
Integrated selective nitrite reduction to ammonia with tetrahydroisoquinoline semi-dehydrogenation over a vacancy-rich Ni bifunctional electrode†
Changhong
Wang‡
a,
Wei
Zhou‡
 b,
Zhaojun
Sun
b,
Yuting
Wang
b,
Bin
Zhang
ab and
Yifu
Yu
*a
b,
Zhaojun
Sun
b,
Yuting
Wang
b,
Bin
Zhang
ab and
Yifu
Yu
*a
aInstitute of Molecular Plus, Tianjin University, Tianjin 300072, China. E-mail: yyu@tju.edu.cn
bSchool of Science, Key Laboratory of Molecular Optoelectronic Sciences, Tianjin University, Tianjin 300072, China
First published on 26th November 2020
Abstract
The development of efficient electrocatalysts for nitrite reduction to ammonia, especially integrated with a value-added anodic reaction, is important. Herein, Ni nanosheet arrays with Ni vacancies (Ni-NSA-VNi) were demonstrated to exhibit outstanding electrocatalytic performances toward selective nitrite reduction to ammonia (faradaic efficiency: 88.9%; selectivity: 77.2%) and semi-dehydrogenation of tetrahydroisoquinolines (faradaic efficiency: 95.5%; selectivity: 98.0%). The origin and quantitative analyses of ammonia were performed by 15N isotope labeling and 1H NMR experiments. The decrease in electronic cloud density induced by the Ni vacancies was found to improve the NO2− adsorption and NH3 desorption, leading to high nitrite-to-ammonia performance. In situ Raman results revealed the formation of NiII/NiIII active species for anodic semi-dehydrogenation of tetrahydroisoquinolines on Ni-NSA-VNi. Importantly, a Ni-NSA-VNi‖Ni-NSA-VNi bifunctional two-electrode electrolyzer was constructed to simultaneously produce ammonia and dihydroisoquinoline with robust stability and high selectivity.
Nitrite (NO2−), as one of the most common contaminants in groundwater, can cause serious health problems.1–4 Electro-catalytic reduction has emerged as a promising strategy to remove nitrite.5–8 Simultaneously, ammonia/ammonium has been widely used in agriculture and industry.9–11 Interestingly, nitrite is difficult to be recycled, while ammonia can be easily reclaimed from its aqueous solution.12,13 Thus, adopting nitrite as the nitrogen source and water as the hydrogen source to produce ammonia is promising for the artificial nitrogen cycle. Recently, a series of metal complexes as homogeneous electro-catalysts exhibited high activity for selective nitrite reduction to ammonia.14–16 Considering the separation difficulties of homogeneous electrocatalysts, a heterogeneous electrocatalyst for selective nitrite reduction to ammonia is urgently needed.
The counter reaction to nitrite reduction is the kinetically sluggish oxygen evolution reaction (OER), which requires large overpotential, and its product O2 does not bear a high price tag.17 An alternative strategy is to develop a thermodynamically favorable anodic reaction to produce value-added fine chemicals.18 Dihydroisoquinolines (DHIQs), as an important intermediate in the pharmaceutical industry, display a wide range of bioactivities in anti-tumor, anti-fungal, vasodilation and nonoamine oxidase inhibition.19 DHIQs can be prepared through the dehydrogenation of tetrahydroisoquinolines (THIQs). The challenge of this reaction lies in the controlled semi-dehydrogenation of THIQs to avoid undesirable over-dehydrogenation.20–22 Recently, our group reported that Ni2P could electrocatalyze semi-dehydrogenation of THIQs into DHIQs and the in situ formed NiII/NiIII redox served as the active species.23 But, expensive phosphorus sources or/and dangerous gaseous PH3 byproducts are big concerns for the synthesis of Ni2P nanostructures.23–25 Pure Ni is one of the cheap Ni-based materials, but its electrocatalytic performance is low. Interestingly, introducing anion or cation vacancies into materials could regulate their electronic structures to improve electrocatalytic performances.26–33 However, Ni nanostructures with Ni vacancies are rarely touched due to the wet-chemical synthetic difficulty. Therefore, the development of a facile method to synthesize nickel with rich Ni vacancies as a bifunctional electrocatalyst for integrating semi-dehydrogenation of THIQs with selective nitrite electroreduction to ammonia is significant but still remains a great challenge.
Here, Ni nanosheet arrays with Ni vacancies, denoted as Ni-NSA-VNi, were prepared through plasma-assisted synthesis and exhibited high performance for selective nitrite electroreduction and semi-dehydrogenation of THIQs. The experimental and density functional theory (DFT) calculation results revealed that Ni vacancies promoted the conversion of nitrite to ammonia. In situ Raman spectra confirmed the formation of NiII/NiIII active species for semi-dehydrogenation of THIQs. Ni-NSA-VNi could act as efficient bifunctional electrodes for simultaneously producing ammonia and DHIQs with high stability and selectivity in a Ni-NSA-VNi‖Ni-NSA-VNi two-electrode electrolyzer.
Ni-NSA-VNi were synthesized through a facile H2-plasma chemical conversion (see the ESI† for details). First, Ni(OH)2 nanosheet arrays supported on Ni foam were prepared by a typical solution-chemical method (Fig. S1†) and then reduced to Ni nanosheet arrays (Ni-NSA) using ethylene glycol.34 Finally, Ni-NSA-VNi could be obtained through the H2-plasma treatment of Ni-NSA because of the etching role of plasma. A series of advanced techniques were adopted to characterize Ni-NSA-VNi. Scanning electron micrograph (SEM) images (Fig. S2a and b†) show the nanosheet array morphology of Ni-NSA and Ni-NSA-VNi, suggesting the morphology retention during the plasma-assisted conversion synthesis. The transmission electron micrograph (TEM) image of Ni-NSA-VNi (inset in Fig. 1a) displayed a transparent structure, indicating ultrathin thickness. The typical high-resolution TEM (HRTEM) image of Ni-NSA-VNi demonstrated a crystalline structure with a clear lattice spacing of 0.21 nm belonging to the (111) facet of Ni (Fig. 1a). Some amorphous regions within the yellow circles could be clearly observed, indicating the presence of vacancies.35 To exclude the influence of the Ni substrate on X-ray diffraction (XRD), Ni-NSA-VNi was synthesized on Cu foam. The XRD pattern of Ni-NSA-VNi/Cu (Fig. 1b) exhibited exclusive signals of pure Ni (JCPDS no. 04-0850), which were the same as that of the Ni foam (Fig. S3b†).34 The atomic force microscopy (AFM) image and the height configurations (Fig. S4†) revealed the thickness of Ni-NSA-VNi to be 1.8 nm, confirming their ultrathin thickness. X-ray photoelectron spectroscopy (XPS) and extended X-ray absorption fine structure (EXAFS) were performed to analyze Ni-NSA-VNi. In Ni 2p XPS spectra (Fig. 1c), the peaks at 852.9 and 870.3 eV could be assigned to Ni0, while the two peaks of 855.6 and 873.5 eV arising from Ni2+ could be ascribed to the unavoidable oxidation during the XPS test.36 The similarity of the Ni K-edge radial structure functions to those of the Ni foam verified that Ni-NSA-VNi was metallic nickel (Fig. 1d). The weaker peak intensity of the Ni–Ni shell (2.4 Å) in Ni-NSA-VNi compared to Ni foam arose from its decreased coordination number as a result of the nickel vacancies (Table S1†).35 The aforementioned results demonstrated that the H2-plasma treatment of Ni-NSA could produce abundant nickel vacancies with the maintenance of the morphology and structure.
 | ||
| Fig. 1 (a) HRTEM image of Ni-NSA-VNi. Inset is the corresponding TEM image. (b) XRD pattern of Ni-NSA-VNi/Cu foam. (c) XPS spectra of Ni-NSA-VNi. (d) Ni K-edge radial structure functions of Ni foam and Ni-NSA-VNi. | ||
The performance of nitrite electroreduction was recorded versus the saturated calomel electrode (SCE) with 200 ppm nitrite-N (Fig. 2). All electrochemical tests were performed in a standard three-electrode electrochemical cell by using 0.2 M Na2SO4 solution as the electrolyte. A typical H-type electrolytic cell was used. The as-prepared samples on the Ni foam, Pt plate and SCE were used as the working electrode, the counter electrode and the reference electrode, respectively. The reaction for calculation of conversion and faradaic efficiency was carried out for 2 hours. Colorimetric methods were adopted to determine the concentration of nitrite and ammonia in pre- and post-test electrolytes (Fig. S5†).9–11,37–39 The linear sweep voltammetry (LSV) curves of Ni-NSA-VNi showed obvious enhancement in the current density in the presence of NaNO2, and the current density of Ni-NSA-VNi was much higher than that of Ni foam (Fig. S6† and 2a).
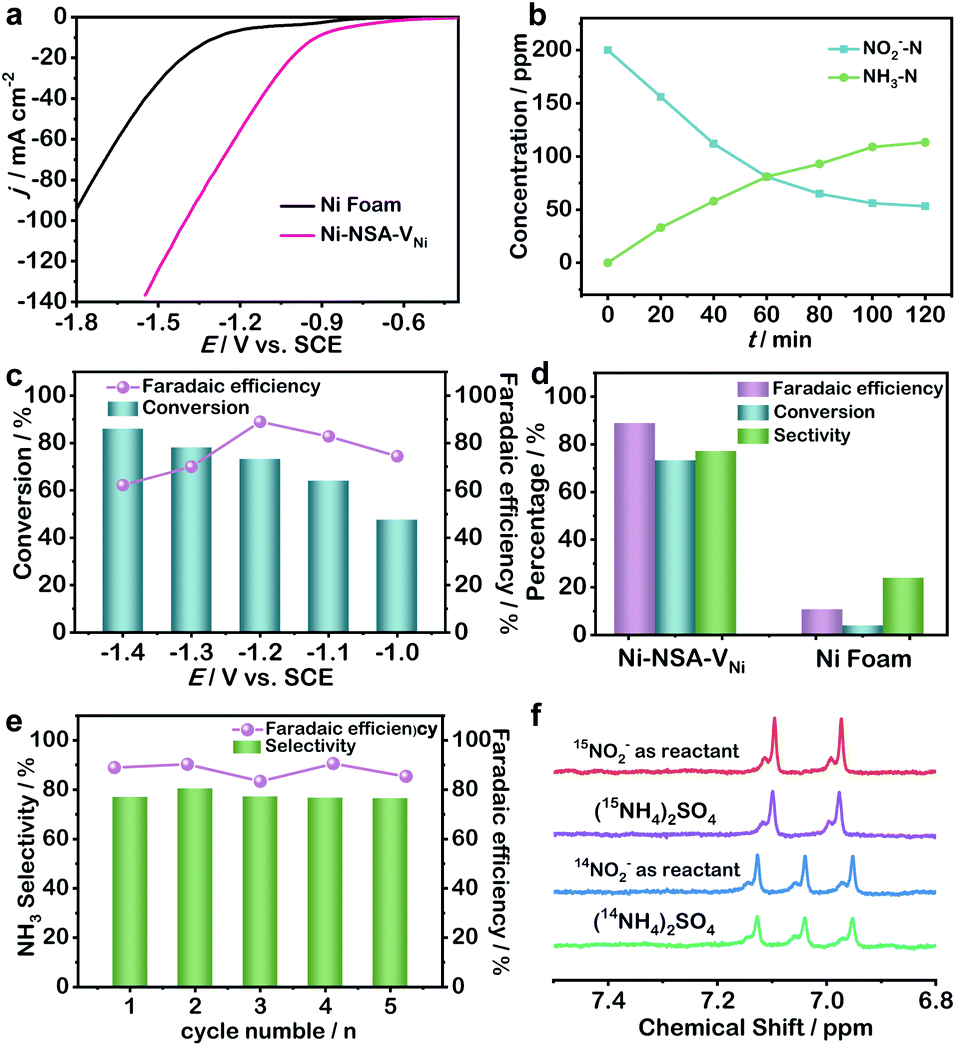 | ||
| Fig. 2 (a) LSV curves of Ni Foam and Ni-NSA-VNi in 0.2 M Na2SO4 with 200 ppm nitrite. (b) Time-dependent concentration of nitrite and ammonia over Ni-NSA-VNi at −1.2 V vs. SCE. (c) Conversion yield of nitrite and faradaic efficiency of ammonia over Ni-NSA-VNi at different potentials. (d) Faradaic efficiency and selectivity of ammonia, and conversion rate of nitrite at −1.2 V vs. SCE. (e) Selectivity and faradaic efficiency of ammonia during consecutive recycling tests for Ni-NSA-VNi at −1.2 V vs. SCE. (f) 1H NMR spectra of standard samples (15NH4)2SO4 and (14NH4)2SO4 and electrolyte after the NO2− reduction using 14NO2− and 15NO2− as the N-source. | ||
With prolonging the reaction time, the concentration of nitrite decreased, while the concentration of ammonia increased, suggesting nitrite reduction to ammonia (Fig. 2b). The selectivity of ammonia over Ni-NSA-VNi kept increasing from −1.0 to −1.4 V, while the faradaic efficiency displayed a volcanic shape curve with a maximum at −1.2 V (Fig. 2c). Thus, we chose −1.2 V as the operation voltage for investigation of nitrite electroreduction. The electrocatalytic performance of Ni-NSA-VNi (conversion rate: 73.4%, faradaic efficiency: 88.9%, selectivity: 77.2%) was remarkably higher than those of Ni foam (conversion rate: 10.8%, faradaic efficiency: 3.9%, and selectivity: 23.9%) at −1.2 V vs. SCE (Fig. 2d). Notably, the faradaic efficiency and yield rate of ammonia were much higher than all the reported values of nitrogen electroreduction and among the best for electroreduction of nitrite to ammonia (Table S2†). The electroreduction of 200 ppm nitrite in 2 h was regarded as one cycle. The selectivity and faradaic efficiency of ammonia exhibited no decay during five cycling tests (Fig. 2e). Moreover, the morphology and structure of Ni-NSA-VNi were maintained well after the durability test, suggesting the high stability (Fig. S7†). Negligible ammonia was detected without the addition of nitrite (Fig. S8†), indicating that the produced ammonia originated from nitrite reduction. Meanwhile, 15N isotope labelling experiments were conducted and the yield of ammonium was quantified using 1H nuclear magnetic resonance (NMR) spectra.40 The 1H NMR spectra of electrolyte adopting 15NO2− as the reactant showed typical double peaks of 15NH4+, while the 1H NMR spectra of electrolyte adopting 14NO2− as the reactant displayed typical triple peaks of 14NH4+ (Fig. 2f).41,42 The 1H NMR results further confirmed that the produced ammonia was derived from nitrite. For quantification, maleic acid (C4H4O4) was chosen as the 1H NMR external standard. Based on the standard curve of the integral area (NH4+–N/C4H4O4) against the NH4+–N concentration, the ammonia was quantified (Fig. S9 and S10†). The generated 15NH4+–15N and 14NH4+–14N quantified by 1H NMR were very close to the results quantified by colorimetric methods (Table S3†), suggesting the quantitative accuracy.
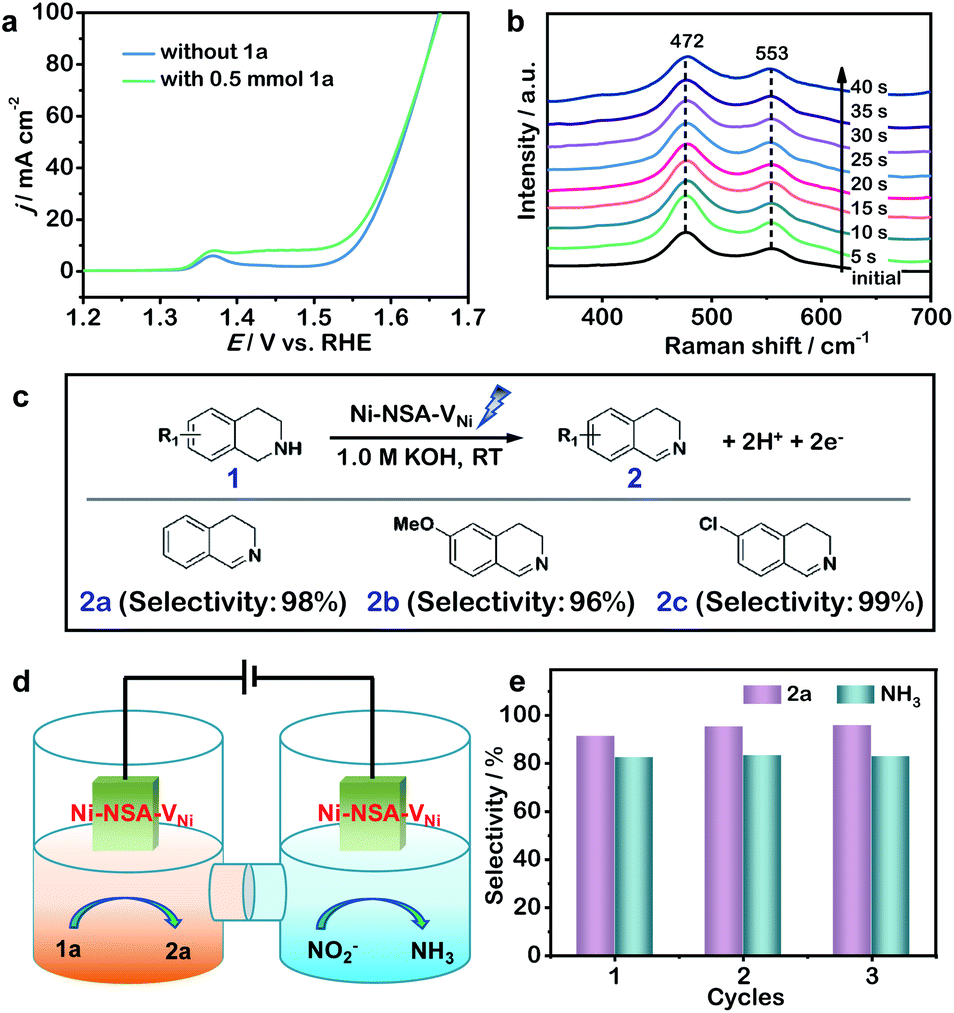
To further probe the origin of high performance of Ni-NSA-VNi for selective nitrite reduction to ammonia, a series of in situ and ex situ experiments and theoretical calculations were performed. First, the ammonia yield rate of Ni-NSA-VNi was much higher than that of Ni foam (Fig. 3a). Furthermore, the electrochemical impedance spectroscopy (EIS) results illustrated that vacancy-rich Ni-NSA-VNi possessed smaller interfacial charge-transfer resistance than Ni foam (Fig. S11†). Online differential electrochemical mass spectrometry (DEMS) measurements were performed for the detection of the molecular intermediate and product over Ni-NSA-VNi (Fig. 3b).43–45 The m/z signals of NO (30) and NH3 (17) combined with the large desorption free energies (ΔGdes) of NO on the surfaces of Ni foam (ΔGdes = 2.53 eV) and Ni-NSA-VNi (ΔGdes = 2.61 eV) obtained by DFT calculations indicated NO as one of the intermediates. To gain insights into the nitrite electroreduction process, the surface electron cloud density and reaction Gibbs free energies (ΔG) were calculated based on DFT. Compared to Ni foam, Ni-NSA-VNi possessed lower surface electron cloud density (Fig. 3c and d). As displayed in Fig. 3e, the adsorption energy of NO2− on Ni-NSA-VNi (−0.44 eV) was much lower than that of Ni foam (0.12 eV). For further hydrogenation processes, the reaction Gibbs free energy of each step was downhill or uphill with a small degree (ΔGmax = 0.25 and 0.16 eV over Ni foam and Ni-NSA-VNi, respectively), guaranteeing the conversion from 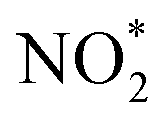 to
to  . The last desorption of NH3 from Ni-NSA-VNi (ΔG = 1.12 eV) was also easier than that on Ni foam (ΔG = 1.34 eV). Thus, the decreased surface electron cloud density in Ni-NSA-VNi improved the adsorption and activation of NO2− and the desorption of NH3, leading to high nitrite-to-ammonia catalytic performance over the Ni-NSA-VNi cathode.
. The last desorption of NH3 from Ni-NSA-VNi (ΔG = 1.12 eV) was also easier than that on Ni foam (ΔG = 1.34 eV). Thus, the decreased surface electron cloud density in Ni-NSA-VNi improved the adsorption and activation of NO2− and the desorption of NH3, leading to high nitrite-to-ammonia catalytic performance over the Ni-NSA-VNi cathode.
 | ||
| Fig. 3 (a) The yield rate of ammonia over Ni foam and Ni-NSA-VNi. (b) DEMS of Ni-NSA-VNi for nitrite electroreduction. Calculated surface electron density distribution of (c) Ni foam and (d) Ni-NSA-VNi. (e) Free energy diagram of the nitrite electroreduction process over Ni foam and Ni-NSA-VNi at 0 V vs. RHE. | ||
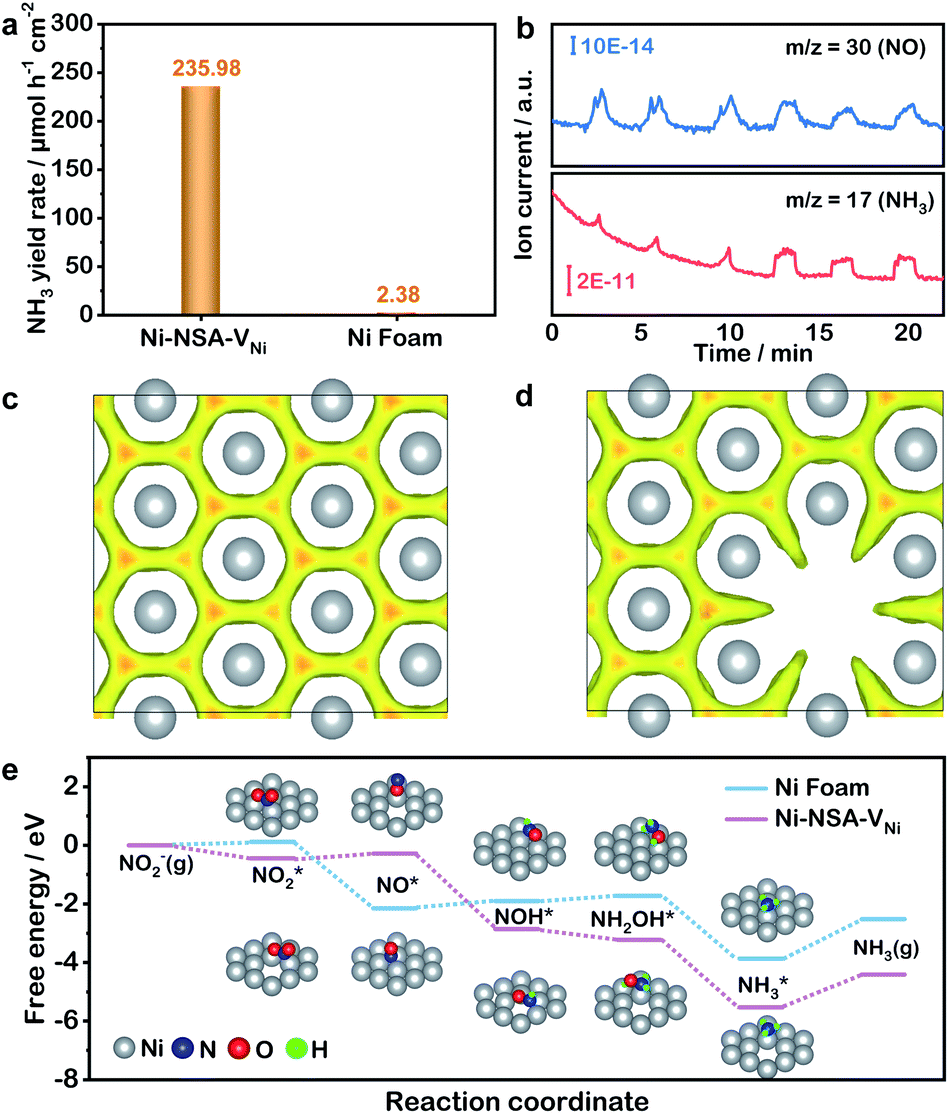
Next, the electrocatalytic semi-dehydrogenation of THIQs over the Ni-NSA-VNi anode was carried out in 1.0 M KOH aqueous solution. The 1,2,3,4-tetrahydroisoquinoline (1a) was selected as a model substrate. Without 1a, the OER proceeded on Ni-NSA-VNi (Fig. 4a). To achieve the current density of 50 mA cm−2 for the OER, Ni-NSA-VNi required 1.61 V vs. the reversible hydrogen electrode (RHE). Notably, the oxidation peak before 1.45 V arose from the NiII/NiIII redox, which was demonstrated as active species for the electrocatalytic semi-dehydrogenation conversion of THIQs.23 After adding 1a, Ni-NSA-VNi, exhibited increased current density, indicating the oxidation of THIQs (Fig. 4a). Compared with Ni foam (Fig. S12†), Ni-NSA-VNi exhibited much higher current density, indicating the superior oxidation ability of THIQs. Time-dependent Raman spectra initially showed two peaks of NiOOH at 472 and 553 cm−1 for Ni-NSA-VNi (Fig. 4b).23,46 The two peaks of NiOOH in Ni-NSA-VNi were maintained well after the addition of 1a. This proved the superior stability of NiII/NiIII species on Ni-NSA-VNi during the electrooxidation process of 1a. Ni-NSA-VNi could efficiently drive a semi-dehydrogenation conversion of 1a into 2a with 98.0% selectivity and 95.5% faradaic efficiency. 6-Methoxy tetrahydroisoquinoline (1b) and hexachlorotetrahydroisoquinoline (1c) were efficiently converted to the corresponding dihydroisoquinolines with 96–99% selectivity, suggesting the good group tolerance of the tetrahydroisoquinoline semi-dehydrogenation over the Ni-NSA-VNi anode (Fig. 4c and S13†). Our results demonstrated that Ni-NSA-VNi could act as an efficient bifunctional electrode material for both cathodic nitrite-to-ammonia reduction and anodic tetrahydroisoquinoline semi-dehydrogenation. Thus, a Ni-NSA-VNi‖Ni-NSA-VNi two-electrode electrolyzer was constructed to simultaneously produce ammonia at the cathode and DHIQs at the anode (Fig. 4d). After adding 1a, the current density increased for Ni-NSA-VNi, indicating that the overall potential can be decreased by adopting oxidation of THIQs to replace the OER (Fig. S14†). The selectivity of 2a and ammonia over the Ni-NSA-VNi bifunctional electrodes achieved 91.5% and 82.7% at the voltage of 1.5 V vs. counter electrode (CE). During three continuous cycles, no decrease in their selectivities was observed (Fig. 4e), reflecting the good durability of the bifunctional Ni-NSA-VNi electrodes. Despite a great advance in using value-added reactions to replace the OER in hydrogen production,18,47–50 this work is the first report on the development of high-value organic oxidation as an alternative anodic reaction at a low voltage for ammonia synthesis.
 | ||
| Fig. 4 (a) LSV curves of the Ni-NSA-VNi anode at a scan rate of 5 mV s−1 in 1.0 M KOH electrolyte with and without 0.5 mmol 1a. (b) Time-dependent in situ Raman spectra of Ni-NSA-VNi recorded at 1.4 V versus RHE in 0.1 M KOH solution without 1a (initial) and after the addition of 1a. (c) The substrate scope of the selective semi-dehydrogenation of THIQs over a Ni-NSA-VNi anode. (d) Schematic illustration for coupling electrocatalytic semi-dehydrogenation of THIQs with nitrite electroreduction to ammonia. (e) Cycle-dependent selectivities of 2a and ammonia over the Ni-NSA-VNi bifunctional electrodes. | ||
Conclusions
In conclusion, we successfully introduced Ni vacancies into Ni nanosheets through the H2-plasma treatment. Ni vacancies could not only promote the cathodic selective nitrite electroreduction to ammonia (faradaic efficiency: 88.9% and selectivity: 77.2%), but also improve the anodic semi-dehydrogenation transformation of THIQs into DHIQs (selectivity: 98.0%; faradaic efficiency: 95.5%) over the Ni-NSA-VNi electrodes. The combined results of in situ and ex situ experiments and theoretical calculations revealed the enhanced mechanism of vacancies. The vacancy-induced decrease in the electronic cloud density of Ni improved the adsorption of NO2− and the desorption of NH3, leading to high catalytic performance for nitrite reduction. Moreover, the NiII/NiIII active species for the semi-dehydrogenation of THIQs were maintained well during the electrooxidation process. Notably, the bifunctional Ni-NSA-VNi electrodes can be assembled into a two-electrode electrolyzer for simultaneously producing ammonia and DHIQs with robust stability and high selectivity. This work opens a facile electrocatalytic strategy to integrate nitrite-to-ammonia reduction with the oxidative transformation of organic compounds with high efficiency.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 22071173 and 21871206).References
- S. G. Lehman, M. Badruzzaman, S. Adham, D. J. Roberts and D. A. Clifford, Water Res., 2008, 42, 969–976 CrossRef CAS PubMed.
- V. Rosca, M. Duca, M. T. de Groot and M. T. M. Koper, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS PubMed.
- H. Shin, S. Jung, S. Bae, W. Lee and H. Kim, Environ. Sci. Technol., 2014, 48, 12768–12774 CrossRef CAS PubMed.
- N. Singh, K. Patel, S. K. Sahoo, R. K. Pati and R. Kumar, J. Mater. Chem. A, 2017, 5, 3389–3403 RSC.
- Y. Matatov-Meytal, Appl. Catal., B, 2003, 45, 127–134 CrossRef CAS.
- M. Duca, S. Khamseh, S. C. Lai and M. T. Koper, Langmuir, 2010, 26, 12418–12424 CrossRef CAS PubMed.
- H. Liu, X. Liu, Y. Yu, W. Yang, J. Li, M. Feng and H. Lia, J. Mater. Chem. A, 2018, 6, 4611–4616 RSC.
- D. He, Y. Li, H. Ooka, Y. K. Go, F. Jin, S. H. Kim and R. Nakamura, J. Am. Chem. Soc., 2018, 140, 2012–2015 CrossRef CAS PubMed.
- P. Wang, F. Chang, W. Gao, J. Guo, G. Wu, T. He and P. Chen, Nat. Chem., 2017, 9, 64–70 CrossRef CAS PubMed.
- J. Wang, L. Yu, L. Hu, G. Chen, H. Xin and X. Feng, Nat. Commun., 2018, 9, 1795 CrossRef PubMed.
- C. Tang and S. Z. Qiao, Chem. Soc. Rev., 2019, 48, 3166–3180 RSC.
- I. Ozturk, M. Altinbas, I. Koyuncu, O. Arikan and C. Gomec-Yangin, Waste Manage., 2003, 23, 441–446 CrossRef CAS PubMed.
- A. Thornton, P. Pearce and S. A. Parsons, J. Hazard. Mater., 2007, 147, 883–889 CrossRef CAS PubMed.
- M. H. Baryley, K. J. Takeuchi and T. J. Meyer, J. Am. Chem. Soc., 1986, 108, 5876–5885 CrossRef PubMed.
- Y. Arikawa, Y. Otsubo, H. Fujino, S. Horiuchi, E. Sakuda and K. Umakoshi, J. Am. Chem. Soc., 2018, 140, 842–847 CrossRef CAS PubMed.
- Y. Guo, J. R. Stroka, B. Kandemir, C. E. Dickerson and K. L. Bren, J. Am. Chem. Soc., 2018, 140, 16888–16892 CrossRef CAS PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- B. You and Y. Sun, Acc. Chem. Res., 2018, 51, 1571–1580 CrossRef CAS PubMed.
- M. Chaumontet, R. Piccardi and O. Baudoin, Angew. Chem., Int. Ed., 2009, 48, 179–182 CrossRef CAS PubMed.
- X. Cui, Y. Li, S. Bachmann, M. Scalone, A. E. Surkus, K. Junge, C. Topf and M. Beller, J. Am. Chem. Soc., 2015, 137, 10652–10658 CrossRef CAS PubMed.
- S. Kato, Y. Saga, M. Kojima, H. Fuse, S. Matsunaga, A. Fukatsu, M. Kondo, S. Masaoka and M. Kanai, J. Am. Chem. Soc., 2017, 139, 2204–2207 CrossRef CAS PubMed.
- M. Zheng, J. Shi, T. Yuan and X. Wang, Angew. Chem., Int. Ed., 2018, 57, 5487–5491 CrossRef CAS PubMed.
- C. Huang, Y. Huang, C. Liu, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2019, 58, 12014–12017 CrossRef CAS PubMed.
- Y. Shi and B. Zhang, Chem. Soc. Rev., 2016, 45, 1529–1541 RSC.
- J. Tian, Q. Liu, A. M. Asiri and X. Sun, J. Am. Chem. Soc., 2014, 136, 7587–7590 CrossRef CAS PubMed.
- T. Mitsui, M. K. Rose, E. Fomin, D. F. Ogletree and M. Salmeron, Nature, 2003, 422, 705–707 CrossRef CAS PubMed.
- D. Yan, Y. Li, J. Huo, R. Chen, L. Dai and S. Wang, Adv. Mater., 2017, 29, 1606459 CrossRef PubMed.
- Q. He, Y. Wan, H. Jiang, Z. Pan, C. Wu, M. Wang, X. Wu, B. Ye, P. M. Ajayan and L. Song, ACS Energy Lett., 2018, 3, 1373–1380 CrossRef CAS.
- C. Lv, Y. Qian, C. Yan, Y. Ding, Y. Liu, G. Chen and G. Yu, Angew. Chem., Int. Ed., 2018, 57, 10246–10250 CrossRef CAS PubMed.
- C. Li, Q. Yuan, B. Ni, T. He, S. Zhang, Y. Long, L. Gu and X. Wang, Nat. Commun., 2018, 9, 3702 CrossRef PubMed.
- Q. Wang, Y. Lei, D. Wang and Y. Li, Energy Environ. Sci., 2019, 12, 1730–1750 RSC.
- Y. Tong, P. Chen, M. Zhang, T. Zhou, L. Zhang, W. Chu, C. Wu and Y. Xie, ACS Catal., 2017, 8, 1–7 Search PubMed.
- Y. Zhu, X. Liu, S. Jin, H. Chen, W. Lee, M. Liu and Y. Chen, J. Mater. Chem. A, 2019, 7, 5875–5897 RSC.
- Y. Kuang, G. Feng, P. Li, Y. Bi, Y. Li and X. Sun, Angew. Chem., Int. Ed., 2016, 55, 693–697 CrossRef CAS PubMed.
- S. He, C. Li, H. Chen, D. Su, B. Zhang, X. Cao, B. Wang, M. Wei, D. G. Evans and X. Duan, Chem. Mater., 2013, 25, 1040–1046 CrossRef CAS.
- P. Prieto, V. Nistor, K. Nouneh, M. Oyama, M. Abd-Lefdil and R. Díaz, Appl. Surf. Sci., 2012, 258, 8807–8813 CrossRef CAS.
- L. S. Clesceri, A. D. Eaton, A. E. Greenberg, A. P. H. Association, A. W. W. Association and W. E. Federation, Standard methods for the examination of water and wastewater, American Public Health Association, 1998 Search PubMed.
- M. Li, H. Huang, J. Low, C. Gao, R. Long and Y. Xiong, Small Methods, 2019, 3, 1800388 CrossRef.
- L. Li, C. Tang, B. Xia, H. Jin, Y. Zheng and S.-Z. Qiao, ACS Catal., 2019, 9, 2902–2908 CrossRef CAS.
- S. Z. Andersen, V. Colic, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. McEnaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh, B. A. Rohr, M. J. Statt, S. J. Blair, S. Mezzavilla, J. Kibsgaard, P. C. K. Vesborg, M. Cargnello, S. F. Bent, T. F. Jaramillo, I. E. L. Stephens, J. K. Norskov and I. Chorkendorff, Nature, 2019, 570, 504–508 CrossRef CAS PubMed.
- Y. Wang, W. Zhou, R. Jia, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 5350–5354 CrossRef CAS PubMed.
- L. Zhang, L. X. Ding, G. F. Chen, X. Yang and H. Wang, Angew. Chem., Int. Ed., 2019, 58, 2612–2616 CrossRef CAS PubMed.
- O. Wolter and J. Heitbaum, Ber. Bunsenges. Phys. Chem., 1984, 88, 2–6 CrossRef CAS.
- X. Wang, J. F. de Araujo, W. Ju, A. Bagger, H. Schmies, S. Kuhl, J. Rossmeisl and P. Strasser, Nat. Nanotechnol., 2019, 14, 1063–1070 CrossRef CAS PubMed.
- C. J. Bondue, F. Calle-Vallejo, M. C. Figueiredo and M. T. M. Koper, Nat. Catal., 2019, 2, 243–250 CrossRef CAS.
- Y. Huang, X. Chong, C. Liu, Y. Liang and B. Zhang, Angew. Chem., Int. Ed., 2018, 57, 13163–13166 CrossRef CAS PubMed.
- Y. Yuan and A. Lei, Acc. Chem. Res., 2019, 52, 3309–3324 CrossRef CAS PubMed.
- S. Barwe, J. Weidner, S. Cychy, D. M. Morales, S. Dieckhofer, D. Hiltrop, J. Masa, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2018, 57, 11460–11464 CrossRef CAS PubMed.
- C. Tang, R. Zhang, W. Lu, Z. Wang, D. Liu, S. Hao, G. Du, A. M. Asiri and X. Sun, Angew. Chem., Int. Ed., 2017, 56, 842–846 CrossRef CAS PubMed.
- Z. Ji, J. Liu, Y. Deng, S. Zhang, Z. Zhang, P. Du, Y. Zhao and X. Lu, J. Mater. Chem. A, 2020, 8, 14680–14689 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, supplementary characterization and electrochemical measurements. See DOI: 10.1039/d0ta09590g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |