
Utilizing spent Li-ion batteries to regulate the π-conjugated structure of g-C3N4: a win–win approach for waste recycling and highly active photocatalyst construction†
Bo
Niu
a,
Jiefeng
Xiao
a and
Zhenming
Xu
 *ab
*ab
aSchool of Environmental Science and Engineering, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai 200240, People's Republic of China. E-mail: zmxu@sjtu.edu.cn; Fax: +86 21 5474495; Tel: +86 21 5474495
bShanghai Institute of Pollution Control and Ecological Security, Shanghai 200092, People's Republic of China
First published on 9th December 2020
Abstract
The recycling of spent lithium-ion batteries (LIBs) has been one of the global environmental concerns due to their huge quantity and hazardous and valuable character. The existing spent LIB processing mainly focuses on element separation, which is usually complex and energy consuming and even causes new environmental issues. This study breaks through the traditional element separation and utilizes spent LIBs for photocatalytic application. Through facile one-pot sintering of a spent LiCoO2 cathode, melamine and NH4Cl at 530 °C, we introduced a Li–Cl–Co anion–cation to synergistically regulate the π-conjugated structure of g-C3N4 for enhancing its photocatalytic activity. The Li–Cl–Co anion–cation was stabilized in g-C3N4 through Li–N, Li–Cl, Cl–C and Co–N bonds. The Co among the adjacent tri-s-triazine units decreased the bandgap to lower the electron transition energy. The Li and Cl intercalation respectively decreases and increases the interlayer spacing, leading to a gradient decrease in the energy barrier for dramatically boosting the charge migration and separation between g-C3N4 layers. These special characteristics synergistically contribute to the superior photocatalytic performance. The simulated sunlight photocatalytic H2 evolution and RhB degradation rates of this special photocatalyst were 12.6 and 15.3 times higher than those of pristine g-C3N4. This study avoids element separation and distinctively utilizes spent LIBs for photocatalytic application, which minimizes the environmental risks associated with recycling. Our study also reveals a synergetic concept of recycling wastes as high-efficiency photocatalysts for environmental protection.
1. Introduction
Currently, the market demand for lithium-ion batteries (LIBs) is rapidly rising year by year with the rapid development of information technology and the electric vehicle industry. However, the cycle life of LIBs is typically 2–4 years for consumer electronics and 5–8 years for electric vehicles. Consequently, large amounts of spent LIBs are discarded each year.1,2 It is estimated that the global number of spent LIBs will reach over 11 million tons by 2030.3 Spent LIBs are considered to be an environmental hazard but also a valuable resource since they contain hazardous substances (such as heavy metals and organic electrolytes) while rich in valuable metals with huge economic value. For instance, the content of lithium (Li) and cobalt (Co) in spent LIBs could reach 7 and 15 wt%, respectively, which are much higher than their respective mined ores.4 Therefore, the recycling of spent LIBs is significant for the sustainable utilization of metal resources, environmental protection and human health.Currently, pyrometallurgy, hydrometallurgy and mechanical processes were applied in the separation of resources from spent LIBs.2,3,5,6 Hydrometallurgy is a traditional commercial application in spent LIB recycling because of its high metal leaching rate and high purity of recovered products. This process usually involves multiple steps and long-time leaching and extraction using a lot of chemicals, even caustic acids, which poses a potential danger to workers and the environment.7 Pyrometallurgy is another commercially viable option for spent LIB recycling due to the simple operation and large amount processing. It usually consumes higher energy from the extreme temperature (above 1400 °C) and produces harmful fumes, which requires safety precautions and pollution gas disposal. Moreover, the residues (mixed slag) make it difficult to fully separate the metals (such as Li) through this process alone.3 To lower the treatment temperature, some advanced methods based on pyrometallurgy such as carbon reduction8,9 and sulfide10,11 and chlorination roasting12–14 have been developed to extract the metal resources from spent LIBs. However, a second separation of the obtained products was still needed.
To avoid the multi-step separation of metal ions and minimize secondary pollution, some researchers innovatively proposed to prepare high-valued functional materials using spent LIBs. Natarajan et al. prepared MnCo2O4 for electrocatalytic oxygen evolution from spent LIBs through an integrated process including chemical leaching with 2 M acetic acid, precipitation with ammonium carbonate, hydrothermal conditioning and calcination at 650 °C.15 Assefi et al. prepared a core–shell NiO@Co3O4 photocatalyst using spent LIBs and a Ni–Cd battery as precursors by NaOH and H2SO4 leaching followed by calcining at 500 °C.16 Dai et al. obtained a defect LiCoO2 catalyst (synergy of Li, Co, and O vacancies) through strict control of HNO3 leaching conditions for airborne benzene oxidation.17 The synthesis of functional materials using spent LIBs currently involves multi-leaching, Li separation, precipitation and sintering. Although the wastes are low-cost and the as-prepared materials exhibited good performance, these preparation processes are still complex and exhibit some environmental risks. Therefore, it is strongly needed to develop a simple and environmentally friendly approach to recycle spent LIBs as environment functional materials.
Based on waste recycling and environmental protection, this study broke through the traditional element separation and utilized spent LIBs for photocatalytic application. Through one-pot sintering of a spent LiCoO2 cathode, melamine and NH4Cl at 530 °C, we introduced the Li–Cl–Co anion–cation to synergistically regulate the π-conjugated structure of g-C3N4 for improving its photocatalytic activity. As is known, g-C3N4 is a very promising visible light photocatalyst due to its lower cost (metal-free), simple preparation and superior photocatalytic stability.18–21 During the one-pot sintering process, melamine acted as the reductant and the precursor of g-C3N4, and NH4Cl acted as the chlorination agent and foaming agent. The LiCoO2 cathode was transformed into LiCl–CoCl2 and directly participated in the g-C3N4 formation, which in situ regulated the π-conjugated structure of g-C3N4. Combined with experiments and DFT calculations, we investigated the π-conjugation regulation properties, including the interlayer spacing, charge density distribution, energy barrier between g-C3N4 layers, charge separation and electronic structure as well as the synergistically enhanced photocatalytic mechanism. And as expected, the special porous photocatalyst exhibited superior photocatalytic performance for H2 evolution and RhB degradation. This study avoided element separation and distinctively utilizes Li and Co in spent LIBs to regulate the π-conjugated structure of g-C3N4 for photocatalytic application, which minimized the environmental risks associated with recycling. Our study also revealed a synergetic approach to recycling wastes as high-efficiency photocatalysts, which realizes an all-win situation for waste recycling and environmental protection.
2. Experimental section
2.1. Materials and synthesis
Spent LiCoO2 batteries were completely discharged and manually dismantled to separate the plastic shell, cathode, anode and separator. Then, the LiCoO2 cathode powder was obtained from Al foil,14 as shown in Scheme 1a. The LiCoO2 was used as a Li and Co source to regulate the π-conjugated structure of g-C3N4 by a simple one-pot sintering process. The schematic illustration of the preparation process is shown in Scheme 1b. Melamine (10 g), NH4Cl (5 g) and a certain amount of spent LiCoO2 powder (0.75, 1.5, 3, and 5 wt%, relative to melamine) were first uniformly mixed. Melamine acted as the reductant and the precursor of g-C3N4. NH4Cl acted both as the chlorination agent and the foaming agent.14 Then, the mixed powders were put into a covered crucible and calcined in a tube furnace under an argon (Ar) atmosphere at 530 °C for 2 h with a heating rate of 3 °C min−1. During the one-pot sintering process, spent LiCoO2 was transformed into LiCl and CoCl2 and directly participated in the g-C3N4 formation, which in situ regulated the π-conjugated structure of g-C3N4. The model for the Li–Cl–Co regulated π-conjugated structure of g-C3N4 is shown in Scheme 1c. In this study, we mainly investigated the π-conjugation regulation properties including the interlayer spacing, charge density distribution, energy barrier between g-C3N4 layers, charge separation and electronic structure, as well as their synergetic mechanism on the enhanced photocatalytic activity. Simultaneously, the excess amount of NH4Cl also acted the foaming agent to produce the porous structure. The excess NH3 was gathered by H2O. After cooling naturally to room temperature, the product was thoroughly washed with deionized water a few times and then was collected by centrifugation and dried at 80 °C. Finally, the special porous photocatalysts with different weight ratios of spent LiCoO2 powder (0.75, 1.5, 3, and 5 wt%) were collected and denoted as 0.75LiClCo–C3N4, 1.5LiClCo–C3N4, 3LiClCo–C3N4 and 5LiClCo–C3N4, respectively. For comparison, the pristine g-C3N4 was prepared without adding LiCoO2 and NH4Cl.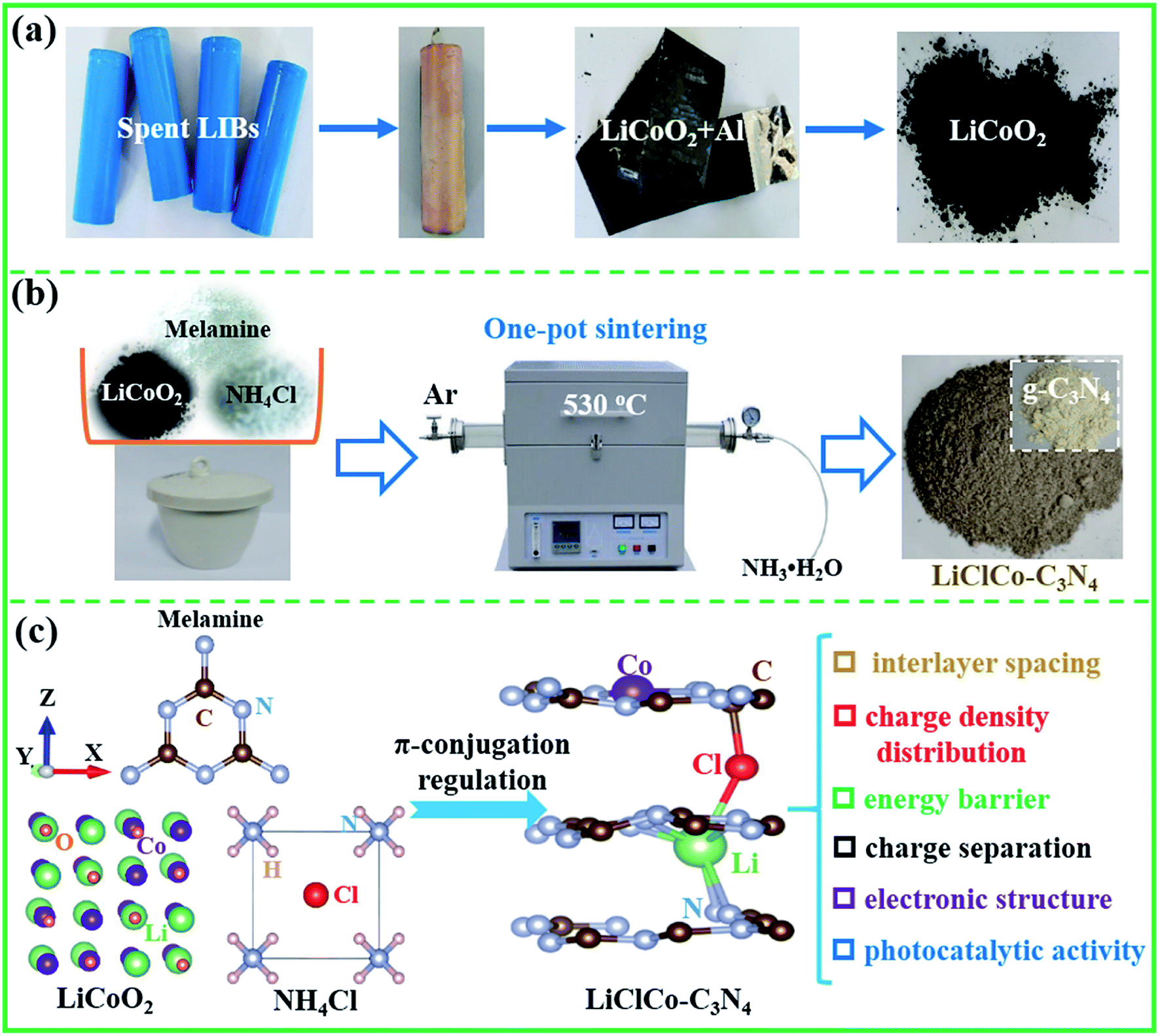 | ||
| Scheme 1 (a) the pretreatment of spent LIBs and collected LiCoO2 powder, (b) the illustration of the one-pot preparation process and (c) the mode for regulating the π-conjugated structure of g-C3N4 using spent LiCoO2 and NH4Cl (g-C3N4 displayed a yellow colour, while the LiClCo–C3N4 products exhibited a brown colour). | ||
2.2. Characterization
The detailed characterization, photoelectrochemical and photocatalytic activity experiments and DFT calculations are provided in the ESI.†3. Results and discussion
3.1. Morphology, microstructure and component analysis
The phase structures of the samples were identified by XRD. As shown in Fig. 1a, the patterns of pristine g-C3N4 displayed two distinct diffraction peaks at about 13.1° and 27.5°, which respectively correspond to the (1 0 0) and (0 0 2) crystal planes of g-C3N4.22 For the LiClCo–C3N4 samples, the main g-C3N4 peaks were maintained and no other Li or Co species was observed. However, with the increase of the spent LiCoO2 addition amount, the intensity of two peaks in LiClCo–C3N4 samples gradually decreased, suggesting that the crystal growth of g-C3N4 is slightly inhibited. Besides, the peak at 27.5° of LiClCo–C3N4 samples shifted toward a higher 2θ value (as shown in Fig. 1b), which was attributed to some metal ions intercalating into the g-C3N4 layers, and the coordination bonds were possibly formed between the interlayer ions and N, thus enhancing the inner interactive force and reducing interlayer spacing, further changing the π-conjugated system of g-C3N4. Similar results have been observed in other cation ions (Fe–Pd and K–Fe) intercalated g-C3N4 photocatalysts.23,24 Furthermore, the cation ions intercalated into g-C3N4 layers and the coordination bonds can form a bridge for charge transfer.23,25,26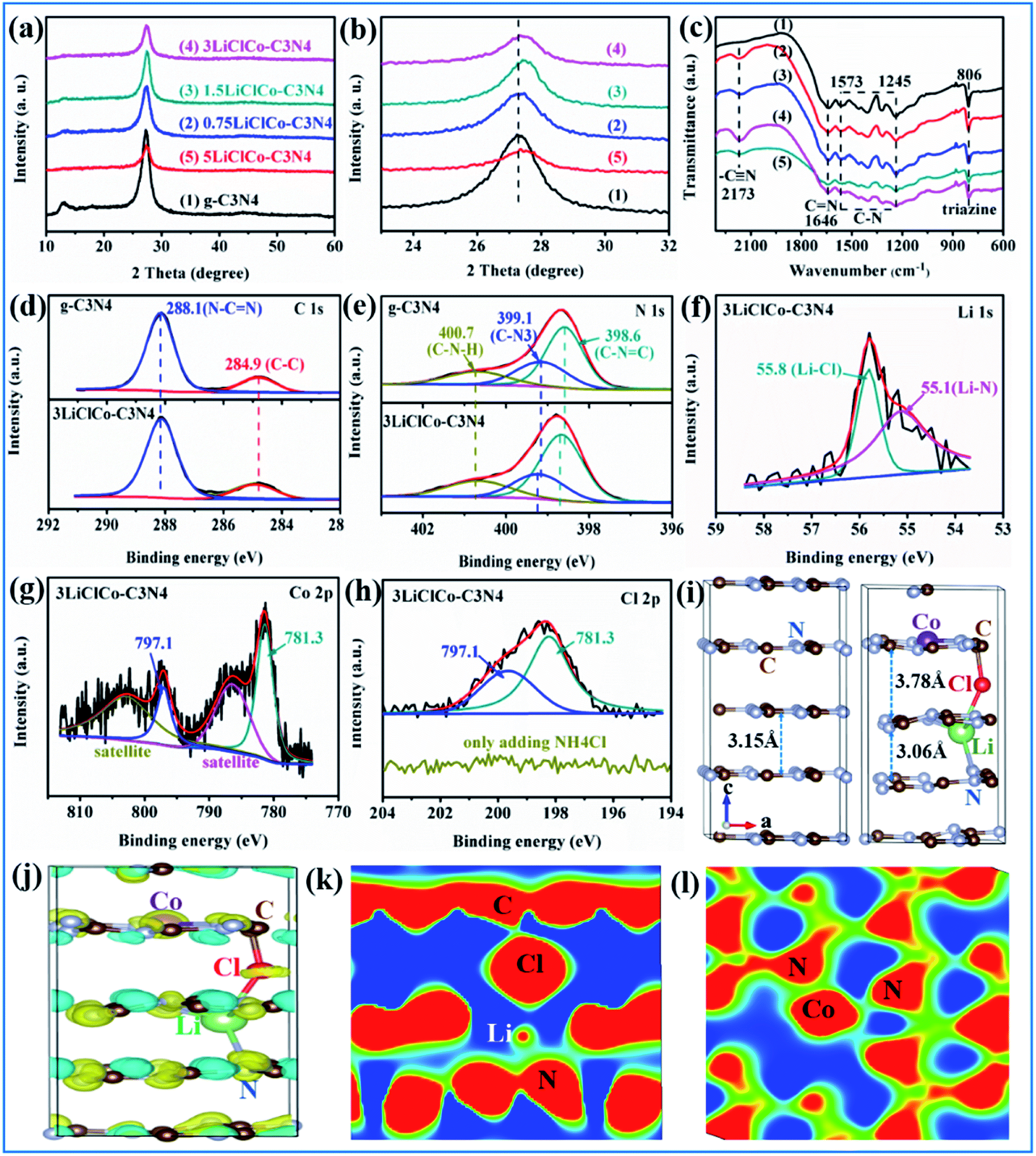 | ||
| Fig. 1 (a) XRD, (b) partial enlarged detail, (c) FT-IR spectroscopy of different samples; (d–h) XPS spectra of C1s, N1s, Li 1s, Co 2p and Cl 2p of 3LiClCo–C3N4; (i) the optimal g-C3N4 and obtained stable LiClCo–C3N4 structure; (j) charge difference distribution; (k) and (l) 2D sectional drawing of LiClCo–C3N4 obtained by DFT calculation. | ||
The structure and functional groups were investigated by FT-IR spectroscopy. As shown in Fig. 1c, g-C3N4 exhibited typical stretching modes of C–N (between 1245 and 1573 cm−1) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (at 1646 cm−1) and breathing modes of triazine units (at 806 cm−1).27,28 After adding spent LiCoO2 and NH4Cl, the FT-IR spectra displayed all the characteristic peaks of g-C3N4, proving that the original framework of g-C3N4 is not changed after regulating the π-conjugated structure. Besides, the new peak at 2173 cm−1 is assigned to the stretching mode of cyano groups (–C
N (at 1646 cm−1) and breathing modes of triazine units (at 806 cm−1).27,28 After adding spent LiCoO2 and NH4Cl, the FT-IR spectra displayed all the characteristic peaks of g-C3N4, proving that the original framework of g-C3N4 is not changed after regulating the π-conjugated structure. Besides, the new peak at 2173 cm−1 is assigned to the stretching mode of cyano groups (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N).24 This was attributed to the fact that while heating melamine, Cl on the surface of chloride (LiCl and CoCl2) could react with H in amino functions (C–NH2 or C–NH) to produce HCl and cyano groups.29 The cyano groups could improve the capture of photogenerated carriers.24
N).24 This was attributed to the fact that while heating melamine, Cl on the surface of chloride (LiCl and CoCl2) could react with H in amino functions (C–NH2 or C–NH) to produce HCl and cyano groups.29 The cyano groups could improve the capture of photogenerated carriers.24
XPS was conducted to further investigate the specific bonds and chemical states of the sample. As shown in Fig. 1d, the C1s peak for pristine g-C3N4 around 284.6 eV was assigned to the graphitic species, and the peak around 287.8 eV was due to the sp2-bonded carbon (N–CN).30 The N1 s spectra of pristine g-C3N4 (Fig. 1e) could be fitted into three peaks at 398.6, 399.9 and 401.1 eV, which were attributed to the (C–NC), (N–C3) and (C–N–H) groups, respectively.31 After adding spent LiCoO2 and NH4Cl, the C 1s peak positions were unchanged, while the N 1s peaks of 3LiClCo–C3N4 shifted toward higher binding energies compared with those of pristine g-C3N4, which indicated the formation of strong interaction between the introduced anion–cation ions and N atoms of g-C3N4. The shifting phenomenon described above has also been observed in other cation (Li, Fe–Pd, K–Fe) intercalated g-C3N4.23,24,32 It was reported that the lone pair of electrons on the N atoms would shift toward the intercalated cation through the metal-N coordination bond, thus leading to the decrease of electron density of N atoms and increase of binding energy.32,33 As shown in Fig. 1f, the Li 1s spectrum for 3LiClCo–C3N4 could be fitted into two peaks at 55.1 and 55.8 eV, which were respectively assigned to the Li–N and Li–Cl bonds.32,34 Li could be inserted at the interstitial position and was stabilized in g-C3N4 through the Li–N coordination bond. In the Co 2p spectrum (Fig. 1g), two peaks at 781.3 and 797.1 eV with satellite peaks at 786.5 and 802.8 eV corresponded to the Co 2p3/2 and Co 2p1/2, which characterized the Co(II) state.35 The sharp peak at 781.3 eV was assigned to the Co–N coordination bond, stabilizing Co in g-C3N4, as reported in previous studies.36,37 As shown in Fig. 1h, the Cl 2p spectrum of 3LiClCo–C3N4 can be fitted to two peaks located at 198.1 and 199.6 eV, corresponding to the Cl 2p5/2 and Cl 2p3/2, respectively, which indicated the presence of Cl in the sample.32 The Cl intercalated g-C3N4 could be obtained by co-pyrolysis of melamine and excessive NH4Cl.26 However, no Cl 2p signal was observed in the g-C3N4/NH4Cl (no addition of LiCoO2) in this study due to the relatively low addition amount of NH4Cl as shown in Fig. 1h. Therefore, the intercalated Cl in LiClCo–C3N4 was mainly attributed to the reactions between LiCl/CoCl2 and g-C3N4. Since the obtained photocatalysts have been thoroughly washed with deionized water, the LiCl and CoCl2 phases cannot exist in the sample. Consequently, the Cl may stabilize in g-C3N4 with some Cl–Li–N, Cl–Co–N or Cl–C bonds. Similarly, for the g-C3N4 treated with LiCl–KCl–NaCl, the Cl bonded with alkali metal ions and balanced partial charges of metal ions, forming a stable alkali metal–Cl intercalated structure in g-C3N4 through alkali metal-N coordination bonds.29 The intercalation could constitutionally change the interlayer spacing and further alter the π-conjugated structure of g-C3N4.
As for the ion doping in g-C3N4, it generally includes three situations, namely, substituted doping, interlayer doping and cave doping (among adjacent tri-s-triazine units on the same π-conjugated planes). The cation doping situations were usually cave and interlayer doping.18,38 It has been proven that Cl was mainly due to the interlayer doping.26,29 In order to study the Li–Cl–Co doping position and its effect on regulating the π-conjugated structure of g-C3N4, we performed some DFT calculations. The Li, Cl and Co atoms were randomly located in cave and interlayer positions of g-C3N4 and then were fully relaxed. After the structure optimization of different situations, the same results were found that Co was stabilized in the cave position, and Li and Cl were intercalated in g-C3N4, forming the Co–N, Cl–C, Li–Cl, and Li–N bonds, as shown in Fig. 1i and S1 of the ESI.† Similar results have also been observed in the previous studies about single Li, Cl and Co doped g-C3N4.26,29,37,39,40 Li decreased the interlayer spacing of g-C3N4 (001), while Cl increased the interlayer spacing, which indicated that the Li and Cl could regulate the π conjugated system and further affect the charge distribution between the g-C3N4 layers. From the XRD results (Fig. 1b), the decreased interlayer spacing of g-C3N4 was mainly due to the stronger Li effect. Then we analyzed the charge difference distribution, as shown in Fig. 1j. The yellow regions denote charge accumulation while the cyan ones represent charge depression. It is obvious that the Li–Cl–Co caused the charge redistribution, suggesting that there existed some chemical interaction between dopant atoms and g-C3N4. The 2D sectional drawing (1k and 1l) demonstrated that the C–Cl, Li–N and Co–N tended to form covalent bonds while Li–Cl could be an ionic bond, which further clarified the XPS results. The regulation of the electronic structure of g-C3N4 by Li–Cl–Co will be investigated in Section 3.3.
SEM and time of flight secondary ion mass spectrometry (TOF-SIMS) were used to observe the morphology and microstructure of the as-prepared photocatalysts. As shown in Fig. 2a, g-C3N4 had a graphite-like bulk structure. After introducing spent LiCoO2 and NH4Cl, the sample exhibited an obvious porous network structure, as shown in Fig. 2b and c (side view). Such a porous character was attributed to the abundant gas soft-template offered by the NH4Cl decomposition and corrosion.26 To investigate the distribution of Li–Cl–Co, TOF-SIMS was conducted. We first randomly selected an area (Fig. 2d) to simultaneously detect the Li and Co elements. Fig. 2e and f show the X–Y and X–Z (perpendicular to X–Y) face views of Li distribution, respectively, demonstrating the uniform Li distribution on both X–Y and X–Z faces. This suggested that Li was uniformly distributed in bulk g-C3N4, instead of surface. The Co distribution was not obviously observed on the X–Z face (Fig. 2h) due to the limit of TOF-SIMS for Co detection. However, from the X–Y face (Fig. 2g) and the above XPS and DFT calculations (Co was among adjacent tri-s-triazine units and forms a Co–N bond, Fig. 1g and i), it can still be clarified that Co was also uniformly distributed in bulk g-C3N4. In addition, because of the destruction of area by TOF-SIMS (Fig. S2 of ESI†), we then randomly selected another area (Fig. 2i) to detect the Cl element (because Cl cannot be simultaneously examined with Li and Co elements). Likewise, the X–Y and X–Z face views (Fig. 2j and k) demonstrate the uniform Cl distribution in g-C3N4. Fig. 2l–n are the mass spectra of Li, Co and Cl when performing TOF-SIMS, which clearly specifies the Li, Co and Cl elements in the sample.
 | ||
| Fig. 2 SEM images of (a) g-C3N4, (b) top and (c) side view for 3LiClCo–C3N4; TOF-SIMS images of (d–n) Li, Co, and Cl element distribution (X–Y, X–Z faces) and the corresponding Li, Co, and Cl mass spectra for the 3LiClCo–C3N4 sample. | ||
The porosity and surface area were investigated by the N2 adsorption isotherm test as shown in Fig. S3 of the ESI.† LiClCo–C3N4 displays a type IV isotherm with an obvious hysteresis loop, demonstrating the formation of a porous structure, which was consistent with the SEM and TEM analysis. The Brunauer–Emmett–Teller surface area (SBET) and pore volume for g-C3N4 and 3LiClCo–C3N4 are listed in Fig. S3 of the ESI.† The SBET and pore volume of 3LiClCo–C3N4 were about 2–3 times higher than those of g-C3N4, which can provide more reactive reaction sites for photochemical reactions.
3.2. Optical and photocatalytic performance
The optical properties of the samples were analyzed by using UV-vis absorption spectra, as shown in Fig. 3a. Pristine g-C3N4 exhibited a primary adsorption edge around 450 nm. After introducing spent LiCoO2 and NH4Cl, the optical absorption was enhanced both in ultraviolet and visible light regions. The enhanced ultraviolet absorption may be attributed to the porous structure, while the orderly strengthened visible absorption was due to the narrowed bandgap derived from Li–Cl–Co regulating the π-conjugated structure of g-C3N4. Especially, the evident absorption band in the visible light region 600–700 nm originated from the Li or Co effect.32,37 The bandgap of the samples was estimated by the Kubelka–Munk method, as shown in Fig. 3b. The band gaps of g-C3N4 and XLiClCo–C3N4 (X = 0.75, 1.5, 3, and 5) were calculated to be 2.82, 2.79, 2.75, 2.72 and 2.67 eV, respectively. It is clear that the band gap of g-C3N4 was continuously narrowed with the increase of the Li–Cl–Co amount, which can facilitate producing more photogenerated carriers.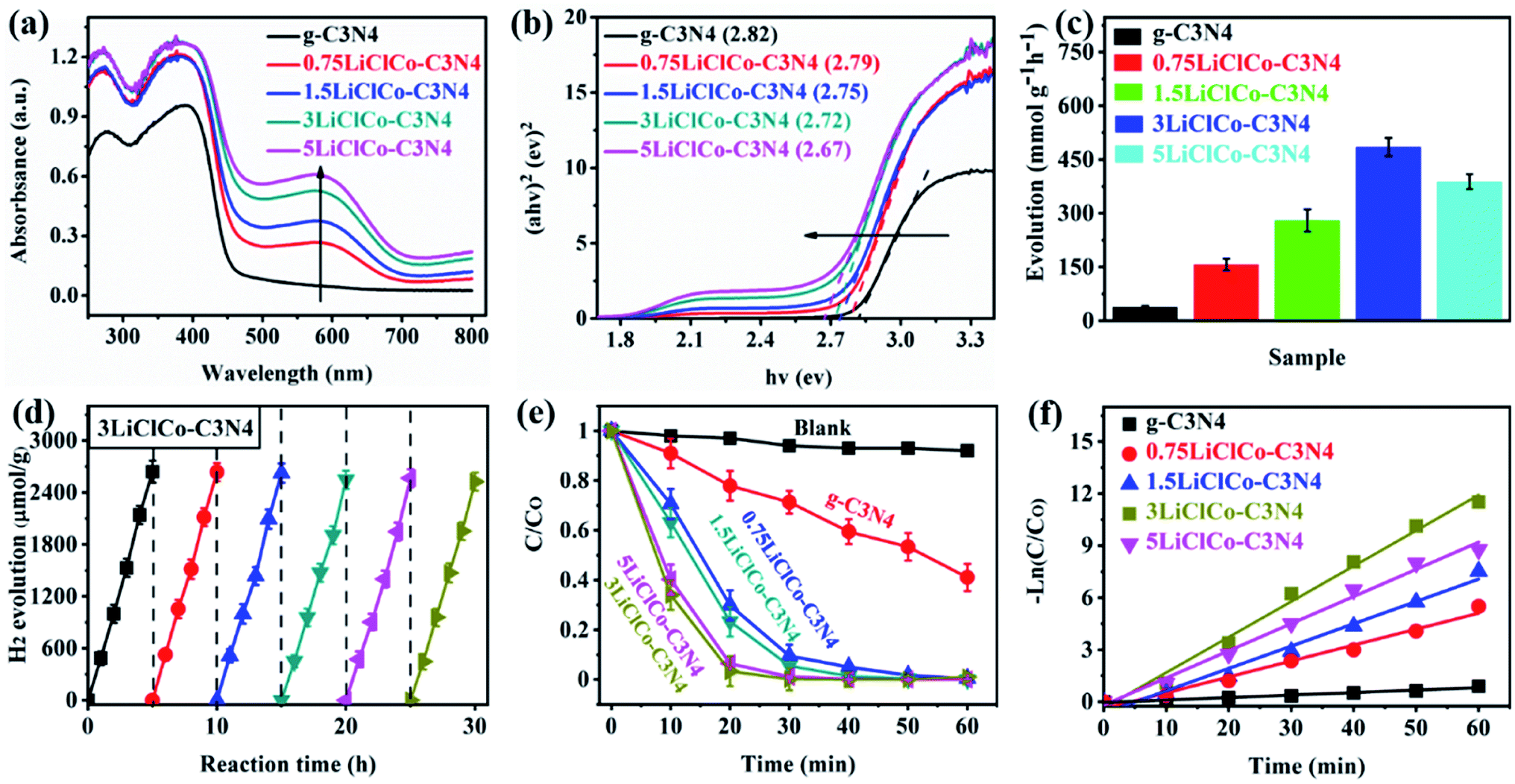 | ||
| Fig. 3 (a) UV-vis absorption spectra; (b) (αhν)2versus photon energy (hν) curves; (c) photocatalytic H2 evolution; (d) the recyclable photocatalytic activity for H2 evolution; (e) photocatalytic RhB degradation; (f) the chemical kinetics curve for RhB degradation of different photocatalysts. | ||
The photocatalytic performance of the samples was evaluated by photocatalytic H2 evolution and RhB degradation under simulated sunlight. As shown in Fig. 3c, pristine g-C3N4 exhibited a photocatalytic H2 evolution rate of 38.5 μmol g−1 h−1. After regulating the π-conjugation of g-C3N4, the H2 evolution performance was significantly enhanced. Among the samples, the 3LiClCo–C3N4 presented the highest H2 evolution rate of 485.1 μmol g−1 h−1, which is almost 12.6 times higher than that of pristine g-C3N4. However, on further increasing the addition amount, the photocatalytic H2 activity decreased due to the increased recombination probability of photoelectrons and holes in the defects. The stability and reproducibility of the optimal 3LiClCo–C3N4 photocatalyst were tested with eight-run tests under simulated sunlight irradiation. As shown in Fig. 3d, the photocatalytic H2 evolution was not obviously decreased, demonstrating a stable photocatalytic activity for H2 production.
The photocatalytic degradation of RhB was also conducted under simulated sunlight, as shown in Fig. 3e. The absence of a catalyst had a negligible effect on the RhB degradation, while g-C3N4 and LiClCo–C3N4 samples exhibited an obvious photocatalytic RhB degradation. Compared to pristine g-C3N4, LiClCo–C3N4 samples could significantly improve the photocatalytic degradation of RhB. The degradation process follows the pseudo-first-order model and the apparent rate constant k was calculated, as shown in Fig. 3f. The k of 3LiClCo–C3N4 was calculated to be 0.214 min−1, which was about 15.3 times higher than that of unmodified g-C3N4 (0.014 min−1). Compared with 3LiClCo–C3N4, the other LiClCo–C3N4 showed a lower photoactivity for RhB degradation. The results accorded with the above photocatalytic H2 evolution. In addition, the recycling runs (Fig. S4 of the ESI†) of 3LiClCo–C3N4 clearly demonstrated good stability for the photocatalytic degradation of RhB. Also, we made a comparison with recent studies about anion or cation doped g-C3N4 photocatalysts, as shown in Tables S1 and S2 of the ESI.† The results suggested that the as-prepared LiClCo–C3N4 photocatalyst in our study was competitive with literature reports.
3.3. Enhanced photocatalytic mechanism
Since the photocatalytic activity is closely related to the band structure and charge separation efficiency, we performed some experiments and DFT calculations to investigate the regulation effect of Li–Cl–Co on the π-conjugated system and electronic structure of g-C3N4. To estimate the band positions, the Mott–Schottky measurements were first conducted. Fig. 4a shows the Mott–Schottky plots for g-C3N4 and LiClCo–C3N4 samples. The positive slope of the curves demonstrated the N-type semiconductor characteristic. The flat-band potential (VFB) of g-C3N4 and X-LiClCo–C3N4 (X = 0.75, 1.5, 3, and 5) samples was estimated to be −1.24, −1.11, −0.97, −0.81, and −0.72 V vs. Ag/AgCl (−1.03, −0.90, −0.76, −0.60 and −0.51 V vs. NHE). In general, the conduction band (CB) edge potential can approximately coincide with the VFB for N-type semiconductors.41 So, based on the Mott–Schottky analysis and the band gaps (Fig. 3b), the schematic diagram of the estimated band positions for C3N4 and LiClCo–C3N4 samples is presented in Fig. 4b. The results suggested that the Li–Cl–Co caused the down-shift of the CB and VB levels in g-C3N4. Similar results have also been reported for the K, Na, Sr and K–NO3 intercalated g-C3N4.20,21,25 To further reveal the effect of Li–Cl–Co on the electronic structure of g-C3N4, the density of states (DOS) of the samples was calculated by DFT, as shown in Fig. 4c. For g-C3N4, the valence band (VB) maximum and conduction band (CB) minimum are both composed of C and N. The calculated bandgap was 0.92 eV, which was consistent with the previous DFT results.26 After Li–Cl–Co regulation, Li and Cl did not participate in the formation of VB and CB bands, while Co contributed to both VB and CB. The results suggested that Co mainly regulates the band structure of g-C3N4. The bandgap of LiClCo–C3N4 clearly narrowed, indicating the enhanced visible-light adsorption. In addition, the down-shift of CB and VB positions can be observed after Li–Cl–Co regulation. The DOS results verified the above UV-vis and Mott–Schottky experiment results. | ||
| Fig. 4 (a) Mott–Schottky plots; (b) schematic diagram of band positions; (c) the calculated DOS; (d) radical trapping experiments; (e) PL spectra; (f) photocurrent response; (g) electrochemical impedance spectra; and (h) the calculated electrostatic potentials of different samples. | ||
Radical trapping experiments were used to investigate the main reactive species involved during the photodegradation of RhB. Triethanolamine (TEOA), 1,4-benzoquinone (BQ) and isopropyl alcohol (IPA) were added to capture h+, ˙O2−, and ˙OH, respectively. As displayed in Fig. 4d, an obvious decrease in photodegradation efficiency was observed when TEOA was added into the RhB solution. By contrast, BQ and IPA also have a lower impact on the photodegradation process. This suggested that h+ was the main reactive species but ˙O2−and ˙OH were also indispensable for RhB degradation. It is well known that the N-deethylation and cleavage of the chromophore structure of RhB usually occurred in the presence of active species.42–46 The dye molecule in the reaction system was first adsorbed in the photocatalyst surface through the positively charged diethylamine function of RhB. Then, the step-by-step N-deethylation of RhB occurred to produce N,N-diethyl-N′-ethylrhodamine (DER), N,N-diethylrhodamine (DR), N-ethylrhodamine (ER) and rhodamine (R).42–44 These N-deethylation intermediates further produced some small molecule products (such as oxalic acid, malonic acid, succinic acid, and phthalic acid, etc.) during the decolorization process.42–44 On the other hand, the rather facile and highly efficient cleavage of the RhB chromophore structure simultaneously occurred through the whole photo-degradation process. Such cleavage of the chromophore structure could directly produce the small molecule products.42,44,46 Finally, these small molecules were mineralized into CO2 and H2O.
To study the transfer and separation efficiency of photogenerated carriers, photoluminescence (PL) spectra were recorded with an excitation wavelength of 330 nm. As shown in Fig. 4e, g-C3N4 exhibited a strong emission peak at about 460 nm, which was attributed to the radiative recombination of self-trapped excitation on the g-C3N4 surface.47 After introducing spent LiCoO2 and NH4Cl, the PL intensity of LiClCo–C3N4 samples was lower than that of pristine g-C3N4 at the same position. Theoretically, a smaller PL intensity represents a lower recombination rate of photogenerated charger carriers, leading to a higher photocatalytic performance.48,49 The PL results suggested that the π-conjugation regulation by Li–Cl–Co could inhibit the charge carrier recombination in g-C3N4. Moreover, 3LiClCo–C3N4 possessed the lowest PL intensity and the highest charge carrier separation efficiency. However, with further increasing the spent LiCoO2 amount (5 wt%), the PL intensity began to increase due to the excess Li–Cl–Co acting as the recombination center, which was consistent with the above photocatalytic activity experiments.
To further investigate the charge separation and charge transfer resistance of the photocatalysts, photocurrent response (I–t) curves and electrochemical impedance spectra (EIS) were recorded. Generally, a higher photocurrent intensity and smaller arc radius signify a higher charger separation efficiency and lower charge transfer resistance.50 As shown in Fig. 4f, all the samples showed a reproducible photocurrent response during the light on/off cycles. Compared with pristine g-C3N4, the photocurrent of the LiClCo–C3N4 samples was enhanced. 3LiClCo–C3N4 exhibited the highest photocurrent, indicating the highest charger separation. Fig. 4g shows the EIS Nyquist plots of photocatalysts. The arc radius of LiClCo–C3N4 samples was smaller than that of g-C3N4, indicating a lower charger transfer resistance of the LiClCo–C3N4 samples. It was attributed to the fact that Li–Cl–Co regulation could help the charge separation between the g-C3N4 interlayers. Moreover, 3LiClCo–C3N4 exhibited the smallest charge transfer resistance. The above PL and photoelectrochemical results confirmed that Li–Cl–Co could serve as a channel to efficiently separate and transport the charge carriers between g-C3N4 interlayers, thus promoting the photochemical reactions.
Furthermore, to study the charger transfer path, we calculated the electrostatic potentials of g-C3N4 and LiClCo–C3N4 between adjacent g-C3N4 layers by DFT, as presented in Fig. 4h. The bottom represents the position of the g-C3N4 layer. g-C3N4 showed the same electrostatic potential between the adjacent layers. Compared with g-C3N4, the electrostatic potential of LiClCo–C3N4 first decreased then gradually increased from the bottom to top layers (potential gradient), which was consistent with the change of interlayer spaces caused by the Li and Cl intercalated in g-C3N4 (the Z-axis direction in Fig. 4h, also can be clearly seen in Fig. 1i). Therefore, we can conclude that the Li and Cl intercalation could synergistically regulate the π conjugated structure and further adjust the electrostatic potential of g-C3N4. Such electrostatic potential differences could lead to the gradient decrease in the energy barrier for dramatically boosting the charge migration and separation between g-C3N4 layers.20,21 This phenomenon further verified the above PL and photoelectrochemical results.
Based on the above experiment and DFT results, we successfully utilized spent Li-ion batteries to regulate the π-conjugated structure of g-C3N4 for enhancing its photocatalytic activity. During the one-pot sintering of melamine, NH4Cl and spent LiCoO2, the Li, Cl and Co participated in the g-C3N4 formation and thus regulated the π-conjugated structure of g-C3N4. The π-conjugation regulation was characterized by the changes of interlayer spacing (Fig. 1b and i), charge density distribution (Fig. 1j–l), energy barrier between layers (Fig. 4h), charge transfer and separation (Fig. 4e–g), and electronic structure of g-C3N4 (Fig. 4b and c). These π-conjugation regulation properties synergistically contributed to the improvement of the photocatalytic activity of g-C3N4 (Fig. 3). The π-conjugation regulation of g-C3N4 and the enhanced photocatalytic mechanism are summarized and illustrated in Fig. 5. Firstly, the Co among the adjacent tri-s-triazine units narrowed the bandgap of g-C3N4, which enhanced the visible light absorption and produced more photoinduced charges participating in the photochemical reactions. Secondly, the Li and Cl intercalation respectively decreased and increased the interlayer spacing and caused a gradient decrease in the energy barrier between g-C3N4 layers, acting as the charge transport channel, which remarkably improved the charge transfer and separation between g-C3N4 layers. Thirdly, the Li–Cl–Co caused the charge redistribution and the formation of Li–Cl, C–Cl, Li–N and Co–N chemical bonds (see the XPS results). These chemical bonds contributed to the good stability for the photocatalytic performance. Finally, the porous structure of LiClCo–C3N4 caused by the excess NH4Cl (foaming agent) exhibited a larger specific surface area and provided more reactive reaction sites for photochemical reactions.
 | ||
| Fig. 5 Schematic diagram of utilizing spent LIBs to regulate the π-conjugated structure of g-C3N4 and the Li–Cl–Co synergistic effect on the enhanced photocatalytic mechanism. | ||
4. Conclusions
This study utilizes spent LIBs to regulate the π-conjugated structure of g-C3N4 for enhancing its photocatalytic activity. The Li–Cl–Co changed the π-conjugation of g-C3N4, including the interlayer spacing, charge density distribution, energy barrier between layers, charge transfer and separation and electronic structure. To be specific, the Li–Cl–Co caused the charge redistribution and formed Li–N, Li–Cl, Cl–C and Co–N chemical bonds, which stabilized the Li–Cl–Co in g-C3N4. Co among the adjacent tri-s-triazine units decreased the bandgap to lower the electron transition energy. The Li and Cl intercalation respectively decrease and increase the interlayer spacing, leading to a gradient decrease in the energy barrier for significantly improving the charge migration and separation between g-C3N4 layers. These π-conjugation regulations synergistically contributed to the superior photocatalytic activity. The simulated sunlight photocatalytic H2 evolution and RhB degradation rates of the Li–Cl–Co regulated g-C3N4 were 12.6 and 15.3 times higher than those of pristine g-C3N4. Furthermore, the special photocatalyst exhibited good photocatalytic stability due to the stable structure. In short, this study avoids element separation and utilizes spent LIBs for photocatalytic application, which minimizes the environmental risks associated with recycling. Our study reveals a synergetic approach to recycling wastes as high-efficiency photocatalysts, which realizes an all-win situation for waste recycling and environmental protection.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2019YFC1904400), the Postdoctoral Innovative Talent Support Program (BX20190200), the China Postdoctoral Science Foundation (2020M671129) and the National Natural Science Foundation of China (51534005).References
- B. Wang, X.-Y. Lin, Y. Tang, Q. Wang, M. K. H. Leung and X.-Y. Lu, J. Power Sources, 2019, 436, 226828 CrossRef CAS.
- J. Xiao, J. Li and Z. Xu, Environ. Sci. Technol., 2019, 54, 9–25 Search PubMed.
- S. Natarajan and V. Aravindan, Adv. Energy Mater., 2018, 8, 1802303 CrossRef.
- S. Wang, Z. Zhang, Z. Lu and Z. Xu, Green Chem., 2020, 22, 4473–4482 RSC.
- Y. Zhao, X. Yuan, L. Jiang, J. Wen, H. Wang, R. Guan, J. Zhang and G. Zeng, Chem. Eng. J., 2020, 383, 123089 CrossRef CAS.
- X. Zhang, L. Li, E. Fan, Q. Xue, Y. Bian, F. Wu and R. Chen, Chem. Soc. Rev., 2018, 47, 7239–7302 RSC.
- M. K. Tran, M.-T. F. Rodrigues, K. Kato, G. Babu and P. M. Ajayan, Nat. Energy, 2019, 4, 339–345 CrossRef CAS.
- J. Xiao, J. Li and Z. Xu, J. Hazard. Mater., 2017, 338, 124–131 CrossRef CAS PubMed.
- J. Xiao, J. Li and Z. Xu, Environ. Sci. Technol., 2017, 51, 11960–11966 CrossRef CAS PubMed.
- J. Lin, C. Liu, H. Cao, R. Chen, Y. Yang, L. Li and Z. Sun, Green Chem., 2019, 21, 5904–5913 RSC.
- J. Lin, L. Li, E. Fan, C. Liu, X. Zhang, H. Cao, Z. Sun and R. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 18482–18489 CrossRef CAS PubMed.
- X. Qu, H. Xie, X. Chen, Y. Tang, B. Zhang, P. Xing and H. Yin, ACS Sustainable Chem. Eng., 2020, 8, 6524–6532 CrossRef CAS.
- E. Fan, L. Li, J. Lin, J. Wu, J. Yang, F. Wu and R. Chen, ACS Sustainable Chem. Eng., 2019, 7, 16144–16150 CrossRef CAS.
- J. Xiao, B. Niu and Z. Xu, J. Clean. Prod., 2020, 227, 122718 CrossRef.
- S. Natarajan, S. Anantharaj, R. J. Tayade, H. C. Bajaj and S. Kundu, Dalton Trans., 2017, 46, 14382–14392 RSC.
- M. Assefi, S. Maroufi, Y. Yamauchi and V. Sahajwalla, ACS Sustainable Chem. Eng., 2019, 7, 19005–19014 CrossRef CAS.
- T. Dai, H. Zhou, Y. Liu, R. Cao, J. Zhan, L. Liu and B. W. L. Jang, ACS Sustainable Chem. Eng., 2019, 7, 5072–5081 CrossRef CAS.
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- J. Zhang, Y. Chen and X. Wang, Energy Environ. Sci., 2015, 8, 3092–3108 RSC.
- X. Zou, Z. Sun and Y. H. Hu, J. Mater. Chem. A, 2020, 8, 21474–21502 RSC.
- N. Tian, H. Huang, X. Du, F. Dong and Y. Zhang, J. Mater. Chem. A, 2019, 7, 11584–11612 RSC.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS PubMed.
- B.-W. Sun, H.-J. Li, H.-y. Yu, D.-J. Qian and M. Chen, Carbon, 2017, 117, 1–11 CrossRef CAS.
- W. Guo, J. Zhang, G. Li and C. Xu, Appl. Surf. Sci., 2019, 470, 99–106 CrossRef CAS.
- T. Xiong, W. Cen, Y. Zhang and F. Dong, ACS Catal., 2016, 6, 2462–2472 CrossRef CAS.
- C. Liu, Y. Zhang, F. Dong, A. H. Reshak, L. Ye, N. Pinna, C. Zeng, T. Zhang and H. Huang, Appl. Catal. B: Environ., 2017, 203, 465–474 CrossRef CAS.
- J. Fu, B. Chang, Y. Tian, F. Xi and X. Dong, J. Mater. Chem. A, 2013, 1, 3083–3090 RSC.
- Y. Jiang, Z. Sun, C. Tang, Y. Zhou, L. Zeng and L. Huang, Appl. Catal. B: Environ., 2019, 240, 30–38 CrossRef CAS.
- H. Gao, S. Yan, J. Wang, Y. A. Huang, P. Wang, Z. Li and Z. Zou, Phys. Chem. Chem. Phys., 2013, 15, 18077–18084 RSC.
- Z. Zhang, J. Huang, Y. Fang, M. Zhang, K. Liu and B. Dong, Adv. Mater., 2017, 29, 1606688 CrossRef PubMed.
- L. Cui, J. Song, A. F. McGuire, S. Kang, X. Fang, J. Wang, C. Yin, X. Li, Y. Wang and B. Cui, ACS Nano, 2018, 12, 5551–5558 CrossRef CAS PubMed.
- G. Gu, K. Wang, N. Xiong, Z. Li, Z. Fan, S. Hu and X. Zou, Dalton Trans., 2019, 48, 5083–5089 RSC.
- K. Artyushkova, B. Kiefer, B. Halevi, A. Knop-Gericke, R. Schlogl and P. Atanassov, Chem. Commun., 2013, 49, 2539–2541 RSC.
- W. Ma, X. Wang, F. Zhang, X. Fei, X. Zhang, H. Ma and X. Dong, Mater. Res. Bull., 2017, 86, 72–79 CrossRef CAS.
- W. Liu, L. Cao, W. Cheng, Y. Cao, X. Liu, W. Zhang, X. Mou, L. Jin, X. Zheng, W. Che, Q. Liu, T. Yao and S. Wei, Angew. Chem. Int. Edit., 2017, 56, 9312–9317 CrossRef CAS PubMed.
- Z. Zhu, X. Tang, T. Wang, W. Fan, Z. Liu, C. Li, P. Huo and Y. Yan, Appl. Catal. B: Environ., 2019, 241, 319–328 CrossRef CAS.
- L. Wang, X. Guo, Y. Chen, S. Ai and H. Ding, Appl. Surf. Sci., 2019, 467–468, 954–962 CrossRef CAS.
- J. Wen, J. Xie, X. Chen and X. Li, Appl. Surf. Sci., 2017, 391, 72–123 CrossRef CAS.
- M. Xie, J. Tang, L. Kong, W. Lu, V. Natarajan, F. Zhu and J. Zhan, Chem. Eng. J., 2019, 360, 1213–1222 CrossRef CAS.
- W. Zhang, Z. Zhang, S. H. Choi and W. Yang, Catal. Today, 2019, 321–322, 67–73 CrossRef CAS.
- T. Giannakopoulou, I. Papailias, N. Todorova, N. Boukos, Y. Liu, J. Yu and C. Trapalis, Chem. Eng. J., 2017, 310, 571–580 CrossRef CAS.
- K. Yu, S. Yang, H. He, C. Sun and Y. Ju, J. Phy. Chem. A, 2009, 113, 10024 CrossRef CAS PubMed.
- H. E. Zhong, Y. Shaogui, J. U. Yongming and S. Cheng, J. Environ. Sci., 2009, 268–272 Search PubMed.
- Y. Zhang, J. Zhou, W. Cai, J. Zhou and Z. Li, Appl. Surf. Sci., 2018, 430, 549–560 CrossRef CAS.
- Z. He, C. Sun, S. Yang, Y. Ding, H. He and Z. Wang, J. Hazard. Mater., 2009, 162, 1477–1486 CrossRef CAS PubMed.
- F. U. Hongbo, S. Zhang, X. U. Tongguang, Y. Zhu and J. Chen, Environ. Sci. Technol., 2008, 42, 2085–2091 CrossRef PubMed.
- M. Ou, S. Wan, Q. Zhong, S. Zhang, Y. Song, L. Guo, W. Cai and Y. Xu, Appl. Catal. B: Environ., 2018, 221, 97–107 CrossRef CAS.
- Y. Jiang, P. Liu, L. Yan, X. F. Liu, L. Fan, N. Liang, Y. Yan and P. Huo, Sep. Purif. Technol., 2016, 170, 10–21 CrossRef CAS.
- Z. Chen, S. Peng, F. Bing, Z. Zhang and X. Fang, J. Phys. Chem. C, 2014, 118, 7801–7807 CrossRef CAS.
- L. Liu, Y. Qi, J. Lu, S. Lin, W. An, Y. Liang and W. Cui, Appl. Catal. B: Environ., 2016, 183, 133–141 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta10881b |
| This journal is © The Royal Society of Chemistry 2021 |