
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn unexpected non-conjugated AIEgen with a discrete dimer for pure intermolecular through-space charge transfer emission†
Xiujie
Jiang‡
a,
Wei
Tao‡
a,
Cheng
Chen
a,
Guoyong
Xu
a,
Haoke
Zhang
 *bcd and
Peifa
Wei
*ad
*bcd and
Peifa
Wei
*ad
aInstitutes of Physical Science and Information Technology, Key Laboratory of Structure and Functional Regulation of Hybrid Materials of Ministry of Education, Anhui Graphene Engineering Laboratory, Anhui University, Hefei, China. E-mail: pfwei@ahu.edu.cn
bMOE Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou, China
cZJU-Hangzhou Global Scientific and Technological Innovation Center, Hangzhou 311215, China. E-mail: zhanghaoke@zju.edu.cn
dGuangdong Provincial Key Laboratory of Luminescence from Molecular Aggregates, South China University of Technology, Guangzhou, China
First published on 22nd November 2021
Abstract
Manipulation of the charge transfer in donor–acceptor-type molecules is essential for the design of controllable aggregate luminescent materials. Apart from the traditional through-bond charge transfer (TBCT) systems which suffer from complicated structural design, poor tunability and low quantum efficiency, through-space charge transfer (TSCT) has been proved as an alternative yet facile strategy in tuning photophysical processes. In this work, by simply changing nucleophilic reaction bases, a traditional conjugated acrylonitrile AP1 and an unexpected non-conjugated AP2 with a carboxamide-functionalized oxirane linker could be obtained. The long-range π–π stacking in conjugated AP1 results in mixed intramolecular TBCT plus intermolecular TSCT emission. However, facilitated by the steric hindrance effect of the big oxirane connector and the unique discrete dimer packing, non-conjugated AP2 exhibits pure and efficient intermolecular TSCT emission in both aggregate and crystalline states. The flexibility of the non-conjugated character further leads to better reversible stimuli-responsiveness to mechanical force for AP2 than for the rigid AP1.
Introduction
Organic photoluminescent materials are widely used in numerous fields such as optoelectronic devices,1 bioimaging,2 medical diagnosis,3 chemical sensing,4etc. due to their favorable flexibility, biocompatibility, processability and structural diversity. Compared with a molecule in a molecularly dispersed state, its aggregate state exhibits macroscopic optical properties and functions that are completely different from those of a single molecule owing to a variety of intermolecular interactions. For example, aggregation-caused quenching (ACQ) is a typical representative,5 which is characterized by high luminescence in solution, but weak or no emission in the aggregate state. This is unfavorable for the study of the luminescence mechanism and the regulation of luminescence behavior in the aggregate state. The aggregation-induced emission (AIE) phenomenon proposed by Tang is completely the opposite: fluorescent molecules do not emit light in dilute solution but exhibit significant fluorescence enhancement in the aggregate state.6,7 This brings the research on photoluminescent materials from molecular science to aggregate science.8–11From the perspective of aggregate science, how to achieve high-efficiency and controllable luminescence in aggregates through structural design is of great significance for the understanding of the luminescence mechanism and the extension of luminescence systems. Constructing charge transfer (CT) systems is a common and effective method to control the emission wavelength and efficiency.12–14 So far, most of the luminescence systems with CT characteristics have been based on through-bond charge transfer (TBCT) emission.15–17 The organic fluorophores mainly connect the electron donors (D) and electron acceptors (A) through double bonds, triple bonds, heteroatoms, or aromatic rings to form structures with larger π conjugation, which enables strong electron coupling between the donor and acceptor through covalent bonds.18,19 However, the strong TBCT effect in the conjugate system often leads to redshift emission accompanied by a significant decrease in quantum efficiency, which is unfavorable for practical applications.20,21 Therefore, the development of new methods to balance the CT intensity is essential for the design of controllable aggregate luminescent materials.
In recent years, through-space charge transfer (TSCT), which connects donors and acceptors in a spatially separate manner, has been widely adopted to effectively avoid the negative effects of strong CT.22–27 In this motif, donors and acceptors are separated but spatially close to each other, which allows the through-space charge transfer to occur.28–30 The currently studied TSCT system mainly relies on the intramolecular through-space interaction between the donor and acceptor.31 However, this type of material extremely relies on a tailored structure design, especially the spatial distance of the donor and acceptor, which has a great influence on the photophysics.32 This also brings multiple synthetic steps, which not only leads to sophisticated and tedious purification but is also time-consuming and economically unfriendly. What is worse, most of the TSCT systems still rely on large D–A conjugated π structures, which lead to high rigidity of the structures, and further bring poor solubility and difficulties in luminescence modulation.33 Though the D–A structure connected by non-conjugated bonds can increase the molecular flexibility, it cannot regulate the intramolecular TSCT precisely, and some of them cannot even form stable intramolecular TSCT. However, in the aggregate state, the flexibility of the molecules can endow the system with enhanced intermolecular TSCT effect.34,35 Therefore, constructing intermolecular TSCT is an effective strategy for regulating the photophysical properties of luminescent materials in aggregate states, which is easier and more controllable than the manipulation of intramolecular TSCT in the molecular state.
In this work, by simply changing the nucleophilic reaction conditions, specifically, by only tuning the base via the transition metal-free, nonhazardous, nontoxic, and atom-economic synthetic method, we were able to simply access conjugated acrylonitriles AP1 and non-conjugated AP2 based on anthracene (D) and pyridinium salt (A), as shown in Scheme 1. It is found that AP2 exhibits unique and pure intermolecular TSCT emission in both aggregate and crystalline states based on the formation of a discrete dimer. The existence of a big carboxamide-functionalized oxirane linker in AP2 prevents neighboring molecules from approaching the dimer, while long-range π–π stacking is observed in the conjugated AP1, leading to the higher emission efficiency of AP2 than AP1. Besides, the synergy of flexibility of the non-conjugated linker and the comparative weak intermolecular interactions between the discrete dimer endows AP2 with reversible stimuli-responsiveness to mechanical force, which is absent in the conjugated AP1.
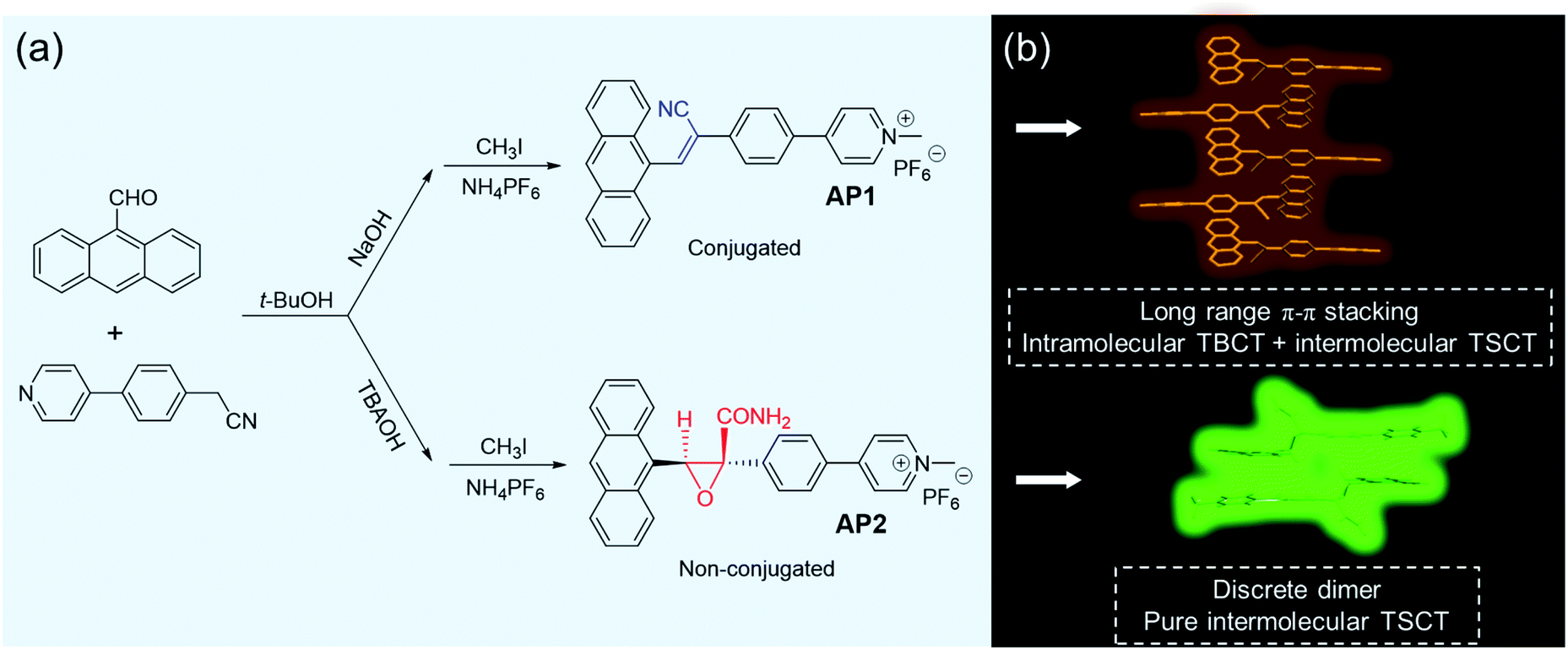 | ||
| Scheme 1 (a) Synthesis of AP1 and AP2 and (b) their emission illustrations in the aggregate state. | ||
Results and discussion
The Knoevenagel condensation is a very important transformation in organic synthesis catalyzed by bases and is widely employed for carbon–carbon bond formation, which has been used in connecting donors and acceptors to deliver conjugated AIE-active molecules.36–39 In this work, anthracene was used as the donor while a typical pyridine salt was chosen as the acceptor. Considering that the base may have a strong effect on the efficiency of the reaction, we chose two different bases, that is, an organic base tetrabutylammonium hydroxide (TBAOH) and an inorganic base NaOH, to catalyze the reaction. However, to our great surprise, though both of the reactions proceeded in t-BuOH at room temperature with different bases, their reaction phenomena were quite different. The NaOH system yielded yellow precipitates which are often observed in Knoevenagel condensation reactions, indicating that we obtained a traditional π-conjugated acrylonitrile derivative, while the TBAOH system did not generate the expected precipitates but produced a clear light yellow solution instead, which was proved later to be a non-conjugated structure (Scheme 1). The difference between the two structures is the connectors between the donor and acceptor. The conjugated one was bridged by a cyanostilbene while the non-conjugated system was linked by a carboxamide-functionalized oxirane. It is noteworthy that both compounds can be obtained easily with satisfactory yields. As far as we know, it is scarcely reported that different linkages (conjugated or non-conjugated) between donors and acceptors can be realized by simply tuning the nucleophilic reaction base. Both of the compounds were further functionalized with pyridine salts, which were named AP1 and AP2, respectively. The detailed synthetic procedures, characterizations and thermal stability are summarized in the ESI (Fig. S1–S14, Schemes S1 and S2†).It is reasonable that AP1 can be formed by a direct nucleophilic reaction under the NaOH condition. Why was the non-conjugated AP2 obtained under the condition of TBAOH? After dissolving π-conjugated 1 in solvents and adding TBAOH as the base, we observed that 1 could be transformed into 2 which was similar to the alkaline epoxidation of acrylonitrile as most of the references reported.40–42 However, a key oxidant was missing in this system. The anthracene substrate would likely produce reactive oxygen species (ROS) in the presence of light which may be the oxidant.43 This hypothesis was confirmed by repeating the reaction in the absence of light and oxygen which resulted in the expected π-conjugated acrylonitrile derivative 1 (Fig. S7†). The whole process can thus be divided into two steps: the first step involved Knoevenagel condensation to yield 1 as an intermediate; in the second step, 1 would hydrolyze to form acrylamide 3 with a cis configuration (Scheme S3†). The anthracene part would produce ROS in the presence of light which can further oxidize 3 to deliver the target cis-2 with the amide and anthracene groups on the same face of the oxirane.
After confirming the molecular structure, we then investigated the photophysics of AP1 and AP2. The color of AP1 in CH3CN was yellow while AP2 was colorless, which suggests better conjugation in AP1. Absorption spectra of AP1 and AP2 were further studied in CH3CN to confirm their electronic structures. AP1 in the crystalline state showed one structureless broad peak located at 408 nm corresponding to the extended through-bond conjugation (TBC), while AP2 exhibited one finely structured peak at 379 nm which almost overlapped with the maximum absorption of anthracene, suggesting the non-conjugated nature of AP2 (Fig. 1A and S15†). The absorption maximum of anthracene was hypsochromically shifted by 10 nm from that of AP2 and was located at 369 nm, which may be due to the hyperconjugation between the benzene ring and the methyl groups in AP2.44,45 Similarly, though the molecule contains a donor and an acceptor, its CH3CN solution only shows the characteristic fine structure emission of monomeric anthracene ranging from 370 nm to 500 nm (Fig. 1B),46 which indicates that the two non-conjugated units of AP2 cannot form inter- or intramolecular D–A interactions in the solution state.
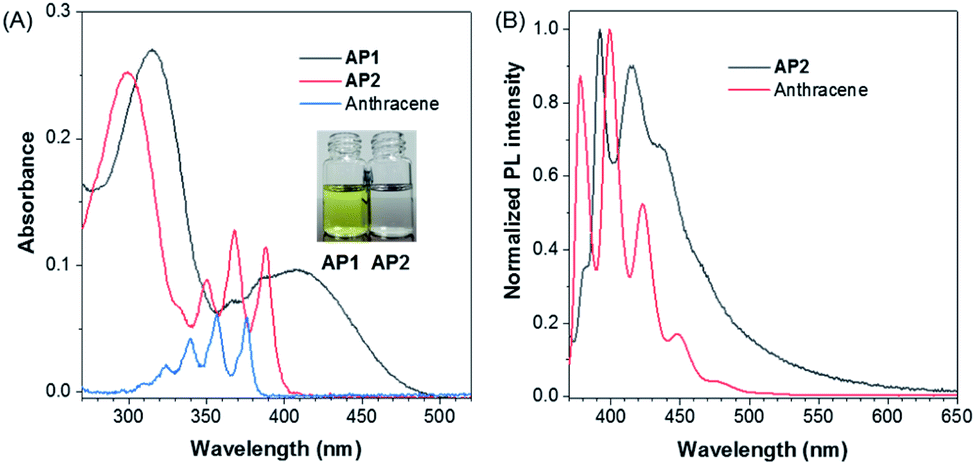 | ||
| Fig. 1 (A) UV/Vis absorption spectra of AP1, AP2 and anthracene in CH3CN solution (c = 10 μM), respectively. Insets in (A): photos of AP1 (left) and AP2 (right) in CH3CN solution under daylight. (B) Normalized emission spectra of anthracene and AP2 in CH3CN solution, λex = 350 nm. | ||
To investigate their photophysical properties in the aggregate state, the PL spectra were recorded in mixed solvents through the addition of the poor solvent hexane to the THF solution of AP2. It can be seen from the UV spectra that the profile of AP2 in THF/hexane mixtures with hexane fraction (fHex) of 99% was slightly red-shifted compared to that of AP2 in THF (Fig. 2A), suggesting the existence of weak intermolecular interaction in AP2 aggregates. The absorption spectrum of AP2 in the crystalline state shows an intense, broad absorption with λmax = 420 nm which is redder than that of the aggregate at fHex = 99%, indicating a different packing behavior in the crystalline state compared to aggregates. Considering the non-conjugated donor and acceptor character of AP2, it is speculated that intermolecular TSCT may lead to a broad redder absorption as the intramolecular D–A distance is too large for intramolecular TSCT. Its fluorescence spectra provide more detailed information. Fig. 2B shows that the PL spectrum in THF/hexane mixtures with low fHex exhibited a broad emission with multiple peaks at 391, 415 and 437 nm, which was assigned to anthracene emission. The intensity of the shorter-wavelength peak decreased gradually when fHex increased from 0% to 50%, which is ascribed to the polarity effect. However, a marvelous new emission band at a longer wavelength ranging from 500 to 700 nm intensified progressively with increasing the hexane fraction at fHex ≥ 60%, indicating the AIE nature of this longer-wavelength emission. The maximum intensity enhancement was 28-fold at fHex = 80% (Fig. 2C). The second-stage intensity annihilation of the short-wavelength at fHex > 50% should be ascribed to the energy transfer from TBC to intermolecular TSCT.
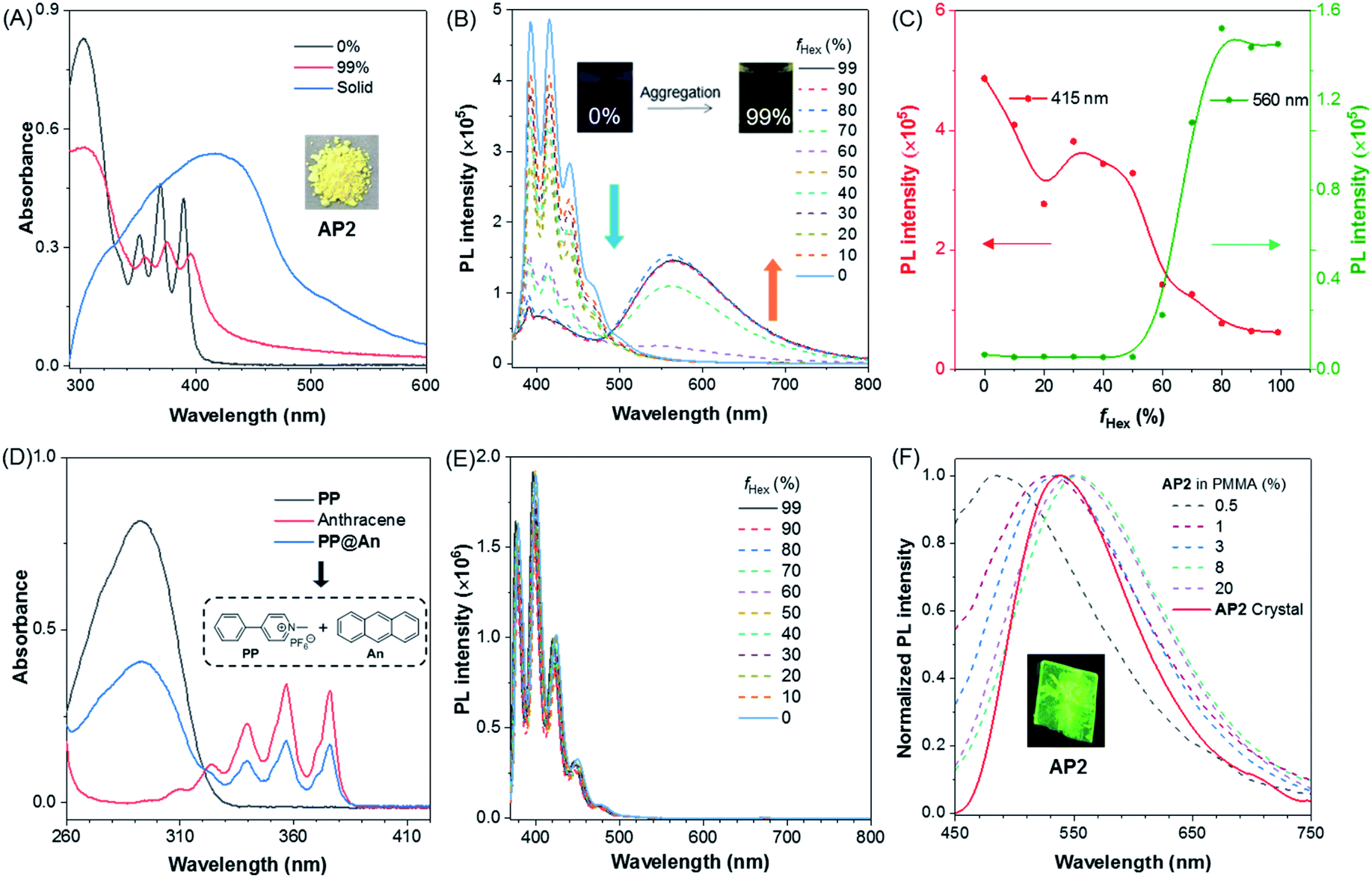 | ||
| Fig. 2 (A) Absorption spectra of AP2 in different fHex and crystalline states, c = 10 μM. Inset: roomlight image of AP2 powder. (B) Emission spectra of AP2 at different fHex, λex = 350 nm. Inset: fluorescence photo of AP2 at fHex = 0% and 99%, respectively. (C) Emission intensity of AP2 at λ = 415 nm and 560 nm with gradually increasing fHex. (D) Absorption spectra of PP, anthracene, and PP@An in CH3CN solution, c = 10 μM. (E) Emission spectra of PP@An at different fHex, λex = 350 nm. An = anthracene. (F) Emission spectra of AP2 in the crystal state and at different concentrations (wt%) in PMMA thin film, λex = 350 nm. Inset: fluorescence photo of AP2 taken under a UV lamp. | ||
As mentioned above, the intermolecular TSCT in AP2 aggregates was proposed as the cause of longer-wavelength emission around 560 nm. However, the polarity effects,47,48 and intramolecular TSCT between anthracene and pyridinium salt are two more possibilities that induced the redder emission. The solvent polarity effect that may contribute to the longer-wavelength emission was first ruled out by comparing the emissions in solvents with different polarities (Fig. S16†). The emission maximum of AP2 shows almost no change in high polarity MeOH and low polarity THF solvents. The characteristic red-shifted emission band observed in the aggregate state is not detected. To disclose the possibility of intramolecular TSCT, the PL spectrum of AP2 in dilute THF solution (c = 10−6 mol L−1) was measured at room temperature (rt) and 77 K, respectively. As shown in Fig. S17,† from rt to 77 K, the emission intensity corresponding to the anthracene moiety is increased as nonradiative deactivation pathways are restricted, while the wavelength of the maxima emission remains unchanged and no new peak around 560 nm is observed, suggesting the negligible contribution of intramolecular TSCT to the longer-wavelength emission of AP2.
To explore the role of the non-conjugated linker in generating efficient intermolecular TSCT, the donor and acceptor were physically separated instead of chemically isolating them. Then, the new compound PP was designed as the donor (Scheme S4†). The 1H NMR spectrum of AP2 shows that it is not a simple superposition of that of anthracene and PP (Fig. S18A, C and D†). In contrast, the 1H NMR spectrum of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio mixture of anthracene and PP, named PP@An, is exactly the superposition of the two compounds. The above results provide two conclusions: the first one is that oxirane plays a very important role in the properties of AP2; the second one is that simple physical mixing of anthracene and PP cannot produce any intermolecular interactions. In addition, the UV absorption spectra of the model compound PP, anthracene and PP@An, as well as their corresponding fluorescence emission spectra, were recorded (Fig. 2D and S19†). We found that both the absorption and fluorescence spectra of PP@An were simply superimposition of that of PP and anthracene. Neither tuning the molar ratio of PP and anthracene nor changing the concentration of PP@An from 10−7 to 10−3 mol L−1 could generate any new absorption or emission peaks in the solution state (Fig. S20†). Meanwhile, the PL spectra of PP@An in THF/hexane mixtures with different fHex were also measured but without longer-wavelength emission in the aggregate state (Fig. 2E). All these results prove the vital role of the non-conjugated oxirane linker in the generation of intermolecular TSCT for AP2. Physical blending of the donor and acceptor is incapable of producing efficient TSCT.
:1 molar ratio mixture of anthracene and PP, named PP@An, is exactly the superposition of the two compounds. The above results provide two conclusions: the first one is that oxirane plays a very important role in the properties of AP2; the second one is that simple physical mixing of anthracene and PP cannot produce any intermolecular interactions. In addition, the UV absorption spectra of the model compound PP, anthracene and PP@An, as well as their corresponding fluorescence emission spectra, were recorded (Fig. 2D and S19†). We found that both the absorption and fluorescence spectra of PP@An were simply superimposition of that of PP and anthracene. Neither tuning the molar ratio of PP and anthracene nor changing the concentration of PP@An from 10−7 to 10−3 mol L−1 could generate any new absorption or emission peaks in the solution state (Fig. S20†). Meanwhile, the PL spectra of PP@An in THF/hexane mixtures with different fHex were also measured but without longer-wavelength emission in the aggregate state (Fig. 2E). All these results prove the vital role of the non-conjugated oxirane linker in the generation of intermolecular TSCT for AP2. Physical blending of the donor and acceptor is incapable of producing efficient TSCT.
The aggregation behavior is replicated for AP2 in five different films which were fabricated by spin-coating of the solution with AP2 and poly(methyl methacrylate) (PMMA), the obtained films possess different doping ratios of 0.3%, 1%, 3%, 8% and 20% (AP2/PMMA, wt%). With the gradual addition of AP2, the emission wavelength of the thin film gradually redshifts from 486 nm to 550 nm (Fig. 2F), which further confirms the formation of intermolecular TSCT at high AP2 concentration. In the crystalline state, AP2 exhibited one comparatively sharp and green emission peak at 537 nm (Fig. 2F). The resulting long lifetime of the crystal (45.30 ns) was a possible characteristic of TSCT emission (Fig. S21† and Table 1). Variation in excitation could change only the emission intensity but not the wavelength of the luminophore (Fig. S22†), which suggests the uniform and stabilized TSCT structure in the AP2 crystal.
| Comp. | λ ab,soln (nm) | λ ab,crystal (nm) | λ em,crystal/ground (nm) | Φ crystal (%) | τ crystal (ns) |
|---|---|---|---|---|---|
| a Abbreviation: λab,soln = absorption maximum in CH3CN solution, λab,crystal = absorption maximum in crystalline powder, λem,crystal/ground = emission maximum in crystal and ground powder states, Φcrystal = fluorescence quantum yield of the crystal measured with an integrating sphere, τcrystal = fluorescence lifetime of the crystal. | |||||
| AP1 | 408 | 482 | 623/620 | 1.13 | 2.29 |
| AP2 | 379 | 420 | 537/581 | 18.45 | 45.3 |
The detailed structure–property relationship was revealed by single-crystal analysis. Yellow crystals were obtained by slow vapor diffusion of acetone into a n-pentane solution of AP2 over a period of night at room temperature (see Table S2† for details). The anthracene and pyridine salt units are nearly spatially parallel in the crystal, and arrange into step-like offset columnar stacks of head-to-tail antiparallel dimers with close intermolecular π–π stacking (3.442 Å) and high degree of molecular overlap (Fig. 3A–F). The multiple hydrogen bonds resulting from carboxamides between the adjacent discrete dimer and column together with the nearest C–H⋯π (≈3.257 and 3.635 Å) of adjacent columns should be the main driving force for the formation of a 3D structure (Fig. S23 and S24†). Within each column, the slip angle of the dimer along the long molecular axis is 47.8°, while the similar slip angle of 54.6° along the short axis is also observed, resulting in a relatively large distance between the two dimers (5.445 Å). This suggests that each TSCT dimer in the crystal lattice is discrete. The crystal structure indicates that the oxirane linker and its big carboxamide substituent prevent neighboring molecules from approaching the dimer and forming the detrimental long-range π–π stacking, which results in the formation of a discrete dimer and considerable quantum efficiency (18.45%) in the crystalline state. It is noteworthy that AP2 is racemic as R and S configurations coexist in one unit cell, suggesting the absence of stereospecificity during the reaction (Fig. S25†). The two racemic structures benefit the close arrangement of two chromophores to form the discrete dimer and thus the generation of intermolecular TSCT. Electronic excited-state delocalization of the donor and acceptor through spatial interactions results in the significantly red-shifted emission of AP2 crystal compared to its solution.
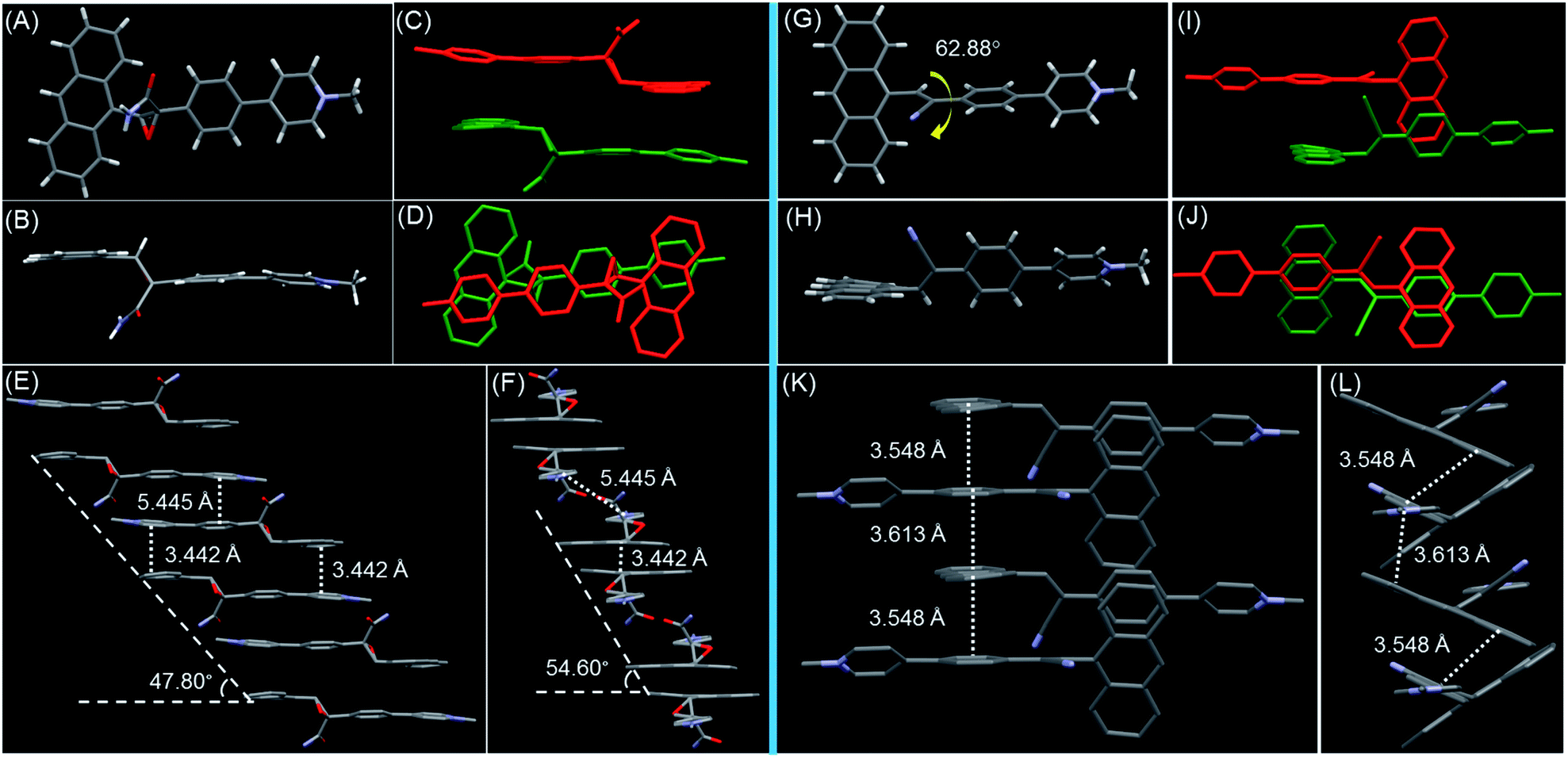 | ||
| Fig. 3 AP2 crystal analysis (left column). The molecular conformation in (A) top and (B) side view. (C) The dimer formed through TSCT viewed along the long molecule axis. (D) The overlapping in the dimer. Partial view of the molecular packing along the (E) long and (F) short molecule axes. AP1 crystal analysis (right column). The molecular conformation in (G) top and (H) side view. The molecular overlap between adjacent molecules viewed in (I) side and (J) top view. Partial view of the molecular packing along the (K) long and (L) short molecule axes. Hydrogen atoms in (C)–(F) and (I)–(L) are omitted for the sake of clarity. The slip angles and the intermolecular distance are listed. | ||
Considering the better flexibility of non-conjugated bonds in AP2 than the conjugated AP1, the TSCT emission of AP2 is anticipated to exhibit more obvious reversible response to external stimuli. Single crystals of AP1 were also obtained by slow vapor diffusion of i-Pr2O into its CH3CN solution as a comparison (see Table S1† for details). The dihedral angle between anthracene and the cyanostilbene approaches 62.88° (Fig. 3G). Different from the formation of the discrete dimer in AP2, infinite stacks with a small slip along both the short and long axes in AP1 were observed in which offset head-to-tail antiparallel π-overlap of neighboring molecules by intermolecular π–π interactions (3.548 and 3.613 Å) occurred (Fig. 3K and L). The counterions in AP1 and AP2 play the role of a linker between two adjacent columns during the crystal packing (Fig. S26†). We can thus conclude that the reddish orange emission with much lower quantum efficiency (1.13%) in the crystalline state for AP1 than that for AP2 should be ascribed to the mixed intramolecular TBCT plus intermolecular TSCT effect. Considering the possibility of photoreaction of anthracene, we record the emission profiles of AP1 and AP2 in fHex = 99% (aggregate state) and crystalline state with different scan times (Fig. S27†). The slight or no change of profiles indicates that its influence on TSCT emission during the fluorescence study is almost negligible.
As per our expectation, the intermolecular interactions in conjugated AP1 were stronger than those in non-conjugated AP2. Their stimulus-responsive behavior under mechanical force was thus investigated. After grinding, the emission of AP1 shows almost no change (Fig. 4A and B). The powder X-ray diffraction (PXRD) measurement confirmed that the grinding powder of AP1 was still crystalline (Fig. 4D), indicating that the strong π–π interaction prevented the mechanical force from destroying the intermolecular forces. However, the stimulus-response behavior of AP2 was completely different from that of AP1. The different emission of the original crystal and the grinding powder is large enough to be easily distinguished by the naked eye with fluorescence changes from green to yellow (Fig. 4A and C). The broad and featureless PXRD pattern for the ground samples of AP2 reflects its amorphous characteristic (Fig. 4E). On further fumigation with acetone, both the PXRD and emission of the ground powder returned to their original state. This indicates that the synergy of flexibility of the non-conjugated linker and the comparative weak intermolecular interactions between the discrete dimer leads to better stimuli-responsiveness of AP2, which makes the manipulation of the aggregate structures and properties realizable.
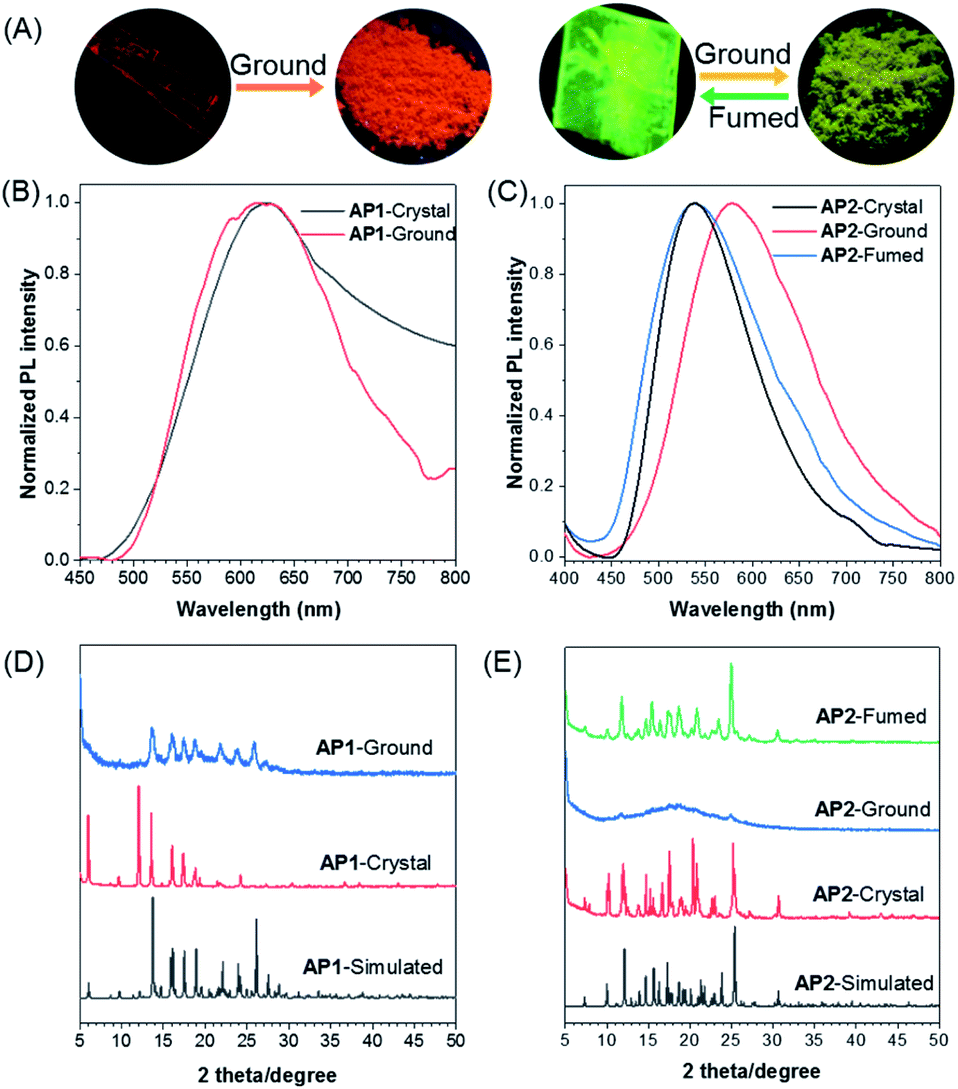 | ||
| Fig. 4 (A) Fluorescence photographs of the crystal and ground powder of AP1 (left); fluorescence photographs of the crystal, ground powder and the fumed powder of AP2 (right). Normalized emission spectra of (B) AP1 and (C) AP2 crystals upon external stimuli, λex = 360 nm. PXRD patterns of (D) AP1 and (E) AP2 upon external stimuli. | ||
Conclusions
In this work, we found that through simply tuning the nucleophilic reaction bases, a traditional conjugated acrylonitrile AP1 and an unexpected non-conjugated AP2 with carboxamide-functionalized oxirane linkage could be obtained. Interestingly, the long-range π–π stacking in conjugated AP1 results in mixed intramolecular TBCT plus intermolecular TSCT emission, while AP2 with discrete dimer packing could exhibit pure and efficient intermolecular TSCT emission in both aggregate and crystalline states. The physical mixing of anthracene and model compound PP does not work, indicating the vital role of the non-conjugated oxirane linker in producing efficient TSCT. The synergy of flexibility of the non-conjugated linker and the comparative weak intermolecular interactions between the discrete dimer led to better reversible stimuli-responsiveness to mechanical force for AP2 than that for the rigid AP1. This work not only provides a simple and smart way for regulating photophysical properties in the aggregate state but also introduces an efficient method to develop stimuli-responsive luminogens.Experimental section
Materials and methods, synthetic procedures, crystallographic data and characterizations are available in the ESI.†Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
X. J., W. T., H. Z. and P. W. conceived and designed the experiments. X. J. and W. T. performed the experiments. X. J., W. T., H. Z. and P. W. analyzed the data. W. T. and C. C. contributed the single crystal analysis. G. X. contributed the 1H NMR experiments. X. J., H. Z. and P. W. co-wrote the paper.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China Grants (22001006), the Open Fund of Guangdong Provincial Key Laboratory of Luminescence from Molecular Aggregates and South China University of Technology (2019B030301003). P. W. is thankful for the support from the Innovation and Entrepreneurship Project of Overseas Returnees in Anhui Province (2020LCX017) and the Open Project of Key Laboratory of Structure and Functional Regulation of Hybrid Materials of Anhui University, Ministry of Education. H. Z. is thankful for the support from the Fundamental Research Funds for the Central Universities (2021QNA4032).Notes and references
- J. X. Wang, B. Y. Liang, J. B. Wei, Z. Q. Li, Y. C. Xu, T. Yang, C. L. Li and Y. Wang, Angew. Chem., Int. Ed., 2021, 60, 15335–15339 CrossRef CAS PubMed.
- E. H. K. Stelzer, Nat. Methods, 2015, 12, 23–26 CrossRef CAS PubMed.
- M. R. Loizzo, R. Tundis, F. Conforti, G. A. Statti and F. Menichini, Pharm. Biol., 2009, 47, 516–520 CrossRef.
- L. Chen, M. Nakamura, T. D. Schindler, D. Parker and Z. Bryant, Nat. Nanotechnol., 2012, 7, 252–256 CrossRef CAS PubMed.
- W. Watson and R. Livingston, Nature, 1948, 162, 452–453 CrossRef CAS PubMed.
- J. Mei, Y. N. Hong, J. W. Y. Lam, A. J. Qin, Y. H. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- J. D. Luo, Z. L. Xie, J. W. Y. Lam, L. Cheng, H. Y. Chen, C. F. Qiu, H. S. Kwok, X. W. Zhan, Y. Q. Liu, D. B. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- Z. Zhao, H. K. Zhang, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9888–9907 CrossRef CAS PubMed.
- H. K. Zhang, Z. Zhao, A. T. Turley, L. Wang, P. R. McGonigal, Y. J. Tu, Y. Y. Li, Z. Y. Wang, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Adv. Mater., 2020, 32, 2001457 CrossRef CAS PubMed.
- Q. Peng and Z. Shuai, Aggregate, 2021, 2, e91 Search PubMed.
- S. Ma, S. Du, G. Pan, S. Dai, B. Xu and W. Tian, Aggregate, 2021, 2, e96 Search PubMed.
- J.-L. Brédas, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971–5004 CrossRef PubMed.
- P. Roy, A. Jha, V. B. Yasarapudi, T. Ram, B. Puttaraju, S. Patil and J. Dasgupta, Nat. Commun., 2017, 1716 CrossRef PubMed.
- C. J. Chen, R. J. Huang, A. S. Batsanov, P. Pander, Y. T. Hsu, Z. G. Chi, F. B. Dias and M. R. Bryce, Angew. Chem., Int. Ed., 2018, 57, 16407–16411 CrossRef CAS PubMed.
- M. Numata, T. Yasuda and C. Adachi, Chem. Commun., 2015, 51, 9443–9446 RSC.
- K. Shizu, H. Tanaka, M. Uejima, T. Sato, K. Tanaka, H. Kaji and C. Adachi, J. Phys. Chem. C, 2015, 119, 1291–1297 CrossRef CAS.
- Q. Xue and G. H. Xie, Adv. Opt. Mater., 2021, 9, 2002204 CrossRef CAS.
- Y. Yamaguchi, Y. Matsubara, T. Ochi, T. Wakamiya and Z.-i. Yoshida, J. Am. Chem. Soc., 2008, 130, 13867–13869 CrossRef CAS PubMed.
- B.-K. An, J. Gierschner and S. Y. Park, Acc. Chem. Res., 2012, 45, 544–554 CrossRef CAS PubMed.
- J. Z. Zhao, W. H. Wu, J. F. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC.
- S. J. Liu, H. K. Zhang, Y. Y. Li, J. K. Liu, L. L. Du, M. Chen, R. T. K. Kwok, J. W. Y. Lam, D. L. Phillips and B. Z. Tang, Angew. Chem., Int. Ed., 2018, 57, 15189–15193 CrossRef CAS PubMed.
- Y. J. Luo, Z. G. Pang, C. Li, K. Chen, X. J. Zheng, Y. Huang and Z. Y. Lu, J. Mater. Chem. C, 2020, 8, 11603–11609 RSC.
- S. Y. Shao and L. X. Wang, Aggregate, 2020, 1, 45–56 CrossRef.
- M. K. Etherington, N. A. Kukhta, H. F. Higginbotham, A. Danos, A. N. Bismillah, D. R. Graves, P. R. McGonigal, N. Haase, A. Morherr, A. S. Batsanov, C. Pflumm, V. Bhalla, M. R. Bryce and A. P. Monkman, J. Phys. Chem. C, 2019, 123, 11109–11117 CrossRef CAS PubMed.
- L. Salah, M. K. Etherington, A. Shuaib, A. Danos, A. A. Nazeer, B. Ghazal, A. Prlj, A. T. Turley, A. Mallick, P. R. McGonigal, B. F. E. Curchod, A. P. Monkman and S. Makhseed, J. Mater. Chem. C, 2021, 9, 189–198 RSC.
- H. Tsujimoto, D.-G. Ha, G. Markopoulos, H. S. Chae, M. A. Baldo and T. M. Swager, J. Am. Chem. Soc., 2017, 139, 4894–4900 CrossRef CAS PubMed.
- B. Li, Z. Yang, W. Q. Gong, X. H. Chen, D. W. Bruce, S. Y. Wang, H. L. Ma, Y. Liu, W. G. Zhu, Z. G. Chi and Y. F. Wang, Adv. Opt. Mater., 2021, 9, 2100180 CrossRef CAS.
- B. Z. He, J. Zhang, J. Y. Zhang, H. K. Zhang, X. Y. Wu, X. Chen, K. H. S. Kei, A. J. Qin, H. H. Y. Sung, J. W. Y. Lam and B. Z. Tang, Adv. Sci., 2021, 8, 2004299 CrossRef CAS PubMed.
- H. K. Zhang, X. Y. Zheng, N. Xie, Z. K. He, J. K. Liu, N. L. C. Leung, Y. L. Niu, X. H. Huang, K. S. Wong, R. T. K. Kwok, H. H. Y. Sung, I. D. Williams, A. J. Qin, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 16264–16272 CrossRef CAS PubMed.
- J. Y. Zhang, L. R. Hu, K. H. Zhang, J. K. Liu, X. G. Li, H. R. Wang, Z. Y. Wang, H. H. Y. Sung, I. D. Williams, Z. B. Zeng, J. W. Y. Lam and H. K. Zhang, J. Am. Chem. Soc., 2021, 143, 9565–9574 CrossRef CAS PubMed.
- S. Kumar, L. G. Franca, K. Stavrou, E. Crovini, D. B. Cordes, A. M. Z. Slawin, A. P. Monkman and E. Z-Colman, J. Phys. Chem. Lett., 2021, 12, 2820–2830 CrossRef CAS PubMed.
- J. Sturala, M. K. Etherington, A. N. Bismillah, H. F. Higginbotham, W. Trewby, J. A. Aguilar, E. H. C. Bromley, A.-J. Avestro, A. P. Monkman and P. R. McGonigal, J. Am. Chem. Soc., 2017, 139, 17882–17889 CrossRef CAS PubMed.
- Z. J. Zhao, S. M. Chen, J. W. Y. Lam, Z. M. Wang, P. Lu, F. Mahtab, H. H. Y. Sung, I. D. Williams, Y. G. Ma, H. S. Kwok and B. Z. Tang, J. Mater. Chem., 2011, 21, 7210–7216 RSC.
- S. Y. Shao, J. Hu, X. D. Wang, L. X. Wang, X. B. Jing and F. Wang, J. Am. Chem. Soc., 2017, 139, 17739–17742 CrossRef CAS PubMed.
- G. L. Niu, X. L. Zheng, Z. Zhao, H. K. Zhang, J. G. Wang, X. W. He, Y. C. Chen, X. J. Shi, C. Ma, R. T. K. Kwok, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, K. S. Wong, P. F. Wang and B. Z. Tang, J. Am. Chem. Soc., 2019, 141, 15111–15120 CrossRef CAS PubMed.
- G. P. Bartholomew and G. C. Bazan, J. Am. Chem. Soc., 2002, 124, 5183–5196 CrossRef CAS PubMed.
- H. J. Kim, P. C. Nandajan, J. Gierschner and S. Y. Park, Adv. Funct. Mater., 2018, 28, 1705141 CrossRef.
- S. Lee, C.-H. Chen and A. H. Flood, Nat. Chem., 2013, 5, 704–710 CrossRef CAS PubMed.
- C. D. Fiandra, M. Moccia, V. Cerulli and M. F. Adamo, Chem. Commun., 2016, 52, 1697–1700 RSC.
- G. B. Payne and P. H. Williams, J. Org. Chem., 1961, 26, 651–659 CrossRef CAS.
- M. Miyashita, T. Suzuki and A. Yoshikoshi, Chem. Lett., 1987, 285–287 CrossRef CAS.
- S. Sugiyama, S. ÔHigashi, R. Sawa and H. Hayashi, Bull. Chem. Soc. Jpn., 1989, 62, 3202–3206 CrossRef CAS.
- H. Bouas-Laurent, A. Castellan, J.-P. Desvergne and R. Lapouyade, Chem. Soc. Rev., 2000, 29, 43–55 RSC.
- A. Pross, L. Radom and N. V. Riggs, J. Am. Chem. Soc., 1980, 102, 2253–2259 CrossRef CAS.
- I. V. Alabugin, K. M. Gilmore and P. W. Peterson, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2011, 1, 109–141 CrossRef CAS.
- T. Seko, K. Ogura, Y. Kawakami, H. Sugino, H. Toyotama and J. Tanaka, Chem. Phys. Lett., 1998, 291, 438–444 CrossRef CAS.
- D. S. Karpovich and G. J. Blanchard, J. Phys. Chem., 1995, 99, 3951–3958 CrossRef CAS.
- D. Oelkrug, A. Tompert, J. Gierschner, H.-J. Egelhaaf, M. Hanack, M. Hohloch and E. Steinhuber, J. Phys. Chem. B, 1998, 102, 1902–1907 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Other experimental procedures, compound characterization, other supplementary figures and crystal data. CCDC 2090628 and 2090629. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc05426k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |