
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric synthesis of dibenzo[b,d]azepines by Cu-catalyzed reductive or borylative cyclization†
Patricia
Rodríguez-Salamanca
 a,
Rocío
Martín-de la Calle
a,
Verónica
Rodríguez
b,
Pedro
Merino
c,
Rosario
Fernández
*b,
José M.
Lassaletta
*a and
Valentín
Hornillos
*ab
a,
Rocío
Martín-de la Calle
a,
Verónica
Rodríguez
b,
Pedro
Merino
c,
Rosario
Fernández
*b,
José M.
Lassaletta
*a and
Valentín
Hornillos
*ab
aInstituto Investigaciones Químicas (CSIC-US), C/Américo Vespucio, 49, 41092 Sevilla, Spain. E-mail: jmlassa@iiq.csic.es
bDepartamento de Química Orgánica, Universidad de Sevilla, C/Prof. García González, 1, 41012 Sevilla, Spain
cInstituto de Biocomputación y Física de Sistemas Complejos (BIFI), Universidad de Zaragoza, 50009 Zaragoza, Spain
First published on 10th November 2021
Abstract
A copper-catalyzed asymmetric intramolecular reductive cyclization for the synthesis of dibenzo[b,d]azepines is described. Use of 2′-vinyl-biaryl-2-imines as substrates and in situ formed [CuI/(Ph-BPE)] as the catalyst enables the synthesis of 7-membered bridged biarylamines containing both central and axial stereogenic elements in high yields (up to 98%) and with excellent diastereo- and enantioselectivities (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 d.r., up to 99% ee). Moreover, the same catalyst was found to facilitate a related borylative cyclization to afford versatile boronic ester derivatives. Both reactions proceed under mild conditions (rt) and are applicable to a variety of substituted aromatic and heterocyclic derivatives.
:1 d.r., up to 99% ee). Moreover, the same catalyst was found to facilitate a related borylative cyclization to afford versatile boronic ester derivatives. Both reactions proceed under mild conditions (rt) and are applicable to a variety of substituted aromatic and heterocyclic derivatives.
Introduction
The asymmetric synthesis of biaryl atropisomers has evolved into a stimulating research field in organic synthesis owing to the ubiquitous occurrence of this structural motif in a variety of natural products and bioactive substances, and for their wide-ranging utilities in catalyst design and material science.1 Most of these compounds comprise conformers with restricted rotation around a single bond whose configurational stability is generally determined by the number and size of the substituents at the ortho positions relative to the stereogenic axis. A less common class of biaryl atropisomers consist of those where the biaryl unit is incorporated into a cyclic system, with the typical ortho, ortho′ substituents being replaced by a bridge (Fig. 1).2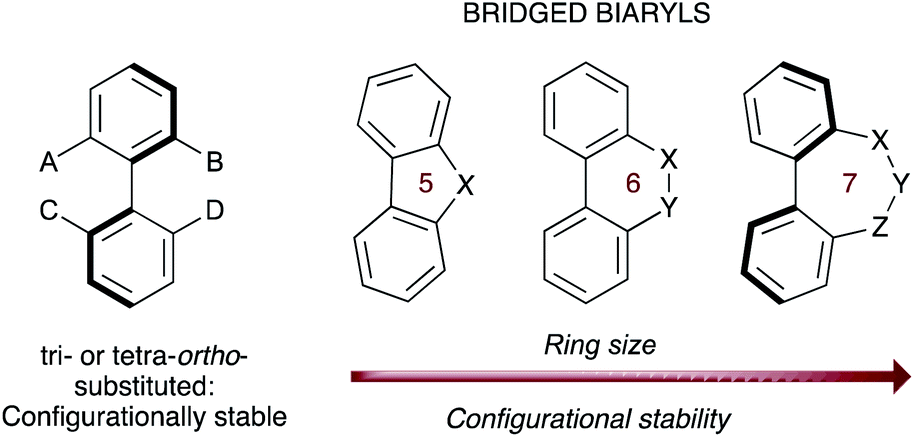 | ||
| Fig. 1 Configurational stability of acyclic and bridged biaryls. | ||
In these systems, the configurational stability directly correlates with the ring size: in 5- and 6-membered rings the rotation around the stereogenic axis is usually not hindered, but 7-membered bridged biaryls exhibit a higher configurational stability owing to conformational reasons and can be often resolved as atropisomers. Additionally, the introduction of sp2 hybridized atoms increases the rigidity in the bridging cycle, and therefore the configurational stability,3 while the presence of stereogenic centers in the bridge is known to impose a specific configuration on the biaryl axis by a central to axial chirality relay event.4 Notably, these properties have been recently exploited for the construction of a unidirectional rotary molecular motor based on the formation of biaryl structures featuring a seven-membered lactone bridge.5
The most distinctive class of bridged biaryls presenting this relay phenomenon comprises chiral dibenzoazepines. These 7-membered cyclic bridged biaryl amines and related analogues have received considerable attention due to their presence in several natural substances and drugs. Selected examples shown in Fig. 2 include erythrivaeine B (A), a dimeric Erythrina alkaloid isolated from E. variegate,6 dipeptide LY-411575 (B) which has demonstrated effectivity as γ-secretase inhibitor for the treatment of melanoma and Alzheimer's disease,7 RO4929097 (C), another potent and selective γ-secretase which targets Notch signaling in tumor cells,8 indolobenzoazepinone D, a tubulin polymerization inhibitor exhibiting antiproliferative activities in a variety of cancer cell lines,9 and paullones E, a family of cytotoxic compounds which efficiently inhibit cyclin-dependent kinases (CDKs).10
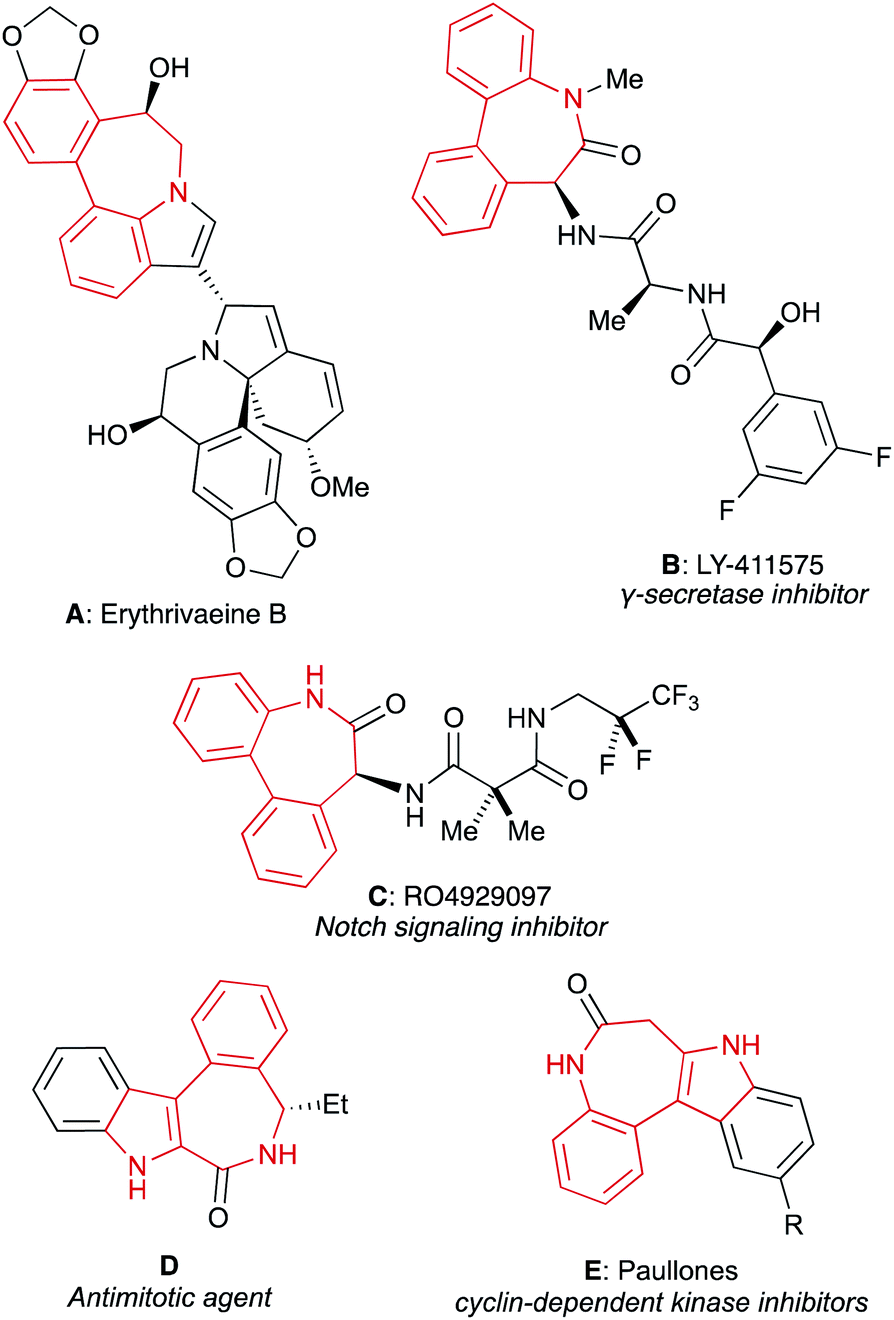 | ||
| Fig. 2 Selected bioactive dibenzoazepines and related analogues. | ||
The asymmetric synthesis of axially chiral 7-membered bridged biaryl amines has traditionally relied on chiral auxiliaries or starting materials from the chiral pool, generally requiring multistep procedures.3,11 The control of both central and axial chirality elements in the most interesting dibenzo[b,d]azepine derivatives constitutes a challenge: to date, only a handful of methods have been described for their catalytic asymmetric synthesis (Scheme 1).
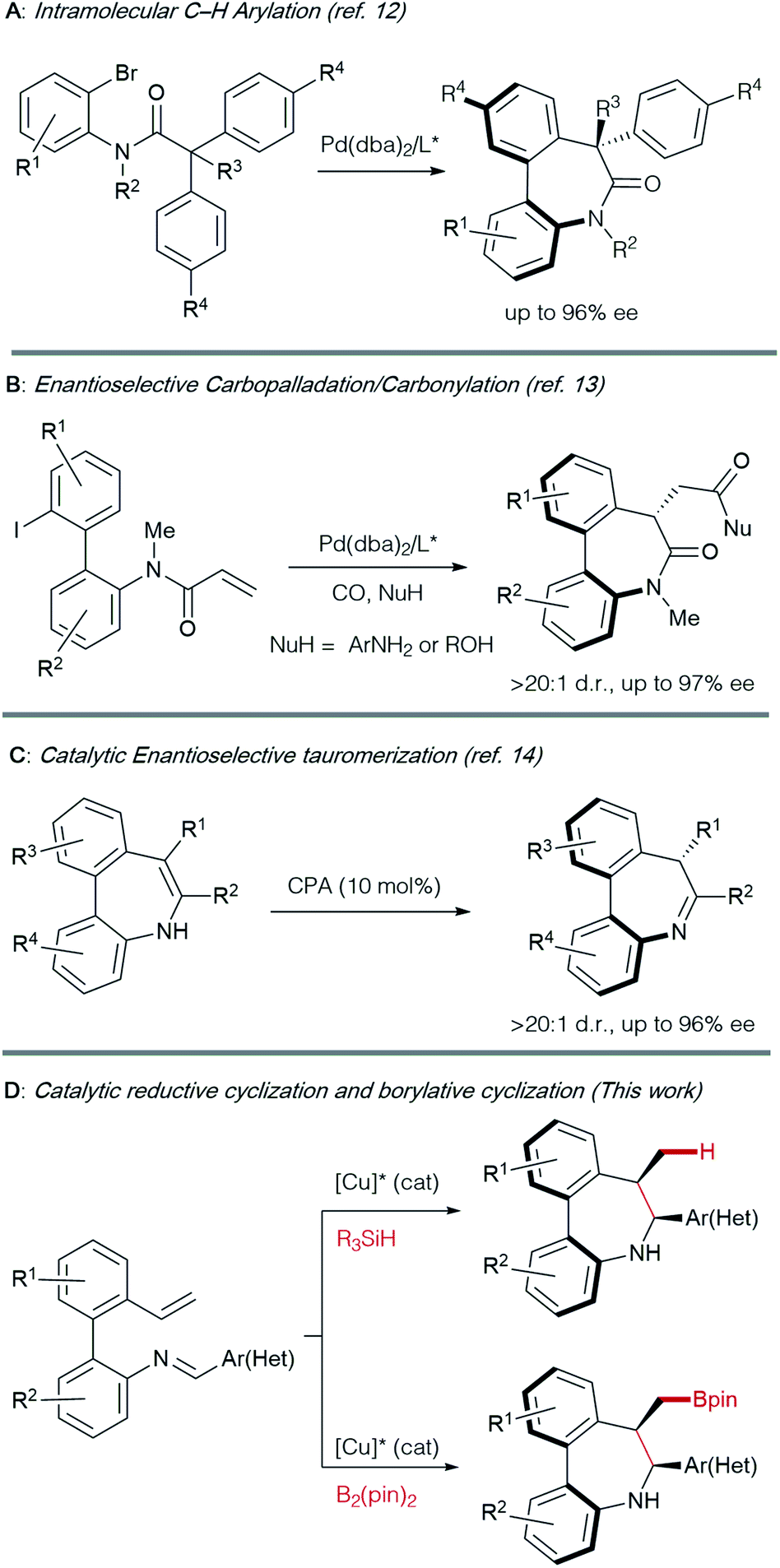 | ||
| Scheme 1 Catalytic asymmetric synthesis of axially chiral 7-membered cyclic bridged biaryl amines. | ||
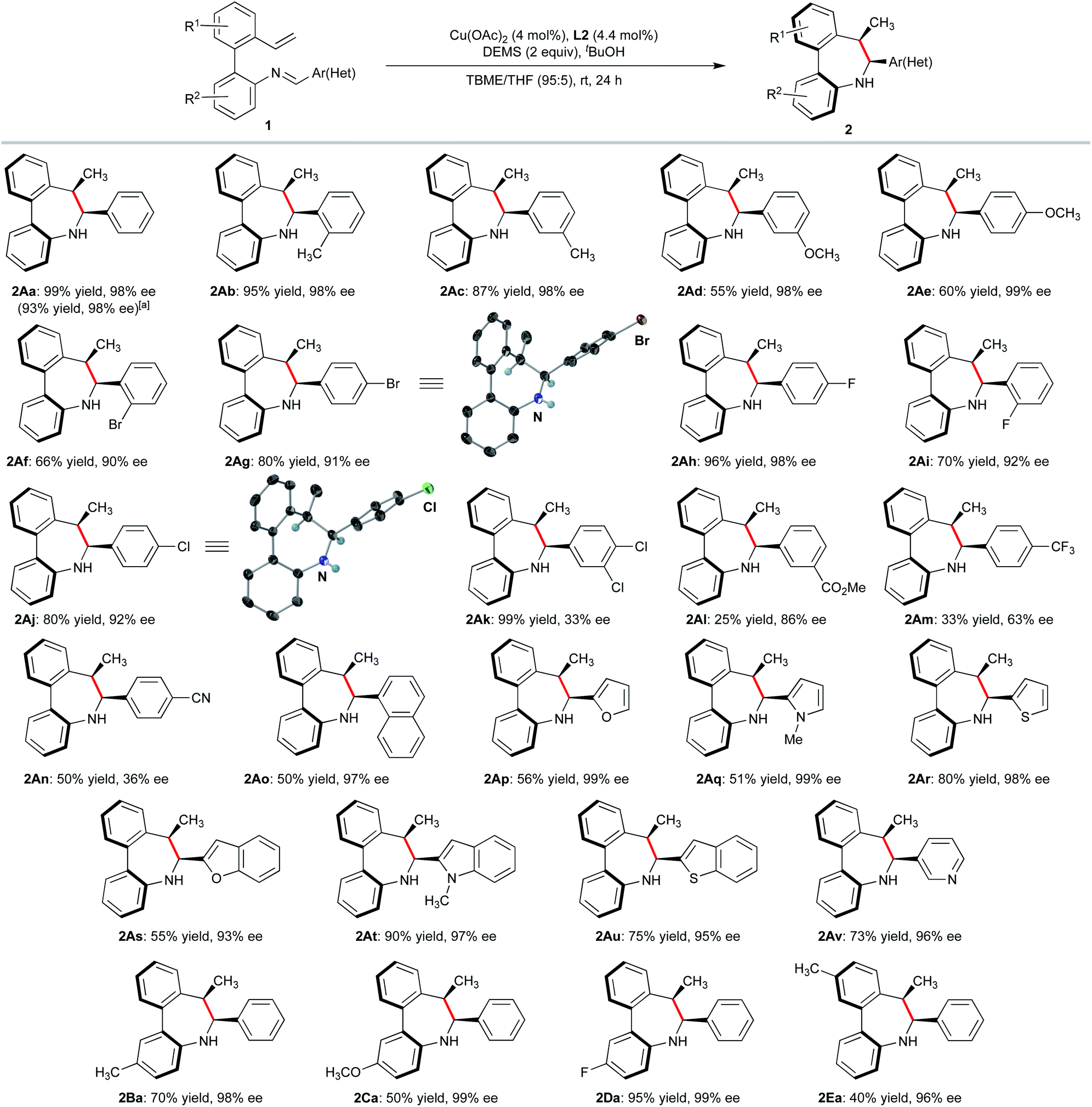 | ||
| Scheme 2 Substrate scope. Reactions performed on 0.2 mmol scale for a 36 h period at rt. Yields of isolated product after chromatography. A single diastereomer was observed by 1H NMR in the crude reaction mixtures. Ee's were determined by HPLC on chiral stationary phases. aReaction performed on 2 mmol (566 mg) scale. | ||
Axially chiral dibenzoazepinones with amide bridges have been prepared by desymmetrization via Pd-catalyzed C–H arylation12 (Scheme 1A) and, very recently, by a cyclocarbopalladation-carbonylation cascade reaction using alcohols or anilines as nucleophiles13 (Scheme 1B). To our knowledge, the tautomerization of metastable enamines promoted by a chiral phosphoric acid catalyst appears as the only catalytic method reported to obtain axially chiral dibenzo[b,d]azepines (Scheme 1C).14,15 Hence, the development of a modular and straightforward catalytic enantioselective method, providing access to these structural motifs remains as a desirable goal.
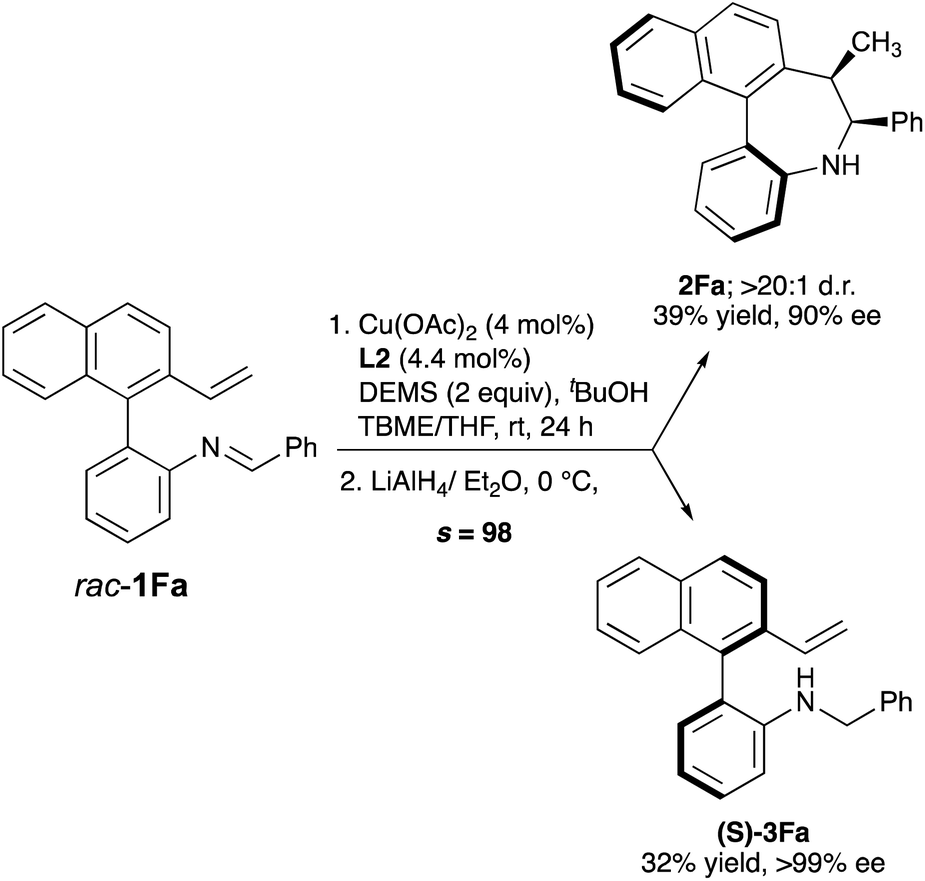 | ||
| Scheme 3 Kinetic resolution of rac-1Fa. Reaction performed on 0.2 mmol scale for a 36 h period at rt. Yields of isolated product after chromatography. Ee's were determined by HPLC on chiral stationary phases. | ||
Results and discussion
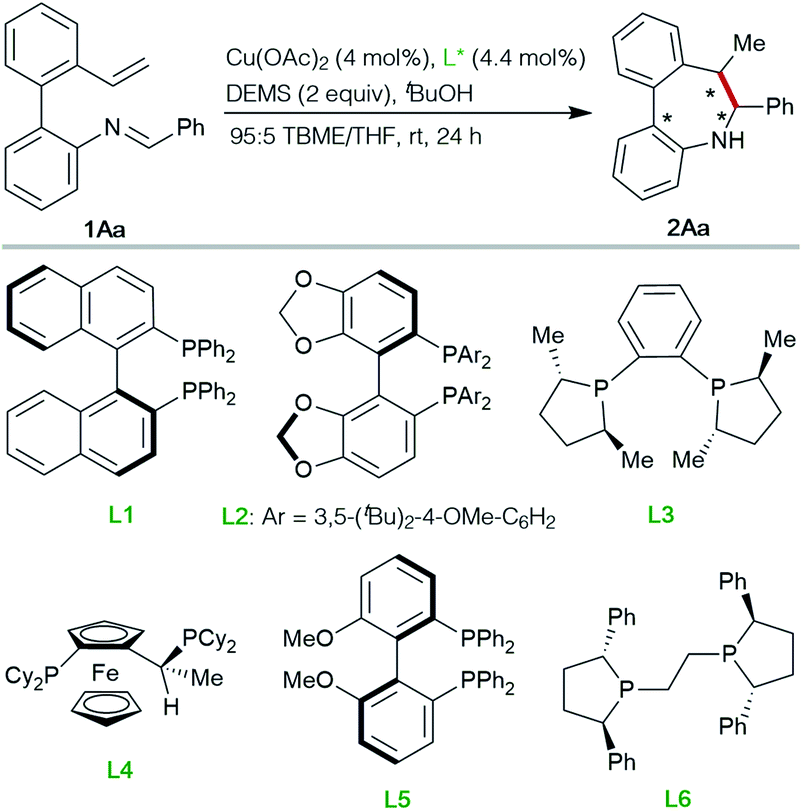
Inspired by the work of Buchwald16 and Yun17 on Cu-catalyzed cyclizations using aldimines as electrophiles, we envisioned that Schiff bases from ortho-vinyl, ortho′-amino biaryls could also be suitable substrates to perform reductive or borylative cyclizations for the construction of axially chiral dibenzo[b,d]azepine derivatives featuring a stereogenic axis and two contiguous stereogenic centers (Scheme 1D).Preliminary studies were performed with compound 1Aa, readily obtained by condensation of 2′-vinyl-biphenyl-2-amine with benzaldehyde, as a model substrate (Table 1). Using Cu(OAc)2 as the precatalyst, methyldiethoxysilane (DEMS) as the hydride source and anhydrous TBME:THF (95:5) mixture as the solvent at room temperature, representatives of commercially available chiral biphosphine ligands L1–L6 were tested for the synthesis of the desired azepine 2Aa. Poor reactivities were observed with BINAP L1, SEGPHOS L2, DUPHOS L3 or the JOSIPHOS representative L4 (entries 1–4), while moderate catalytic activity and enantioselectivity were observed with MeO-SEGPHOS L5 (entry 5). Finally, (R,R)-Ph-BPE L6 proved to be the ligand of choice, affording optimal results in terms of reactivity, diastereo- and enantioselectivity (99% yield, >20:1 d.r., 99% ee, entry 6).
|
|
|||
|---|---|---|---|
| Entrya | Ligand | Conv.b (%) | eec (%) |
| a Reactions performed on 0.2 mmol scale. b Estimated by 1H-NMR spectroscopy. c Determined by HPLC on chiral stationary phases. d A single diastereomer was observed by 1H NMR in the crude reaction mixture. | |||
| 1 | L1, (S)-BINAP | <5 | nd |
| 2 | L2, (R)-DTBM-SEGPHOS | <10 | nd |
| 3 | L3, (S,S)-Me-DUPHOS | <20 | nd |
| 4 | L4, (R)-(S)-JOSIPHOS | <20 | nd |
| 5 | L5, (R)-MeO-BIPHEP | 42 | 77 |
| 6d | L6, (R,R)-Ph-BPE | 99 (99 yield) | 99 |
With the optimized conditions established, we examined the substrate scope of the 2′-vinyl-biphenyl-2-imine precursors to explore the generality of this asymmetric reductive cyclization. Substrates with electron-rich methyl and methoxy substituents on the aldimine aryl ring were transformed into the corresponding dibenzoazepines 2Ab–e in high yields and excellent enantioselectivities. The steric effect of placing substituents on the ortho position of the ring has a negligible effect on reactivity and enantioselectivity as demonstrated for the synthesis of 2Ab, 2Af and 2Ai. Electron-deficient substituents on the phenyl ring of the aldimine were also tolerated. Thus, fluorinated substrates furnished the corresponding dibenzoazepines 2Ah and 2Ai with excellent selectivities. However, the enantioselectivity drops when a CF3 group is located at the para position of the phenyl ring (2Am). Importantly, products bearing halide (2Af, 2Ag, 2Aj and 2Ak), ester (2Al) and nitrile (2An) functionalities were also obtained in good to high yields (66–99%) and moderate to high enantioselectivities (up to 92%), providing synthetically useful functionalities for further transformations. Moreover, the method also tolerates a variety of heterocyclic substrates, leading to furan (2Ap), N-methyl pyrrole (2Aq), thiophene (2Ar), pyridine (2Av) and fused heterocyclic derivatives (benzofuran 2As, N-methylindole 2At and thianaphthene 2Au) in good yields and excellent enantioselectivities. The Sa,6S,7R absolute configuration of products 2Ag, and 2Aj was determined by X-ray diffraction analysis,18 while that of other products 2 was assigned by analogy. Moreover, the central to axial chirality relay phenomenon was confirmed by a dihedral angle of 39° between the two aryl groups in both compounds. It should be noted that the cis-diastereomers were exclusively formed in all cases. Biaryls decorated with several electron-donating or withdrawing groups (e.g. Me, OMe, or F) were also suitable substrates for this transformation, affording the desired products in moderate to good yields with high enantioselectivities (2Ba–2Ea, 94–99% ee).
We next explored the kinetic resolution (KR) of a trisubstituted, hence configurationally stable, biarylimine 1Favia Cu-catalyzed hydrocupration/cyclization followed by reduction of the unreacted, enantioenriched imine (Scheme 3). The reaction stopped at 50% conversion, despite the presence of a large excess (2 equiv.) of silane. In this way, dibenzoazepine 2Fa could be obtained in high enantioselectivity under the previously optimized conditions, while the enantioenriched starting material was transformed into amine (S)-3Fa, obtained in nearly enantiopure form (>99% ee) after LiAlH4 reduction (s = 98). The biaryl-2-amine 3Fa, featuring only axial chirality, shows an appealing structure with potential applications in asymmetric catalysis. Reasoning that substrates 1 should form very similar organocopper intermediates after insertion of the vinyl group into L*Cu–Bpin catalysts, we decided to explore also the Cu-catalyzed enantioselective borylative cyclization of the same substrates 1 as an alternative approach to axially chiral dibenzo[b,d]azepines, in this case decorated with a boryl group that should be useful for further derivatization or bioconjugation (Scheme 4). Using again 1A as a model substrate in the reaction with bis(pinacolato)diboron [B2(pin)2], the catalyst formed by combination of [Cu(MeCN)4]PF6 with biphosphine L2, KOtBu as the base, iPrOH as the proton source and anhydrous THF as the solvent were identified as the best conditions, leading to the borylated axially chiral dibenzoazepine (Sa,6S,7R)-4Aa in 85% yield with excellent regio-, diastereo- and enantioselectivity (>20:1 d.r. 98% ee). Single crystal X-ray analysis of this compound18 confirmed the assigned absolute configuration. Borylative cyclization of other 2′-vinyl-biphenyl-2-imines proceeded efficiently under the same conditions to afford products 4 with excellent enantiocontrol (>20:1 d.r., 92–98% ee). Again, electron-rich and -deficient substituents as well as ortho-substitution in the phenyl ring were tolerated. The reaction also accepts heteroarenes as illustrated for compound 4At.
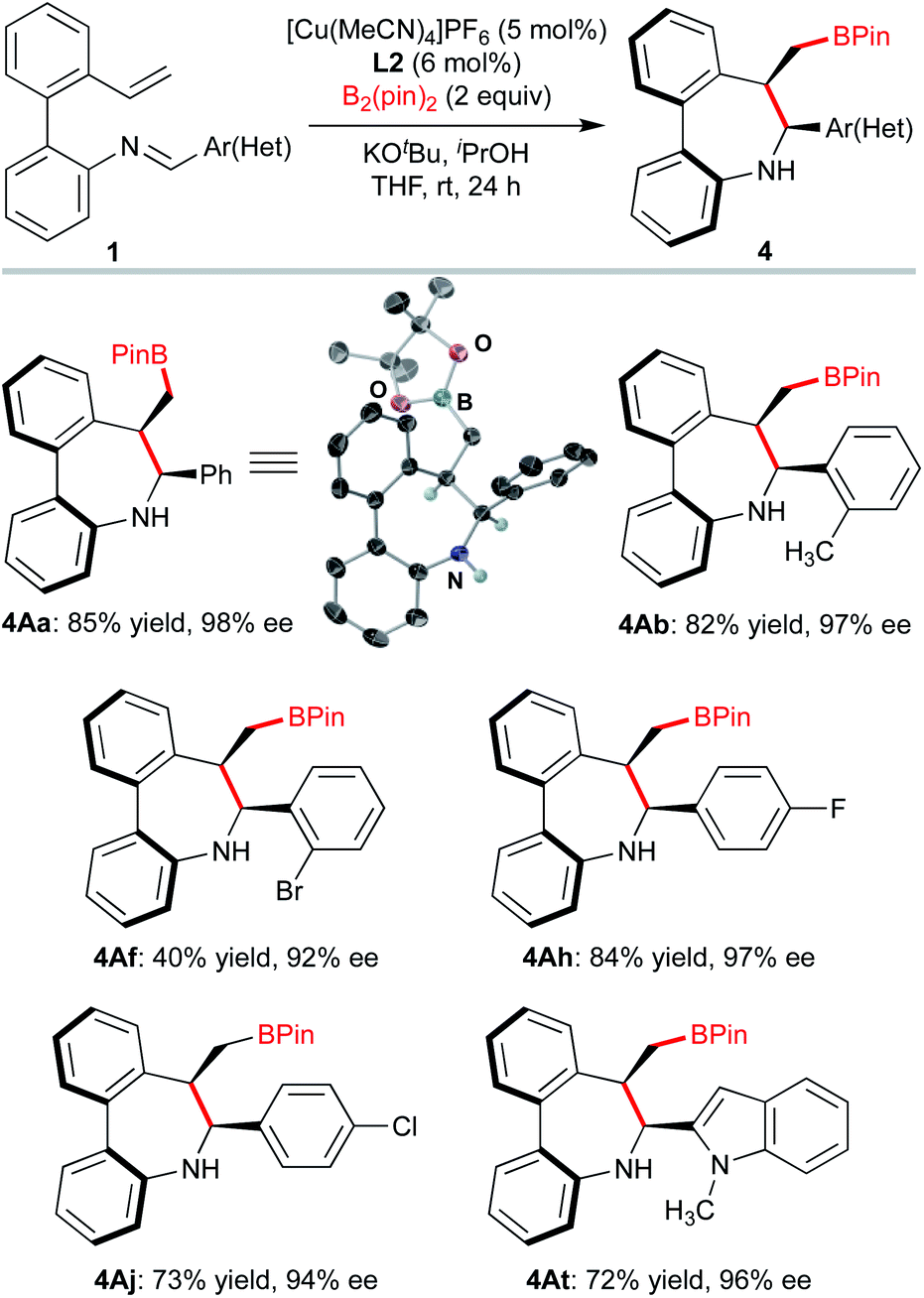 | ||
| Scheme 4 Copper-catalyzed borylative cyclization. Reactions performed on 0.2 mmol scale. Yields of isolated product after chromatography. A single diastereomer (>20:1 d.r.) was observed as determined by 1H NMR in the crude reaction mixtures. Ee's were determined by HPLC analysis. | ||
To explore the synthetic potential of the methodology, gram-scale synthesis and derivatizations were performed. Asymmetric intramolecular reductive cyclization of 1Aa on a 2 mmol scale afforded the desired product 2Aa in 93% yield and 98% ee (Scheme 2). As shown in Scheme 5, demethylation of 2Ae offered the corresponding phenol product 5 which can be used as synthetic handle for further transformations. Moreover, fluorescent labelling of 2Ag was accomplished via Suzuki−Miyaura coupling providing pyrene substituted compound 6 with emitting properties for their potential use in biological studies.19 On the other hand, oxidation of 4Ah with sodium perborate led to chiral primary alcohol 7 in high yield while intramolecular Suzuki coupling of 4Af affords cis-tetrahydrodibenzoindenoazepine 8 in 56% yield.
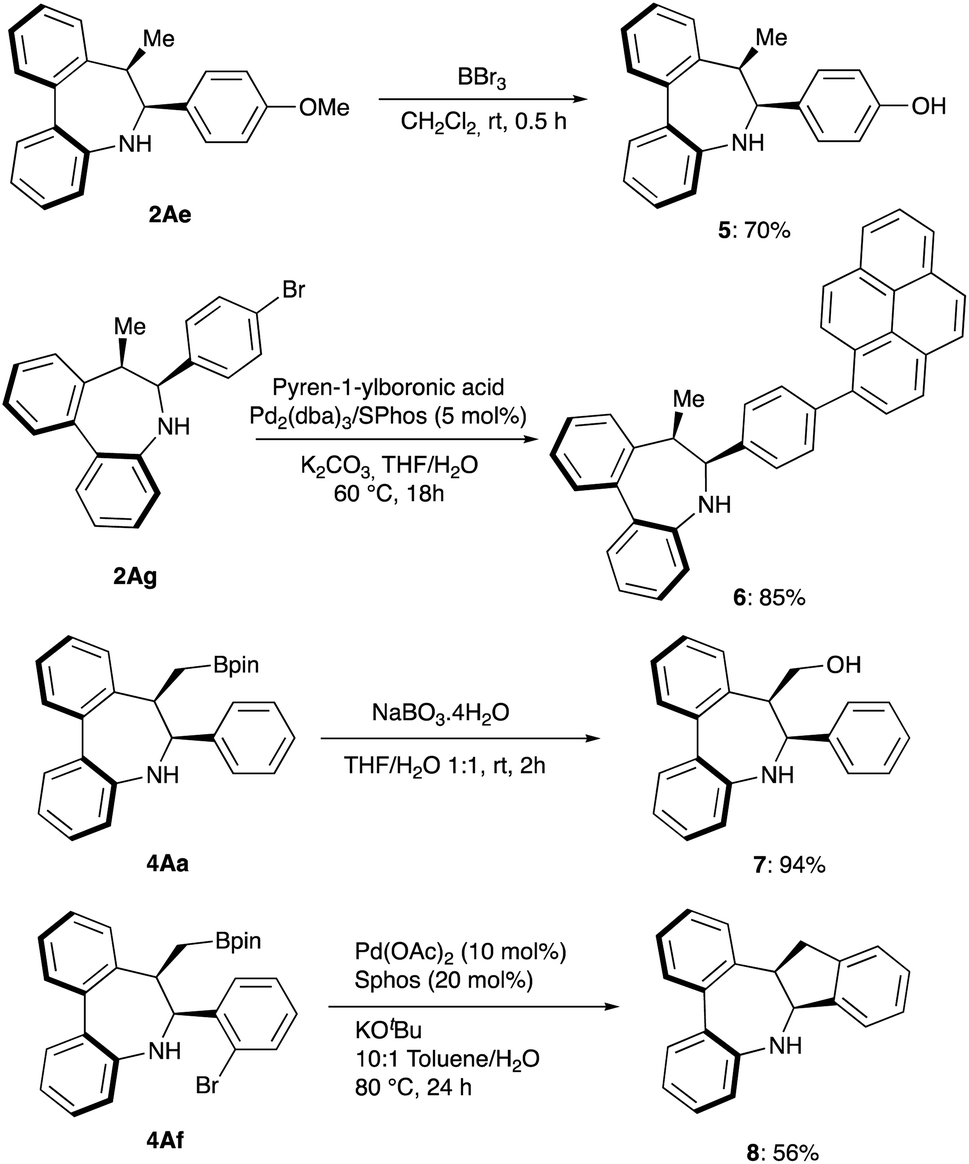 | ||
| Scheme 5 Derivatization reactions. | ||
To assess the configurational stability of the biaryl axis of products 2, we calculated the rotation about the axis of the biphenyl moiety for compound 2Aa. The dibenzoazepine ring can adopt boat a and half-chair b conformations (Scheme 6).20 A third conformation can be located but at a considerable higher energy.21 The most stable conformation for 2Aa was found to be C1a, matching the X-ray structures of analogs 2ag, 2Aj and 4Aa. Remarkably, no interconversion between C1a and C2a conformations is possible due to steric reasons. Thus, axial epimerization by biphenyl bond rotation requires a previous change from a to b conformations. The interconversion of the Sa-C1a/Sa-C1b and Ra-C2a/Ra-C2b conformers have free energy barriers of 15.6 and 14.0 kcal mol−1, respectively and the difference between free energy of conformers accounts for the preferred C1a and C2b conformations in each case. The interconversion between Sa-C1b/Ra-C2b conformations involving epimerization has free energy barriers of only 2, 6 and 8.1 kcal mol−1. Those values clearly show that a fast equilibrium is established at 25 °C (a barrier of 15.6 kcal mol−1 corresponds, approximately, to a kinetic constant of 22.4 s−1 with t1/2 of 0.031) and, consequently, the observed population of conformers depends on their relative stability. The calculated relative energies for the most stable Sa-C1a/Ra-C2b conformers correspond to a 2.4 kcal difference, corresponding to a 98:2 ratio. Considering these values and the assumed DFT experimental error it is possible to conclude that, essentially, only Sa-C1a will be observed. Low temperature 1H-NMR experiments provided further support to this conclusion: a single set of peaks is observed at temperatures as low as −60 °C.
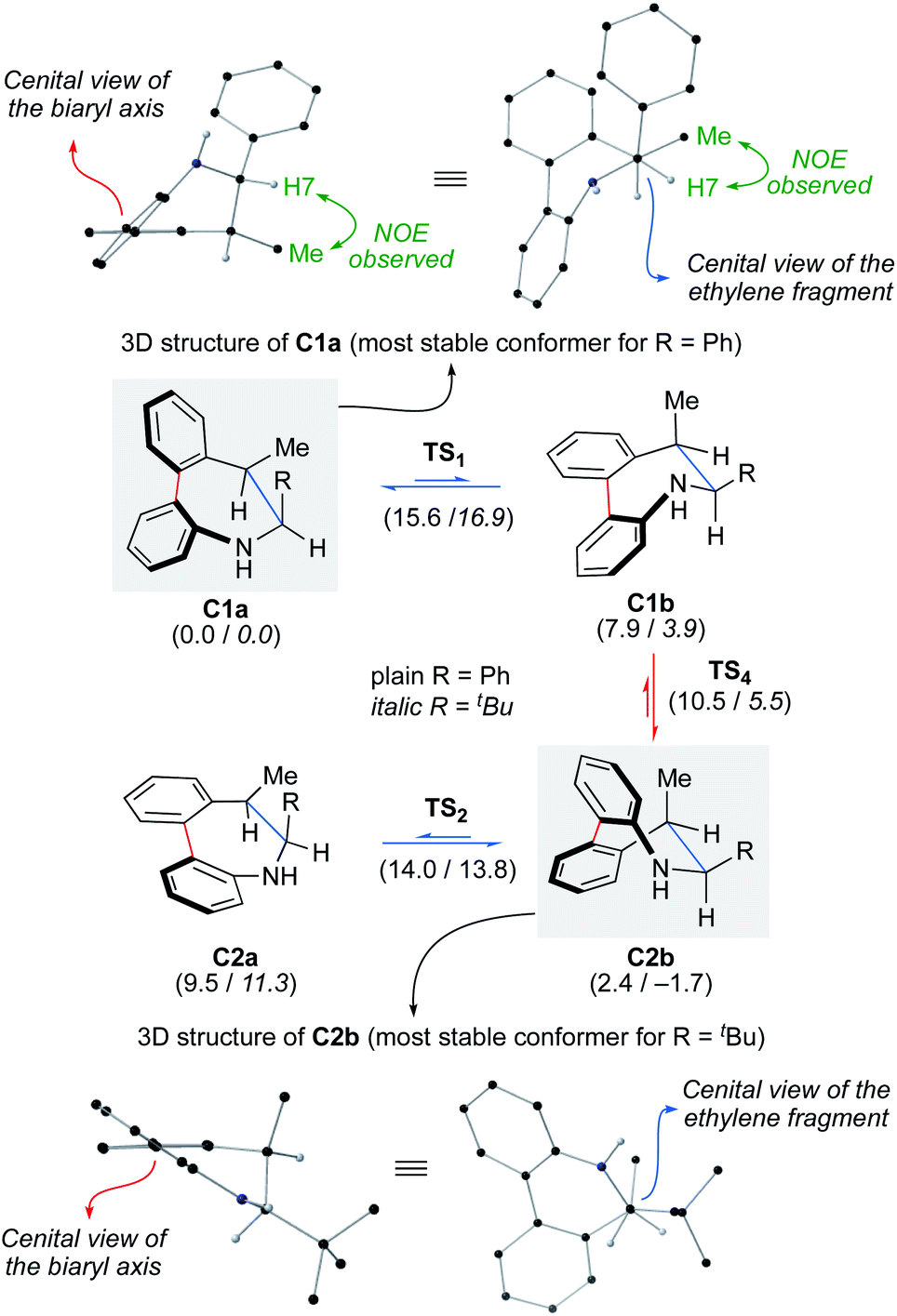 | ||
| Scheme 6 Computational analysis for the axial epimerization for dibenzoazepines 2A (wb97xd/def2tzvp//wb97xd/def2svp/cpcm = diisopropylether). Relative free energies are given in brackets in kcal mol−1. Data for R = Ph in plain text, data for R = tBu in italics. For details see the ESI.† | ||
On the light of these calculations, we speculated on the possibility of freezing or shifting the conformational equilibrium by introducing a bulkier tBu group instead of the Ph group. Interestingly, the calculation predicts that Ra-C2b is the lowest energy conformation in this case (1.7 lower than Sa-C1a). This result can be rationalized by the lower impact of the steric effects in the Sa-C1a conformer as a result of the two gauche interactions of the R group, while there is only one in Ra-C2b. In order to check whether a shift of the axial chirality could be indeed experimentally achieved, pivalaldehyde derivative 1Aw was prepared and subjected to the reductive cyclization protocol to achieve product 2Aw in 85% yield and 97% ee (Scheme 7). The Ra,6S,7R configuration is in this case tentatively assigned on the basis of the above calculations and the absence of a NOE observed between H-7 and the Me group, clearly observed for 2Aa (R = Ph) due to their relative gauche disposition (supported by the computational analysis of non-covalent interactions, NCI; see ESI†). As in the precedent case, a single set of signals is visible in the 1H-NMR spectrum upon cooling to −60 °C, indicating the absence of any significant amounts of minor conformers.
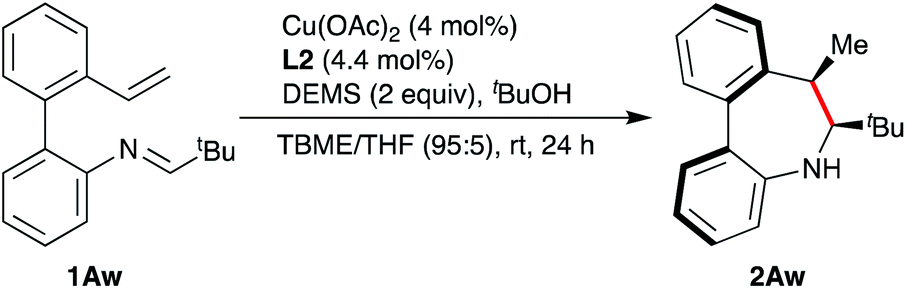 | ||
| Scheme 7 Synthesis of tert-butyl-substituted dibenzoazepine 2Aw. | ||
Based on previous mechanistic studies by Buchwald22 and Hartwig23 laboratories, we assume that the hydrocupration of substrates 1, generating intermediate Ia (Scheme 8) is the stereodetermining step. Ensuing cis-selective cyclization to afford complex IIa should then proceed via a well-organized transition state TS with the assistance provided by coordination of the imine nitrogen. Then, the resulting intermediate IIa reacts with the silane reagent to regenerate the CuH catalyst. A similar model and catalytic cycle can be also proposed for the borylative cyclization (X = Bpin).
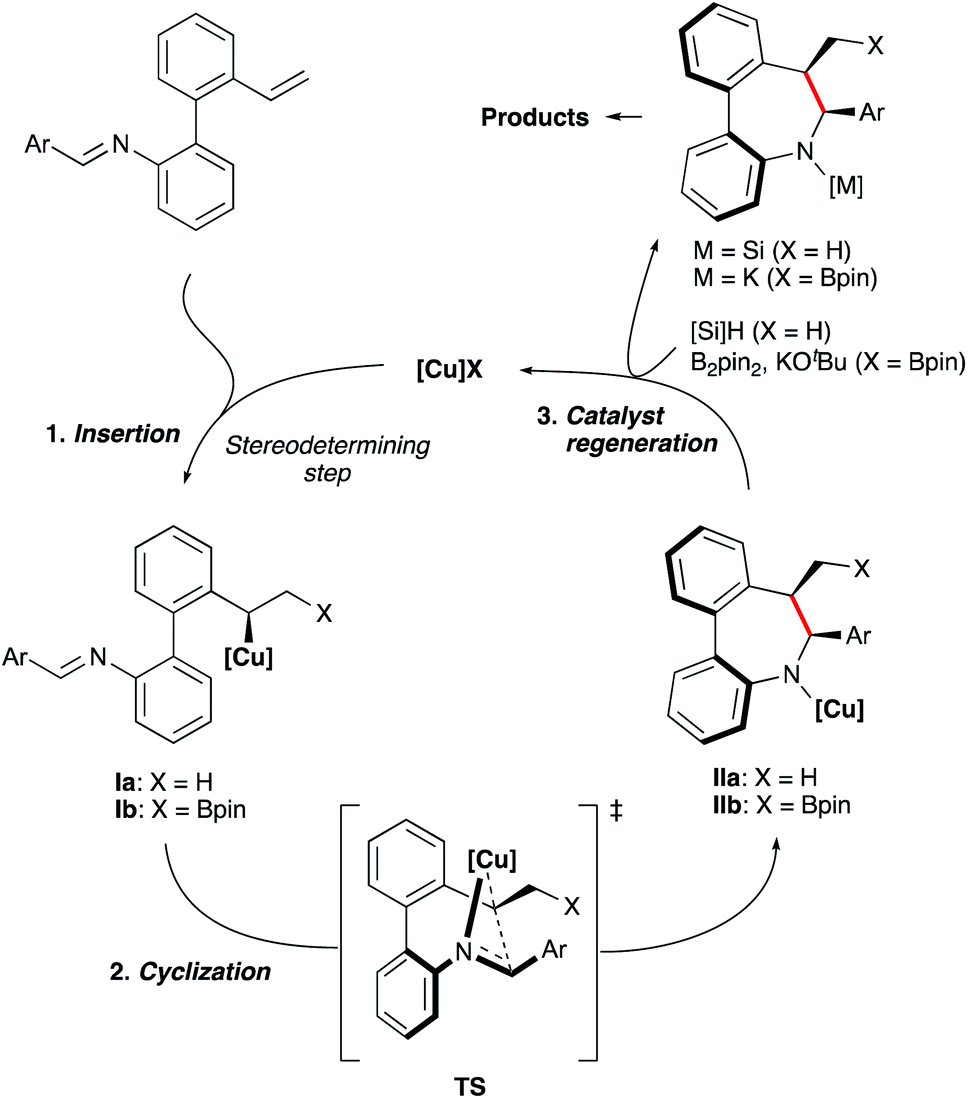 | ||
| Scheme 8 Catalytic cycle and stereochemical model. | ||
Conclusions
We have successfully developed a straightforward approach for the enantioselective synthesis of dibenzo[b,d]azepines featuring central and axial chirality elements by means of Cu-catalyzed asymmetric intramolecular reductive or borylative cyclizations. Both reactions proceed with good yields and excellent diastereo- and enantioselectivities under mild conditions. Axially chiral biaryl-2-amines could also be obtained in nearly perfect enantioselectivity through a kinetic resolution process. Computational work indicates that the axial chirality is thermodynamically controlled, and that the sense of the axial chirality depends on the nature of the imine R group.Data availability
All experimental and computational data associated with this study can be found in the article or in the ESI.†Author contributions
R. F., J. M. L. and V. H. conceived and supervised the study. P. R.-S., R. M. C. and V. R. performed the experiments and analyzed the data. V. H. and J. M. L. wrote the manuscript. P. M. performed the computational studies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Spanish Ministerio de Ciencia e Innovación (Grants PID2019-106358GB-C21, PID2019-106358GB-C22, PID2019-104090RB-100, contracts RYC-2017-22294 for V.H. and BES-2017-081561 for P. R-S), European funding (ERDF), Junta de Andalucía (Grants P18-FR-3531, P18-FR-644, US-1262867, and US-1260906), and Aragon Government (Grupos E34_20R). Computational resources from the super-computers “Memento” and “Cierzo” provided by BIFI-ZCAM (University of Zaragoza, Spain) are also acknowledged. We also thank Dr Francisco José Fernández de Córdova for X-ray structure determination of compounds 2Aj, 2Ag and 4Aa.Notes and references
- (a) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563–639 CrossRef CAS PubMed; (b) J. E. Smyth, N. M. Butler and P. A. Keller, Nat. Prod. Rep., 2015, 32, 1562–1583 RSC; (c) J. Wencel-Delord, A. Panossian, F. R. Leroux and F. Colobert, Chem. Soc. Rev., 2015, 44, 3418–3430 RSC; (d) Y.-B. Wang and B. Tan, Acc. Chem. Res., 2018, 51, 534–547 CrossRef CAS PubMed; (e) J. K. Cheng, S.-H. Xiang, S. Li, L. Ye and B. Tan, Chem. Rev., 2021, 121, 4805–4902 CrossRef CAS PubMed; (f) J. M. Lassaletta, Atropisomerism and Axial Chirality, World Scientific, New Jersey, 2019 CrossRef.
- G. Bringmann, A. J. P. Mortimer, P. A. Keller, M. J. Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384–5427 CrossRef CAS PubMed.
- S. L. Pira, T. W. Wallace and J. P. Graham, Org. Lett., 2009, 11, 1663–1666 CrossRef CAS PubMed.
- (a) S. Superchi, D. Casarini, A. Laurita, A. Bavoso and C. Rosini, Angew. Chem., Int. Ed., 2001, 40, 451–454 CrossRef CAS; (b) Y. Zhang, Y.-Q. Liu, L. Hu, X. Zhang and Q. Yin, Org. Lett., 2020, 22, 6479–6483 CrossRef CAS PubMed; (c) Y. Guo, M.-M. Liu, X. Zhu, L. Zhu and C. He, Angew. Chem., Int. Ed., 2021, 60, 13887–13891 CrossRef CAS PubMed.
- Y. Zhang, Z. Chang, H. Zhao, S. Crespi, B. L. Feringa and D. Zhao, Chem, 2020, 6, 2420–2429 CAS.
- B. Zhang, M. Bao, C. Zeng, X. Zhong, L. Ni, Y. Zeng and X. Cai, Org. Lett., 2014, 16, 6400–6403 CrossRef CAS PubMed.
- G. T. Wong, D. Manfra, F. M. Poulet, Q. Zhang, H. Josien, T. Bara, L. Engstrom, M. Pinzon-Ortiz, J. S. Fine, H. J. Lee, L. Zhang, G. A. Higgins and E. M. Parker, J. Biol. Chem., 2004, 279, 12876–12882 CrossRef CAS PubMed.
- J. S. Nair, T. Sheikh, A. L. Ho and G. K. Schwartz, Anticancer Res., 2013, 33, 1307–1316 CAS.
- L. Keller, S. Beaumont, J.-M. Liu, S. Thoret, J. S. Bignon, J. Wdzieczak-Bakala, P. Dauban and R. H. Dodd, J. Med. Chem., 2008, 51, 3414–3421 CrossRef CAS PubMed.
- W. Zaharevitz, R. Gussio, M. Leost, A. M. Senderowicz, T. Lahusen, C. Kunick, L. Meijer and E. A. Sausville, Cancer Res., 1999, 59, 2566–2569 Search PubMed.
- (a) L. A. Saudan, G. Bernardinelli and E. P. Kündig, Synlett, 2000, 483–486 CAS; (b) C. A. Cheetham, R. S. Massey, S. L. Pira, R. G. Pritchard and T. W. Wallace, Org. Biomol. Chem., 2011, 9, 1831–1838 RSC; (c) S. Postikova, M. Sabbah, D. Wightman, I. T. Nguyen, M. Sanselme, T. Besson, J.-F. Briere, S. Oudeyer and V. Levacher, J. Org. Chem., 2013, 78, 8191–8197 CrossRef CAS PubMed; (d) P. C. Bulman Page, C. A. Pearce, Y. Chan, P. Parker, B. R. Buckley, G. A. Rassias and M. R. Elsegood, J. Org. Chem., 2015, 80, 8036–8045 CrossRef CAS PubMed.
- (a) T. Saget and N. Cramer, Angew. Chem., Int. Ed., 2013, 52, 7865–7868 CrossRef CAS PubMed; (b) C. G. Newton, E. Braconi, J. Kuziola, M. D. Wodrich and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 11040–11044 CrossRef CAS PubMed.
- H. Hu, Y. Peng, T. Yu, S. Cheng, S. Luo and Q. Zhu, Org. Lett., 2021, 23, 3636–3640 CrossRef CAS PubMed.
- J. Liu, X. Yang, Z. Zuo, J. Nan, Y. Wang and X. Luan, Org. Lett., 2018, 20, 244–247 CrossRef CAS PubMed.
- For the synthesis of related dibenzo[c,e]azepine derivatives (N atom not attached to the biaryl moiety) see: (a) S. P. France, G. A. Aleku, M. Sharma, J. Mangas-Sanchez, R. M. Howard, J. Steflik, R. Kumar, R. W. Adams, I. Slabu, R. Crook, G. Grogan, T. W. Wallace and N. J. Turner, Angew. Chem., Int. Ed., 2017, 56, 15589–15593 CrossRef CAS PubMed; (b) T. Yang, X. Guo, Q. Yin and X. Zhang, Chem. Sci., 2019, 10, 2473–2477 RSC; (c) S. Zhang, F. Chen, Y.-M. He and Q.-H. Fan, Org. Lett., 2019, 21, 5538–5541 CrossRef CAS PubMed; (d) T. Kano, H. Sugimoto and K. Maruoka, J. Am. Chem. Soc., 2011, 133, 18130–18133 CrossRef CAS PubMed . See also ref. 4b..
- E. Ascic and S. L. Buchwald, J. Am. Chem. Soc., 2015, 137, 4666–4669 CrossRef CAS PubMed.
- D. Li, J. Kim, J. W. Yang and J. Yun, Chem.–Asian J., 2018, 13, 2365–2368 CrossRef CAS PubMed.
- CCDC 2095490 [(Sa,6S,7R)-2Aj], 2095492 [(Sa,6S,7R)-2Ag] and 2095491 [(Sa,6S,7R)-4Aa].†.
- R. W. Sinkeldam, N. Greco and Y. Tor, Chem. Rev., 2002, 110, 2579–2619 CrossRef PubMed.
- J. Messinger and V. Buss, J. Org. Chem., 1992, 57, 3320–3328 CrossRef CAS.
- S. Saebo and J. E. Boggs, J. Mol. Struct.: THEOCHEM, 1982, 87, 365–373 CrossRef.
- Y. Yang, S.-L. Shi, D. Niu, P. Liu and S. L. Buchwald, Science, 2015, 349, 62–66 CrossRef CAS PubMed.
- Y. Xi and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 12758–12772 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental protocols, characterization data. CCDC 2095490–2095492. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04980a |
| This journal is © The Royal Society of Chemistry 2021 |