
Labile oxygen participant adsorbate evolving mechanism to enhance oxygen reduction in SmMn2O5 with double-coordinated crystal fields†
Li
Wang‡
a,
Hui
Li‡
b,
Jieyu
Liu
a,
Xiuyao
Lang
a and
Weichao
Wang
 *a
*a
aIntegrated Circuits and Smart System Lab (Shenzhen), Renewable Energy Conversion and Storage Center, Tianjin Key Laboratory of Photo-Electronic Thin Film Device and Technology, College of Electronic Information and Optical Engineering, Nankai University, Tianjin, 300071, China. E-mail: weichaowang@nankai.edu.cn
bCollege of Mechanical and Electrical Engineering, Jiaxing University, Jiaxing, 314001, China
First published on 21st November 2020
Abstract
The current understanding of the oxygen reduction reaction (ORR) mechanism can fall into two categories: (1) the adsorbate evolving mechanism (AEM) over active metallic sites, in which all oxygen-containing intermediates originate from the electrolyte; (2) the lattice oxygen-mediated mechanism (LOM), in which the lattice oxygen in perovskite directly participates in the reaction. For more complicated metallic oxides with multiple ligand fields, these two mechanisms may fail to precisely describe the ORR process, as the local oxygen environment on the terminated surfaces of the catalyst is more variable relative to perovskites with only one type of ligand field. Herein, based on the constructed (SmMn2O5)n (n = 1, 2, 3, 4, 8) clusters and (001) slab model of a Mn-based mullite catalyst with a double-coordinated crystal field (Mn3+-centered square pyramid and octahedral crystal field centered on Mn4+), we discovered a new ORR mechanism, named the labile oxygen participant adsorbate evolving mechanism (LAM), via density functional theory calculations. Compared with the AEM, our proposed LAM further considers the labile oxygen participating in the reactions in the presence of intermediate OOH*, in contrast to the LOM, which does not involve OOH* formation. During the LAM, the formation of OOH* was determined to be the rate-limiting step. The moderate binding strength of the OOH* stems from the reasonable p–d orbital coupling between Mn–O bonds, trigged by the multiple oxygen coordination environments. The proposed LAM provides new insights into oxygen reactions over the more complicated catalysts with multiple ligands.
1. Introduction
Understanding the sluggish oxygen redox reaction enables the development of efficient catalysts and thus promotes their application in oxygen-based energy conversion and storage, such as fuel cells, metal–air batteries, and water electrolyzers.1–3 To date, the main oxygen reaction mechanism has been dominated by the adsorbate evolution mechanism (AEM) historically proposed by Nørskov et al.4–7 The key feature of the AEM is that all the oxygen intermediate species react on the metallic active sites without directly involving the lattice oxygen. For the AEM in alkaline conditions, the elementary steps can be either a 2e− or 4e− reaction process. Taking the 4e− process as an example, O2 first adsorbs on the electrode surface occupied by OH* (* represents the adsorption site of the catalyst) to form OO* prior to being reduced to OOH* by binding protons in solution. Finally, the O–O bond in OOH* breaks to form O*, which is then protonated to form OH* (Scheme 1a). During the 4e− process, no lattice elements are directly involved in the formation of the intermediates. This mechanism has been widely adopted to access the structure–activity relationship.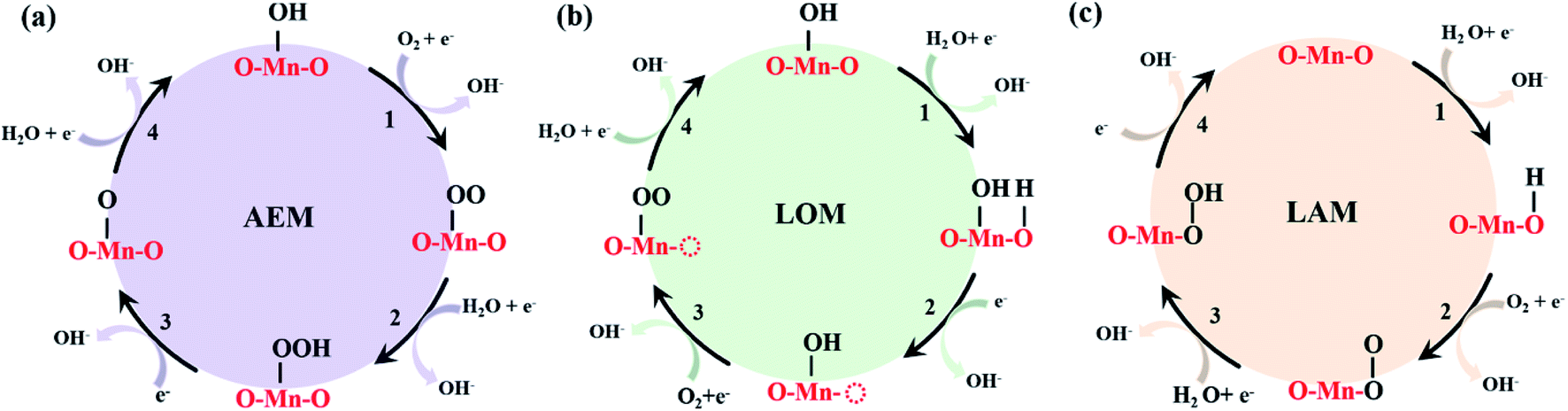 | ||
| Scheme 1 Oxygen reduction reaction (ORR) mechanism diagrams with a comparison between (a) the adsorbate evolution mechanism (AEM), (b) the lattice-oxygen participation mechanism (LOM), and (c) the labile oxygen participant adsorbate evolving mechanism (LAM). The atoms marked in red represent the original atoms of the system, while the adsorbed substances are labelled in black. | ||
However, extensive work has demonstrated a discrepancy between experimental catalytic performance and theoretically predicted results based on the AEM. For instance, perovskites such as Ba0.5Sr0.5Co0.8Fe0.2O3−δ and La0.5Ca0.5CoO3−δ with respective overpotentials of 0.25 V and 0.28 V have lower thermodynamic barriers than pristine LaNiO3 (∼0.33 V), which was predicted to be the optimum compound.8–10 This discrepancy may fundamentally stem from the exclusion of participation of the lattice oxygen in the reactions according to the AEM model. In fact, the high activity in the La1−xSrxCoO3−σ system was derived to be due to the participation of lattice oxygen through a combination of DFT calculations and electrochemical characterization.6 Subsequently, the lattice oxygen-mediated mechanism (LOM) based on the reversible formation of oxygen vacancies was firstly proposed, as shown in Scheme 1b.6,7 Simultaneously, Shao-Horn et al. provided direct experimental evidence to verify that the oxygen generated during the OER process was indeed derived from lattice oxygen through in situ18O-labeled mass spectrometry.10 Successively, JongSuk Yoo and his colleagues confirmed the electronic origin and kinetic feasibility of the participation of lattice oxygen of the perovskite system based on DFT calculations.11 Recently, Xu and Kuznetsov et al. consecutively validated the LOM in ZnxCo1−xOOH and the ruthenium-based pyrochlore Y1.8M0.2Ru2O7−σ (M = Cu, Co, Ni, Fe, Y).12,13 Because the LOM was identified in perovskites, it remains an open question whether the LOM applies to other oxides.
