
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRoom temperature synthesis and characterization of novel lead-free double perovskite nanocrystals with a stable and broadband emission†
Yingying
Tang
a,
Leyre
Gomez
ab,
Marco
van der Laan
a,
Dolf
Timmerman
c,
Victor
Sebastian
 def,
Chia-Ching
Huang
a,
Tom
Gregorkiewicz
a and
Peter
Schall
*a
def,
Chia-Ching
Huang
a,
Tom
Gregorkiewicz
a and
Peter
Schall
*a
aInstitute of Physics, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: P.Schall@uva.nl
bCatalan Institute of Nanoscience and Nanotechnology, CSIC, BIST, and CIBERBBN, 08193 Bellaterra Barcelona, Spain
cGraduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan
dDepartment of Chemical Engineering and Environmental Technology, Universidad de Zaragoza, Campus Río Ebro-Edificio I + D, 50018 Zaragoza, Spain
eInstituto de Nanociencia y Materiales de Aragón (INMA), University of Zaragoza-CSIC, c/María de Luna 3, 50018 Zaragoza, Spain
fNetworking Research Center on Bioengineering Biomaterials and Nanomedicine (CIBER-BBN), Madrid, Spain
First published on 23rd November 2020
Abstract
Low-dimensional and lead-free halide perovskites are of great interest for their wide application potential for optoelectronic applications. We report on the successful synthesis of novel lead-free colloidal Cs3BiBr6 nanocrystals (NCs) with an ultra-small size of ∼1.5–3 nm by a room temperature antisolvent process. From crystallographic characterization we show that it is critical to precisely control the ratio of precursors to obtain the pure 3-1-6 phase. The synthesis process is facile and repeatable and results in Cs3BiBr6 NCs that display stable blue emission around 438 nm with a relatively broad linewidth of 92.1 nm. Low-temperature photoluminescence (PL) measurements displayed a red-shift of bandgap with decreasing temperature, which might be attributed to the thermal expansion of the lattice. In addition, the NCs demonstrate high stability at ambient conditions.
Introduction
Lead-halide perovskite nanocrystals (NCs) have recently emerged in various applications like solar cells1,2 and light-emitting diodes.3,4 The benefits of NCs in this context are the tunability of their optoelectronic properties by variation of their size, composition and surface ligands5 and the high photoluminescence quantum yield (PLQY). Other interesting properties observed in these materials include efficient carrier multiplication,6 activation of a strong light-induced forbidden exciton transition due to the influence of polaron in confined space,7 and an efficient triplet energy-transfer efficiency to pyrene complexes.8 Despite these interesting features, the important limitation of these materials in applications is the toxicity of lead; Pb2+ is soluble in water, and exposure to this has a serious impact on health.9For above reasons, there is a strong research effort into finding lead-free perovskite materials that have similar beneficial optoelectronic properties as their lead-containing counterparts. So far, several works have reported on lead-free perovskites; among these are Bi, Sn, and Ge-based perovskites.10–17 However, oxidation of Sn2+ and Ge2+ in ambient conditions limits their synthesis and application in natural environments. Bismuth-based perovskites do not suffer from this effect and are strong candidates for non-toxic and stable perovskite material. Currently, most of the Bi-based perovskites are low-dimensional structures, which could be accompanied by exotic and novel optical properties. For instance, recent reports in defect-tolerant double perovskites Cs2ABX6 (with A: Ag, Cs, Na; B: In, Bi, Sb; X: Cl, Br, I), showed slow hot-carrier relaxation, dual-color emission, coexistence of direct–indirect bandgap and single emitter based warm-white emission.18–21 Especially, for NCs in the quantum confinement regime, optical and electronic properties are strongly altered. For example, Cs3Bi2I9 NCs showed emission from exciton and defect states inside the bandgap, and induced broadband emission, as well as enhanced Raman scattering due to the strong exciton–ligand electronic coupling.22 Several structures have been reported in the A–Bi–X perovskite series, however, only a few of them are in the form of colloidal NCs. For example, it has been shown that relatively large NCs of Cs3BiCl6 could be obtained by a hot-injection method.23
Here, we report on the synthesis of stable bismuth-based double perovskite NCs, Cs3BiBr6, using an antisolvent precipitation method at room temperature. Cs3BiBr6 features an isolated BiBr6 octahedra structure, which can be considered a double perovskite.24 The synthesized NCs have extremely small particle sizes in the range of 1.5–3 nm, depending on the precursor ratio, and display blue emission around 438 nm, independent of this ratio. Furthermore, they show an excellent stability at room temperature for at least 3 weeks. The synthesis is reproducible, and the NCs are obtained in a wide range of precursor ratios.
Experimental section
Materials
BiBr3 (≥98%), CsBr (99.9%), oleic acid (OA, 90%), hydrobromic acid (HBr, 48%) and dimethyl sulfoxide (DMSO, ≥99.9%) were all purchased from Sigma-Aldrich. Isopropanol (IPA, ≥99.5%) was purchased from EMD Millipore company. All materials were used as received without further purification.Synthesis of Cs3BiBr6 NCs
For the synthesis of ligand-free Cs3BiBr6 NCs, 120 μmol CsBr (25.5 mg) and 30 μmol BiBr3 (13.45 mg) were dissolved in 2 mL DMSO at room temperature or 70 °C to form the precursor solution. Alternatively, 90 μmol CsBr (19.2 mg), 30 μmol BiBr3 (13.45 mg) and HBr (100 μL) were dissolved in 2 mL DMSO to form precursor solution. 200 μL of the above precursor solution was injected into 5 mL IPA, under vigorous stirring for one minute. Then the solution was centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 5 min to discard the large particles. After that, clear colloidal NCs dispersed in IPA were obtained. For the synthesis of OA-capped Cs3BiBr6 NCs, all the procedures were the same with the only difference being the addition of 300 μL OA into the precursor.
000 rpm for 5 min to discard the large particles. After that, clear colloidal NCs dispersed in IPA were obtained. For the synthesis of OA-capped Cs3BiBr6 NCs, all the procedures were the same with the only difference being the addition of 300 μL OA into the precursor.
Measurement and characterization
Optical measurements
The absorption spectra was obtained by measuring the transmission on A LAMBDA 950 UV/vis/NIR spectrophotometer (PerkinElmer). The separately measured absorption spectrum of the solvent (IPA) was subtracted from the NC spectrum. PL spectra were determined with a Jobin Yvon FluoroLog spectrofluorometer (Horiba). As excitation source, a 450 W xenon lamp coupled to a monochromator was used. The PL data was collected for excitation wavelength of 355 nm. All spectra are corrected for the spectral sensitivity of the spectrofluorometer. The determination of the PLQY was based on an integrating sphere methodology.28 Time-resolved PL measurements were performed using a LifeSpecII time correlated single photon counting (TCSPC) spectrometer series, 100 ps was used as excitation source. A right angle between the excitation and emission beam paths was maintained to reduce detection of scattered excitation light. For the low temperature PL setup, a xenon lamp (L2273 Hamamastu) coupled to a Solar MSA 130 double grating monochromator was used as an excitation source. The collected emission was dispersed by an M266 (Solar LS) monochromator coupled to a silicon CCD (Horiba scientific). The sample was cooled in a CCC1104 (Oxford inst.) cryostat, which was used in combination with a closed-cycle helium compressor (Cryomech). The sample was placed on a cold finger in a static exchange gas to ensure the temperature setting was obtained. A 355 nm excitation wavelength was used to collect the low temperature PL spectra.Results and discussion
The double perovskite Cs3BiBr6 crystallizes in the orthorhombic system in a space group of Pbcm, with a = 8.689(2) Å, b = 13.628(1) Å, c = 27.694(9) Å.24 Each Bi is coordinated with six Br atoms, forming BiBr6 octahedra. BiBr6 octahedra are separated from each other, forming a zero-dimensional perovskite structure, as shown in Fig. 1a. In the present work, the Cs3BiBr6 NCs were synthesized by an antisolvent precipitation method. For the synthesis of the nanomaterials, a precise control of the precursor ratios between BiBr3 and CsBr was required. As control experiments, different ratios of precursors were tried to obtain the NCs. To exclude the effects of excess of Cs+ on the synthesis, HBr was instead used to introduce excess Br− while keeping the stoichiometric ratio between CsBr and BiBr3. Cs3BiBr6 NCs could also be obtained using HBr as precursor (Fig. 1b). Controlling the ratios of the precursors is critical for the formation of the NCs, as we confirmed by a series of control experiments. As displayed in Fig. S1a (ESI†), when the ratio (CsBr/BiBr3) was below 4, Cs3Bi2Br9 NCs or a mixture of Cs3Bi2Br9 and Cs3BiBr6 NCs were obtained. In contrast, for ratios between 4 and 7.5, we clearly observed the Cs3BiBr6 phase alone, as shown by the XRD patterns in Fig. 1c, which match well with its simulated pattern. When the ratios are larger than 7.5, several dominant peaks are displayed, i.e. (136), (410), (331), (130), (025) crystalline planes for Cs3BiBr6.24 Peaks from CsBr and BiBr3 precursor impurities can be excluded by comparison with their XRD peaks. We thus consider that the morphology of the NCs may differ for lower precursor ratios. In order to check this, we characterized the NCs with different ratios of precursors. For convenience we use Cs3BiBr6-4, Cs3BiBr6-5.5, Cs3BiBr6-7.5, Cs3BiBr6-10, Cs3BiBr6-20 to indicate molar ratios of CsBr/BiBr3 precursors of 4, 5.5, 7.5, 10, 20, respectively. Corresponding XRD patterns plotted in Fig. 1c show that the peaks are robust. Furthermore, to investigate if the Cs3BiBr6 phase is still stable at high temperature, the reaction temperature was increased to 70 °C. The corresponding XRD pattern shows that Cs3BiBr6 NCs could also be obtained.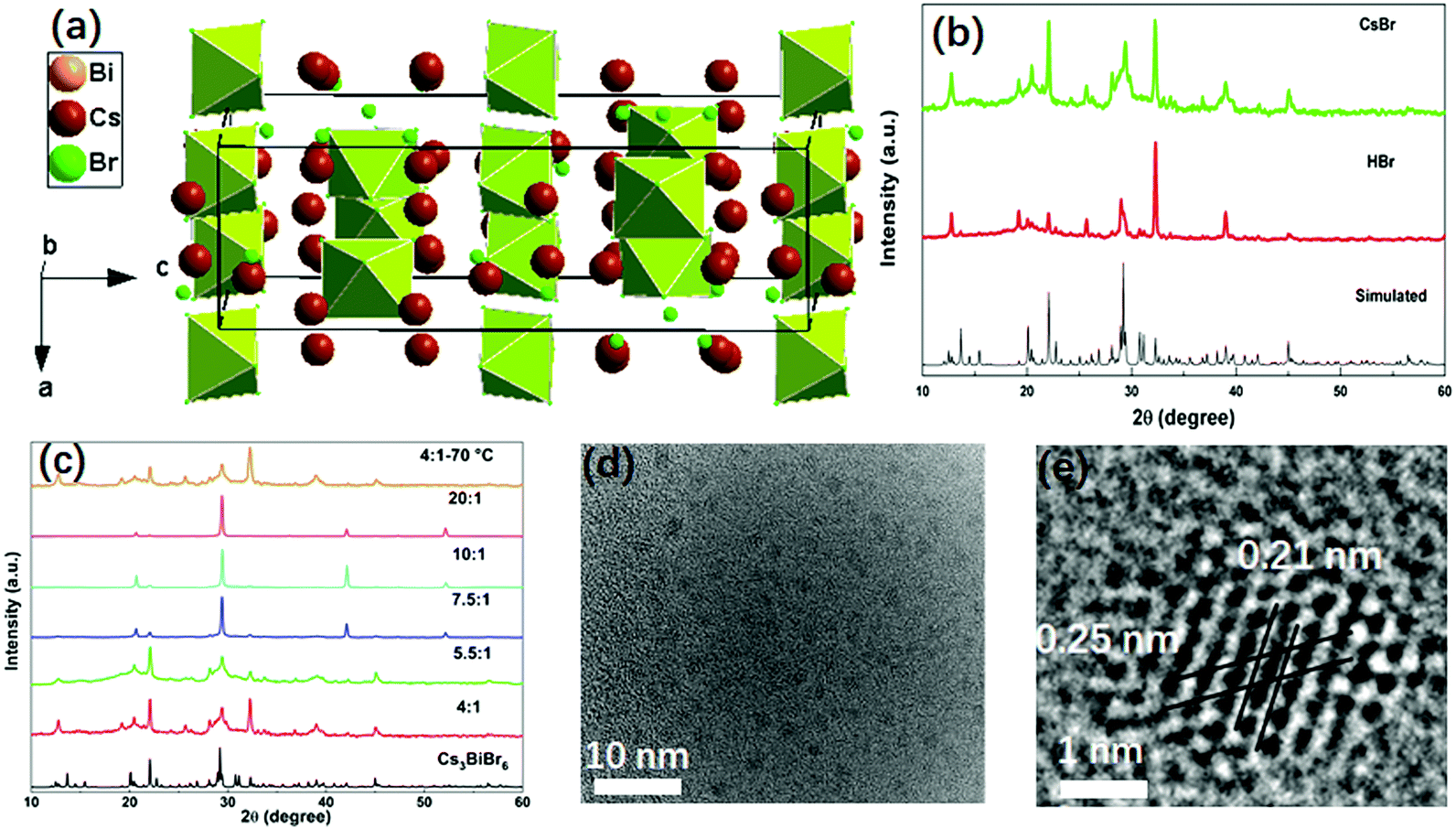 | ||
| Fig. 1 (a) Crystal structure of Cs3BiBr6. (b) XRD pattern of Cs3BiBr6 NCs with addition of CsBr or HBr precursors and the simulated XRD pattern (CCDC, #1877001). (c) XRD pattern of Cs3BiBr6 for several precursor ratios of CsBr/BiBr3 at room temperature and at 70 °C. (d) Representative TEM image for Cs3BiBr6-5.5 NCs. (e) HRTEM image with an atomic resolution for a Cs3BiBr6-5.5 NC. | ||
Fig. 1d and e present HRTEM images of Cs3BiBr6-5.5 NCs. The individual NCs could be observed with an average size of around 1.6 nm (inset of Fig. S2a, ESI†). For Cs3BiBr6-10, the average size NCs is about 2.3 nm (Fig. S2b and d, ESI†). Also, a branch-like morphology of NCs was observed (Fig. S2c, ESI†), which might induce the dominant orientation of NCs corresponding to the pronounced XRD diffraction peaks, for example, the XRD diffraction peak at ∼29° in the XRD pattern. We conclude by comparison with simulated XRD patterns (CsBr, Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m; CsBr, Pmm; and BiBr3, P21/c) and Cs3BiBr6 that these dominant XRD diffraction peaks e.g. XRD diffraction peak at ∼29°, cannot result from the precursors (Fig. S3a, ESI†). Meanwhile, as shown in the zoomed-in XRD for Cs3BiBr6-7.5, Cs3BiBr6-10, Cs3BiBr6-20 NCs (Fig. S3b, ESI†), all the dominant peaks can be well identified by the simulated Cs3BiBr6 XRD pattern. Yet, in Fig. 1c, many of the weak diffraction peaks are hidden by the pronounced peaks with high intensity in these three samples. In the HRTEM image of Cs3BiBr6-5.5 NCs, the clear lattice fringes of 0.21 nm and 0.25 nm in a single NC could be related to the crystalline planes (412) and (244) of Cs3BiBr6, respectively, while a lattice spacing of 0.22 nm corresponds to the (111) plane (Fig. S2d, ESI†). In order to check the chemical composition of these NCs, XPS measurements on Cs3BiBr6-5.5 NCs were conducted, which indeed show characteristic peaks of Cs, Bi and Br (Fig. S4, ESI†). The detailed elemental ratio between Cs, Bi, and Br elements was calculated as Cs2.3BiBr6.1, which is close to the reported chemical formula.
m; CsBr, Pmm; and BiBr3, P21/c) and Cs3BiBr6 that these dominant XRD diffraction peaks e.g. XRD diffraction peak at ∼29°, cannot result from the precursors (Fig. S3a, ESI†). Meanwhile, as shown in the zoomed-in XRD for Cs3BiBr6-7.5, Cs3BiBr6-10, Cs3BiBr6-20 NCs (Fig. S3b, ESI†), all the dominant peaks can be well identified by the simulated Cs3BiBr6 XRD pattern. Yet, in Fig. 1c, many of the weak diffraction peaks are hidden by the pronounced peaks with high intensity in these three samples. In the HRTEM image of Cs3BiBr6-5.5 NCs, the clear lattice fringes of 0.21 nm and 0.25 nm in a single NC could be related to the crystalline planes (412) and (244) of Cs3BiBr6, respectively, while a lattice spacing of 0.22 nm corresponds to the (111) plane (Fig. S2d, ESI†). In order to check the chemical composition of these NCs, XPS measurements on Cs3BiBr6-5.5 NCs were conducted, which indeed show characteristic peaks of Cs, Bi and Br (Fig. S4, ESI†). The detailed elemental ratio between Cs, Bi, and Br elements was calculated as Cs2.3BiBr6.1, which is close to the reported chemical formula.
The optical properties of Cs3BiBr6 NCs are shown in Fig. 2 and Fig. S5 (ESI†). Fig. 2a shows that the absorption peak of the NCs appears around 385 nm, which is similar to the previous result through a halide exchange reaction from Cs3BiCl6,23 and its behaviour is independent of the precursor ratio (Fig. 2b). The absorption spectra for Cs3Bi2Br9 NCs display an absorption onset of around 440 nm (Fig. S1b, ESI†), which is different from the onset at 385 nm for Cs3BiBr6 NCs. This further excludes the possibility of impurities from large amount of Cs3Bi2Br9 NCs. We thus hypothesize that it may be the crystallization pathway that makes it necessary to mix with a molar ratio of 4:1 or more to obtain the pure phase. The PL peak is located at 438 nm and the bandgap was determined from the Tauc plot in Fig. S6 (ESI†) as 2.89 eV (429 nm). Similar robustness as the absorption spectra is observed for the PL spectra of the different materials as shown in Fig. 2c, which are nearly identical. The peak positions display no obvious changes upon increasing CsBr, which is consistent with the TEM results. This can be well explained by the small Bohr radius of Cs3BiBr6 NCs (∼0.5 nm).29 The as-synthesized Cs3BiBr6 NCs with size of 1.5–3 nm are around 3 to 6 times larger than this Bohr radius, and are thus not expected to display any significant quantum confinement effects, resulting in robust PL emission peaks within this size regime. We have determined the PLQY for the Cs3BiBr6 NCs passivated by OA ligands as 22%. However, we found that the OA itself has a strong emission band partly overlapping with the emission of the NCs, as shown in Fig. S7a (ESI†). For comparison, the PL spectra of OA-capped NCs are also shown in Fig. S7b (ESI†), which reveal a strong emission signal in the range of 340–500 nm. A similar phenomenon has also been reported previously.22 We therefore prepared fresh samples without ligands. These demonstrated a considerably lower PLQY, which was difficult to determine accurately.
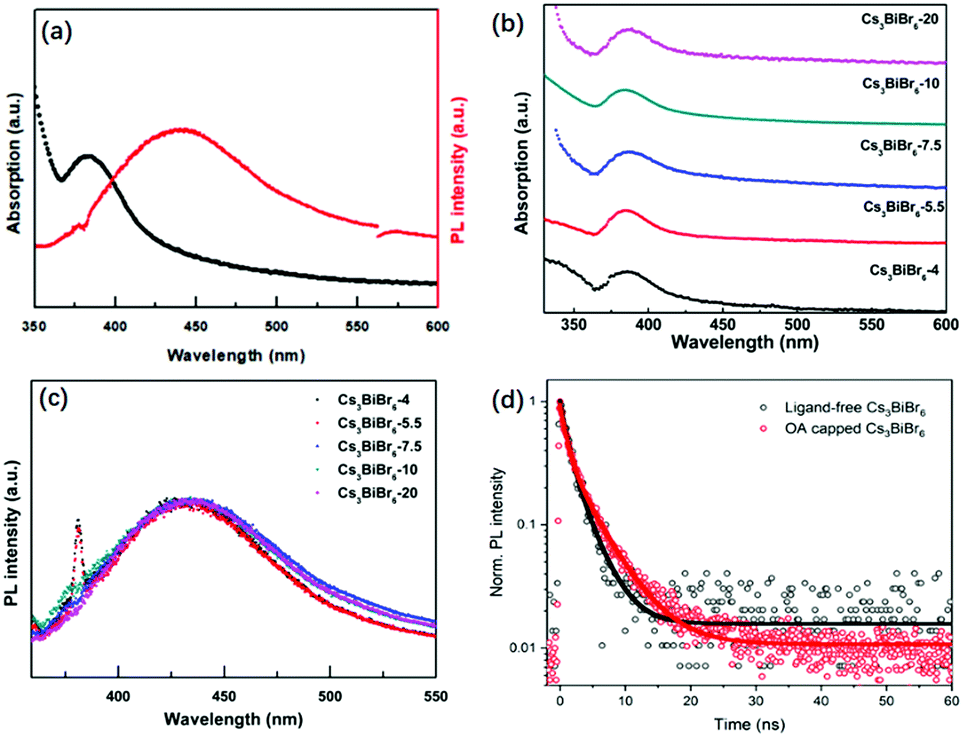 | ||
| Fig. 2 Optical properties for ligand-free Cs3BiBr6 NCs: (a) absorption spectra and PL spectra for Cs3BiBr6-5.5 NCs. (b) Absorption spectra for different ratios of precursors. (c) PL spectra for the addition of different ratios of precursors. (d) TR-PL kinetics of Cs3BiBr6-5.5 NCs with and without OA capping measured with TCSPC (375 nm pump) fitted with a bi-exponential function. | ||
To obtain more insight, we studied the photophysics of both ligand-free and OA-capped Cs3BiBr6-5.5 NCs by time-resolved photoluminescence (TRPL) under excitation of a 375 nm laser. The results are shown in Fig. 2d. The PL decays were fitted with a bi-exponential function, both with a fast component (τ1 = 1.0 ns) and a slower component of 2.8 and 4.3 ns for the ligand-free and OA capped NCs, respectively (ligand-free: A1 = A2 = 0.5; OA-capped: A1 = 0.55, A2 = 0.45). The longer lifetime is comparable to other reported lead-free perovskite NCs: average lifetimes of 4.3 ns in Cs3Sb2Br9 NCs25 and 2.1 ns in Cs3Bi2Br9 NCs30 have been obtained. The increase in lifetime for OA-capped NCs cannot fully explain the much higher PLQY that we observe as compared to ligand-free NCs. It is known from studies on carrier dynamics in other lead-free NCs that they typically show an ultrafast (≪100 ps) carrier trapping process that strongly limits their PLQY.31–34 Passivation by OA alleviates this process and increases the PLQY. Since the time resolution of our system is longer than such and ultrafast trapping process, we do not observe it in the PL dynamics. However, we note that the longer decay time we observe for OA-capped NCs indicates that it is either due to OA-related passivation, or direct emission from the OA ligands. Future studies on carrier dynamics will be necessary to point out the exact origin.
One important characteristic for understanding their photophysical properties, especially those depending on photo-excited charge carriers and thus applicability of photonic or optoelectronic applications, is the absorption cross-section. We have determined the absorption cross-section σ from linear absorption measurements using the following equation:
| T = e(−l×σ×n) | (1) |
Due to the low PLQY of Cs3BiBr6 at room temperature, we also performed PL measurements at low temperatures in order to obtain information about processes that are responsible for PL quenching at higher temperatures. The PL spectra recorded for a temperature range of 50–300 K are displayed in Fig. 3a and Fig. S8 (ESI†). They reveal two important features: an increase of the PL intensity for lower temperatures, accompanied by a red-shift. The integrated PL intensity as a function of reciprocal temperature is depicted in Fig. 3b. We use this to determine the activation energy of the non-radiative process responsible for the thermal quenching, by fitting with the Arrhenius formula:
| PL(T) = I0/(1 + A × exp(−ε/kBT)) | (2) |
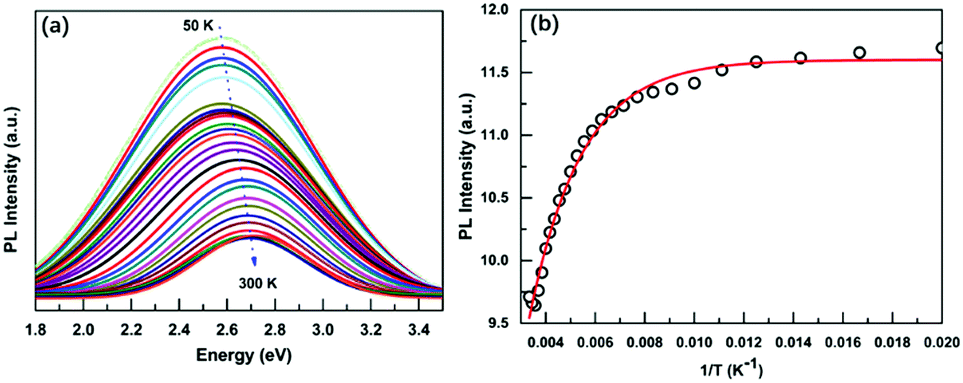 | ||
| Fig. 3 (a) Temperature-dependent PL spectra of OA-capped Cs3BiBr6 NCs at the temperature range of 50–300 K. (b) Integrated PL intensity of Cs3BiBr6 NCs versus inverse temperature and the Arrhenius fitting. | ||
The stability of NCs is critical for their potential applications. We investigated various aspects of their stability which are displayed in Fig. 4. First, the stability against an IPA washing treatment was investigated and as shown in Fig. 4a. Before the treatment, the absorption peak for Cs3BiBr6 NCs appears at around 385 nm. After one additional washing treatment by IPA, the absorption peaks for all samples are still at the same position, consistent with the different kinds of ratios of precursors and also for higher temperature. This demonstrates the stability of Cs3BiBr6 NCs against washing treatment with IPA solvent. Additionally, we used XRD to check the chemical composition after the IPA treatment (Fig. 4b), after single and double washing. Compared to the XRD in Fig. 1b, no impurities or other phases were found in the XRD data, supporting the conclusion that these NCs are stable against IPA solvent washing. Another important factor for applications is the air stability. The XRD spectra for a fresh and aged sample, after storage at ambient condition for 7 days, were investigated as shown in Fig. 4c. In addition, the optical stability was determined after 3 weeks, as shown in Fig. S9 (ESI†). The PL peak and intensity were nearly identical after storing in ambient conditions, demonstrating their air stability.
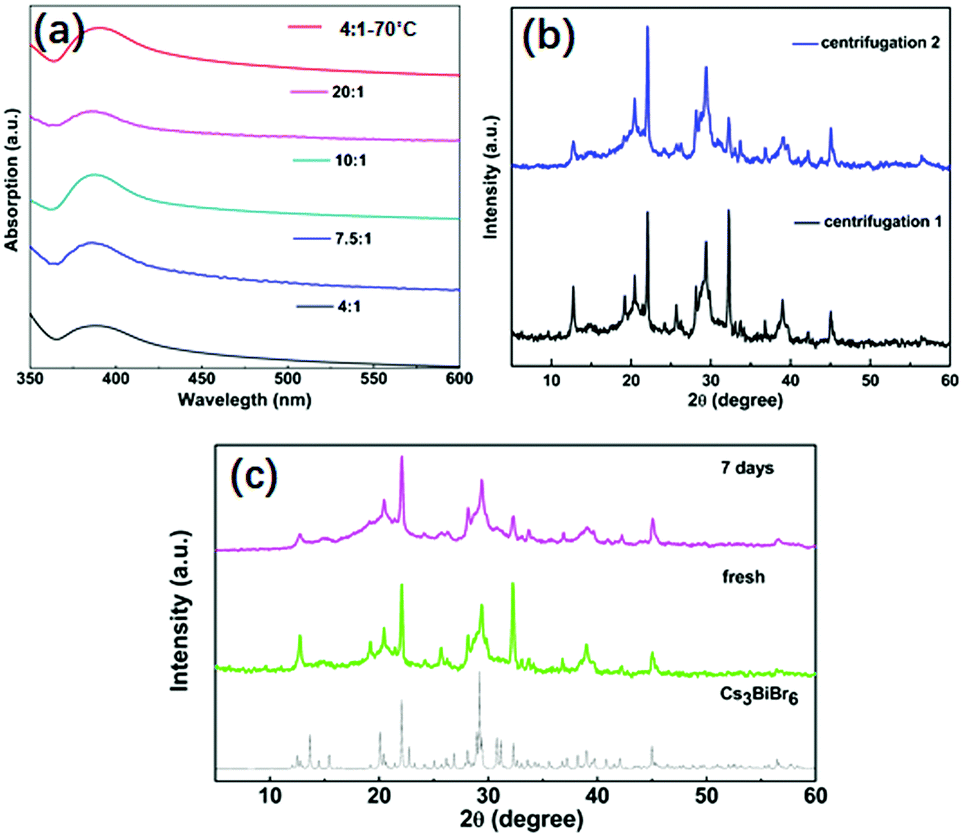 | ||
| Fig. 4 Stability for Cs3BiBr6 ligand-free NCs. (a) UV-vis absorption spectra after wash treatment with IPA twice. (b) XRD pattern after washing treatment by IPA twice. (c) XRD patterns for fresh and aged samples after 7 days. | ||
Conclusions
Here, we report on successful synthesis and characterization of novel lead-free Cs3BiBr6 NCs, obtained by a room temperature synthesis method. The synthesis process is facile and repeatable and generates NCs with an ultra-small size of ∼1.5–3 nm. In order to obtain pure Cs3BiBr6 NCs, an excess amount of Br− is necessary. Otherwise, another bismuth phase, Cs3Bi2Br9, will also be one of the products. The Cs3BiBr6 NCs display stable blue emission, centred at 438 nm, with a broad linewidth of 92.1 nm. At ambient conditions, the NCs display a high stability against the solvent and ambient air. This work proposes a promising low-dimensional candidate for lead-free optoelectronic applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the Dutch Technology Foundation STW, The Netherlands Organization for Scientific Research (NWO), and the Joint Solar Program (JSP III, 680-91-011) of The NWO for financial support. We acknowledge Arnon Lesage and Dido Van der Gon for their kind help. V. S. acknowledges the LMA-INA for offering access to their instruments and expertise.Notes and references
- A. Swarnkar, A. R. Marshall, E. M. Sanehira, B. Chernomordik, D. T. Moore, J. A. Christians, T. Chakrabarti and J. M. Luther, Science, 2016, 354, 92 CrossRef CAS.
- L. M. Wheeler, E. M. Sanehira, A. R. Marshall, P. Schulz, M. Suri, N. C. Anderson, J. A. Christians, D. Nordlund, D. Sokaras and T. Kroll, J. Am. Chem. Soc., 2018, 140, 10504 CrossRef CAS.
- J. Sun, J. Yang, J. I. Lee, J. H. Cho and M. S. Kang, J. Phys. Chem. Lett., 2018, 97, 1573 CrossRef.
- J. Xing, Y. B. Zhao, M. Askerka, L. N. Quan, X. W. Gong, W. J. Zhao, J. X. Zhao, H. R. Tan, G. K. Long, L. Gao, Z. Y. Yang, O. Voznyy, J. Tang, Z.-H. Lu, Q. H. Xiong and E. H. Sargent, Nat. Commun., 2018, 9, 3541 CrossRef.
- Y.-F. Li, J. Feng and H.-B. Sun, Nanoscale, 2019, 11, 19119 RSC.
- C. de Weerd, L. Gomez, A. Capretti, D. M. Lebrun, E. Matsubara, J. H. Lin, M. Ashida, F. C. M. Spoor, L. D. A. Siebbeles, A. J. Houtepen, K. Suenaga, Y. Fujiwara and T. Gregorkiewicz, Nat. Commun., 2018, 9, 4199 CrossRef.
- D. Rossi, H. Wang, Y. T. Dong, T. Qiao, X. F. Qian and D. H. Son, ACS Nano, 2018, 12, 12436 CrossRef CAS.
- X. Luo, R. C. Lai, Y. L. Li, Y. Y. Han, G. J. Liang, X. Liu, T. Ding, J. H. Wang and K. F. Wu, J. Am. Chem. Soc., 2019, 141, 4186 CrossRef CAS.
- G. Flora, D. Gupta and A. Tiwari, Interdiscip. Toxicol., 2012, 5, 47 CAS.
- Y. Q. Hu, T. Qiu, F. Bai, W. Ruan and S. F. Zhang, Adv. Energy Mater., 2018, 8, 1703620 CrossRef.
- B.-W. Park, B. Philippe, X. L. Zhang, H. Rensmo, G. Boschloo and E. M. J. Johansson, Adv. Mater., 2015, 27, 6806 CrossRef CAS.
- T.-B. Song, T. Yokoyama, S. Aramaki and M. G. Kanatzidis, ACS Energy Lett., 2017, 24, 897 CrossRef.
- S. Y. Shao, J. Liu, G. Portale, H. H. Fang, G. R. Blake, G. H. ten Brink, L. J. A. Koster and M. A. Loi, Adv. Energy Mater., 2018, 8, 1702019 CrossRef.
- W. J. Ke and M. G. Kanatzidis, Nat. Commun., 2019, 10, 965 CrossRef.
- S. P. Liu, B. Yang, J. S. Chen, D. H. Wei, D. Y. Zheng, Q. K. Kong, W. Q. Deng and K. L. Han, Angew. Chem., Int. Ed., 2020, 59, 1 CrossRef.
- B. Yang and K. L. Han, Acc. Chem. Res., 2019, 52, 3188 CrossRef CAS.
- M. Y. Cong, B. Yang, F. Hong, T. C. Zheng, Y. B. Sang, J. W. Guo, S. Q. Yang and K. L. Han, Sci. Bull., 2020, 65, 1078 CrossRef CAS.
- J. J. Luo, X. M. Wang, S. R. Li, J. Liu, Y. M. Guo, G. D. Niu, L. Yao, Y. H. Fu, L. Gao, Q. S. Dong, C. Y. Zhao, M. Y. Leng, F. S. Ma, W. X. Liang, L. D. Wang, S. Y. Jin, J. B. Han, L. J. Zhang, J. Etheridge, J. B. Wang, Y. Y. Yan, E. H. Sargent and J. Tang, Nature, 2018, 563, 541 CrossRef CAS.
- B. Yang, X. Mao, F. Hong, W. W. Meng, Y. X. Tang, X. S. Xia, S. Q. Yang, W. Q. Deng and K. L. Han, J. Am. Chem. Soc., 2018, 140, 17001 CrossRef CAS.
- Z. W. Xiao, K.-Z. Du, W. W. Meng, J. B. Wang, D. B. Mitzi and Y. F. Yan, J. Am. Chem. Soc., 2017, 139, 6054 CrossRef CAS.
- F. Locardi, M. Cirignano, D. Baranov, Z. Y. Dang, M. Prato, F. Drago, M. Ferretti, V. Pinchetti, M. Fanciulli, S. Brovelli, L. D. Trizio and L. Manna, J. Am. Chem. Soc., 2018, 140, 12989 CrossRef CAS.
- G. M. Paternò, N. Mishra, A. J. Barker, Z. Y. Dang, G. Lanzani, L. Manna and A. Petrozza, Adv. Funct. Mater., 2018, 29, 1805299 CrossRef.
- H. J. Yang, T. Cai, E. Liu, K. Hills-Kimball, J. B. Gao and O. Chen, Nano Res., 2020, 13, 282 CrossRef CAS.
- Y. Y. Tang, M. L. Liang, B. D. Chang, H. Y. Sun, K. B. Zheng, T. Pullerits and Q. J. Chi, J. Mater. Chem. C, 2019, 7, 3369 RSC.
- CrystalClear, version 1.3.5, Rigaku Corp., The Woodlands, TX, 1999 Search PubMed.
- G. M. Sheldrick, Crystallographic Software Package, SHELXTL, version 5.1, Bruker AXS, Madison, WI, 1998 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- J. Zhang, Y. Yang, H. Deng, U. Farooq, X. K. Yang, J. Khan, J. Tang and H. Song, ACS Nano, 2017, 11, 9294 CrossRef CAS.
- M. N. Tran, I. J. Cleveland and E. S. Aydil, J. Mater. Chem. C, 2020, 8, 10456 RSC.
- M. Y. Leng, Y. Yang, K. Zeng, Z. W. Chen, Z. F. Tan, S. R. Li, J. H. Li, B. Xu, D. B. Li, M. P. Hautzinger, Y. P. Fu, T. Y. Zhai, L. Xu, G. D. Niu, S. Jin and J. Tang, Adv. Funct. Mater., 2018, 28, 1704446 CrossRef.
- M. A. Boles, D. Ling, T. Hyeon and D. V. Talapin, Nat. Mater., 2016, 15, 141 CrossRef CAS.
- B. Yang, J. S. Chen, F. Hong, X. Mao, K. B. Zheng, S. Q. Yang, Y. J. Li, T. Pullerits, W. Q. Deng and K. L. Han, Angew. Chem., Int. Ed., 2017, 56, 12471 CrossRef CAS.
- B. Yang, J. S. Chen, S. Q. Yang, F. Hong, L. Sun, P. G. Han, T. Pullerits, W. Q. Deng and K. L. Han, Angew. Chem., Int. Ed., 2018, 57, 5359 CrossRef CAS.
- B. Yang, F. Hong, J. S. Chen, Y. X. Tang, L. Yang, Y. B. Sang, X. S. Xia, J. W. Guo, H. X. He, S. Q. Yang, W. Q. Deng and K. L. Han, Angew. Chem., Int. Ed., 2019, 58, 2278 CrossRef CAS.
- F. R. Hu, H. C. Zhang, C. Sun, C. Y. Yin, B. H. Lv, C. F. Zhang, W. W. Yu, X. Y. Wang, Y. Zhang and M. Xiao, ACS Nano, 2015, 9, 12410 CrossRef CAS.
- J. Puthenpurayil, O. Hsu-Cheng Cheng, T. Qiao, D. Rossi and D. H. Son, J. Chem. Phys., 2019, 151, 154706 CrossRef.
- T. J. Savenije, C. S. Ponseca Jr, L. Kunneman, M. Abdellah, K. B. Zheng, Y. X. Tian, Q. S. Zhu, S. E. Canton, I. G. Scheblykin, T. Pullerits, A. Yartsev and V. Sundström, J. Phys. Chem. Lett., 2014, 5, 2189 CrossRef CAS.
- S. Tombe, G. Adam, H. Heilbrunner, D. H. Apaydin, C. Ulbricht, N. S. Sariciftci, C. J. Arendse, E. Iwuoha and M. C. Scharber, J. Mater. Chem. C, 2017, 5, 1714 RSC.
- S. Y. Sun, T. Salim, N. Mathews, M. Duchamp, C. Boothroyd, G. C. Xing, T. C. Sum and Y. M. Lam, Energy Environ. Sci., 2014, 7, 399 RSC.
- A. Shinde, R. Gahlaut and S. Mahamuni, J. Phys. Chem. C, 2017, 121, 14872 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tc04394j |
| This journal is © The Royal Society of Chemistry 2021 |