. Mitton and L. Turculet, Mild Reduction of Carbon Dioxide to Methane with Tertiary Silanes Catalyzed by Platinum and Palladium Silyl Pincer Complexes, Chem.–Eur. J., 2012, 18, 15258–15262 CrossRef CAS.
- S. J. Mitton, R. McDonald and L. Turculet, Synthesis and Characterization of Neutral and Cationic Platinum(II) Complexes Featuring Pincer-like Bis(phosphino)silyl Ligands: Si−H and Si−Cl Bond Activation Chemistry, Organometallics, 2009, 28, 5122–5136 CrossRef CAS.
- J. Mullay, Atomic and group electronegativities, J. Am. Chem. Soc., 1984, 106, 5842–5847 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Structural variation in copper(i) complexes with pyridylmethylamide ligands: structural analysis with a new four-coordinate geometry index, τ4, Dalton Trans., 2007, 955–964 RSC.
- A. Okuniewski, D. Rosiak, J. Chojnacki and B. Becker, Coordination polymers and molecular structures among complexes of mercury(II) halides with selected 1-benzoylthioureas, Polyhedron, 2015, 90, 47–57 CrossRef CAS.
- F. R. Hartley, The cis- and trans-effects of ligands, Chem. Soc. Rev., 1973, 2, 163–179 RSC.
- J. Chatt, C. Eaborn and S. Ibekwe, Silicon, a ligand atom of exceptionally high trans-effect, Chem. Commun., 1966, 700–701 RSC.
- T. G. Appleton, H. C. Clark and L. E. Manzer, The trans-influence: its measurement and significance, Coord. Chem. Rev., 1973, 10, 335–422 CrossRef CAS.
- M. Wolfsberg, Theoretical evaluation of experimentally observed isotope effects, Acc. Chem. Res., 1972, 5, 225–233 CrossRef CAS.
- D. G. Churchill, K. E. Janak, J. S. Wittenberg and G. Parkin, Normal and Inverse Primary Kinetic Deuterium Isotope Effects for C−H Bond Reductive Elimination and Oxidative Addition Reactions of Molybdenocene and Tungstenocene Complexes: Evidence for Benzene σ-Complex Intermediates, J. Am. Chem. Soc., 2003, 125, 1403–1420 CrossRef CAS PubMed.
- J. W. Raebiger, A. Miedaner, C. J. Curtis, S. M. Miller, O. P. Anderson and D. L. DuBois, Using Ligand Bite Angles To Control the Hydricity of Palladium Diphosphine Complexes, J. Am. Chem. Soc., 2004, 126, 5502–5514 CrossRef CAS PubMed.
- E. Buncel and T. K. Venkatachalam, Carbanion mechanisms XXI. Solution acidity of triphenylsilane, J. Organomet. Chem., 2000, 604, 208–210 CrossRef CAS.
- S. M. Reid and M. J. Fink, Reductive Routes to Dinuclear d10−d10 Palladium(0) Complexes and Their Redistribution Equilibria in Solution, Organometallics, 2001, 20, 2959–2961 CrossRef CAS.
- G. Parkin, Applications of deuterium isotope effects for probing aspects of reactions involving oxidative addition and reductive elimination of H–H and C–H bonds, J. Labelled Compd. Radiopharm., 2007, 50, 1088–1114 CrossRef CAS.
- F. Abu-Hasanayn, K. Krogh-Jespersen and A. S. Goldman, Theoretical study of primary and secondary deuterium equilibrium isotope effects for hydrogen and methane addition to iridium complex trans-Ir(PR3)2(CO)X, J. Am. Chem. Soc., 1993, 115, 8019–8023 CrossRef CAS.
- M. Gómez-Gallego and M. A. Sierra, Kinetic Isotope Effects in the Study of Organometallic Reaction Mechanisms, Chem. Rev., 2011, 111, 4857–4963 CrossRef PubMed.
- Y. Tsuji and Y. Obora, Structure and fluxional behavior of platinum and palladium complexes having M-Si or M-Sn (M=Pt or Pd) inter-element linkages, J. Organomet. Chem., 2000, 611, 343–348 CrossRef CAS.
- S. F. Vyboishchikov and G. I. Nikonov, Rhodium Silyl Hydrides in Oxidation State +5: Classical or Nonclassical?, Organometallics, 2007, 26, 4160–4169 CrossRef CAS.
- F. P. Gasparro and N. H. Kolodny, NMR determination of the rotational barrier in N,N-dimethylacetamide. A physical chemistry experiment, J. Chem. Educ., 1977, 54, 258 CrossRef CAS.
- K. C. Ramey, D. J. Louick, P. W. Whitehurst, W. B. Wise, R. Mukherjee and R. M. Moriarty, A line width method for determining chemical exchange rates from NMR spectra, Org. Magn. Reson., 1971, 3, 201–216 CrossRef CAS.
- H. Yamashita and Y. Uchimaru, Highly efficient palladium catalyst system for addition of trihydrosilanes to acetylenes and its application to thermally stable polycarbosilane synthesis, Chem. Commun., 1999, 1763–1764 RSC.
- D. Motoda, H. Shinokubo and K. Oshima, Efficient Pd(0)-catalyzed hydrosilylation of alkynes with triorganosilanes, Synlett, 2002, 1529–1531 CAS.
- H. Azizian, K. R. Dixon, C. Eaborn, A. Pidcock, N. M. Shuaib and J. Vinaixa, Dynamic stereochemistry of cis-[PtH(SiR3)(PPh3)2]. Spontaneous ligand interchange with retentilon of nuclear spin–spin correlation, J. Chem. Soc., Chem. Commun., 1982, 1020–1022 RSC.
- J. D. Feldman, G. P. Mitchell, J.-O. Nolte and T. D. Tilley, Synthesis and study of platinum silylene complexes of the type (R3P)2Pt=SiMes2 (Mes = 2,4,6-trimethylphenyl), Can. J. Chem., 2003, 81, 1127–1136 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Figures, schemes, tables and graphs, experimental procedures, characterization, and NMR and IR spectra. CCDC 2086211. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04419b |
| This journal is © The Royal Society of Chemistry 2021 |