Compared with perovskites with one type of ligand unit, oxides with multiple ligand units could in principle hold multiple active oxygen atoms to trigger a different reaction mechanism with respect to the LOM. Taking the Mn-based mullite SmMn2O5 as an example, two types of crystal fields exist in its crystal unit, i.e., Mn3+-centered square pyramid and Mn4+-centered octahedral units. The exposed surface thus contains several types of labile oxygen, such as Oα located at the Mn pyramid (Mnpyr) vertex and Mn octahedral (Mnoct) apex, Oβ coordinated as vertex sites between two Mnpyr, and Oγ sited at the Mnpyr apex and Mnoct vertex.14 In our previous work, we found that for the pristine Mn-based mullite SmMn2O5, the ORR onset potential reached 0.817 V experimentally. However, the calculated overpotential was 0.52 V based on the AEM; even the active stepped surface was included in regard to the flat (001) surface.15 In other work, SmMn2O5 was introduced into nitrogen-doped reduced graphene oxide in situ, and the corresponding theoretical results were reduced to 0.455 V via AEM, which is higher than the value of 0.39 V obtained experimentally.16 Even for the defective SmMn2O5 electrocatalysts, the same gap exists.17 We also found the same phenomenon in spinel oxides, in which there are also two types of coordination crystal fields, i.e., tetrahedron and octahedron.18–20 These discrepancies inspired us to search for new mechanisms in a system with more complex multi-coordination crystal fields.
Herein, we use the particle swarm optimization (CALYPSO) method to construct ground state configurations of Mn-based mullite SmMn2O5 clusters to mimic the local environments of multiple oxygens.21–23 Based on the cluster structures, we found that when introducing labile oxygens into the ORR and OER process, the oxygen intermediate species were different from those obtained via the LOM, and the thermodynamic barrier of the clusters was greatly reduced. We thus propose, for the first time, the labile oxygen participant adsorbate evolving mechanism (LAM) (as shown in Scheme 1c) within the mullite system. The linear relationship between the Bader charge on labile oxygen and the binding energies of the intermediates was established accordingly to relate the ORR (OER) overpotential and Bader charge. Importantly, when further applying the three mechanisms (AEM, LOM, LAM) to the slab model, LAM still showed the lowest overpotential, which is consistent with the experimental results. These findings provide new insights into the reaction path of oxygen catalysis, which is of importance for the development of electrochemical catalysts.
2. Computational methods and details
2.1 Simulation setups
In the present work, all spin-polarized DFT calculations were performed based on the Vienna Ab Initio Simulation Package (VASP).24–26 The exchange and correlation energies were described by generalized gradient approximation (GGA) with the PW91 version. Also, the projector augmented wave (PAW) method was employed to treat the electron–ion interactions.27 An energy cutoff of 400 eV was adopted based on the plane wave functions. The energy convergence criterion between two electronic steps was 10−5 eV, and structural optimization was performed until the maximum Hellmann–Feynman force per atom was less than 0.02 eV Å−1. To avoid periodic cluster–cluster interactions, simple cubic cells with side lengths of 20–30 Å were utilized. For cluster calculations, we used the Γ point for Brillouin zone sampling. For the slab model (a = 14.68 Å, b = 8.58 Å, and c = 25.05 Å), a Γ-centered k-mesh of (1 × 2 × 1) was used. For localized 3d electrons of Mn, a Hubbard U value of 2.7 eV was included to correct the Coulomb repulsion interactions.21 Specifically, with an effective U value of 2.7 eV, the theoretically calculated band gap (1.21 eV) and lattice constants (a = 7.32 Å, b = 8.56 Å, and c = 5.69 Å) of the SmMn2O5 bulk agree well with the experimental band gap (1.23 eV) and lattice constants (a = 7.43 Å, b = 8.59 Å, and c = 5.70 Å, respectively).28–302.2 Establishment of the calculation model and simulation details of the electrochemical reaction
Based on the particle swarm optimization (CALYPSO) method, we constructed the ground state cluster structures of Mn-based mullite SmMn2O5 and oxygen-rich SmMnO5, labelled as (115)n. During the searching process, 60% of the configurations from 50 generations of structural seeds, i.e., at least 1500 structures, were selected for subsequent DFT optimization. Further, the Cambridge Cluster Database was introduced to verify the cluster models obtained from CALYPSO.31From the perspective of thermodynamic stability, in order to explore the feasibility of different mechanisms on the mullite cluster and the slab models, we compared the binding energies of the oxygen-containing intermediates (O*, OH*, OO*, and OOH*) based on eqn (1)–(4):
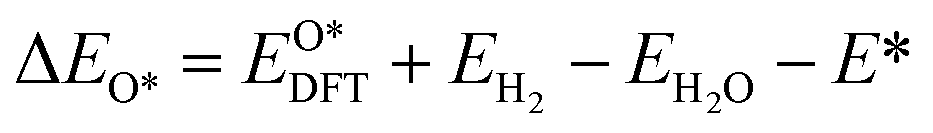 | (1) |
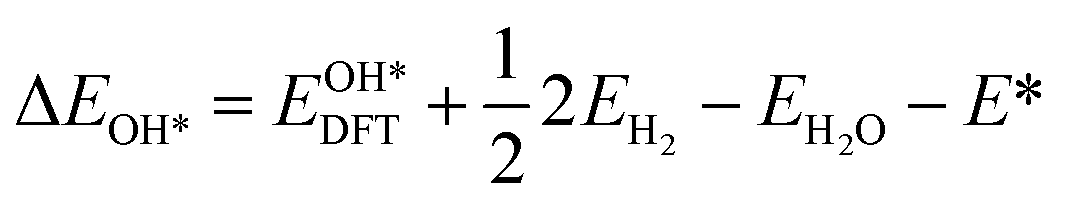 | (2) |
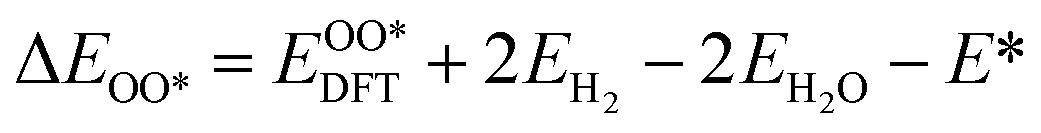 | (3) |
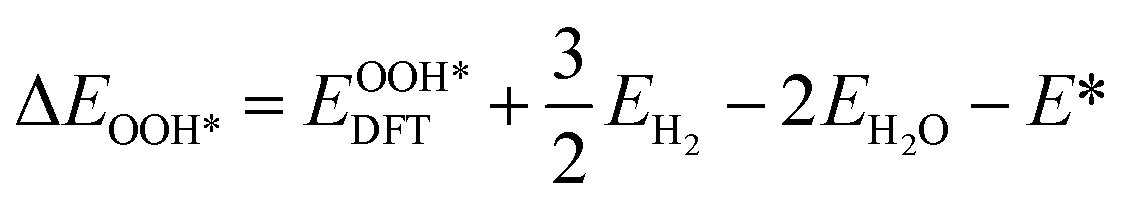 | (4) |
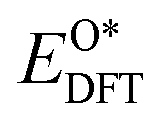 ,
, 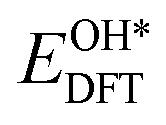 ,
, 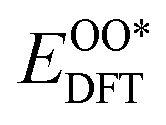 , and
, and  respectively represent the total energies of the system before and after the adsorption of intermediates. EH2 and EH2O are the total energies of H2 and H2O in gas phases. Based on this, the Gibbs free energy change (ΔGi, i = 1, 2, 3, 4) for each ORR step in the different mechanisms was calculated using eqn (5)
respectively represent the total energies of the system before and after the adsorption of intermediates. EH2 and EH2O are the total energies of H2 and H2O in gas phases. Based on this, the Gibbs free energy change (ΔGi, i = 1, 2, 3, 4) for each ORR step in the different mechanisms was calculated using eqn (5)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :32–34
:32–34| ΔGi = ΔEi + Δ(ZPE)i − TΔSi + ΔGU | (5) |
| ηORR = max[ΔG1,ΔG2,ΔG3,ΔG4]/e + 1.23 | (6) |
Bader charge analysis provides reasonable supporting evidence support for the comparison of the electron gain and loss ability of each atom of the cluster, especially for the labile oxygen atoms.35 Simultaneously, to explore the dependence of the binding energy of the oxygen intermediates on the electronic structures of the clusters, the d-band center ξd was calculated based on eqn (7):
| ξd = ∑Ed/∑Nd | (7) |
3. Results and discussion
3.1 Ground state configurations of Mn-based mullite (125)n clusters
In electrocatalysis or heterogeneous catalysis, the reliability of theoretical simulations heavily depends on the constructed calculation models. For a given catalyst, the simulation structures can be built into either slab models or cluster models based on the different simulated environments. Each model has unique advantages over the other. For instance, in the slab model, the exposed surface is large enough for the reactants to react with each other without spurious periodic interference. On the other hand, considering the complex coordination environments of the active sites in experiments, the cluster model can be validated to accommodate multiple coordination. In the following sections, we will discuss the difference between the two models in terms of structures and catalytic performance.During oxygen catalysis, activated oxygen plays a crucial role in determining the catalytic performance.37,38 This specific activated oxygen can be less coordinated than bulk oxygen, which has been identified via combinations of TPD, XPS, in situ DRIFTS, and Raman.14,38,39 In Mn-based mullite SmMn2O5 systems, our recent work also confirmed that labile oxygens are involved in the oxidation process. These labile oxygens can be different from one another in terms of their coordination environments based on experimental observations.14,40 The coordination of active sites, i.e., labile oxygen, is the key to determine the catalytic performance. For oxide catalysts, a common problem is excessively strong bonding between the oxygen species and the substrates. Therefore, a lower coordination environment would be crucial to produce high performance. To explore the reaction mechanism, it is necessary for more active sites to be present simultaneously during the reactions. Therefore, it would be more appropriate to adopt the cluster models herein.
