
Boosting the performance of a silicon photocathode for photoelectrochemical hydrogen production by immobilization of a cobalt tetraazamacrocyclic catalyst†
Chengming
Nie
,
Chang
Liu
,
Lunlun
Gong
and
Mei
Wang
 *
*
State Key Laboratory of Fine Chemicals, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, China. E-mail: symbueno@dlut.edu.cn
First published on 8th November 2020
Abstract
Modification of the surface of semiconductor photoelectrodes with molecular catalysts provides a way to realize atom-efficient catalysis for photoelectrochemical (PEC) water splitting; however, only a few molecular catalysts have been applied for PEC H2 production to date. Herein we present an efficient and stable Si-based photocathode modified with a highly active cobalt tetraazamacrocyclic catalyst (denoted as Co(CR-DCP)) through a 2,6-dicarboxypyridin-4-yl (DCP) anchor. The immobilization of Co(CR-DCP) boosted the photocurrent density of the electrode up to −682 μA cm−2 at 0 V vs. RHE, which was increased by a factor of 31 compared to that of bare Si/TiO2. Meanwhile, the Si/TiO2/Co(CR-DCP) photocathode displayed a steady photocurrent over 10 h of controlled potential photoelectrolysis experiment. In terms of PEC activity and stability, the Si/TiO2/Co(CR-DCP) electrode outperforms previously reported molecular catalyst-modified hybrid photocathodes based on Ga- and In-free semiconductors without an extra TiO2 outer layer. The kinetic studies revealed that the surface-bound cobalt catalyst substantially improved the PEC activity of the Si-based electrode not only by evidently accelerating the electron transfer rate, but also by retarding the surface electron–hole recombination rate.
Hydrogen production by solar-driven water splitting is an ideal approach to obtain a carbon-neutral and sustainable fuel by utilizing renewable energy sources. One of the most promising and practical technologies for large-scale solar-driven hydrogen production is photoelectrochemical (PEC) water splitting. Although the pioneering work on PEC water splitting can be traced back to the early 1970s,1 the development of highly efficient, robust, and inexpensive photoelectrodes for constructing bias-free or low bias total water splitting PEC cells still remains a substantial challenge after half a century.
Semiconductor/molecular catalyst (SC/MC) hybrid photoelectrodes are of great interest for PEC water splitting,2–6 as molecular catalysts can form a light penetrable monolayer on the surface of semiconductors and realize atom-efficient catalysis. Although lots of highly active molecular catalysts were reported for the hydrogen evolution reaction (HER), only a few of them have been applied for PEC H2 production to date, being limited to cobaloximes,7–17 DuBois-type nickel complexes,17–20 diiron hydrogenase models,21 iron and cobalt porphyrin complexes,22 and incomplete cubane-like Mo3S4 clusters.23,24 The cobalt tetraazamacrocyclic complex [Co(CR)X2]+ (CR = 2,12-dimethyl-3,7,11,17-tetraazabicyclo[11.3.1]heptadeca-1(17),2,11,13,15-pentaene; X = Br, Cl) has been demonstrated to be one of the most efficient and stable H2-evolution molecular catalysts,25–27 but it has only been used as a free catalyst in bulk solution for electro- or photochemical H2 evolution and CO2 reduction in the past thirty years.28–35 There has been no report on covalently linking [Co(CR)X2]+ to the surface of (photo)electrodes to date.
Herein we report an efficient and stable Si-based hybrid photocathode, in which the functionalized cobalt tetraazamacrocyclic complex [Co(CR-DCP)Br2]+ (denoted as Co(CR-DCP), DCP = 2,6-dicarboxypyridin-4-yl group at position 15 of the CR ligand) was covalently immobilized on the surface of a TiO2-protected p-Si electrode through the DCP anchor (Fig. 1). The 2,6-dicarboxypyridin-4-yl group has been demonstrated to be an effective anchor for immobilization of molecular catalysts on metal oxide semiconductor photoelectrodes.36–39 The studies on the PEC performance of Si/TiO2/Co(CR-DCP) manifested that modification of the Si/TiO2 electrode with Co(CR-DCP) greatly boosted the PEC activity of the photocathode. The photocurrent density (J(0V)) of the Co(CR-DCP)-decorated photocathode at 0 V (all potentials given in this paper are versus the reversible hydrogen electrode (RHE)) was enhanced by a factor of 31, and the photocurrent onset potential (Eon, designated at J = −0.01 mA cm−2) was positively shifted by 0.32 V, compared to that of bare Si/TiO2 in 0.1 M acetate buffer at pH 4.5 under illumination (100 mW cm−2, λ > 400 nm). Rather appealingly, the J(0V) of −682 μA cm−2 achieved for Si/TiO2/Co(CR-DCP) is higher than those achieved for previously reported molecular catalyst-modified hybrid photocathodes which were based on Ga- and In-free semiconductors without an extra TiO2 outer layer. Meanwhile, Si/TiO2/Co(CR-DCP) displayed good durability for PEC H2-evolution under test conditions. The quantitative kinetic studies on surface electron transfer and charge recombination revealed the origin for the evidently improved PEC activity of the Si-based electrode by the surface-bound Co(CR-DCP). To the best of our knowledge, this is the first example of covalently immobilizing the cobalt tetraazamacrocyclic complex on the surface of electrodes and photoelectrodes.
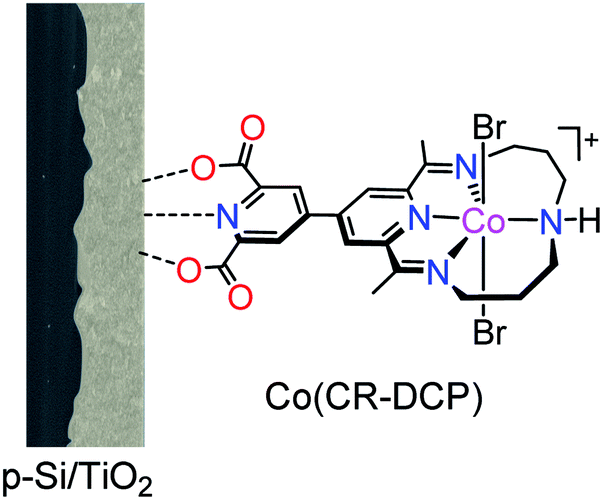 | ||
| Fig. 1 Schematic illustration of the Si/TiO2/Co(CR-DCP) hybrid photocathode. | ||
The DCP-functionalized cobalt tetraazamacrocyclic complex was synthesized according to our previously reported procedure.40 The TiO2 film was coated on the surface of a freshly treated p-Si wafer by doctor-blading of a paste of TiO2 nanoparticles (18–20 nm), and then Si/TiO2 was annealed following a reported controlled sintering procedure up to 450 °C under atmospheric conditions.17 The field-emission scanning electron microscopy (FE-SEM) images of the as-prepared Si/TiO2 electrode showed that the TiO2 film had a porous microstructure (Fig. 2a and S1†) with a thickness of about 5.0 μm (inset in Fig. 2a). Subsequently, the Si/TiO2 electrode was immersed in a methanol solution of Co(CR-DCP) (1.0 mM) for 16 h in the dark. Afterward, the electrode was rinsed with distilled water and MeOH to remove the non-bonded catalyst molecules on the surface of the TiO2 film.
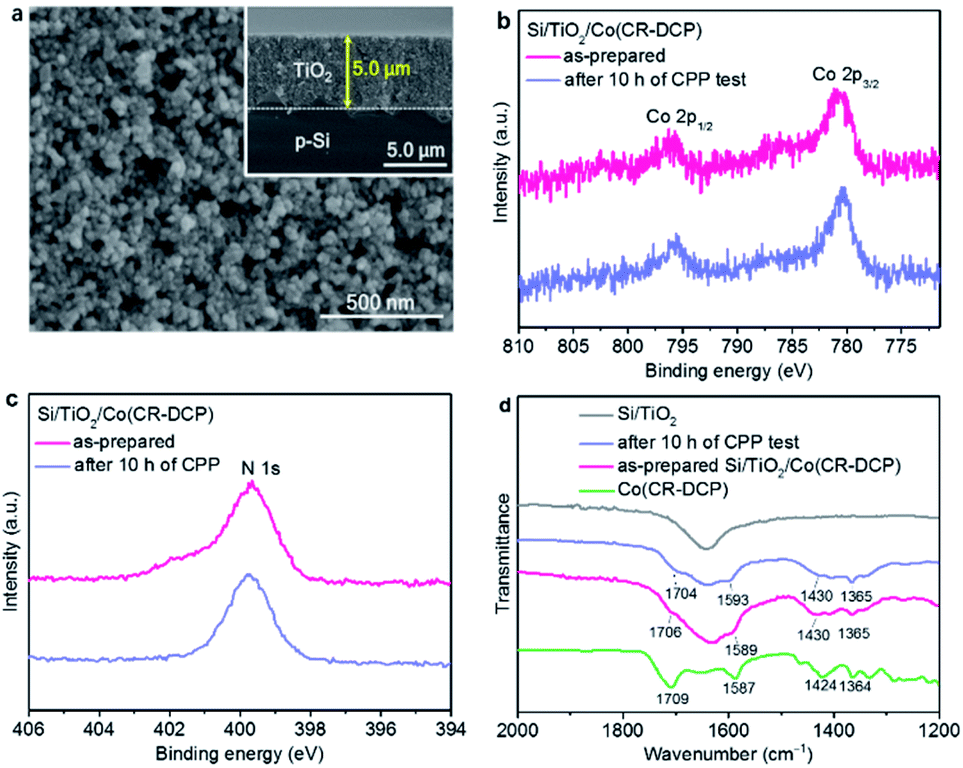 | ||
| Fig. 2 (a) Top-view SEM image of Si/TiO2 (inset: side-view SEM image). XPS spectra of (b) Co 2p and (c) N 1s for Si/TiO2/Co(CR-DCP) before and after its use for 10 h of controlled potential photoelectrolysis (CPP) experiment under chopped illumination. (d) ATR-FTIR spectra of Co(CR-DCP) (KBr disc), Si/TiO2, and Si/TiO2/Co(CR-DCP) before and after its use for 10 h of CPP experiment. | ||
The X-ray photoelectron spectroscopy (XPS) spectrum (Fig. S2†) of the as-prepared Si/TiO2/Co(CR-DCP) photocathode showed characteristic peaks with binding energies of 780.7 and 796.1 eV in the Co 2p core level region (Fig. 2b), which were assigned to Co 2p3/2 and Co 2p1/2 of the Co3+ in the surface-attached cobalt catalyst.16,17 The broad peak around 400 eV in the N 1s region was attributed to the CR ligand and DCP anchor (Fig. 2c). The XPS spectra ascertained the successful immobilization of the cobalt catalyst on the surface of the TiO2 layer. Moreover, the inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis showed that the amount of cobalt catalyst loaded on the Si/TiO2 electrode was 37.8 (± 5.8) nmol cm−2.
The attenuated total reflectance Fourier-transform infrared (ATR-FTIR) spectroscopic characteristics of the as-prepared Si/TiO2/Co(CR-DCP) photocathode provided further evidence for immobilization of the intact cobalt catalyst on the TiO2 surface (Fig. 2d). The characteristic IR absorptions of the free Co(CR-DCP) complex in the region of 1250 to 1750 cm−1 were detected for the Si/TiO2/Co(CR-DCP) electrode.41 The typical ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) absorption of the DCP anchor appeared at about 1706 cm−1, and the ν(CN) absorptions of the pyridyl moieties and the Schiff base ligand were observed at around 1589 cm−1. The vibration bands in the region of 1300–1450 cm−1 arose from the methyl and methylene moieties of the macrocyclic ligand.
O) absorption of the DCP anchor appeared at about 1706 cm−1, and the ν(CN) absorptions of the pyridyl moieties and the Schiff base ligand were observed at around 1589 cm−1. The vibration bands in the region of 1300–1450 cm−1 arose from the methyl and methylene moieties of the macrocyclic ligand.
The PEC performance of Si/TiO2/Co(CR-DCP) was studied in 0.1 M acetate buffer solution at pH 4.5 in a home-made PEC cell with a three-electrode setup under simulated AM 1.5G illumination with a UV light cutoff filter (λ > 400 nm, 100 mW cm−2). Fig. S3† shows that the photocurrent density went up evidently with extending the catalyst soaking time from 0.5 to 16 h, which indicated that the increase of the photocurrent did come from the loading of the cobalt catalyst. The photocurrent density did not rise further with the soaking time longer than 16 h. Linear sweep voltammograms (LSVs, Fig. 3) illustrated that the J(0V) of Si/TiO2/Co(CR-DCP) was greatly boosted to −682 from the −22 μA cm−2 achieved for Si/TiO2, and the Eon of the Co(CR-DCP)-decorated photocathode was shifted to the positive direction by 0.32 V compared to that of bare Si/TiO2 (Eon = 0.05 V). Notably, the photocurrent rose rapidly with scanning to potentials more negative than 0 V. The photocurrent density reached −1.75 mA cm−2 at −0.2 V. In contrast, no current was observed at up to −0.4 V for Si/TiO2/Co(CR-DCP) in the dark. We are pleased to note that the J(0V) achieved with Si/TiO2/Co(CR-DCP) is twice as high as that reported for Si/mesoTiO2/NiP (NiP = phosphonate-functionalized P2N2-coordinated molecular nickel catalyst, J(0V) = −340 μA cm−2) and Si/mesoTiO2/CoP (CoP = phosphonate-functionalized cobaloxime catalyst, J(0V) = −330 μA cm−2), and it is even higher than that reported for Si/mesoTiO2/Pt (J(0V) = −430 μA cm−2) under identical test conditions (Table S1†).17
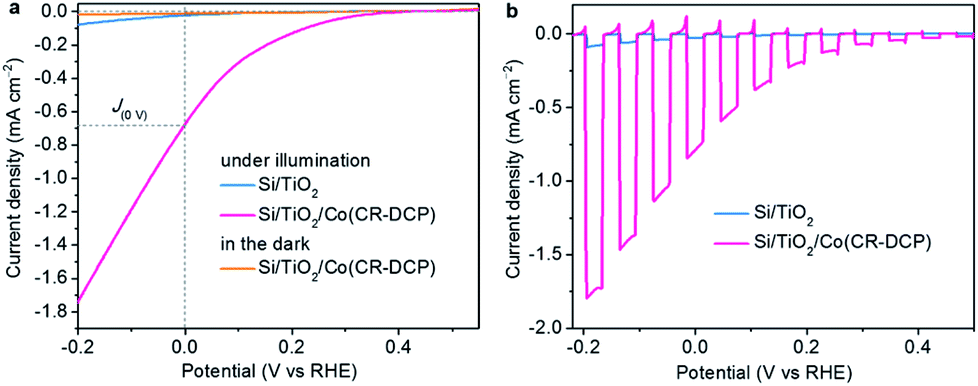 | ||
| Fig. 3 LSVs of Si/TiO2/Co(CR-DCP) and bare Si/TiO2 in 0.1 M acetate buffer at pH 4.5 under (a) continuous and (b) chopped illumination, at a scan rate of 10 mV s−1. | ||
Besides, the type of buffer and the pH value of electrolyte had an apparent influence on the PEC performance of the hybrid photocathode. When the LSVs were measured in phosphate buffer at pH 7 and borate buffer at pH 9, the J(0V) of the photocathode was apparently decreased (Fig. S4†). Replacement of the 5 μm TiO2 film with a porous microstructure by a 5 nm TiO2 film deposited by atomic layer deposition resulted in a large decrease of photocurrent density to 90 μA cm−2 at 0 V with large anodic dark current in the LSV (Fig. S5†), because of the very low achievable loading amount of the molecular catalyst on the 5 nm TiO2 film. The LSVs of Si/TiO2/Co(CR-DCP) scanned successively in reduction and oxidation directions under chopped illumination demonstrated that the photoexcited electrons in the TiO2 film could be effectively extracted by the surface-attached molecular catalysts (Fig. S6†).
To understand the effect of the surface-bound cobalt tetraazamacrocyclic catalyst on the kinetics of photogenerated electron transfer, the interfacial electron-transfer resistance of Si/TiO2/Co(CR-DCP) was studied by electrochemical impedance spectroscopy (EIS) under illumination at a bias of 0 V. The smaller semicircle displayed by the hybrid photocathode than the one exhibited by bare Si/TiO2 illustrated that the immobilization of Co(CR-DCP) effectively reduced the interfacial electron-transfer resistance between the Si/TiO2 surface and electrolyte. From data fittings of the Nyquist impedance plots (Fig. 4a) using a Randles equivalent circuit (inset in Fig. 4a), the calculated value of Rct was dramatically decreased to 646 Ω for Si/TiO2/Co(CR-DCP) from 3336 Ω for bare Si/TiO2. The observations demonstrate that the DCP group with three potential dentates is an effective anchor for electron transfer from the semiconductor to the surface-bound molecular catalyst to be used for the HER. Furthermore, the quantitative kinetic information on surface charge transfer and electron–hole recombination was probed by intensity modulated photocurrent spectroscopy (IMPS) to understand how the surface-bound cobalt catalyst improved the PEC performance of the TiO2-protected Si electrode. Although the IMPS technique has been widely used to study photoelectrodes that were modified by inorganic catalysts, it has been seldom applied for SC/MC hybrid photoelectrodes. To our knowledge, there is only one report that deeply probed the charge transfer kinetics at the surface of a SC/MC hybrid photoanode by the IMPS technique,42 while there is currently no report on IMPS studies of SC/MC hybrid photocathodes in the literature. For a photocathode, the charge transfer efficiency (TE = ktrans/(ktrans + krec)) can be extracted from the low frequency end (Im(Jphoto/Jh) = 0) of the semicircle in the negative imaginary quadrant, and the sum of rate constants (ktrans + krec) for surface electron transfer and charge recombination can be obtained from the frequency at the apex of the semicircle.43
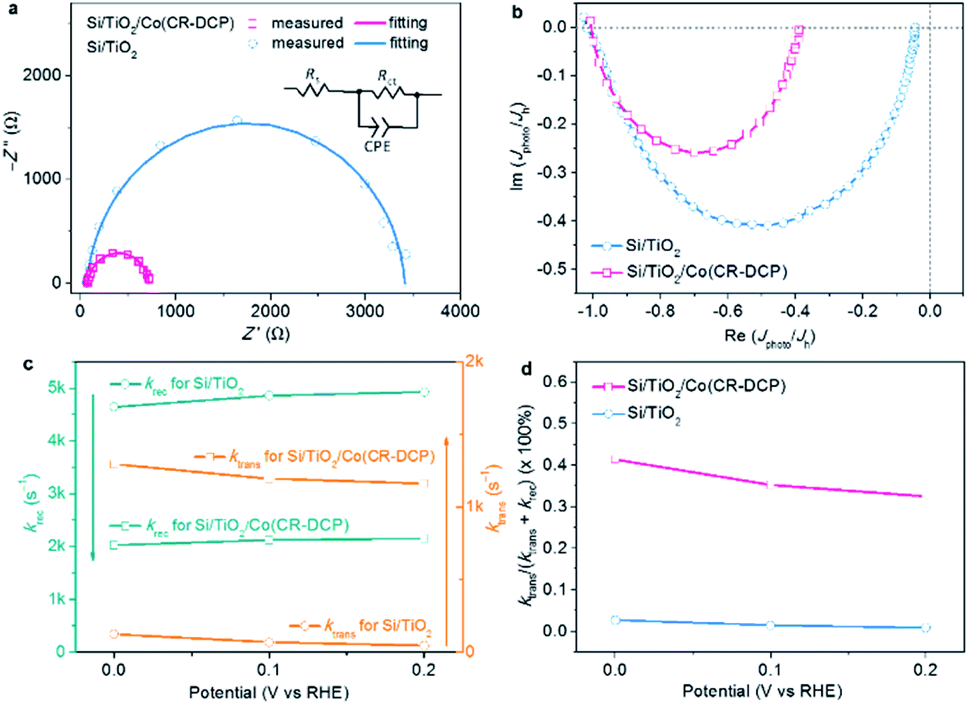 | ||
| Fig. 4 (a) Nyquist plots of Si/TiO2/Co(CR-DCP) and Si/TiO2 under illumination at zero bias (inset: Randles equivalent circuit used for fitting of EIS data). (b) IMPS responses of Si/TiO2/Co(CR-DCP) and bare Si/TiO2 at zero bias. (c) Plot of ktrans and krec against applied potential. (d) Charge transfer efficiency calculated from ktrans and krec for Si/TiO2/Co(CR-DCP) and bare Si/TiO2. | ||
Fig. 4b shows the normalized IMPS responses of Si/TiO2/Co(CR-DCP) and bare Si/TiO2 at zero bias. The values of ktrans and krec were extracted from Fig. 4b (Table S2†), which showed that with immobilization of Co(CR-DCP) on the surface of Si/TiO2, ktrans was increased by a factor of 10 and krec was decreased by a factor of 2 at 0 V. Accordingly, the TE of Si/TiO2/Co(CR-DCP) was enhanced to 39%, which was about 15-fold higher than the surface TE at bare Si/TiO2. Furthermore, the values of ktrans and krec extracted from the IMPS responses of Si/TiO2/Co(CR-DCP) and bare Si/TiO2 at different potentials (Fig. S7†) showed that the slight deceleration of ktrans concurred with the small elevation of krec when the applied potential was changed from 0 to 0.2 V (Fig. 4c). As such, the TE at the surface of Si/TiO2/Co(CR-DCP) decreased from 39% at 0 V to 35% at 0.2 V (Fig. 4d). The results revealed that the surface-bound Co(CR-DCP) substantially improved the PEC HER activity not only by evidently accelerating the interfacial electron transfer rate but also by retarding the surface electron–hole recombination rate. Interestingly, this observation is different from the previous findings for an Fe2O3/molecular Ir catalyst photoanode, in which the surface-bound Ir catalyst primarily played the role of speeding up the charge transfer rate while it did little to reduce the surface recombination.42
To assess the stability of Si/TiO2/Co(CR-DCP) for PEC H2-evolution, CPP experiments were carried out for Si/TiO2/Co(CR-DCP) and bare Si/TiO2 in pH 4.5 acetate buffer solution at 0 V under illumination with a 2 min dark chop every hour. Fig. 5a shows that the Si/TiO2/Co(CR-DCP) electrode displays a steady photocurrent of 527 (± 35) μA cm−2 over 10 h of CPP experiment. The long durability of Si/TiO2/Co(CR-DCP) indicates the superior attaching stability of the DCP anchor to the TiO2 surface under test conditions, because DCP can not only covalently bond to the Brønsted-acid sites (Ti–OH) of the TiO2 surface through two carboxyl groups but also coordinately attach to the Lewis-acid sites (exposed Tin+ cations) of unsaturated surface titanium ions through the pyridyl N atom.44
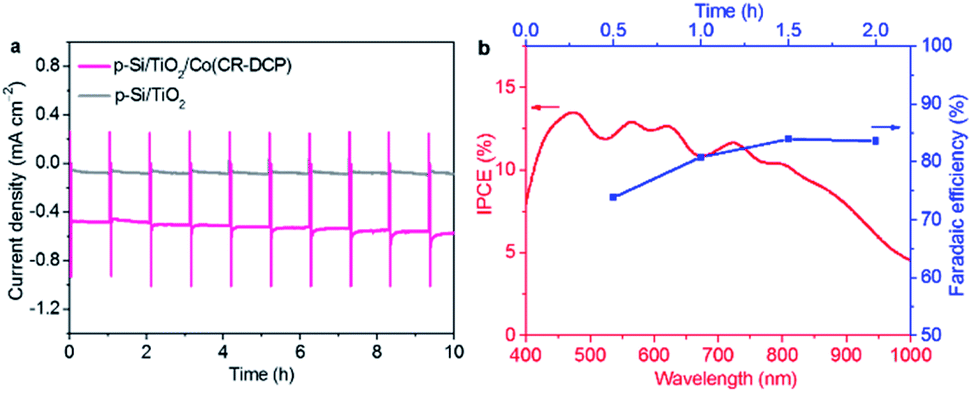 | ||
| Fig. 5 (a) Long-time CPP curves for Si/TiO2/Co(CR-DCP) and Si/TiO2 at 0 V under illumination with a 2 min dark chop every hour. (b) Plots of IPCE and faradaic efficiency (FE) for Si/TiO2/Co(CR-DCP) at 0 V. | ||
As shown in Fig. 5b, the incident photon-to-current efficiency (IPCE) of Si/TiO2/Co(CR-DCP) was 10.34–13.50% across the wavelengths of 420–800 nm. In addition, the cumulative charge passing through the external circuit was 3.4C and the amount of H2 evolved was 14.9 μmol in the first 2 h of the CPP experiment (Fig. S8†). The evolved H2 was determined by gas chromatography analysis of the gaseous phase in the sealed PEC cell. Based on these data, the FE of H2 evolution was calculated to be 80 (± 4)% (Fig. 5b), which is the low-limit of FE for H2 production on Si/TiO2/Co(CR-DCP) as the H2 dissolved in the electrolyte was ignored for the calculation of FE.
After 10 h of CPP experiments, the resulting electrolytes were analyzed by an ICP method and the used electrodes were detected by XPS and ATR-FTIR spectroscopy. The ICP analysis showed that before the start of the CPP experiment a small amount of cobalt catalyst (∼0.12 nmol mL−1 in 40 mL of electrolyte) had been dissolved in the electrolyte (Table S3†), most probably due to the desorption of the physisorbed catalyst from the electrode surface. During 10 h of CPP experiment, about 5.6 nmol of the cobalt catalyst dropped into the electrolyte, and thus 83% of the loaded cobalt catalyst still remained on the TiO2 surface of the hybrid photocathode. The XPS spectra of the used Si/TiO2/Co(CR-DCP) electrode did not show noticeable changes in the Co 2p and N 1s core level regions (Fig. 2b and c). Moreover, the ATR-FTIR spectra of Si/TiO2/Co(CR-DCP) before and after the CPP experiment were essentially identical, illustrating that the immobilized cobalt catalyst remained intact on the electrode after 10 h of CPP test (Fig. 2d).
In summary, the cobalt tetraazamacrocyclic complex, one of the most efficient and stable HER molecular catalysts, was successfully immobilized on the surface of Si/TiO2 through the tridentate anchor of DCP. The surface-bound Co(CR-DCP) significantly improved the performance of the TiO2-protected Si electrode, with a J(0V) of −0.68 mA cm−2 and an Eon of 0.37 V in 0.1 M acetate buffer under simulated 1 sun illumination (λ > 400 nm). Meanwhile, the Co(CR-DCP)-modified electrode displayed a steady photocurrent over at least 10 h of CPP experiment. The good durability of Si/TiO2/Co(CR-DCP) is due to the robustness of the cobalt tetraazamacrocyclic catalyst and the superior attaching stability of the DCP anchor under test conditions. The quantitative kinetic studies on surface charge transfer by EIS and IMPS revealed that the surface-bound Co(CR-DCP) played a role not only in greatly reducing the electron transfer resistance and evidently accelerating the electron transfer rate, but also in retarding the charge recombination rate at the electrode/electrolyte interface. As such, the surface TE for Si/TiO2/Co(CR-DCP) was 15-fold higher than that for bare Si/TiO2. This is the first time it has been demonstrated that surface-bound molecular catalysts can suppress the interfacial electron–hole recombination at hybrid photoelectrodes. The results show the remarkable merits of the cobalt tetraazamacrocyclic complex as a surface-immobilized HER catalyst with DCP as an anchor, which will encourage researchers in the field of solar fuel production to use functionalized [Co(CR)X2]+ complexes in the future to develop highly active and stable hybrid photocathodes for efficient H2 generation and CO2 reduction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the National Natural Science Foundation of China (no. 21673028) for financial support of this work.Notes and references
- A. Fujishima and K. Honda, Nature, 1970, 238, 37–38 CrossRef
.
- N. Queyriaux, N. Kaeffer, A. Morozan, M. Chavarot-Kerlidou and V. Artero, J. Photochem. Photobiol., C, 2015, 25, 90–105 CrossRef CAS
- J. Willkomm, K. L. Orchard, A. Reynal, E. Pastor, J. R. Durrant and E. Reisner, Chem. Soc. Rev., 2016, 45, 9–23 RSC
- M. Wang, Y. Yang, J. Shen, J. Jiang and L. Sun, Sustainable Energy Fuels, 2017, 1, 1641–1663 RSC
- H.-L. Wu, X.-B. Li, C.-H. Tung and L.-Z. Wu, Adv. Sci., 2018, 5, 1700684 CrossRef
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 4, 2752–2875 CrossRef
- J. Gu, Y. Yan, J. L. Young, K. X. Steirer, N. R. Neale and J. A. Turner, Nat. Mater., 2016, 15, 456–460 CrossRef CAS
- A. Krawicz, J. Yang, E. Anzenberg, J. Yano, I. D. Sharp and G. F. Moore, J. Am. Chem. Soc., 2013, 135, 11861–11868 CrossRef CAS
- D. Cedeno, A. Krawicz, P. Doak, M. Yu, J. B. Neaton and G. F. Moore, J. Phys. Chem. Lett., 2014, 5, 3222–3226 CrossRef CAS
- A. Krawicz, D. Cedeno and G. F. Moore, Phys. Chem. Chem. Phys., 2014, 16, 15818–15824 RSC
- A. Reynal, J. Willkomm, N. M. Muresan, F. Lakadamyali, M. Planells, E. Reisner and J. R. Durrant, Chem. Commun., 2014, 50, 12768–12771 RSC
- Y. Chen, H. Chen and H. Tian, Chem. Commun., 2015, 51, 11508–11511 RSC
- A. M. Beiler, D. Khusnutdinova, S. I. Jacob and G. F. Moore, Ind. Eng. Chem. Res., 2016, 55, 5306–5314 CrossRef CAS
- A. M. Beiler, D. Khusnutdinova, S. I. Jacob and G. F. Moore, ACS Appl. Mater. Interfaces, 2016, 8, 10038–10047 CrossRef CAS
- L. Gong, H. Yin, C. Nie, X. Sun, X. Wang and M. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 34010–34019 CrossRef CAS
- S. Chandrasekaran, N. Kaeffer, L. Cagnon, D. Aldakov, J. Fize, G. Nonglaton, F. Baleras, P. Mailley and V. Artero, Chem. Sci., 2019, 10, 4469–4475 RSC
- J. J. Leung, J. Warnan, D. H. Nam, J. Z. Zhang, J. Willkomm and E. Reisner, Chem. Sci., 2017, 8, 5172–5180 RSC
- J. Seo, R. T. Pekarek and M. J. Rose, Chem. Commun., 2015, 51, 13264–13267 RSC
- H. J. Kim, J. Seo and M. J. Rose, ACS Appl. Mater. Interfaces, 2016, 8, 1061–1066 CrossRef CAS
- B. Shan, M. K. Brennaman, L. Troian-Gautier, Y. Liu, A. Nayak, C. M. Klug, T.-T. Li, R. M. Bullock and T. J. Meyer, J. Am. Chem. Soc., 2019, 26, 10390–10398 CrossRef
- T. Nann, S. K. Ibrahim, P. M. Woi, S. Xu, J. Ziegler and C. J. Pickett, Angew. Chem., Int. Ed., 2010, 49, 1574–1577 CrossRef CAS
- D. Khusnutdinova, A. M. Beiler, B. L. Wadsworth, S. I. Jacob and G. F. Moore, Chem. Sci., 2017, 8, 253–259 RSC
- Y. Hou, B. L. Abrams, P. C. Vesborg, M. E. Bjorketun, K. Herbst, L. Bech, A. M. Setti, C. D. Damsgaard, T. Pedersen, O. Hansen, J. Rossmeisl, S. Dahl, J. K. Norskov and I. Chorkendorff, Nat. Mater., 2011, 10, 434–438 CrossRef CAS
- B. Seger, K. Herbst, T. Pedersen, B. Abrams, P. C. K. Vesborg, O. Hansen and I. Chorkendorff, J. Electrochem. Soc., 2014, 161, H722–H724 CrossRef
- C. Gimbert-Surinach, J. Albero, T. Stoll, J. Fortage, M. N. Collomb, A. Deronzier, E. Palomares and A. Llobet, J. Am. Chem. Soc., 2014, 136, 7655–7661 CrossRef CAS
- M. Sandroni, R. Gueret, K. D. Wegner, P. Reiss, J. Fortage, D. Aldakov and M. N. Collomb, Energy Environ. Sci., 2018, 11, 1752–1761 RSC
- S. Roy, M. Bacchi, G. Berggren and V. Artero, ChemSusChem, 2015, 8, 3632–3638 CrossRef CAS
- C.-F. Leung, Y.-Z. Chen, H.-Q. Yu, S.-M. Yiu, C.-C. Ko and T.-C. Lau, Int. J. Hydrogen Energy, 2011, 36, 11640–11645 CrossRef CAS
- S. Varma, C. E. Castillo, T. Stoll, J. Fortage, A. G. Blackman, F. Molton, A. Deronzier and M. N. Collomb, Phys. Chem. Chem. Phys., 2013, 15, 17544–17552 RSC
- R. Gueret, L. Poulard, M. Oshinowo, J. Chauvin, M. Dahmane, G. Dupeyre, P. P. Lainé, J. Fortage and M.-N. Collomb, ACS Catal., 2018, 8, 3792–3802 CrossRef CAS
- M. Zhang, M. El-Roz, H. Frei, J. L. Mendoza-Cortes, M. Head-Gordon, D. C. Lacy and J. C. Peters, J. Phys. Chem. C, 2015, 119, 4645–4654 CrossRef CAS
- C. C. McCrory, C. Uyeda and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 3164–3170 CrossRef CAS
- D. C. Lacy, C. C. McCrory and J. C. Peters, Inorg. Chem., 2014, 53, 4980–4988 CrossRef CAS
- H. Sheng and H. Frei, J. Am. Chem. Soc., 2016, 138, 9959–9967 CrossRef CAS
- D. Moonshiram, C. Gimbert-Suriñach, A. Guda, A. Picon, C. S. Lehmann, X. Zhang, G. Doumy, A. M. March, J. Benet-Buchholz, A. Soldatov, A. Llobet and S. H. Southworth, J. Am. Chem. Soc., 2016, 138, 10586–10596 CrossRef CAS
- K. Fan, F. Li, L. Wang, Q. Daniel, E. Gabrielsson and L. Sun, Phys. Chem. Chem. Phys., 2014, 16, 25234–25240 RSC
- F. Li, K. Fan, B. Xu, E. Gabrielsson, Q. Daniel, L. Li and L. Sun, J. Am. Chem. Soc., 2015, 137, 9153–9159 CrossRef CAS
- C. D. Windle, H. Kumagai, M. Higashi, R. Brisse, S. Bold, B. Jousselme, M. Chavarot-Kerlidou, K. Maeda, R. Abe, O. Ishitani and V. Artero, J. Am. Chem. Soc., 2019, 141, 9593–9602 CrossRef CAS
- C. Tapia, E. Bellet-Amalric, D. Aldakov, F. Boudoire, K. Sivula, L. Cagnone and V. Artero, Green Chem., 2020, 22, 3141–3149 RSC
- C. Nie, W. Ni, L. Gong, J. Jiang, J. Wang and M. Wang, J. Mater. Chem. A, 2019, 7, 27432–27440 RSC
- K. M. Long and D. H. Busch, Inorg. Chem., 1970, 9, 505–512 CrossRef CAS
- W. Li, D. He, S. W. Sheehan, Y. He, J. E. Thorne, X. Yao, G. W. Brudvig and D. Wang, Energy Environ. Sci., 2016, 9, 1794–1802 RSC
- J. E. Thorne, Y. Zhao, D. He, S. Fan, S. Vanka, Z. Mi and D. Wang, Phys. Chem. Chem. Phys., 2017, 19, 29653–29659 RSC
- Y. Harima, T. Fujita, Y. Kano, I. Imae, K. Komaguchi, Y. Ooyama and J. Ohshita, J. Phys. Chem. C, 2013, 117, 16364–16370 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Materials, experimental procedures, LSVs, IMPS responses, current efficiency plot, and Tables S1–S3. See DOI: 10.1039/d0ta09942b |
| This journal is © The Royal Society of Chemistry 2021 |