
Formation of β-Ga2O3 nanorings from metal–organic frameworks and their high catalytic activity for epoxidation of alkenes†
Wei Ping
Wang
a,
Le Xin
Song
 *ab,
Yao
Li
a,
Yue
Teng
c,
Juan
Xia
a,
Fang
Wang
a and
Nan Ning
Liu
a
*ab,
Yao
Li
a,
Yue
Teng
c,
Juan
Xia
a,
Fang
Wang
a and
Nan Ning
Liu
a
aDepartment of Chemistry, University of Science and Technology of China, Jin Zhai Road 96, Hefei 230026, P. R. China. E-mail: solexin@ustc.edu.cn; Fax: +86-551-63601592
bNational Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei 230026, P. R. China
cState Grid Auhui Electric Power Research Institute, Zi Yun Road 299, Hefei 230601, P. R. China
First published on 25th November 2020
Abstract
Here we report a novel synthesis of hollow high-quality β-Ga2O3 nanorings based on an interesting structural evolution from concave Ga-MOF nanodisks. The concave low-crystallinity nanodisks were constructed by non-classical crystallization with particle attachment aid. Further, we propose the evolution mechanism from the Ga-MOF nanostructure to the gallium oxide nanostructure based on the outward contraction of nanoparticles. This synthesis method is highly straightforward and provides high yields. It is important to note that the β-Ga2O3 nanorings exhibit good heterogeneous catalytic activity for epoxidation of alkenes. A new oxygen transfer pathway was illustrated to describe the catalytic process based on density functional theory.
1. Introduction
β-Ga2O3 as the most stable phase in gallium oxides is a wide bandgap semiconductor with a band gap of about 4.8 eV.1 Recently, it has been attracting much attention for its potential use in power electronics devices2 and solar-blind UV photodetectors,3,4 due to its large band gap and high structural stability even at high temperature. Meanwhile, a large number of novel synthetic methods have been developed to obtain β-Ga2O3 nanostructures with various morphologies, such as nanospheres,5 nanowires,6 nanorods7 and nanoplates.8 However, to the best of our knowledge, until now, there are still no synthesis reports of gallium oxide nanoring structures.In the past few years, several novel methods were proposed to synthesize toroid/ring-like nanostructures, such as ZnO nanorings prepared using an etching effect with the aid of the Ostwald ripening process,9 and TiO2 toroids obtained using the Kirkendall effect.10 In addition, it was reported that a simple calcination process can be easily used to fabricate hollow nanostructures using amorphous or low-crystallinity metal-carbonates, glycerates, glycolates, and especially metal–organic frameworks (MOFs) as templates.11–13 This may be related to the fact that massive grain boundaries in these templates may strongly promote the generation of rigid shells during calcination.14
On this basis, here, we would like to report an interesting synthetic process of hollow β-Ga2O3 nanorings (β-Ga2O3-nrs) through a simple calcination treatment using Ga-MOF nanodisks (Ga-MOF-nds) as the template. It is worth highlighting that the low crystallinity Ga-MOF formed through particle attachment, namely the crystallization by particle attachment strategy (CPA).15,16
Alkene epoxidation is an important chemical reaction both in academic research and in industrial production, and epoxides are very valuable intermediates (IMs). Hydrogen peroxide (H2O2) is usually used as the oxidant for epoxidation of alkenes.17,18 The key to successful epoxidation is to realize activation of the O–O bond in H2O2.19 Countless examples of homogeneous or heterogeneous catalysts based on transition metal nanostructures have proved that catalysts with H2O2 can form metal–OOH or metal–OO–metal coordination IMs, thereby activating the O–O bond.20–23 However, among all reported metal coordination activation examples, the examples of main group metals (such as aluminum and gallium) are much less than those of transition metals.
In recent years, Pescarmona and his co-authors demonstrated that ε-Ga2O3, a very rare and difficult to prepare crystalline phase of Ga2O3, exhibited excellent catalytic performance for the epoxidation of alkenes.24 The result is quite interesting and encouraging, inspiring us to do research using other more readily available crystalline phases of Ga2O3 such as β-Ga2O3. This is an important objective of this work.
As illustrated in Scheme 1, we first obtained a thermodynamically unstable concave disk-like Ga-MOF nanostructure by kinetically controlled crystallization by particle attachment. To our knowledge, this is the first example of concave Ga-MOF nanostructures. This is also the first attempt to characterize a concave Ga-MOF nanodisk structure. Further, we obtained a hollow β-Ga2O3-nr through a structural evolution of the Ga-MOF-nd. Importantly, the β-Ga2O3-nr shows the same high heterogeneous catalytic performance as the ε-Ga2O3 mentioned above. Additionally, a new mechanism was proposed for the catalytic epoxidation process. In short, this study achieves two main goals. One is controllable synthesis of high-quality ring-like nanostructures of β-Ga2O3. The other is to propose a probable mechanism to explain the high catalytic performance of the β-Ga2O3 catalyst.
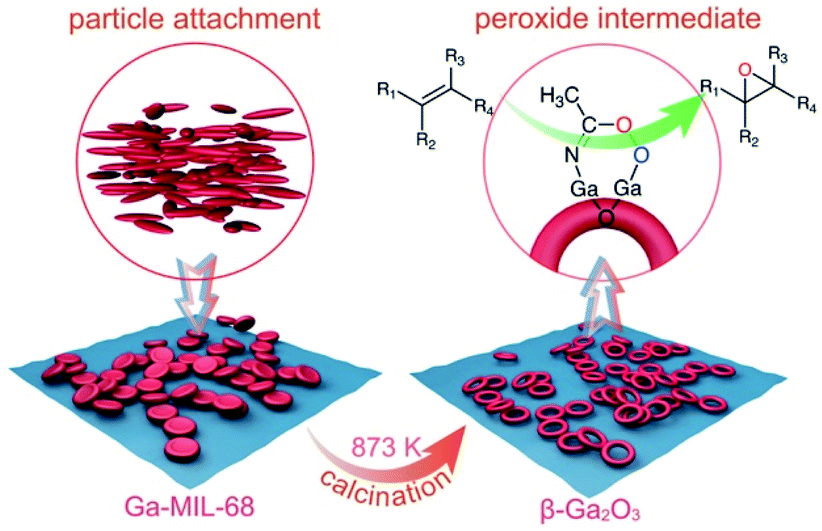 | ||
| Scheme 1 A schematic diagram illustrating the structural evolution from the concave Ga-MOF-nd to the hollow β-Ga2O3-nr with high catalytic activity for epoxidation of alkenes. | ||
2. Experimental section
2.1. Materials
All reagents were purchased from commercial suppliers and used without further purification. Gallium nitrate was purchased from Shanghai Aladdin Chemistry Co. Ltd. H2BDC, β-CD, 30 wt% H2O2, and DMF were purchased from Sinopharm Chemical Reagent Co. Ltd. All the alkenes used in experiments were purchased from Shanghai Aladdin Chemistry Co. Ltd. All the solvents used in epoxidation experiments were purchased from Sinopharm Chemical Reagent Co. Ltd.2.2. Synthesis of the concave Ga-MOF-nd
Gallium nitrate (125 mg, 0.5 mmol), H2BDC (88.0 mg, 0.5 mmol) and β-CD (567 mg, 0.5 mmol) were dissolved in a sealed vial with 5.0 mL DMF at room temperature. The vial was sonicated at room temperature for 30 minutes until all the solid reagents were completely dissolved. Then, the vial was heated in an oil bath at 333 K for 2 h without stirring. After the reaction, the vial was cooled to room temperature naturally. A white solid was collected by centrifugation, and washed three times with DMF and ethanol. Then the solid was suspended in methanol overnight. Finally, a white powder was dried at 393 K for 12 h.2.3. Synthesis of the hollow β-Ga2O3-nr
The as-obtained Ga-MOF sample was placed in a muffle furnace when the temperature reached 873 K. The Ga-MOF-nd was calcined for 1 h under an air atmosphere. After calcination, the crude product was cooled to room temperature naturally. The white powder was washed several times with ethanol and deionized water and dried at 333 K in an oven overnight.2.4. Synthesis of the β-Ga2O3-mr
Gallium nitrate (125 mg, 0.5 mmol), carbamide (1.0 g, 16.7 mmol) and deionized H2O (30 mL) were mixed in a flask and vigorously stirred until all the reagents were dissolved. The mixed solution was transferred into a sealed Teflon-lined stainless steel autoclave (50 mL) and heated at 393 K for 10 h. After the reaction, the autoclave was cooled to room temperature naturally. The white precipitate was collected and washed three times with deionized water and ethanol, and dried at 333 K in an oven overnight. Next, when the temperature reached 873 K, the dried precipitate was placed in a muffle furnace and was calcined for 1 hour under an air atmosphere. After calcination, the crude product was cooled to room temperature naturally. The white powder was washed several times with ethanol and deionized water and dried at 333 K in an oven overnight.2.5. Epoxidation experiments
10 mg β-Ga2O3, 1 mmol selected alkene, 3 mmol 30 wt% H2O2, and 1.0 g acetonitrile as a solvent were added into a sealed vial with full stirring for 4 h at 363 K in an oil bath. After reaction, the vial was cooled to room temperature naturally. The solution was extracted three times with ethyl acetate. The upper layer was dried with anhydrous sodium sulfate. The reaction products were determined by gas chromatography–mass spectrometry (GC-MS), and the reaction yields were determined by gas chromatography (GC) analysis, using 50 mg of toluene as an internal standard. The values of Yepo and Sepo were calculated based on the amount of alkenes using the ratio of the actual production of epoxides to the theoretical production of epoxides, and determined based on the ratio of the actual production of epoxides to the total amount of epoxides and by-products, respectively.2.6. Epoxidation recycling experiments
COE was used as the substrate alkene. All reaction conditions were the same as for the epoxidation process. After the epoxidation reaction, the catalysts used were washed with water and ethanol, dried overnight at 333 K, and reused for the next catalytic reaction. The yield was determined by GC analysis, using 50 mg of toluene as an internal standard.2.7. Computational details
Geometry optimizations were performed with the Gaussian09 package25 at the DFT level by means of the B3LYP function.26 For Ga, the LANL2DZ27 pseudopotential was used. The 6-311G (d,p) basis set was used for the C, H, O and N atoms. Single point energy calculations were performed by means of the M06-2X functional.28 The LANL2DZ pseudopotential was used for Ga atoms and the 6-311+G (d,p) basis set was used for the C, H, O, and N atoms. In all reactants, IMs and TSs, solvent effects were included by means of IEF-PCM (acetonitrile, ε = 36.64).29 The intrinsic reaction coordinate (IRC) method was applied to all TSs.302.8. Instruments and methods
All solid samples were measured under the same drying conditions. The morphologies of the samples were determined using a SU8220 FE-SEM operated at 1 kV. TEM and HR-TEM images and SAED patterns were obtained on a JEF 2100F field-emission transmission electron microscope operated at 200 kV. EDS mapping was performed on a Talos F200X HADDF-STEM.XRD measurements were obtained on a Philips X'Pert Pro X-ray diffractometer (PANalytical, Netherlands) with a scintillation counter detector using a monochromatized Cu Kα radiation source (tube voltage and current were 40 kV and 40 mA) with a wavelength of 0.1542 nm and analyzed in the range of 5° ≤ 2θ ≤ 80°. XPS measurements were carried out on an ESCALAB 250 spectrometer with Al Kα radiation (1486.6 eV) in an ultra-high vacuum (2.00 × 10−9 torr). And all of the values of binding energy were referenced to the C 1s peak (284.8 eV) with an energy resolution of 0.16 eV.
FT-IR spectra were recorded on a Bruker Equinox 55 spectrometer with KBr pellets in the range of 400–4000 cm−1 with a resolution of less than 0.09 cm−1. Raman spectra were recorded with a Renishaw InVia Raman microscope at room temperature with 532 nm laser excitation in the range 100–1800 m−1, with a resolution of 0.6 cm−1.
Nitrogen adsorption/desorption isotherms were obtained using a Micromeritics ASAP-2000 Surface Area and Porosimetry System at 77 K. The 20 mg β-Ga2O3 samples were loaded into a glass tube for electron paramagnetic resonance (EPR) spectra on a JES-FA200 spectrometer at 140 K.
13C nuclear magnetic resonance (NMR) spectra were recorded on a Bruker AC-400FT spectrometer (400 MHz) at room temperature using dimethyl sulfoxide (DMSO-d6) as the solvent.
Thermogravimetric analysis was performed on a Perkin Elmer TL-9000 in air at a 20 K min−1 heating rate. NH3-TPD measurements were carried out on a homemade instrument. First, β-Ga2O3 samples were pretreated in a N2 stream at 673 K for 2 h. Thereafter, the temperature was cooled to 313 K, and then the samples were saturated with NH3/N2 for 60 min, and purged with N2 for 30 min to purge physisorbed NH3. Finally, the catalysts were heated from 313 K to 1273 K at a heating rate of 10 K min−1 with flowing N2 (200 mL min−1).
3. Results and discussion
3.1. Characterization of the Ga-MOF-nd
The field emission scanning electron microscopy (FE-SEM) image in Fig. 1a shows that the obtained Ga sample at 333 K for 2 h (see Synthesis of the concave Ga-MOF-nd) displays a disk-like structure with a diameter of 300 nm and a thickness of about 80 nm (Fig. S1, ESI†). Interestingly, the nanodisks exhibit a concave structure as described in Fig. 1b. The transmission electron microscopy (TEM) image in Fig. 1c reveals that the concave nanodisk structure has high monodispersity and good uniformity. The energy dispersive spectroscopy (EDS) elemental mapping (Fig. S2, ESI†) illustrates the uniform overlapping of all the major elements (C, O and Ga) throughout the entire sample, highlighting the uniform distribution of each element.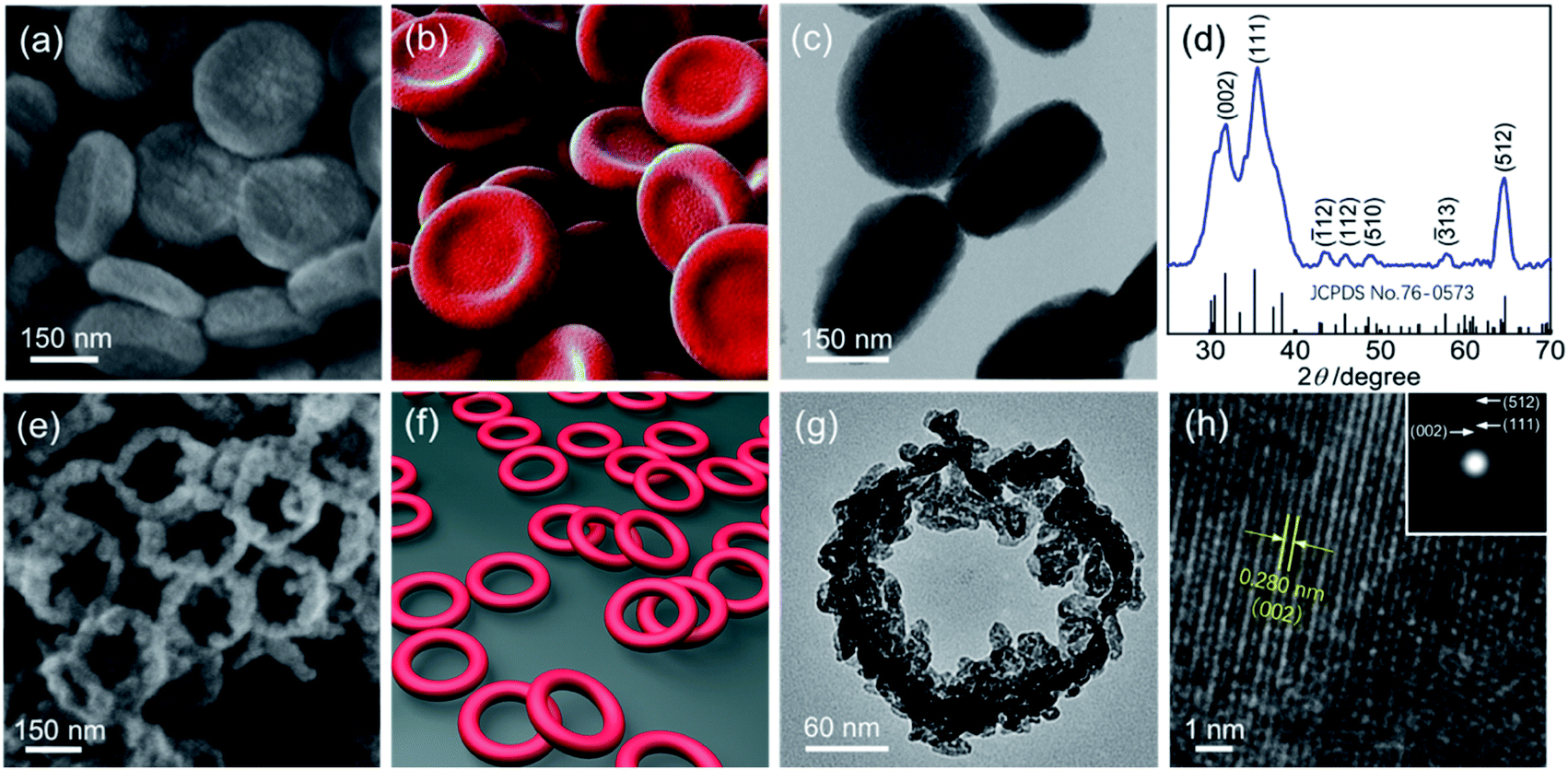 | ||
| Fig. 1 (a) The FE-SEM image of the concave Ga-MOF-nd obtained at 333 K for 2 h; (b) the visual image of the Ga-MOF-nd; (c) the TEM micrograph of the concave Ga-MOF-nd; (d) the XRD pattern of the β-Ga2O3-nr; (e) the FE-SEM image of the β-Ga2O3-nr; (f) the visual image of the β-Ga2O3-nr; (g) the TEM micrograph of a single β-Ga2O3-nr; (h) the HR-TEM image of a single β-Ga2O3-nr. The SAED pattern of a single β-Ga2O3-nr is inserted in the upper right corner of (h). | ||
Fourier transform infrared (FT-IR) spectra and X-ray photoelectron spectra (XPS) of the concave nanodisk structure in Fig. 2 clearly reveal the coordination interaction between the carboxylic acid groups in 1,4-benzenedicarboxylic acid (H2BDC) and Ga(III) ions. As shown in Fig. 2a, the stretching vibration frequency of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in carboxylic acid moves from the 1686 cm−1 of the free H2BDC to the 1588 cm−1 of the BDC in the concave nanodisk structure. Such a big red shift is a very clear indication that the CO bonds in the BDC units were seriously weakened, thereby signifying that there is a strong coordination interaction between the carboxylate groups and gallium ions. The Raman spectra (Fig. S3, ESI†) further confirm the significant effect of the coordination interaction on the aromatic carbon ring. The CCC bending vibration peak of the BDC in the concave nanodisk structure occurs at 802 cm−1, instead of a bimodal band at 816 and 826 cm−1 for free H2BDC.31
O bond in carboxylic acid moves from the 1686 cm−1 of the free H2BDC to the 1588 cm−1 of the BDC in the concave nanodisk structure. Such a big red shift is a very clear indication that the CO bonds in the BDC units were seriously weakened, thereby signifying that there is a strong coordination interaction between the carboxylate groups and gallium ions. The Raman spectra (Fig. S3, ESI†) further confirm the significant effect of the coordination interaction on the aromatic carbon ring. The CCC bending vibration peak of the BDC in the concave nanodisk structure occurs at 802 cm−1, instead of a bimodal band at 816 and 826 cm−1 for free H2BDC.31
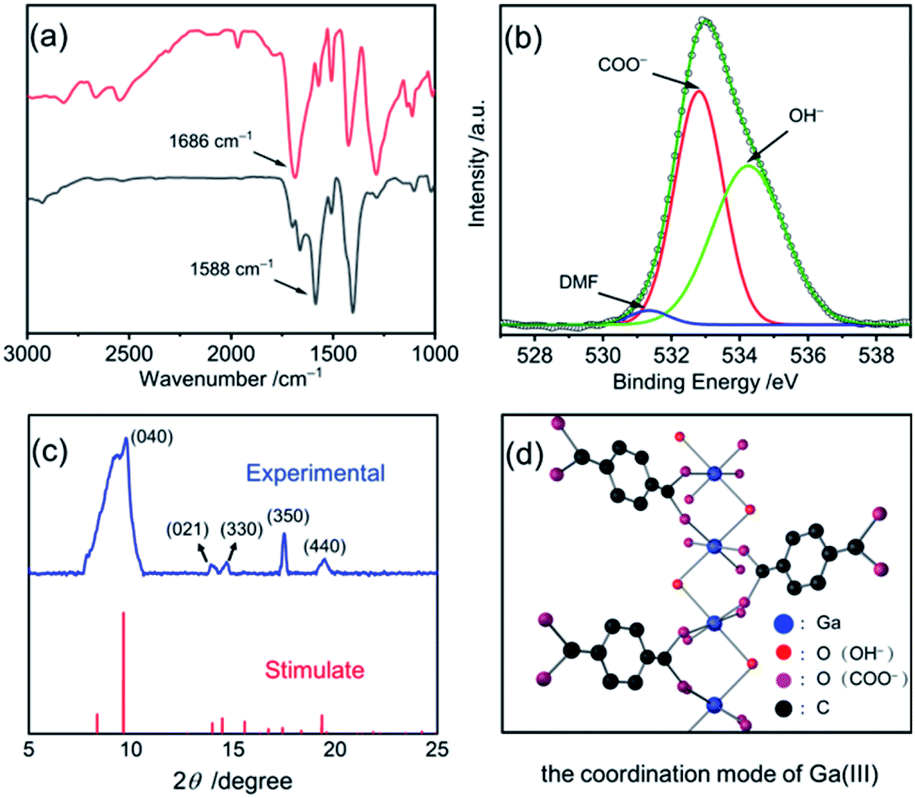 | ||
| Fig. 2 (a) The FTIR spectra of H2BDC and the concave nanodisk structure; (b) the O 1s binding energies of the concave nanodisk structure; (c) the XRD pattern of the concave nanodisk structure (blue line) and XRD data of the Ga-MIL-68 (red line);33 (d) the coordination mode of Ga(III) ions in the concave nanodisk structure. | ||
As shown in Fig. 2b, the O 1s binding energies at 531.3, 532.8 and 534.3 eV for the concave nanodisk structure were clearly attributed to the adsorption of N,N-dimethylformamide (DMF) molecules, the bidentate coordination of COO− ions and the coordination of OH− ions, respectively.32 Furthermore, Fig. 2c shows the powder X-ray diffraction (XRD) pattern of the concave nanodisk structure. Several major peaks at 9.6° (d, 9.18 Å), 13.98° (d, 6.33 Å), 14.5° (d, 6.11 Å), 17.4° (d, 5.10 Å) and 19.3° (d, 4.59 Å) are well indexed to the diffraction data of Ga-MIL-68, an octahedral complex reported by Loiseau and his collaborators.33 Nevertheless, compared with that of the reported Ga-MIL-68, the obviously wide diffraction pattern indicates that the synthesized Ga-MOF-nd has poor crystallization properties.33
Based on these observations, we conclude that the chemical formula of the concave nanodisk structure is Ga(OH)BDC with a spatial structure as shown in Fig. 2d. In the structure, the oxygen atoms of COO− units and OH− ions are directly coordinated to two adjacent gallium ions as bridging oxygen atoms, forming a Ga-MOF structure. And no chelate ring is formed in the polynuclear Ga-MOF.
3.2. Formation of the Ga-MOF-nd
MOFs with good crystallization are usually prepared under hydrothermal conditions, but this is not the best way to prepare low-crystallinity MOF precursors. Here, we report the formation of the Ga-MOF-nd using a facile crystallization by particle attachment (CPA) strategy in solution. It is because that rapid CPA progress usually leads to amorphous or low-crystallinity structures with disorders, dislocations and defects,15,16 thereby facilitating the formation of hollow nanostructures.The Ga-MOF-nd was constructed through a one-pot method from gallium nitrate, H2BDC, and β-cyclodextrin (β-CD) in dimethylformamide at 333 K for 2 h. Also, a series of Ga-MOF samples (Ga-MOF-t) were prepared at other crystallization times (t, 0.5, 1, 10 and 24 h) to understand the formation mechanism of the MOF nanostructure. The time-dependent growth of the Ga-MOF-t reveals that such a nanodisk structure with a concave surface was created through the CPA process. It is clear that the crystallization time of 0.5 h seemed to be a critical point based on the results of Fig. 3a–c, at which the prototype of Ga-MOF nanocrystals began to form. As seen from the red frames in Fig. 3a–c, countless ultrathin shuttle-like nanograins were gradually disappearing with the gradual formation of the Ga-MOF-nd. In Fig. 3a, a series of periodically arranged nanoshuttle layers were obvious when viewed from the lateral side of the nanodisks. Further, the TEM image in Fig. 3d clearly depicts the outward protruding angle of the nanoshuttles.
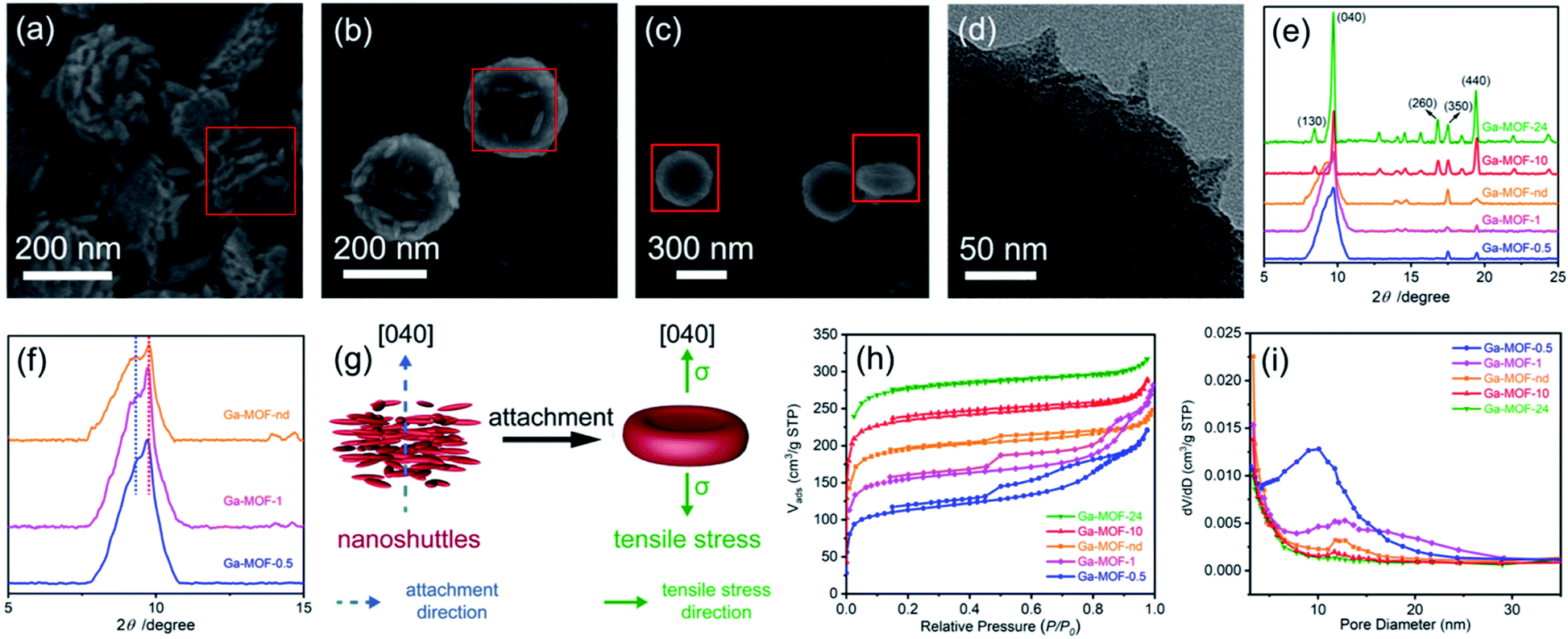 | ||
| Fig. 3 The FE-SEM images with regard to the time-dependent growth of Ga-MOFs at (a) 0.5, (b) 1 and (c) 2 h. (d) The TEM micrograph of the concave Ga-MOF at 0.5 h; (e) the XRD patterns of the Ga-MOF samples at different crystallization times; (f) the magnified XRD patterns of the Ga-MOFs at 0.5, 1 and 2 h and low diffraction angles; (g) the out-of-plane uniaxial tensile stress diagram in a concave nanodisk and the attachment mode of the nanoshuttles. (h) The nitrogen adsorption–desorption isotherms of Ga-MOFs at different crystallization times; and (i) the pore size distribution of Ga-MOFs at different crystallization times. | ||
A more detailed time-dependent study at 15, 20, 25, 30 and 35 min was performed. As shown in Fig. S4 (ESI†), the solutions were clear and no precipitation could be observed before 25 minutes. When the reaction time reached 25 minutes, the solution became turbid. At this time, the yield of the Ga-MOF was very low, about 3%. Additionally, the FE-SEM image shows that at 25 min, although a large number of nanoshuttles are attached to the surface of the nanodisks, there are still many free nanoshuttles at the same time.
The broad (040) diffraction peak at about 9.6° for the Ga-MOFs-0.5, -1 and -nd in Fig. 3e is a clear indication of low crystallinity caused by CPA at an early stage. In consideration of the high intensity and great breadth of the diffraction peak and the 2D-like structure of the nanodisks, we therefore suggest that the (040) planes correspond to the concave surface of the nanodisks and that the nanoshuttles attach together along the [040] direction (Fig. 3g). The magnified image (Fig. 3f) at low diffraction angles shows the presence of splitting shoulder peaks at these crystallization times. The small shoulder peak indicates an expansion in interplanar spacing along the [040] direction (Fig. 3f). An XRD peak splitting usually implies the existence of tensile stress (σ).34 This expansion may be an important cause of uniaxial tensile stress along the [040] direction in the nanodisks, as illustrated in Fig. 3g.
As the crystallization time increased further to 10 and 24 h, the broad (040) peak was replaced by a sharp peak. This emphasizes increased crystallinity and elimination of tensile stress due to Oswald ripening.35 At the same time, irregular microcubes are observable on the edge of the disk-like structures (Fig. S5, ESI†).
Comparative experiments were conducted to examine the role of β-CD. Our results reveal that the Ga-MOF sample in the absence of β-CD (Fig. S6, ESI†) obtained under the same preparation conditions as the Ga-MOF-nd but without β-CD, shows an irregular spherical morphology and a similar crystallinity to the Ga-MOF-nd. These significant changes in the Ga-MOF morphology enable us to consider that β-CD may act as a morphological modulator.
The N2 adsorption isotherms and pore size distribution of the Ga-MOFs in Fig. 3h and i support the structural formation progress described above. The H4 hysteresis loops clearly reveal the formation of hierarchical and mesopore structures of the Ga-MOF-0.5, -1 and -nd. The Barrett–Joyner–Halenda36 model indicates that most of the mesopores are around 10–11 nm. Obvious layer or mesoporous features cannot be found in Ga-MOF-10 and -24. It turns out that the CPA progression may produce disturbances and mesopores early in Ga-MOF formation. Also, with the increase of the crystallization time, Oswald ripening will attempt to eliminate the disorders.
3.3. Characterization of the β-Ga2O3-nr
The XRD analysis (Fig. 1d, JCPDS card no: 76-0573)37 of the sample obtained by structural evolution of the concave nanodisks at 873 K (see Synthesis of the hollow β-Ga2O3-nr) shows that it is pure polycrystalline β-Ga2O3.The SEM image (Fig. 1e) clearly shows that it has a ring-like structure as depicted in Fig. 1f. The TEM image (Fig. 1g) indicates that the ring structure has a height of about 50 nm, an inner diameter of 180 nm and an outer diameter of 230 nm (Fig. S7, ESI†). The nanorings connect with each other to form a honeycomb network, as seen in Fig. 1e. The inner and outer ring walls exhibit a considerably rough surface, on which a large number of tiny nanoparticles are mounted.
The high resolution transmission electron microscope (HR-TEM) image in Fig. 1h shows that the lattice fringes are structurally uniform with a spacing of 0.280 nm, which is in good agreement with the d value of the (002) planes of monoclinic β-Ga2O3.
The ring-like diffraction spot via the selected area electron diffraction (SAED) pattern (see the inset of Fig. 1h) clearly proves the polycrystalline structure of the pure β-Ga2O3. It is worth noting that the three clear diffraction rings observed in the SAED pattern correspond to the three strong peaks (002), (111) and (512) in the XRD pattern of the β-Ga2O3-nr in Fig. 1d, respectively. The trace element distribution (Fig. S8, ESI†) demonstrates the uniform distribution of each element, which is indicative of a single phase microstructure with a homogeneous chemical composition.
3.4. From the Ga-MOF-nd to the β-Ga2O3-nr
The FE-SEM and TEM images in Fig. 4 clearly depict a time-dependent growth of nanoring structures, allowing us to describe the structural evolution process from the Ga-MOF-nd to the β-Ga2O3-nr.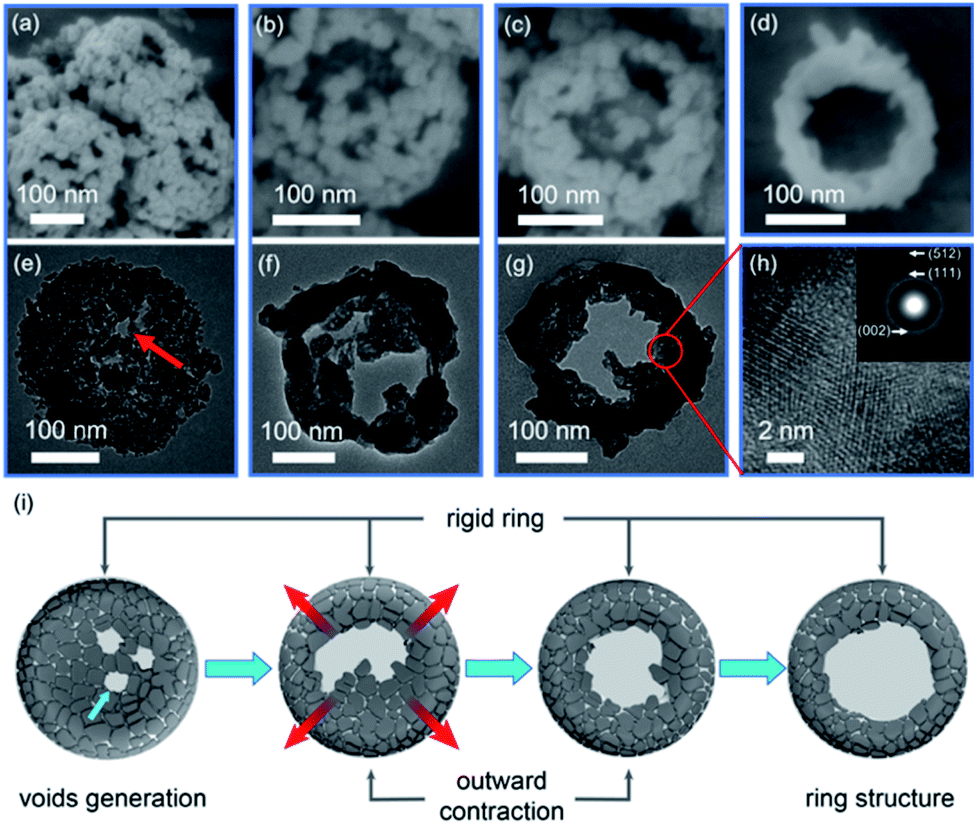 | ||
| Fig. 4 The time-dependent growth of β-Ga2O3 nanoring structures: FE-SEM images at 873 K and (a) 15, (b) 30, (c) 45 and (d) 60 min, TEM images at 873 K and (e) 15, (f) 30, (g) 45 min, (h) HR-TEM image (SAED pattern is placed as an internal illustration in (h)) at 45 min, and (i) schematic illustration of the outward-contraction structural evolution from a nanodisk to a nanoring. | ||
Initially, after 15 min of calcination at 873 K, the mass loss due to release of gases such as CO2 and H2O (Fig. S9 and S10, ESI†) led to the generation of mesopores in the nanodisk structure. The Ga-MOF with low-crystallinity rapidly produced a layer of shell in situ owing to low thermal conductivities derived from effective phonon scatterings at grain boundaries and interfaces,38 as shown in Fig. 4e and f. Concurrently, the uniaxial tensile stress along the [040] direction induced the generation of large voids and openings at the central depression of a nanodisk during the calcination treatment. No doubt the volume loss caused by the oxygenolysis of organic ligands would promote the inward contraction as usual. However, once the central voids and openings were created, the tiny particles at the depression would contract outward instead of inward. Besides, the rapid heating rate led to rapid gas diffusion along the outward direction, promoting the outward migration of particles.11 As a result, the central voids became larger after 30 min of calcination, showing an obvious cavity structure.
Meanwhile, the central particles shrank rapidly due to Ostwald ripening, and were transported to the inner wall of the surface ring.
Subsequently, the outward contraction continued and the cavity size increased further after 45 min of calcination, displaying an initial ring structure.
In other words, these graphs (Fig. 4a–g) clearly highlight the fact that due to outward contraction, voids are produced and dense and rigid ring structures are formed. The HR-TEM image (Fig. 4h) at 45 min gives us an impression that there is an irregular crystal orientation. The polycrystalline morphology (see the SAED pattern, the inset of Fig. 4h) may be a characteristic reflection caused by grain migration.
Also, an almost complete ring structure appears after 60 min of calcination (Fig. 1g). At the same time, most particles were adhered on the outer ring, leaving a cavity in the center. A schematic diagram describing the structural evolution from a nanodisk to a nanoring caused by outward contraction is shown in Fig. 4i.
Further, we study how the (040) peak evolves during calcination. After 5 minutes of calcination, we can see a slight decrease in the relative intensity of the (040) peak as shown in Fig. S11 (ESI†). And there is a significant fall in the intensity and several new peaks appear after 10 minutes of calcination. When the Ga-MOF-nd was calcined for 15 min, we note that the (110), (002) and (111) peaks of β-Ga2O3 began to appear and the peaks of the Ga-MOF-nd disappeared completely at the same time. When the Ga-MOF-nd was calcined for 60 min, the β-Ga2O3-nr finally formed. Given these results, we assume that the evolution of the (040) peak may include two main steps. Firstly, the decrease in the intensity of the (040) peak can be attributed to the conversion of the Ga-MOF into a temporary intermediate between the Ga-MOF and β-Ga2O3 in consideration of the decomposition of organic ligands. In the following step, this intermediate can quickly transform into β-Ga2O3, and the (040) peak disappears along with the appearance of peaks of β-Ga2O3.
To confirm the cavity-ring-induced outward contraction, two new β-Ga2O3 samples (named β-Ga2O3-10 and -24) were prepared via the calcination of the Ga-MOFs-10 and -24 under the same conditions as that of the Ga-MOF nanodisks. Our results show the structural integrity of the two samples without producing a similar hollow morphology (Fig. S12, ESI†). This may be because the elimination of uniaxial stress also eliminates the formation of voids and openings.
3.5. Alkene epoxidation results
The catalytic epoxidation results of several selected alkenes are summarized in Table 1. From these data, we can clearly see that the β-Ga2O3-nr exhibits highly catalytic activity in the epoxidation reaction of the series of alkenes.| Entry | Alkenes | Catalysts | Y epo/% | S epo/% |
|---|---|---|---|---|
| 1 | COE | β-Ga2O3-nr | 95 | 99 |
| 2 | COE | Blank | 20 | 99 |
| 3 | COE | ε-Ga2O3-nr24 | 84 | >99 |
| 4 | COE | Ga2O3 (ref. 39) | 51 | 80 |
| 5 | COE | β-Ga2O3-nr | 82 | 92 |
| 6 | 2,3-Dimethyl-2-butene | β-Ga2O3-nr | 76 | 70 |
| 7 | Norbornylene | β-Ga2O3-nr | 52 | 99 |
| 8 | Cyclohexene | β-Ga2O3-nr | 52 | 99 |
| 9 | Cyclohexene | ε-Ga2O3-nr24 | 45 | >99 |
| 10 | 1-Methylcyclopentene | β-Ga2O3-nr | 53 | 90 |
| 11 | α-Methylstyrene | β-Ga2O3-nr | 41 | 99 |
| 12 | Styrene | β-Ga2O3-nr | 15 | 70 |
| 13 | Styrene | ε-Ga2O3-nr24 | 20 | 58 |
| 14 | COE | WO3 (ref. 40) | 11 | 44 |
| 15 | COE | TiO2 (ref. 41) | 28 | 78 |
| 16 | COE | Fe2O3 (ref. 42) | 43 | 71 |
First, for the catalytic epoxidation conversion of cyclooctene (COE), the epoxidation yield (Yepo, %) and epoxidation selectivity (Sepo, %) reached 95 and 99%, respectively. Also, we prepared the other β-Ga2O3 sample with a rod-like microstructure by a hydrothermal-calcination route (Fig. S13 and S14, ESI†). The β-Ga2O3 microrods (β-Ga2O3-mrs) gave the same high catalytic activity (Yepo 88%; Sepo 99%) as the nanorings for the epoxidation reaction of COE. Next, it is apparent that the catalytic activity of the nanorings in the epoxidation reaction of alkenes is comparable to and better than that of reported transition metal oxides,40–42 ε-Ga2O3-nanorods (ε-Ga2O3-nrs)24 and other Ga2O3 catalysts39 in light of the data of Table 1. Furthermore, both β-Ga2O3-nrs and ε-Ga2O3-nanorods exhibit almost similar catalytic properties for epoxidation of several different structural alkenes. In other words, the catalytic activity of the Ga2O3 catalysts is closely related to the structure of alkenes. Finally, our results show that the β-Ga2O3-nr maintained its original catalytic activity after eight catalytic cycles (Fig. S15, ESI†).
3.6. Catalytic epoxidation mechanism
Fig. 5a and b indicate that acetonitrile as a solvent plays a very important role in the catalytic epoxidation of alkenes, because when ethyl acetate, benzene, chloroform and ethanol were used as solvents, similar epoxidation reactions only provided very low yields under the same conditions.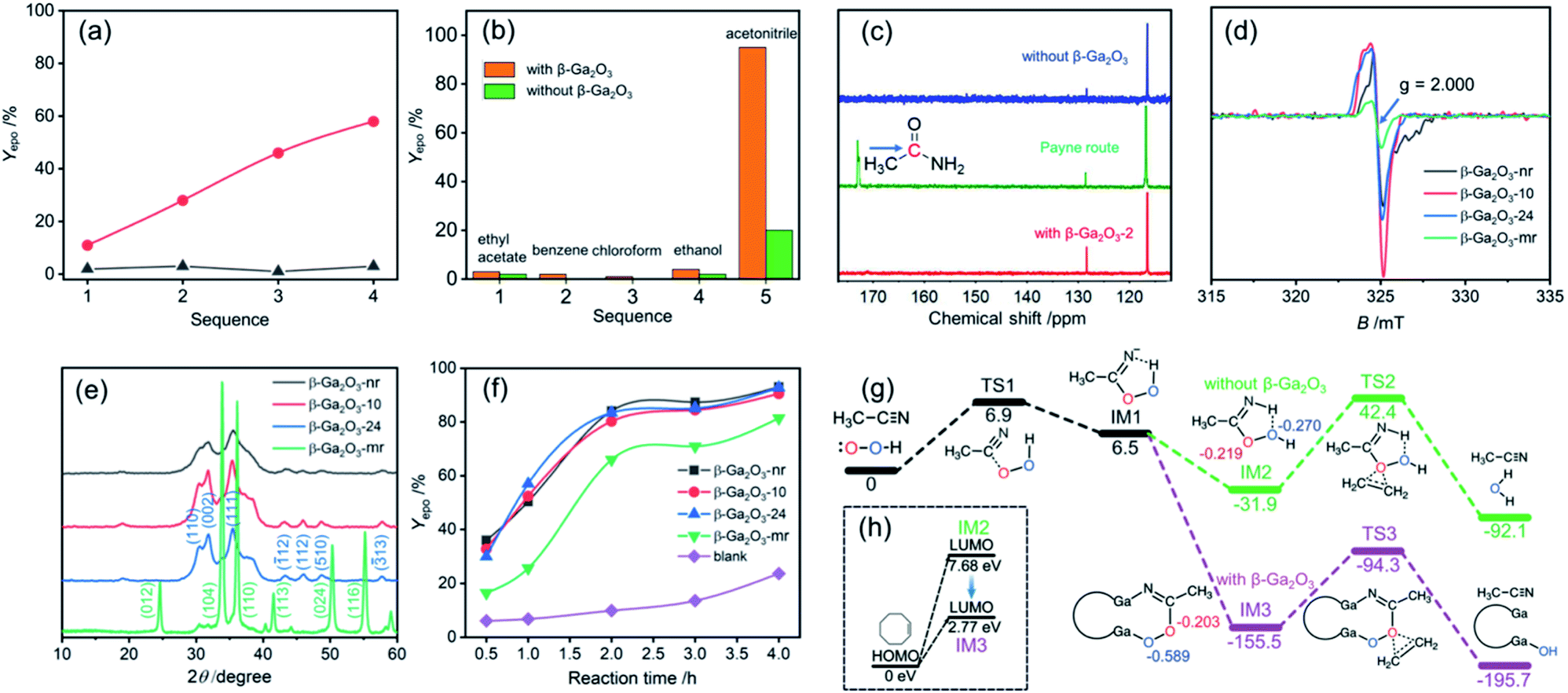 | ||
| Fig. 5 (a) The epoxidation yields of COE under the reaction conditions: 1 mmol COE, 3 mmol 30% H2O2 and 10 mg β-Ga2O3 nanorings at 363 K for 4 h using ethanol as the solvent in the presence of acetonitrile (red dots: 1, 0.1 g acetonitrile; 2, 0.2 g acetonitrile; 3, 0.3 g acetonitrile and 4, 0.4 g acetonitrile) and in the absence of acetonitrile (gray triangles); (b) the epoxidation yields of different solvents under the reaction conditions: 1 mmol COE and 3 mmol 30% H2O2 with or without the β-Ga2O3-nr at 363 K for 4 h; (c) the 13C NMR spectra of epoxidation reaction products: 1 mmol COE and 3 mmol 30% H2O2 at pH = 7.0 (blue line), 1 mmol COE and 3 mmol 30% H2O2 at pH = 8.0 (green line), 34 and 1 mmol COE and 3 mmol 30% H2O2 in the presence of the β-Ga2O3-nr (10 mg) at pH = 7.0 (red line). (d) The EPR spectra of the β-Ga2O3-nr, −10, −24, and -mr; (e) the XRD patterns of the β-Ga2O3-nr, −10, −24, and -mr; (f) the time-dependent epoxidation yields of COE using the β-Ga2O3-nr, −10, −24, and -mr as catalysts; (g) the calculated Gibbs free–energy profiles (kcal·mol−1) for the epoxidation reaction of COE without/with β-Ga2O3, and TSs represent transition states; and (h) the calculated LUMO and HOMO energy levels (eV) of COE, IM2 and IM3. | ||
According to Payne and his collaborators,43 nitrile compounds and H2O2 could act as catalysts in the epoxidation reaction process. It is likely that under alkaline conditions (pH, 8.0), the disassociated OOH group initially would attack the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond of acetonitrile to form the IM peroxycarboximidic acid and simultaneously produce amides as a by-product of the reaction (named the Payne route,43Fig. 5c and S16, ESI†). Nevertheless, under current neutral conditions (pH, 7.0) no chemical shift signal of amides at about 170 ppm was detected (see the blue line for the blank route and the red line for the β-Ga2O3 route in Fig. 5c). This fact allows us to propose a new oxygen transfer pathway for describing the process of alkene epoxidation in both cases, namely, blank route and β-Ga2O3 route under neutral conditions.
N bond of acetonitrile to form the IM peroxycarboximidic acid and simultaneously produce amides as a by-product of the reaction (named the Payne route,43Fig. 5c and S16, ESI†). Nevertheless, under current neutral conditions (pH, 7.0) no chemical shift signal of amides at about 170 ppm was detected (see the blue line for the blank route and the red line for the β-Ga2O3 route in Fig. 5c). This fact allows us to propose a new oxygen transfer pathway for describing the process of alkene epoxidation in both cases, namely, blank route and β-Ga2O3 route under neutral conditions.
The electron paramagnetic resonance (EPR) analysis shows that a strong oxygen defect signal (g = 2.000) appears for the β-Ga2O3-nr, −10 and −24 while the β-Ga2O3-mr exhibits a much weaker signal at the same position (Fig. 5d). This is a clear indication that there is a significant structural difference in the β-Ga2O3 samples obtained by the two synthetic methods. A series of XRD patterns in Fig. 5e confirm this, since there are two different crystal forms, one synthesized by the MOF transformation method (β-Ga2O3-nr, −10 and −24) and the other by the hydrothermal method (β-Ga2O3-mr). The former show a much stronger (002) surface signal then the latter. It was reported that the unstable and polar (002) surface of β-Ga2O3 is more defective and electron-rich than other surfaces such as (111) and (110) surfaces.44 The appearance of this EPR signal may be attributed to shallow trapping donor defects on the (002) surface. Fig. 5f shows a marked difference in the epoxidation yields of COE using different β-Ga2O3 catalysts. When the β-Ga2O3-nr, −10 and −24 were used as catalysts, a similar epoxidation yield much higher than that of the β-Ga2O3-mr was shown. Moreover, we found that the β-Ga2O3 obtained through the calcination of the Ga-MOF in the absence of β-CD at 873 K for 1 h under an air atmosphere also has almost comparable catalytic performance (Yepo 92%; Sepo 99%) to the β-Ga2O3-nr for the epoxidation of COE under the same catalytic conditions. Based on the EPR analysis in Fig. 5d, we infer that the catalytic performance of the β-Ga2O3 catalysts seems to be closely related to the oxygen vacancy concentration. In order to make a further comparison, we prepared the same pure phase β-Ga2O3 material (β-Ga2O3-m) based on a recent report.45 Our results show that the β-Ga2O3-m also has a good catalytic performance for the epoxidation of COE (Yepo 88%; Sepo 99%) under the same catalytic conditions. The EPR analysis (Fig. S17, ESI†) clearly reveals the strong signal of oxygen vacancies, and the signal intensity is highly similar to those of the β-Ga2O3-nr, −10 and −24 of the same phase. This observation further demonstrates that the presence of oxygen vacancies in the catalysts may play a greater role in the epoxidation process than the morphology of the materials. Additionally, comparison of data in Table 1 shows that transition metal oxides may not be an ideal choice for alkene epoxidation catalysts.
By combining these results, we conclude that oxygen defects on the (002) surface led to unsaturated Ga(III) sites becoming main potential catalytically active sites. Further, the characteristic of these Ga(III)active sites with weak acidic sites has been confirmed from the ammonia temperature-programmed desorption (NH3-TPD) curve (Fig. S18, ESI†). Based on these experimental observations, we hope to further utilize density functional theory (DFT) to explain the catalytic epoxidation mechanism.
As far as the blank route is concerned, since no β-Ga2O3 catalyst exists, we speculate that the IM2 peroxycarboximidic acid tended to transfer its α-O (marked in red, Fig. 5g) to the CC bond of alkenes under neutral conditions. This is because the Mulliken charge analysis shows that α- and β-O charges (marked in blue, Fig. 5g) in this figure are −0.219 and −0.270 eV, respectively, supporting that electrophilic reactions are more likely to occur in α-O. Due to geometric and electronic properties, the N–H and β-O in the IM2 had the possibility of forming intramolecular hydrogen bonds (H⋯O bond length, 2.18 Å).
However, as seen from Fig. 5g, with the addition of the β-Ga2O3-nr, due to the weak acidity of Ga(III) and the weak alkaline characteristic of CN, the CN in the IM3 enters the Ga(III) site and coordinates with the Ga(III) ion. At this point, the hydrogen bond interaction in the above blank route is replaced by more stable coordination bonds such as Ga–O and Ga–N bonds, which greatly stabilizes the IMs. Upon completion of the α-O transfer, the remaining β-O on the lattice may recombine to become a surface hydroxy. It is certain that the formation of Ga–O coordination bonds can effectively enhance the polarity of O–O covalent bonds and thus improve the polarization of O–O bonds. This may be one of the reasons for the increase of epoxidation yield. Besides, on the basis of the frontier molecular orbital theory, it is reasonable to assume that the IM3 in the Ga2O3 coordination route exhibits higher activity of the epoxidation reaction than the IM2 in the blank route, because there is a significant decrease in the energy gap of HOMOCOE − LUMOIMs from 7.68 eV to 2.77 eV (see Fig. 5h).
In short, several issues related to the catalytic process need to be highlighted. First, the β-Ga2O3-nr shows good dispersion in acetonitrile solution (Fig. S19, ESI†), which ensured good contact between the β-Ga2O3 catalyst and substrate. Second, acetonitrile not only plays a role of a solvent, but may also act as a cocatalyst. As seen from Fig. 5, the cocatalysis of acetonitrile is associated with its special molecular structure. Third, both the β-Ga2O3 catalysts (nanorings and microrods) show good catalytic activity for the epoxidation reaction of alkenes although they have different surface morphologies. In addition, it is worth noting that the catalytic process has high structural selectivity for epoxidation of alkenes, as shown in Table 1. This may be related to the structure of alkenes because different reactant structures will lead to different coordination IMs and TSs with different stabilities.
4. Conclusions
In conclusion, we successfully constructed a novel concave Ga-MOF nanodisk structure in the present work. The formation mechanism of this structure is attributed to the crystallization by particle attachment strategy. Moreover, a very interesting structural evolution from a concave Ga-MOF nanodisk structure to a hollow β-Ga2O3 nanoring structure was clearly observed. The possible mechanism for such a structural transformation is proposed. In particular, the β-Ga2O3 nanorings exhibit high catalytic activity for epoxidation of alkenes. Two different reaction routes are suggested to explain the epoxidation process of alkenes in the absence and presence of catalysts. We believe that this study can attract a lot of attention because it is closely related to the development of coordination chemistry, materials chemistry, organic synthesis, and heterogeneous catalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Synchrotron Radiation Laboratory (No. KY2060000091) and the Natural Science Foundation of Anhui Province (No. 1508085MB30). The supercomputing center of University of Science and Technology of China (USTC) is greatly acknowledged for the computational support.Notes and references
- H. H. Tippins, Phys. Rev., 1965, 140, A316 CrossRef.
- J. Kim, M. A. Mastro, M. J. Tadjer and J. Kim, ACS Appl. Mater. Interfaces, 2018, 10, 29724 CrossRef CAS.
- W. Y. Kong, G. A. Wu, K. Y. Wang, T. F. Zhang, Y. F. Zou, D. D. Wang and L. B. Luo, Adv. Mater., 2016, 28, 10725 CrossRef CAS.
- D. Y. Guo, Z. P. Wu, Y. H. An, X. C. Guo, X. L. Chu, C. L. Sun, L. H. Li, P. G. Li and W. H. Tang, Appl. Phys. Lett., 2014, 105, 023507 CrossRef.
- X. Sun and Y. Li, Angew. Chem., Int. Ed., 2004, 43, 3827 CrossRef CAS.
- K. W. Chang and J. J. Wu, Adv. Mater., 2004, 16, 545 CrossRef CAS.
- S. C. Vanithakumari and K. K. Nanda, Adv. Mater., 2009, 21, 3581 CrossRef CAS.
- S. Yan, L. Wan, Z. Li, Y. Zhou and Z. Zou, Chem. Commun., 2010, 46, 6388 RSC.
- F. Li, Y. Ding, P. Gao, X. Xin and Z. L. Wang, Angew. Chem., Int. Ed., 2004, 43, 5238 CrossRef CAS.
- J. H. Han, S. Lee, D. Yoo, J.-H. Lee, S. Jeong, J.-G. Kim and J. Cheon, J. Am. Chem. Soc., 2013, 135, 3736 CrossRef CAS.
- L. Zhang, H. B. Wu, S. Madhavi, H. H. Hng and X. W. Lou, J. Am. Chem. Soc., 2012, 134, 17388 CrossRef CAS.
- L. Shen, L. Yu, X. Y. Yu, X. Zhang and X. W. Lou, Angew. Chem., Int. Ed., 2015, 54, 1868 CrossRef CAS.
- G. Zhang, L. Yu, H. B. Wu, H. E. Hoster and X. W. Lou, Adv. Mater., 2012, 24, 460913 Search PubMed.
- L. Yu, X. Y. Yu and X. W. Lou, Adv. Mater., 2018, 30, 1800939 CrossRef.
- D. Li, et al. , Science, 2012, 336, 1014 CrossRef CAS.
- M. H. Nielsen, S. Aloni and J. J. De Yoreo, Science, 2014, 345, 1158 CrossRef CAS.
- C. Aprile, E. Gobechiya, J. A. Martens and P. P. Pescarmona, Chem. Commun., 2010, 46, 7712 RSC.
- K. N. Sands, E. Mendoza Rengifo, G. N. George, I. J. Pickering, B. S. Gelfand and T. G. Back, Angew. Chem., Int. Ed., 2020, 59, 4283 CrossRef CAS.
- N. S. Antonova, J. J. Carbó, U. Kortz, O. A. Kholdeeva and J. M. Poblet, J. Am. Chem. Soc., 2010, 132, 7488 CrossRef CAS.
- N. E. Thornburg, S. L. Nauert, A. B. Thompson and J. M. Notestein, ACS Catal., 2016, 6, 6124 CrossRef CAS.
- D. A. Ruddy and T. D. Tilley, J. Am. Chem. Soc., 2008, 130, 11088 CrossRef CAS.
- J. Zhang, W.-J. Wei, X. Lu, H. Yang, Z. Chen, R.-Z. Liao and G. Yin, Inorg. Chem., 2017, 56, 15138 CrossRef CAS.
- C. Dinoi, M. Ciclosi, E. Manoury, L. Maron, L. Perrin and R. Poli, Chem.-Eur. J., 2010, 16, 9572 CrossRef CAS.
- W. Lueangchaichaweng, N. R. Brooks, S. Fiorilli, E. Gobechiya, K. Lin, L. Li, S. Parres-Esclapez, E. Javon, S. Bals, G. Van Tendeloo, J. A. Martens, C. E. Kirschhock, P. A. Jacobs and P. P. Pescarmona, Angew. Chem., Int. Ed., 2014, 53, 1585 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, et al., Gaussian 09, revision B.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 284 CrossRef.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157 CrossRef CAS.
- J. Tomasi, B. Mennucci and E. Cancès, J. Mol. Struct., 1999, 464, 211 CrossRef CAS.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363 CrossRef CAS.
- H. Embrechts, M. Kriesten, K. Hoffmann, W. Peukert, M. Hartmann and M. Distaso, J. Phys. Chem. C, 2018, 122, 12267 CrossRef CAS.
- X. Quan, Z. Sun, J. Xu, S. Liu, Y. Han, Y. Xu, H. Meng, J. Wu and X. Zhang, Inorg. Chem., 2020, 59, 2667 CrossRef CAS.
- C. Volkringer, M. Meddouri, T. Loiseau, N. Guillou, J. Marrot, G. Férey, M. Haouas, F. Taulelle, N. Audebrand and M. Latroche, Inorg. Chem., 2008, 47, 11892 CrossRef CAS.
- J. Fang, J. Li, C. Tian, Q. Gao, X. Wang, N. Gao, X. Wen, C. Ma, H. You, Z. Yang, Q.-H. Xu, Q. Xiong and Z. Li, NPG Asia Mater., 2016, 8, e323 CrossRef CAS.
- A. I. Lupulescu and J. D. Rimer, Science, 2014, 344, 729 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373 CrossRef CAS.
- H. Yang, R. Shi, J. Yu, R. Liu, R. Zhang, H. Zhao, L. Zhang and H. Zheng, J. Phys. Chem. C, 2009, 113, 21548 CrossRef CAS.
- X. Shi, A. Wu, W. Liu, R. Moshwan, Y. Wang, Z.-G. Chen and J. Zou, ACS Nano, 2018, 12, 11417 CrossRef CAS.
- P. P. Pescarmona, K. P. Janssen and P. A. Jacobs, Chem.-Eur. J., 2007, 13, 6562 CrossRef CAS.
- A. T. Bolsoni, J. S. dos Santos, M. D. Assis and H. P. Oliveira, J. Non-Cryst. Solids, 2011, 357, 3301 CrossRef CAS.
- C. J. Yang, X. J. Lang, W. H. Ma, C. C. Chen, H. W. Ji and J. C. Zhao, Chem.-Eur. J., 2014, 20, 6277 CrossRef CAS.
- Y. Zamani, M. Bakavoli, A. Mohajeri and S. M. Seyedi, J. Nanoanal., 2016, 3, 1 Search PubMed.
- G. B. Payne and P. H. Williams, J. Org. Chem., 1961, 26, 651 CrossRef CAS.
- J. Qu, X. Zhou, F. Xu, X.-Q. Gong and S. C. E. Tsang, J. Phys. Chem. C, 2014, 118, 24452 CrossRef CAS.
- B. Das, B. Das, N. Sankar Das, S. Pal, B. Kumar Das, S. Sarkar and K. K. Chattopadhyay, Appl. Surf. Sci., 2020, 515, 145958 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, PXRD, FE-SEM, Raman spectroscopy, EDS, computational details and Cartesian coordinates. See DOI: 10.1039/d0ta09005k |
| This journal is © The Royal Society of Chemistry 2021 |