In this work, we adopted a combination of manual construction and a particle swarm optimization (CALYPSO) method to obtain the ground-state structure of mullite SmMn2O5 (125)n (n = 1, 2, 3, 4, 8) clusters from ∼1500 structures (as shown in Fig. 1). For the (125)1 cluster, its ground state structure (Fig. 1a) contains a Mn–Mn dimer with a bond length of 2.75 Å, where the atoms connect to each other through two bridging oxygen atoms. Around the Mn–Mn dimer, there are four types of oxygen in different coordination modes (monocoordinated O, Mn–O–Mn, Mn–O–Sm, 2Mn–O–Sm) among five oxygen atoms, providing a complex oxygen environment for the catalytic reaction.
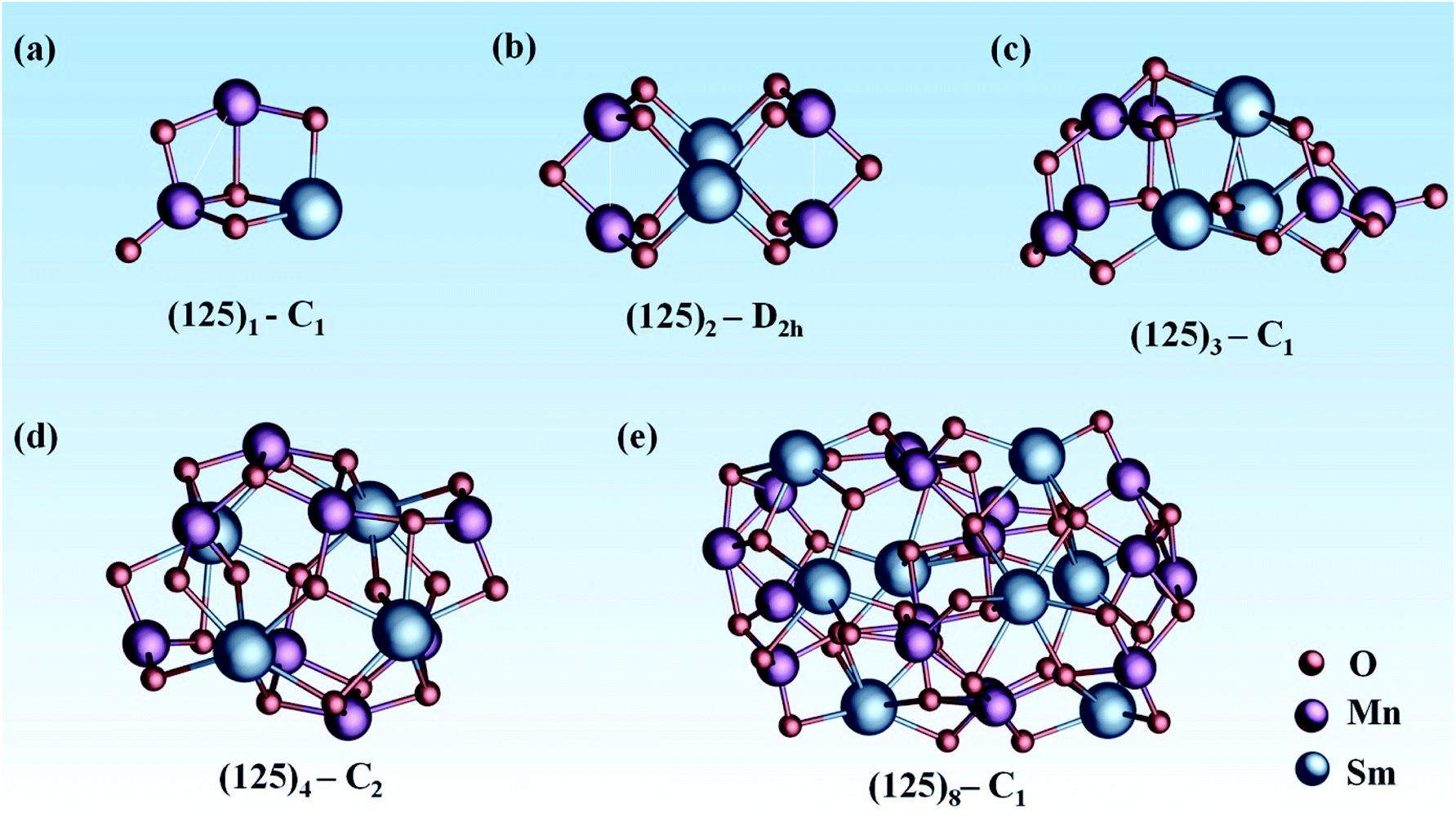 | ||
| Fig. 1 The ground state structures of Mn-based mullite SmMn2O5 (125)n (n = 1, 2, 3, 4, 8) clusters. (a)–(e) present the cluster models with various sizes from (125)1 to (125)8, respectively. O, Mn and Sm atoms are labelled as red, purple, and blue, respectively. The space group of each structure is also marked in the figure. | ||
Compared with the (125)1 cluster with C1 symmetry, the (125)2 cluster (Fig. 1b) consisting of 4 planar MnO3 units has a higher D2h symmetry. Moreover, the bond length of two equivalent Mn–Mn dimers is reduced to 2.52 Å. Interestingly, Mn atoms prefer to locate on the surface of the cluster and Sm atoms tend to move into the interior of the cluster with increasing cluster size. This trend leads to better catalytic potential due to the excellent activity of the facial Mn–Mn dimer.41 Furtherly, our previous electronic structure analysis of frontier orbitals shows that the HOMO and LOMO are mainly dominated by dz2 and dx2–y2 orbitals from the Mn–Mn dimer, leading to high chemical activity.21 For n = 3 (Fig. 1c), the number of exterior Mn–Mn dimers is further increased to three, with bond lengths of 2.52 Å, 2.63 Å, and 2.75 Å. Moreover, there are at least 6 types of oxygen in different coordination environments among 15 oxygen atoms. This complex coordination environment can provide diverse active sites for the catalytic process.42
Starting from the (125)4 cluster (Fig. 1d), MnO5 units with bulk-like square-pyramid coordination fields began to appear; meanwhile, the number of Mn–Mn dimers further increased (four for (125)4 and seven for (125)8). For the larger (125)8 (Fig. 1e), the cluster includes both square-pyramid-coordinated MnO5 units and octahedral-coordinated MnO6 units. Consequently, the coordination environment of oxygen becomes more complicated. In these cluster systems, Sm atoms act as the nucleation centers, and MnOx units tend to stay at the exterior of the surface of the clusters.
3.2 Calculation of the reaction barriers
For electrochemical oxygen catalysis, overpotential is the key factor to evaluate the catalytic performance. A low overpotential is crucial to ensure high performance of oxygen-based energy conversion and storage.43,44 Upon the series of mullite (125)n (n = 1, 2, 3, 4, 8) clusters, we carried out electrochemical reactions towards the ORR and OER processes.We first started the ORR 4e− process over the (125)1 cluster under the traditional adsorbate evolving mechanism (AEM). Based on the adsorption energy of the four oxygen intermediate species on the (125)1 cluster, we derived the corresponding Gibbs free energy and plotted the reaction paths (as shown in Fig. 2a; see Fig. S1† for the details of the AEM reactions). It can be seen that the rate-limiting step occurs during the hydrogenation of OO* to OOH* with an overpotential of 0.805 V. Upon increasing the cluster size to (125)2, the overpotential did not decrease but increased to 1.163 V. Furthermore, the rate-limiting step transformed into the generation of OH* (Fig. 2b). For the (125)3 cluster (Fig. 2c) with one more Mn–Mn dimer, the rate-limiting step returned to the OOH* generation step and the corresponding overpotential was reduced to 0.540 V, which is still higher than the experimental overpotential of 0.413 V.15
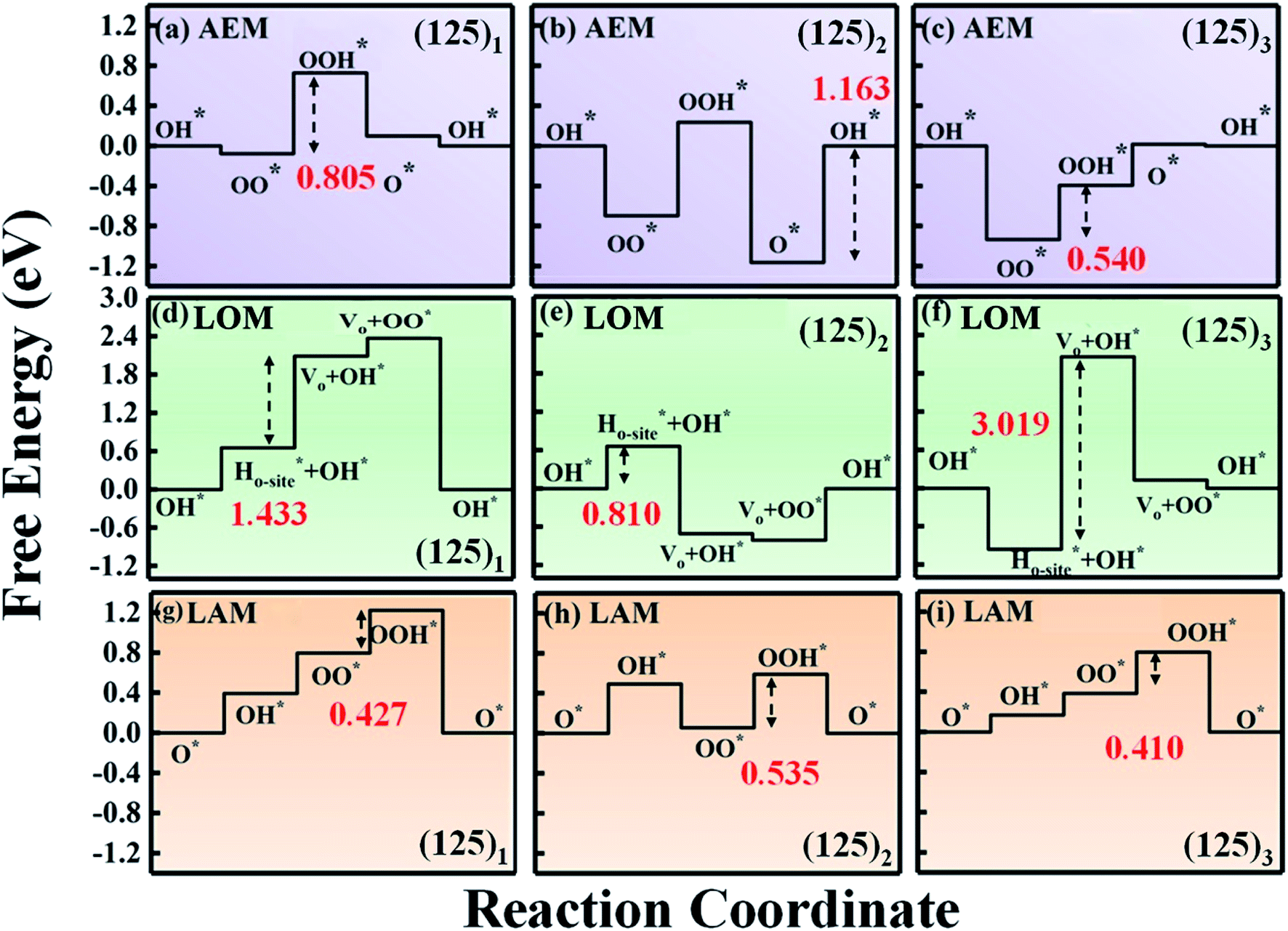 | ||
| Fig. 2 Free energy diagrams for the oxygen reduction reaction (ORR) based on (a–c) the adsorbate evolution mechanism (AEM), (d–f) the lattice-oxygen participation mechanism (LOM), and (g–i) the labile oxygen participant adsorbate evolving mechanism (LAM) for SmMn2O5 (125)n (n = 1, 2, 3) clusters at the equilibrium potential (U) of 1.23 V. The black arrows show the potential-determining step of each system, and the corresponding overpotential is marked in red. | ||
Because the AEM fails to describe the ORR process over mullite oxides, it is rational to reconsider the reaction path and the structure uniquity, which may be out of the scope of the AEM. In fact, lattice oxygen is involved in the reactions. A reaction mechanism excluding the lattice oxygen could be somehow biased. We thus adopted the lattice oxygen mechanism (LOM), in which lattice oxygen participates in the reaction of the perovskites (see Fig. S2† for the elementary steps of the LOM).6,7,11,45 As shown in Fig. 2d–f, under the LOM, the overpotentials of the systems are even higher than those under the AEM. Also, these values are higher than 1.23 V (1.433 V for the (125)1 cluster and 3.019 V for the (125)3 cluster). This clearly indicates that the LOM cannot provide a reasonable reaction path for the mullite oxide.
Both the AEM and LOM produce unreasonable ORR performance over the mullite cluster in regard to experimental activity. This discrepancy inspires us to believe that a new reaction path could exist due to the existence of the multiple activated oxygens in the mullite system. From the structural aspect, there is only one type of ligand unit, i.e., octahedron, in perovskite. As a comparison, there are two types of ligand coordination fields in mullite. As a result, more activated oxygen should be present on the activated surfaces. Therefore, it would be meaningful to investigate the ORR process by a reaction path that is different from the AEM and LOM.
Comparing the OH and H adsorption on the active Mn and O (Table 1), we found that the adsorption strength of H on O is quite strong, ∼1 eV higher than that of OH on the Mn site for each case of SmMn2O5 (125)n (n = 1, 2, 3, 4, 8). Therefore, it would be reasonable to start the reaction from the H adsorption on the active O site.
| (125)1 | (125)2 | (125)3 | (125)4 | (125)8 | |
|---|---|---|---|---|---|
| ΔEads(OH*) (eV) | −0.126 | 0.389 | −0.963 | 0.598 | −1.137 |
| ΔEads(H*) (eV) | −1.174 | −1.087 | −1.400 | −1.758 | −3.273 |
For each cluster, differing from the first step of the AEM, where O2 replaces OH* to form OO*, we first considered the adsorption of proton H on the lattice oxygen of the cluster to form OH*. Subsequently, O2 adsorbs on the Mn site and replaces the newly formed OH* to form OO*; then, OO* combines with the proton in the solution to form OOH*. This oxygen specie does not appear during the LOM; instead, a surface oxygen vacancy is formed. Finally, the O–O bond breaks and produces O* to complete a 4e− cycle (the mechanism diagram is shown in Scheme 1c). In Fig. 2g–i, simultaneously, we provide the corresponding Gibbs free energy diagram for each cluster.
It can be seen that the corresponding overpotentials of (125)n (n = 1, 2, 3) are 0.427 V, 0.535 V and 0.410 V, respectively. Compared with the AEM and LOM, the above reaction mechanism enables further reduction of the overpotential of the mullite cluster system, which is more consistent with the experimental results.15 In order to distinguish it from the conventional AEM and LOM, we defined the aforementioned four-electron reaction path as the labile oxygen participant adsorbate evolving mechanism (LAM), as shown in eqn (8)–(11):
| O* + e− + H2O → OH* + OH− | (8) |
| O2 + e− + OH* → OO* + OH− | (9) |
| OO* + e− + H2O → OOH* + OH− | (10) |
| OOH* + e− → O* + OH− | (11) |
Under the LAM, the rate-limiting steps of these three systems are the generation of OOH*, which is mainly due to the weak bonding strength of OOH* to the cluster. In other words, the binding energy of OOH* can be increased by a modification to further reduce the overpotential of the system. In addition to these three systems, Fig. S1† shows the Gibbs free energy diagrams of the (125)4 cluster under the AEM and LAM. As we predicted, the (125)4 cluster has a lower overpotential under the LAM (0.448 V for the LAM and 0.456 for the AEM).
Comparing the processes of the LOM and LAM, we discovered that the symmetry and stability of the crystal structure essentially determine the reaction mechanism of the ORR or the OER. In addition to mullite, the LAM might apply to other crystal structures with low symmetry and corresponding multiple ligand coordination fields, such as spinel and layered double hydroxides (LDHs). Therefore, it would be interesting to explore the LAM process over these catalysts both theoretically and experimentally.
3.3 The fundamentals of the LAM
The above thermodynamic analysis shows that the LAM is a reasonable reaction path compared to the AEM and LOM in the Mn-based mullite cluster systems. As both Mn and O act as active sites, it is important to probe the Bader charge variations during catalysis, which are certainly linked to their electronic structures. In the present work, the Bader charge difference is defined as the difference between the number of valence electrons of the atoms in the clusters and the number of valence electrons of the element. Moreover, the specific Bader charge of each atom in the stable structure of the (125)n (n = 1, 2, 3, 4, 8) clusters before the intermediates are adsorbed is listed in S4.† In each cluster, O and Mn are labeled with Ox and Mnx (where x is a digital number), respectively, to examine their charge states. It can be seen that in the (125)1 cluster (Fig. 3a), labile O5 with a single coordination gains ∼0.66 electrons, while labile O2 at the bridge site between Mn1, Mn2, and Sm has the most electrons (1.05). In summary, the labile oxygen obtains electrons in the range from 0.6e to 1.3e in the Mn-based mullite cluster (125)n (n = 1, 2, 3, 4, 8) systems.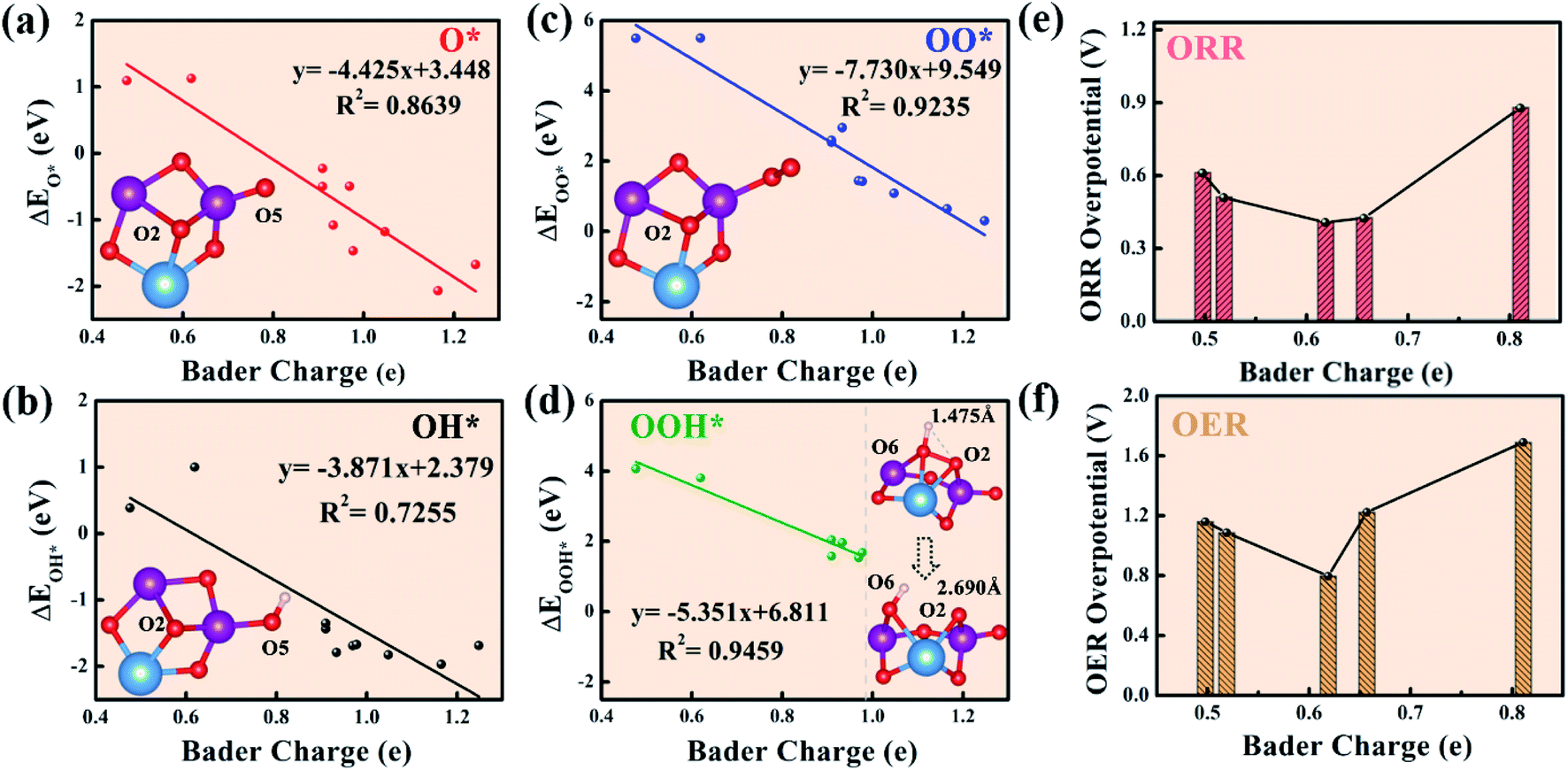 | ||
| Fig. 3 Linear relationships between the binding energies of the oxygen-containing intermediates (a) O*, (b) OH*, (c) OO*, and (d) OOH* in the oxygen reduction reaction (ORR) and the number of electrons obtained in the labile oxygen (Bader charge) for different (125)n (n = 1, 2, 3, 4, 8) clusters and the oxygen-rich (115)n (n = 1, 2, 3) clusters under the labile oxygen participant adsorbate evolving mechanism (LAM). Taking the (125)1 cluster as an example, the illustrations show the structure model with adsorption of intermediates. Blue, purple, red, and white represent Sm, Mn, O, and H atoms, respectively. (e) and (f) Show the inverted volcanic relationship between the ORR and the oxygen evolution reaction (OER) overpotential and Bader charge. | ||
Generally, for polarized covalent bonding, the greater the charge transfer, the stronger the bond that forms. To quantify the relation between the Bader charge of the labile oxygen over all the cluster models and the binding energies of the intermediate species, we present the four oxygen intermediate species versus the Bader charges of the labile oxygens over each cluster, as shown in Fig. 3. The Bader charge of labile oxygen depends on its coordination environment. In other words, as long as the labile oxygen is in the same coordination environment, the Bader charge is equal even if the oxygens are in different clusters. Therefore, we only considered labile oxygen with different Bader charges. Additionally, in order to complete the whole picture, when the Bader charge on the labile oxygen is less than 0.619e, the oxygen-rich (115)n (n = 1, 2, 3) clusters were included in our work (the specific Bader charge of each atom is listed in S5†). From the statistical results between the binding energies of the intermediates O*, OH*, and OO* and the Bader charge of labile oxygen (Fig. 3a–c), there is a linear relationship between the binding energies of these three intermediates and the Bader charge. As the number of electrons on the labile oxygen increases, the interaction between the intermediates and the clusters increases accordingly. Unlike the above three intermediates, the linear relationship between the binding energy of OOH* as the rate-limiting step and the Bader charge of labile oxygen (Fig. 3d) stops before 0.98e. This is because for the labile oxygen with more than 0.98e gained (from 0.98e to 1.3e), the O–O bond is easily activated, resulting in the separation of O and OH. Consequently, the OOH* intermediate fails to form. In the right section of Fig. 3d, we take (125)1 as an example to demonstrate the O–O separation when the labile O2 obtains more electrons than 0.98 eV (1.05e). Clearly, the O–O bond has been elongated from the initial 1.475 Å to 2.690 Å; therefore, stable OOH* fails to remain. Therefore, labile oxygen with electron numbers higher than 0.98e is unsuitable for the adsorption of intermediates.
Fig. 3e shows the inverted volcanic relationship between the optimal ORR overpotential for each cluster (i.e., (125)1, (125)3, (125)8, (115)1, (115)2) and the corresponding labile oxygen Bader charge. Likewise, Fig. 3f describes the OER overpotentials versus the Bader charges of the labile oxygen over all the explored clusters. Alternatively, as the number of electrons obtained by labile oxygen increases, the overpotential of the system tends to reduce to reach the valley point and then increase. When the number of electrons obtained reaches 0.619e, the overpotentials of ORR and OER are 0.41 V and 0.796 V, respectively, upon the (125)3 cluster. As the number of electrons obtained is less than 0.619e, the binding energy is too weak to trigger the reaction. On the other side, because the number of electrons obtained gradually becomes higher than 0.619e, the bonding strength increases accordingly, leading to an increase in the thermodynamic barrier. Therefore, moderate bonding strength is the decisive factor to promote the electrochemical performance of a cluster.
Under the LAM, O2 firstly replaces OH* to form OO* after the proton H is adsorbed on the labile oxygen. In this process, oxygen vacancies are generated prior to O2 adsorption. The neighboring Mn atom connected to the specific labile oxygen can also affect the adsorption strength of the intermediates. Based on the Newns–Anderson model,46,47 the d-band theory developed by Hammer and Nørskov indicates that the bonding strength of adsorbates to the metal surface depends on the electronic structure of the metal surface.48–50 Inspired by the d-band theory, the electronic structures of metal atoms in the mullite clusters were also studied to check the influence of the d-band center on the catalytic performance.
Fig. 4a–d shows the changing trend of the adsorption energies of the intermediates O*, OH*, OO*, and OOH* versus the manganese d-band center. In the SmMn2O5 (125)n (n = 1, 2, 3, 4, 8) clusters, the position of the d-band center of the Mn atom is in the range of −2.5 eV to −1.0 eV below the Fermi level. Specifically, the (125)8 and (125)4 clusters correspond to the lowest and highest d-band centers of −2.303 eV and −1.077 eV, respectively. The d-band centres of the remaining three clusters are located at about −1.4 eV (−1.469 eV for (125)1, −1.437 eV for (125)2 and −1.375 eV for (125)3). Different from the linear relationship between the Bader charge and the binding energies of the intermediates, the change of the d-band center of Mn atom does not lead to a linear trend in the binding energies of the oxygen-containing intermediates on the different clusters. Although the d-band center of the Mn atom is located in the same position, the adsorption strengths of the intermediates on the Mn atom differ (as shown in the shaded parts in Fig. 4a–d). For (125)2 and (125)3, their corresponding d-band centers are close to each other (differing by 0.062 eV); however, the binding energies of O*, OH*, OO*, and OOH* with the clusters deviate by 2.597 eV, 2.672 eV, 4.073 eV and 2.129 eV, respectively. In contrast, the (125)4 and (125)8 clusters have a larger difference (1.226 eV) in terms of the position of the d-band center; however, the difference in the binding ability to the intermediates is relatively small, i.e., 1.282 eV, 0.05 eV, 0.691 eV and 0.029 eV for O*, OH*, OO*, and OOH*, respectively. Therefore, the ORR and OER overpotentials may fail to linearly correlate with the d-band centres. This is because for the oxide catalysts, the degenerations of the d-orbitals are normally suppressed owing to the existence of the ligand fields. Therefore, d-band center theory may not be sufficient to describe the adsorption trend of the oxygen-containing intermediate species for oxides.
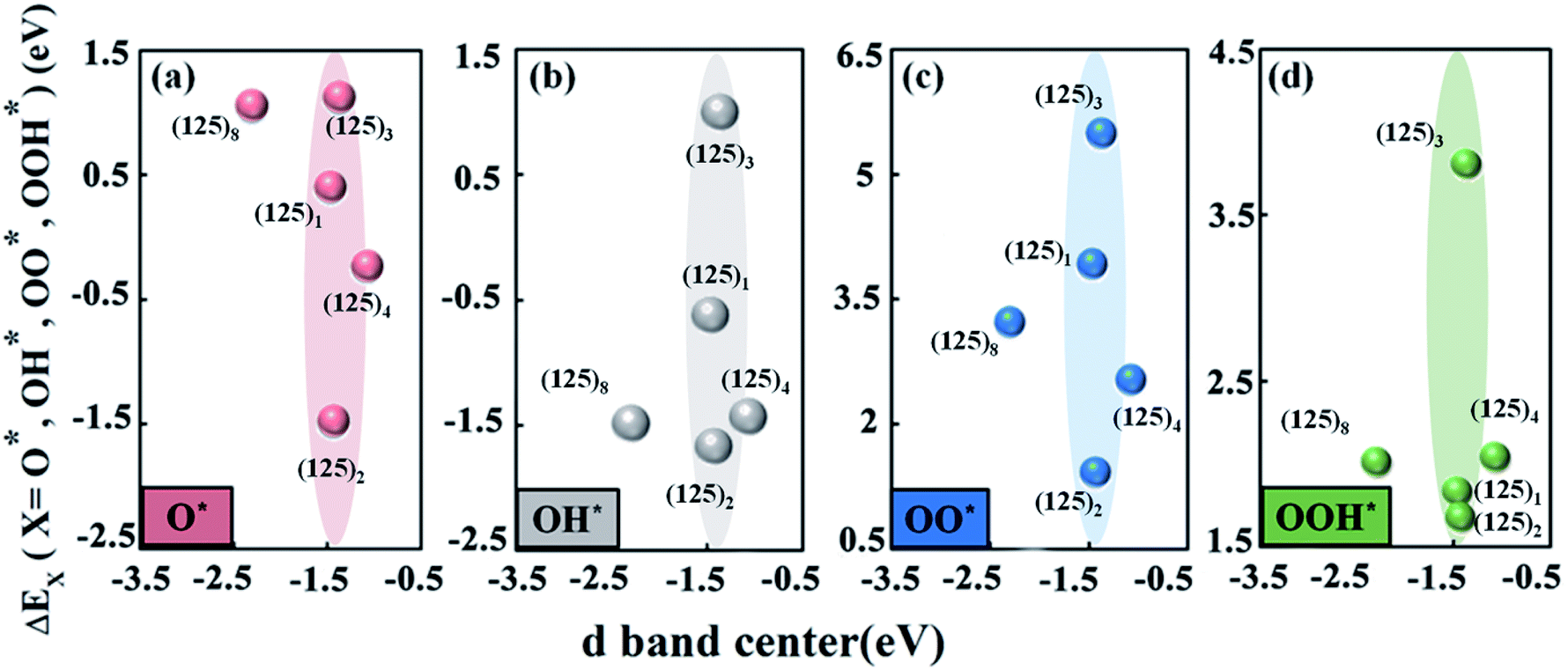 | ||
| Fig. 4 Trend diagrams of the binding energies of the ORR intermediates (via LAM) (a) O*, (b) OH*, (c) OO*, and (d) OOH* as the d band centre of Mn atom moves for the Mn-based mullite SmMn2O5 (125)n (n = 1, 2, 3, 4, 8) clusters. | ||
According to the relationship between the Bader charge of labile oxygen and the binding energies of the intermediates, we found that the labile oxygen with a Bader charge of 0.619e produces a moderate bonding strength between the intermediates and the cluster and leads to optimal electrochemical performance for the SmMn2O5 clusters. It was also noted that the relationship between the d-band centers of the metal atoms and the binding energies of the intermediates fails to describe the catalytic performance.
3.4 The feasibility and universality of the LAM
The above analysis displays that the LAM can reduce the ORR overpotential of mullite clusters to ∼0.4 V, which is superior to the AEM or LOM to reasonably describe the ORR process. The difference between the LAM and traditional AEM lies in the first step of the reactions. Specifically, O2 replaces OH* to become adsorbed OO* during the AEM; however, for the LAM, the first step is the process of proton H adsorbing on labile oxygen to form OH*. Therefore, in order to verify the feasibility of the LAM, it is important to compare their adsorption feasibilities via the binding energies of H adsorbed on labile O atoms and OH adsorbed on Mn atoms (Table 1). Taking (125)n (n = 1, 2, 3) as examples in Fig. 5a–c, we provide the adsorption configurations of the first step states (left side: AEM; right side: LAM) under the two mechanisms. | ||
| Fig. 5 (a–c) The adsorption configuration diagrams and binding energy of OH adsorbed on Mn atom and proton H adsorbed on labile oxygen corresponding to the first step of AEM and LOM over (125)n (n = 1, 2, 3) clusters. (d) The ORR overpotential on the (001) surface (as the illustration shows) under different reaction mechanisms. Sm, Mn, and O atoms are marked as blue, purple, and red, respectively. Particularly, Mn3+ with a pyramidal crystal field in the slab model is labelled as light green. | ||
According to the binding energies, for the (125)n (n = 1, 2, 3) systems, the adsorption strengths of proton H on labile oxygen (−1.174 eV, −1.087 eV and −1.400 eV for (125)1, (125)2, and (125)3, respectively) are much higher than those of OH on Mn atoms (−0.126 eV, 0.389 eV and −0.963 eV for (125)1, (125)2, and (125)3, respectively). These results indicate the feasibility of the first step of the LAM in the clusters, in which proton H is adsorbed on labile oxygen to replace the traditional AEM (the process of replacing OH* with O2).
The LAM is proposed via the small (125)n (n = 1, 2, 3, 4, 8) clusters. Thus, a question remains of whether the LAM applies to larger systems. In order to extend the LAM to other systems, we introduce the slab model of mullite, as follows.
Based on our previous experimental characterization of the mullite exposed surface (001), a slab model with lattice constants of a = 14.68 Å, b = 8.58 Å, and c = 25.05 Å (Fig. S2†) was constructed.15,51 First, we simulated the 4e− reaction steps of the AEM on the pristine (001) surface (Fig. S3† shows the Gibbs free energy diagram). Based on previous work,17 we further generated oxygen vacancies to calculate their thermodynamic barriers (Fig. S4†) and found that the overpotential of the mullite (001) surface with oxygen vacancies was reduced by 177 meV. Also, the LOM was then applied to the SmMn2O5 (001) surface (Fig. S5†). Different from the clusters, the LOM on the slab model displays better performance than the AEM. At zero cell potential (U = 0), all reaction steps are exothermic and the rate-limiting step occurs in the process of OH* desorption; its corresponding overpotential is 0.649 V, which is better than that of the structure with oxygen vacancies under the AEM. Over the same slab, we proposed the LAM (as shown in Fig. S6†). Compared to the AEM and LOM, the LAM again shows more reasonable ORR performance.
More intuitively, as shown in Fig. 5d, the ORR overpotential of the pristine (001) slab under the AEM was 0.950 V. In the presence of the oxygen defects, it was reduced to 0.773 V. Switching the reaction mechanism to the LOM, the overpotential became 0.649 V. This was further reduced to 0.499 V when following our proposed LAM, which is close to the predicted lowest overpotential of 0.43 V based on the scaling relationship over the Mn-based mullite slab models.15 This comparison indicates that in the mullite slab model, the LAM is more energetically favorable than the AEM or LOM.
Together, following the LAM in both the mullite cluster systems and the slab models, we achieved better ORR performance, indicating the greater feasibility of the LAM to describe Mn-based mullite with dual coordination crystal fields. General speaking, for a catalyst with multiple coordination fields, the variations of the activated oxygen can act as multiple active sites. Therefore, the LAM could be further validated in low-symmetric systems such as mullite and spinel.
4. Conclusions
In summary, density functional theory calculations were carried out over Mn-based mullite SmMn2O5 clusters to propose the labile oxygen participant adsorbate evolving mechanism (LAM) in regard to the AEM and LOM. Compared with the LOM without OOH* intermediate during the reactions, in which the lattice oxygen directly participates in the catalytic process, the LAM considered the labile oxygen; also, like the AEM, the intermediate OOH* forms during the catalysis, which acts as the rate-limiting step. Our calculations indicate that for both the Mn-based mullite (125)n (n = 1, 2, 3, 4, 8) clusters and slab model, the binding strength of OOH* is optimal and thus leads to a reasonable thermodynamic barrier. Based on the calculated linear relationship between the Bader charge on the labile oxygen and the binding energies of the intermediates and the inverted volcanic relationship between the ORR and OER overpotentials and Bader charge, we discovered that the labile oxygen gaining 0.619e electrons leads to moderate binding strength and eventually produces optimal performance. Our findings provide new insight into oxygen-related catalytic reactions based on the symmetries of catalyst structures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by National Key Research and Development Program (Grant No. 2016YFB0901600), Tianjin City Distinguish Young Scholar Fund (17JCJQJC45100), National Natural Science Foundation of China (21975136), Tianjin Key Research and Development Program (Grant No. 18ZXSZSF00060), Open funds from National Engineering Lab for Mobile Source Emission Control Technology (NELMS2018A01), Shenzhen Science, Technology and Innovation Committee under the project contract (No. JCYJ20190808151603654).References
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS.
- J. Fu, Z. P. Cano, M. G. Park, A. Yu, M. Fowler and Z. Chen, Adv. Mater., 2017, 29, 1604685 CrossRef.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2011, 11, 19–29 CrossRef.
- J. Rossmeisl, Z.-W. Qu, H. Zhu, G.-J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- I. C. Man, H. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martinez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- J. T. Mefford, X. Rong, A. M. Abakumov, W. G. Hardin, S. Dai, A. M. Kolpak, K. P. Johnston and K. J. Stevenson, Nat. Commun., 2016, 7, 11053 CrossRef CAS.
- X. Rong, J. Parolin and A. M. Kolpak, ACS Catal., 2016, 6, 1153–1158 CrossRef CAS.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS.
- J. R. Petrie, V. R. Cooper, J. W. Freeland, T. L. Meyer, Z. Zhang, D. A. Lutterman and H. N. Lee, J. Am. Chem. Soc., 2016, 138, 2488–2491 CrossRef CAS.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS.
- J. S. Yoo, Y. Liu, X. Rong and A. M. Kolpak, J. Phys. Chem. Lett., 2018, 9, 1473–1479 CrossRef CAS.
- Z.-F. Huang, J. Song, Y. Du, S. Xi, S. Dou, J. M. V. Nsanzimana, C. Wang, Z. J. Xu and X. Wang, Nat. Energy, 2019, 4, 329–338 CrossRef CAS.
- D. A. Kuznetsov, M. A. Naeem, P. V. Kumar, P. M. Abdala, A. Fedorov and C. R. Muller, J. Am. Chem. Soc., 2020, 142, 7883–7888 CrossRef CAS.
- A. Dong, S. Gao, X. Wan, L. Wang, T. Zhang, L. Wang, X. Lang and W. Wang, Appl. Catal., B, 2020, 271, 118932 CrossRef CAS.
- J. Liu, M. Yu, X. Wang, J. Wu, C. Wang, L. Zheng, D. Yang, H. Liu, Y. Yao, F. Lu and W. Wang, J. Mater. Chem. A, 2017, 5, 20922–20931 RSC.
- M. Yu, L. Wang, J. Liu, H. Li, X. Lang, C. Zhao, Z. Hong and W. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 17482–17490 CrossRef CAS.
- X. Zhao, L. Wang, X. Chen, W. Wang, H. L. Xin, X. Du and J. Yang, J. Power Sources, 2020, 449, 227482 CrossRef CAS.
- C. Wei, Z. Feng, G. G. Scherer, J. Barber, Y. Shao-Horn and Z. J. Xu, Adv. Mater., 2017, 29(23), 1606800 CrossRef.
- C. Li, X. Han, F. Cheng, Y. Hu, C. Chen and J. Chen, Nat. Commun., 2015, 6, 7345 CrossRef CAS.
- X.-R. Wang, J.-Y. Liu, Z.-W. Liu, W.-C. Wang, J. Luo, X.-P. Han, X.-W. Du, S.-Z. Qiao and J. Yang, Adv. Mater., 2018, 30, 1800005 CrossRef.
- H. Li, K. Cho, S. Li and W. Wang, Phys. Chem. Chem. Phys., 2018, 20, 16151–16158 RSC.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Comput. Phys. Commun., 2012, 183, 2063–2070 CrossRef CAS.
- J. Lv, Y. Wang, L. Zhu and Y. Ma, J. Chem. Phys., 2012, 137, 084104 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- H. Li, W. Wang, X. Qian, Y. Cheng, X. Xie, J. Liu, S. Sun, J. Zhou, Y. Hu, J. Xu, L. Li, Y. Zhang, X. Du, K. Gao, Z. Li, C. Zhang, S. Wang, H. Chen, Y. Zhao, F. Lu, W. Wang and H. Liu, Catal. Sci. Technol., 2016, 6, 3971–3975 RSC.
- J. Alonso, M. Casais and M. Martínez-Lope, J. Phys.: Condens. Matter, 1997, 9, 8515 CrossRef CAS.
- I. Kagomiya, K. Kohn and T. Uchiyama, Ferroelectrics, 2002, 280, 131–143 CrossRef CAS.
- D. Wales, J. Doye, A. Dullweber, M. Hodges, F. Naumkin, F. Calvo, J. Hernández-Rojas and T. Middleton, The Cambridge Cluster Database, http://www.wales.ch.cam.ac.uk/CCD.html, 2005 Search PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- J. Rossmeisl, J. K. Nørskov, C. D. Taylor, M. J. Janik and M. Neurock, J. Phys. Chem. B, 2006, 110, 21833–21839 CrossRef CAS.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- J. Kitchin, J. K. Nørskov, M. Barteau and J. Chen, J. Chem. Phys., 2004, 120, 10240–10246 CrossRef CAS.
- V. S. Chernyavsky, L. V. Pirutko, A. K. Uriarte, A. S. Kharitonov and G. I. Panov, J. Catal., 2007, 245, 466–469 CrossRef CAS.
- M. S. Palmer, M. Neurock and M. M. Olken, J. Am. Chem. Soc., 2002, 124, 8452–8461 CrossRef CAS.
- F. Han, M. Yuan, S. Mine, H. Sun, H. Chen, T. Toyao, M. Matsuoka, K. Zhu, J. Zhang and W. Wang, ACS Catal., 2019, 9, 10398–10408 CrossRef CAS.
- T. Zhang, X. Lang, A. Dong, X. Wan, S. Gao, L. Wang, L. Wang and W. Wang, ACS Catal., 2020, 10(13), 7269–7282 CrossRef CAS.
- W. Wang, G. McCool, N. Kapur, G. Yuan, B. Shan, M. Nguyen, U. M. Graham, B. H. Davis, G. Jacobs and K. Cho, Science, 2012, 337, 832–835 CrossRef CAS.
- K. Yamamoto, T. Imaoka, W.-J. Chun, O. Enoki, H. Katoh, M. Takenaga and A. Sonoi, Nat. Chem., 2009, 1, 397–402 CrossRef CAS.
- W. Xia, A. Mahmood, Z. Liang, R. Zou and S. Guo, Angew. Chem., Int. Ed. Engl., 2016, 55, 2650–2676 CrossRef CAS.
- A. A. Gewirth and M. S. Thorum, Inorg. Chem., 2010, 49, 3557–3566 CrossRef CAS.
- J. S. Yoo, X. Rong, Y. Liu and A. M. Kolpak, ACS Catal., 2018, 8, 4628–4636 CrossRef CAS.
- P. W. Anderson, Phys. Rev., 1961, 124, 41 CrossRef CAS.
- D. Newns, Phys. Rev., 1969, 178, 1123 CrossRef CAS.
- J. Liu, H. Liu, H. Chen, X. Du, B. Zhang, Z. Hong, S. Sun and W. Wang, Adv. Sci., 2020, 7, 1901614 CrossRef CAS.
- B. Hammer and J. K. Norskov, Nature, 1995, 376, 238–240 CrossRef CAS.
- B. Hammer and J. K. Nørskov, Adv. Catal., 2000, 45, 71–129 CAS.
- M. Yu, Q. Wei, M. Wu, J. Wu, J. Liu, G. Zhang, S. Sun and W. Wang, J. Power Sources, 2018, 396, 754–763 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta09537k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |