
Role of Ln type in the physical mechanisms of defect mediated luminescence of Li, Ln–SnO2 nanoparticles†
Bogdan
Cojocaru
a,
Claudiu
Colbea
bc,
Daniel
Avram
 b,
Cosmin
Istrate
d,
Laura
Abramiuc
d and
Carmen
Tiseanu
*b
b,
Cosmin
Istrate
d,
Laura
Abramiuc
d and
Carmen
Tiseanu
*b
aDepartment of Chemistry, University of Bucharest, B-dul Regina Elisabeta, Nr. 4-12, 030018 Bucharest, Romania
bNational Institute for Laser, Plasma and Radiation Physics, RO 76900 Bucharest-Magurele, Romania. E-mail: carmen.tiseanu@inflpr.ro
cScientific Center for Optical and Electron Microscopy, ETH Zürich, Zürich, Switzerland
dNational Institute of Materials Physics, 405A Atomistilor Street, 077125 Magurele-Ilfov, Romania
First published on 26th November 2020
Abstract
Doping SnO2 with trivalent lanthanide (Ln) metals aiming at optical applications faces several challenges. The elastic and electrostatic misfit between bulkier Ln activators and Sn host cation induces strain in the lattice as well as defects as a result of charge-compensation. These effects can be partially healed by thermal annealing. However, dopant segregation which occurs above a certain temperature drives quenching of Ln emission. In this work, we explore Li co-doping as a vehicle to improve the luminescence of lanthanide (Eu, Sm, Er, Dy and, Tb) doped SnO2 nanoparticles. In case of substitutional Ln dopants (Eu, Sm and Er), Li enhances significantly the Ln luminescence up to 40–46 times. The luminescence enhancement induced by Li co-doping is explained by an interplay of removal of nearby oxygen vacancies (Eu, Sm), improved Ln doping homogeneity (Er) and, improved crystallinity (Eu, Sm, Er). The improved crystallinity caused by Li co-doping accounts for less than 30% of the total enhancement. In the case of surface Ln dopants (Dy and Tb), Li co-doping does not alter the Ln emission, either in shape or intensity. Only a few Dy dopants succeed to substitute for Sn in the rutile lattice as shown by single-photon counting investigations. Collectively, our results show that the extent of luminescence enhancement induced by Li co-doping depend strongly on the Ln type. In SnO2, the common mechanisms that explain the Li induced enhancement of Ln luminescence in various hosts, either contribute partially (improved crystallization) or do not contribute at all (local structure distortion).
I. Introduction
There has been considerable research over the past decades on the n-type SnO2 semiconductor, due to its broad spectrum of applications such as lithium-ion batteries, gas sensors, sensitized solar cells and, catalysts.1–3 SnO2 is a low cost, chemically and thermally stable wide gap (3.5–3.8 eV![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4,5) semiconductor with a rutile-type tetragonal structure belonging to the P42/mnm space group (lattice parameters a = b = 4.738 Å and c = 3.187 Å). The luminescence of pristine SnO2, observed in the UV to Vis region (350–550 nm) is generally correlated with the presence of intrinsic defects resulting from the various synthesis processes.6,7 Of the various transition metals, Mn2+,6,7 Co2+,8,9 Ni2+10 or Cr3+,11 proved to be successful dopants for tailoring their electrical, optical, and microstructural properties of doped SnO2. Among the lanthanide (Ln) series, trivalent Ce,6,7 Nd,12–14 Eu,15–21 Sm,15,22 Yb,23 and Er24–26 are frequently employed as dopants, aiming at solar cells, sensors, and catalytic applications.1–3 On the other side, alkali metal (Li) doping attracted interest not only for the battery-related applications but also to impart magnetic behavior27 by non-magnetic doping as it may reduce the formation energies of various types of defects.28 In principle, in SnO2 both Li and Ln can act as interstitial (shallow donor level following the conduction band) but, more probably, substitutional (shallow acceptor level above the valence band), inducing oxygen vacancies due to the charge compensation effect.29 For Li and Ln p-type dopants (valence of both dopants is less than 4), the substitution of Sn4+ is accompanied by introducing of excess holes, mainly as doubly ionized oxygen vacancies
4,5) semiconductor with a rutile-type tetragonal structure belonging to the P42/mnm space group (lattice parameters a = b = 4.738 Å and c = 3.187 Å). The luminescence of pristine SnO2, observed in the UV to Vis region (350–550 nm) is generally correlated with the presence of intrinsic defects resulting from the various synthesis processes.6,7 Of the various transition metals, Mn2+,6,7 Co2+,8,9 Ni2+10 or Cr3+,11 proved to be successful dopants for tailoring their electrical, optical, and microstructural properties of doped SnO2. Among the lanthanide (Ln) series, trivalent Ce,6,7 Nd,12–14 Eu,15–21 Sm,15,22 Yb,23 and Er24–26 are frequently employed as dopants, aiming at solar cells, sensors, and catalytic applications.1–3 On the other side, alkali metal (Li) doping attracted interest not only for the battery-related applications but also to impart magnetic behavior27 by non-magnetic doping as it may reduce the formation energies of various types of defects.28 In principle, in SnO2 both Li and Ln can act as interstitial (shallow donor level following the conduction band) but, more probably, substitutional (shallow acceptor level above the valence band), inducing oxygen vacancies due to the charge compensation effect.29 For Li and Ln p-type dopants (valence of both dopants is less than 4), the substitution of Sn4+ is accompanied by introducing of excess holes, mainly as doubly ionized oxygen vacancies 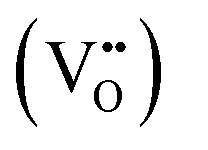 and acceptor-like species
and acceptor-like species 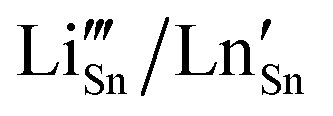 .30
.30
Due to low solubility, increasing the Ln concentration above ca. 1 at% does not enhance the luminescence intensity of Ln–SnO2.31 The trivalent Ln present ionic radii that surpass, on average, that of Sn4+ by 39% (tetrahedral, 6-fold coordination).32 Therefore, Ln induces elastic strain in the lattice along with the oxygen vacancies induced by the valence mismatch. The elastic strain can be partially healed by thermal annealing.15 However, the thermal annealing above a certain temperature induces segregation21,33 which is detrimental for the optical performance.
Here, we exploit the role of Li as a luminescence enhancer of lanthanide-based materials which is much explored in materials science.34,35 The mechanisms responsible for the luminescence enhancement of various Ln based materials are usually assigned to local symmetry distortion or/and improved crystallization,20 but also changes of morphology, reduction of surface OH defects, or sensitization via oxygen vacancies induced by charge compensation.34,35 According to the literature, for the case of SnO2, the emission enhancement was attributed undecidedly to improved crystallization, increased solubility of Ln into the lattice, lower local symmetry around Ln, or enhanced sensitization by SnO2 absorption.20,36 We investigate the physical mechanisms by which Li co-doping enhances the Ln emission in connection with the role of Ln type, location (substitutional or surface centre), and the defects generated by heterovalent doping. A wide range of Ln, such as Eu, Sm, Dy, Tb, and Er, with characteristic f–f luminescence spanning the Vis to NIR (450–1700 nm) range was selected. The Ln, Li–SnO2 nanoparticles were grown by coprecipitation and citrate complexation methods. The structure and morphology were studied by X-ray diffraction and transmission electron microscopy. The electronic and vibrational properties were assessed by diffuse reflectance, X-ray photoelectron spectroscopy, and Raman spectroscopy. Extensive low temperature, site-selective, time-gated luminescence measurements were performed using excitation above band gap of SnO2 and into Ln f–f absorptions into the visible and near-infrared ranges. Using time-gated single-photon counting emission, the emission characteristic of substitutional Dy was identified for the first time in SnO2. The effects of Li co-doping on Ln emission were described in terms of emission intensity, spectral shape, and excited-state dynamics. Finally, structural and luminescence data were correlated to identify the physical processes leading to different luminescence response of Ln to Li co-doping.
II. Results
II. 1. Impact of Li co-doping on structure, morphology, lattice dynamics, and bandgap of Ln–SnO2
Ln, Li–SnO2 nanoparticles were synthesized by both coprecipitation and citrate complexation methods and exhibited different luminescence properties, with the coprecipitated nanoparticles showing superior characteristics. Therefore, the following discussion will focus on those obtained by coprecipitation method. The concentration of Ln was fixed at 1 at% while Li concentration was set at 5, 10, and 15 at%. All samples were annealed at 800 °C to account for Li volatility37 and labeled through the text as Ln, xLi–SnO2. Additional Li free, Ln–SnO2 samples annealed at a higher temperature of 900, 1000, and 1100 °C were characterized by X-ray diffraction. Of these, samples annealed at 1000 °C, which showed similar crystallite size to Ln, 15Li–SnO2 were further characterized in detail and labeled through the text as reference samples or Ln–SnO2 (R).
Fig. 1 illustrates the effects of Li addition on the structural and morphology of selected Ln, xLi–SnO2 (Ln![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Eu, partially Er) determined by XRD (see also Fig. S1, ESI†), Raman spectroscopy, Diffuse Reflectance Infrared Fourier Transform Spectroscopy (DRIFT) and transmission electron microscopy (TEM). In general, similar properties were found for all Ln investigated. Fig. 1a illustrates the X-ray diffraction (XRD) patterns of Eu,xLi–SnO2 cell parameters and volume, lattice strain, D-spacing, and crystallite size are summarized in Table S1 (ESI†). The X-ray diffraction patterns indicate the presence of pure rutile phase (space group P42/mnm – JCPDS card 01-079-5607), irrespective of the Ln type and Li content with no observation of impurity phases, such as stannate (Li2SnO3, JCPDS-310761) or Ln stannate pyrochlore (Ln2Sn2O7, JCPDS-880457). Li co-doping leads to narrowing of XRD patterns and shifting towards lower angles, which may indicate the substitutional doping of the octahedral coordinated Sn4+ (ionic radius of 0.69 Å) by the bulkier Li+ (ionic radius of 0.76 Å).27 The calculated lattice volumes indicate both slightly smaller and higher values with Li addition than those of undoped SnO2 (71.59 ± 0.02) Å3. A definite trend cannot be advanced. Crystallite size of Li free Ln–SnO2 calcined at 1000 °C (R sample) come close to values measured for Ln, 10/15Li samples (Table S1, ESI†). Fig. 1b shows the Raman spectra of Eu,0Li–SnO2, Eu,15Li–SnO2 as well the reference, R, sample. Li free sample presents a more intense broadband at 570 cm−1, which is typically attributed to surface effects (small particle size, around 11 nm estimated from XRD, Table S1, ESI†). All three samples present phonon mode characteristic of rutile SnO2 at 270–302, 474–510 (Eg), 626–641 (A1g), and 680–700 and 768–782 cm−1 (B2g) in good agreement with literature.38 It is evident that the phonon bands narrow with Li addition as a result of improved crystallization determined by XRD and replicate almost entirely the spectrum of reference (R) sample. However, in Eu,15Li sample, weak phonon bands related to Li co-dopant can be observed at 157 and 193 cm−1 (tetrahedral coordinated Li+ ions arising from the presence of LiOH39). Phonon bands at 1091 cm−1 and 587 cm−1 are associated with Li2CO3 (symmetric stretching vibrations).40
Eu, partially Er) determined by XRD (see also Fig. S1, ESI†), Raman spectroscopy, Diffuse Reflectance Infrared Fourier Transform Spectroscopy (DRIFT) and transmission electron microscopy (TEM). In general, similar properties were found for all Ln investigated. Fig. 1a illustrates the X-ray diffraction (XRD) patterns of Eu,xLi–SnO2 cell parameters and volume, lattice strain, D-spacing, and crystallite size are summarized in Table S1 (ESI†). The X-ray diffraction patterns indicate the presence of pure rutile phase (space group P42/mnm – JCPDS card 01-079-5607), irrespective of the Ln type and Li content with no observation of impurity phases, such as stannate (Li2SnO3, JCPDS-310761) or Ln stannate pyrochlore (Ln2Sn2O7, JCPDS-880457). Li co-doping leads to narrowing of XRD patterns and shifting towards lower angles, which may indicate the substitutional doping of the octahedral coordinated Sn4+ (ionic radius of 0.69 Å) by the bulkier Li+ (ionic radius of 0.76 Å).27 The calculated lattice volumes indicate both slightly smaller and higher values with Li addition than those of undoped SnO2 (71.59 ± 0.02) Å3. A definite trend cannot be advanced. Crystallite size of Li free Ln–SnO2 calcined at 1000 °C (R sample) come close to values measured for Ln, 10/15Li samples (Table S1, ESI†). Fig. 1b shows the Raman spectra of Eu,0Li–SnO2, Eu,15Li–SnO2 as well the reference, R, sample. Li free sample presents a more intense broadband at 570 cm−1, which is typically attributed to surface effects (small particle size, around 11 nm estimated from XRD, Table S1, ESI†). All three samples present phonon mode characteristic of rutile SnO2 at 270–302, 474–510 (Eg), 626–641 (A1g), and 680–700 and 768–782 cm−1 (B2g) in good agreement with literature.38 It is evident that the phonon bands narrow with Li addition as a result of improved crystallization determined by XRD and replicate almost entirely the spectrum of reference (R) sample. However, in Eu,15Li sample, weak phonon bands related to Li co-dopant can be observed at 157 and 193 cm−1 (tetrahedral coordinated Li+ ions arising from the presence of LiOH39). Phonon bands at 1091 cm−1 and 587 cm−1 are associated with Li2CO3 (symmetric stretching vibrations).40
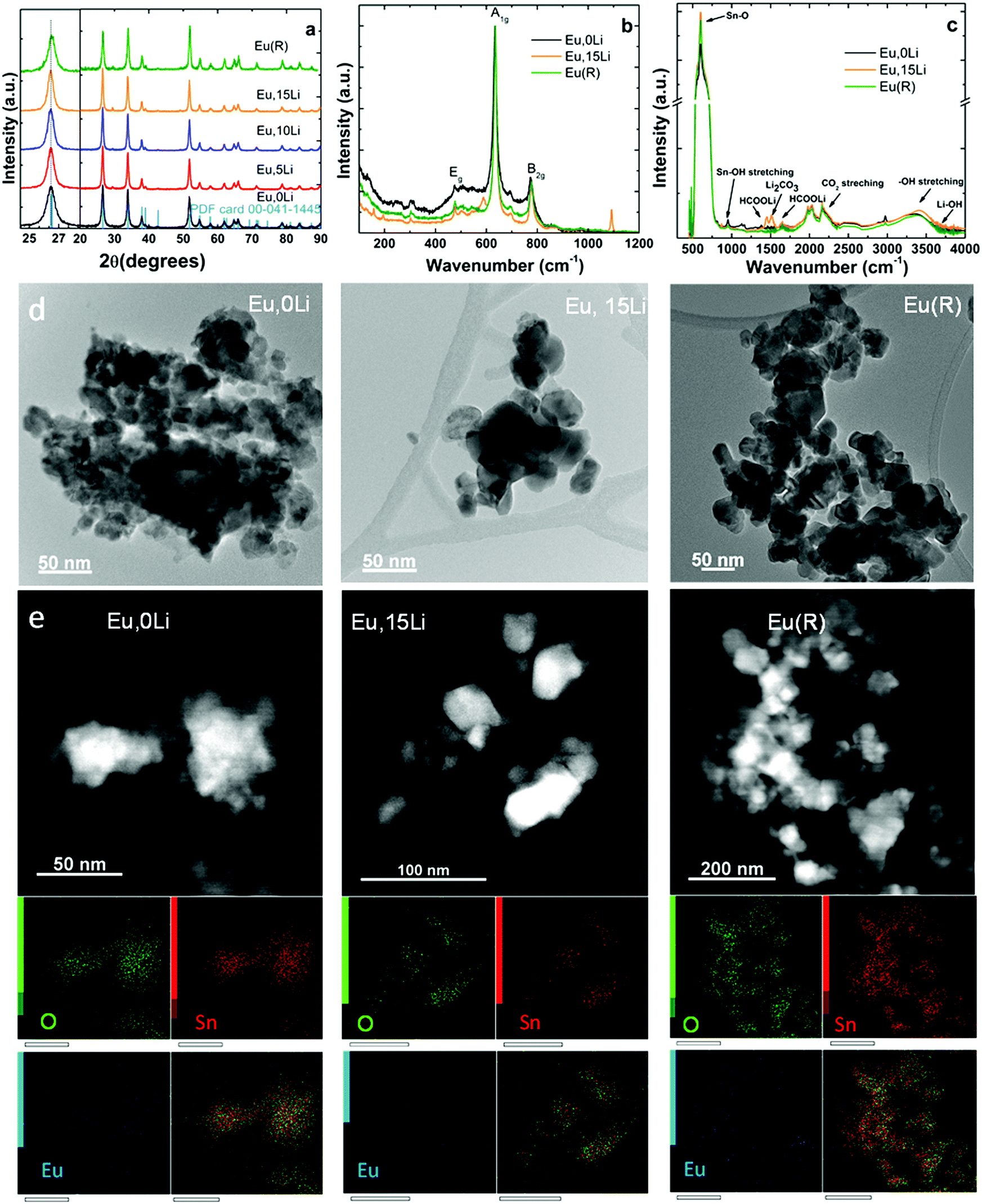 | ||
| Fig. 1 Effects of Li co-doping on structure and morphology of Eu–SnO2. For comparison, results obtained with Eu–SnO2 annealed at 1000 °C and labeled as reference (R) sample were included (see text). (a) XRD patterns, (b) Raman spectra (c) infrared Fourier transform (DRIFT) spectra; (d) TEM images and (e) EDX mapping. | ||
According to DRIFT spectra illustrated in Fig. 1c, the antisymmetric stretching vibrations of Sn–O terminal bond, antisymmetric/symmetric stretching vibrations of Sn–O lattice at 521, 614 and 701 cm−141,42 are increasing with Li content, consistent with improved crystallization (see also Fig. 1a and Table S1, ESI†). The band peaked at 946 cm−1 is characteristic of the asymmetric stretching vibration of Sn–OH terminal bond.21,43 Several overtones and combinations of lattice vibrations were observed in the spectral region between 1350–1650 cm−1.21 Bands at 2342 and 2362 cm−1 are characteristic of the in-plane bending of Sn–OH terminal bond. A noticeable enhancement of OH stretching modes in the range of 2700–3674 cm−1 region is observed with the increase of Li content and the increase of annealing temperature (R sample).21 The presence of Li is signaled by weak bands around 3690 cm−1 (LiOH), 1380 and 1630 cm−1 (HCOOLi), and 1518 cm−1 (Li2CO344,45).
TEM images of Eu,0Li–SnO2, Eu,15Li–SnO2, and Eu–SnO2(R) show mildly agglomerated nanoparticles whose irregular shape remained unperturbed by Li addition (Fig. 1d). The nanoparticle size distribution across the three sample types displays size distributions as 10–24, 20–50, and 18–55 nm, respectively. On one side, the data show the superior crystallinity achieved by Li co-doping in agreement with XRD patterns and, on the other side, the crystallinity similarity between Li co-doped SnO2 and reference (R) sample. EDX mapping images taken on Eu,0Li–SnO2, Eu,15Li–SnO2, and Eu–SnO2(R) show a homogenous distribution of Eu dopant across nanoparticles within the detector's detection limit (Fig. 1e).
The effects of Li addition on the bandgap of Ln–SnO2 were studied by diffuse reflectance and exemplified for Er,xLi–SnO2 in Fig. S2 (ESI†). With the increase of Li concentration the bandgap of Er,xLi–SnO2 enlarges from 2.41 (0Li) to 2.65 eV (15Li) (Table S1, ESI†). This may be the consequence of the complex balance between the increase of crystallite size (which increases from 14 (0Li) to 23 nm (15Li) according to XRD data), defect levels introduced by monovalent Li acceptor28,46 and improved substitutional incorporation of Ln onto Sn lattice sites.
Fig. 2 presents X-ray photoemission spectroscopy (XPS) spectra corresponding to Sn 3d, O 1s, and Eu 3d levels of Eu,0Li–SnO2; Eu,15Li–SnO2 and Eu–SnO2(R). Additional details regarding the deconvolution method are gathered in Supplementary Note S1, ESI.† The energy positions of the lower binding energy line (Note S1, ESI†) is high enough and can be attributed to Eu3+, being close to 1135.6 eV for Eu2O3.47
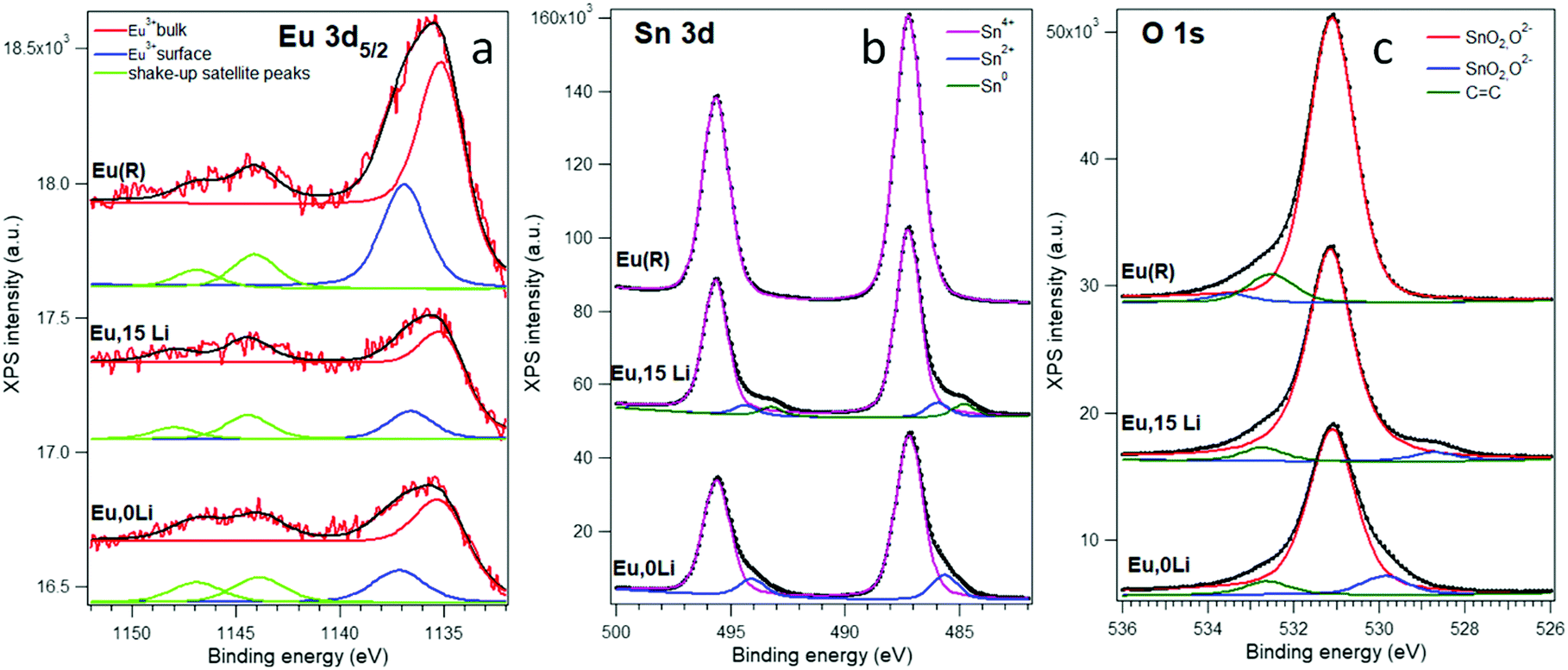 | ||
| Fig. 2 X-ray photoelectron spectra of Eu,0Li–SnO2, Eu,15Li–SnO2 and Eu–SnO2(R). (a) Eu 3d5/2 electron distribution curves (EDCs); (b) Sn 3d EDCs; (c) O 1s EDCs. Spectra are ‘deconvoluted’ with Voigt lineshapes and individual inelastic backgrounds associated with each different chemical state. | ||
The surface Eu3+ has a binding energy 2.0 ± 0.2 eV higher, whose origin may be traced in downward band bending near-surface48 or to under-coordination with oxygen.49 Of all three Eu 3d5/2 spectra analyzed, the spectrum of Eu,15Li–SnO2 sample has the lowest surface/volume ratio, of 44% close to 46% measured Eu,0Li–SnO2. In contrast, the ratio reaches the greatest value for the reference sample, of 57%, suggesting Eu segregation onto SnO2 surface with an increase of annealing temperature.15 However, we should remark that these ratios are difficult to be translated in a real proportion of atoms in the absence of a detailed mechanism for inelastic scattering, giving rise to the background. The Sn 3d and O 1s spectra (Fig. 2b and c) may be interpreted as being dominated by the signal of Sn(IV) oxide, or SnO2.47 The spectra obtained for Eu,0Li, and Eu,15Li exhibit low binding energy peaks both for Sn 3d and O 1s, which are well in the range of the reported data for Sn2+ oxide, SnO.47 The Sn 3d spectrum for Eu,15Li present an additional peak of even lower binding energy, 484.8 eV for Sn 3d5/2, which frames nicely within the actual data for metal Sn. The integral amplitude of this peak is about 7% of the main peak, due to Sn4+ oxide, while the amplitude of the Sn2+ oxide peak is about 8% of the main peak. In other words, co-doping Eu–SnO2 with 15% Li promotes about 8% of Sn as a suboxide and 7% of the Sn as Sn metal. Also, the low binding energy of oxygen represents about 4% of the main peak; considering the stoichiometry, this again points to about 8% of SnO embedded in SnO2. With the exception of the highest binding energy O 1s structures at around 532–533 eV which are due to carbonyl or carboxylic contaminants,49 the spectra for the Eu(R) sample exhibit just structures due to SnO2. Finally, the XPS survey spectra of Eu,0Li–SnO2, Eu,15Li–SnO2, and Eu–SnO2(R) show only the C, Sn, O, and Eu elements sustaining the purity of the samples (Fig. S3, ESI†).
II. 2. Impact of Li co-doping on Ln luminescence
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) Eu.
Fig. 3 summarizes the Li effects on Eu luminescence (PL) properties, such as emission shape, intensity, excitation path and excited-state dynamics. As shown in Fig. 3a, Li addition leads to a significant enhancement, of Eu luminescence, by a factor of 46 (10%Li), as compared to Li free, Eu–SnO2.
Eu.
Fig. 3 summarizes the Li effects on Eu luminescence (PL) properties, such as emission shape, intensity, excitation path and excited-state dynamics. As shown in Fig. 3a, Li addition leads to a significant enhancement, of Eu luminescence, by a factor of 46 (10%Li), as compared to Li free, Eu–SnO2.
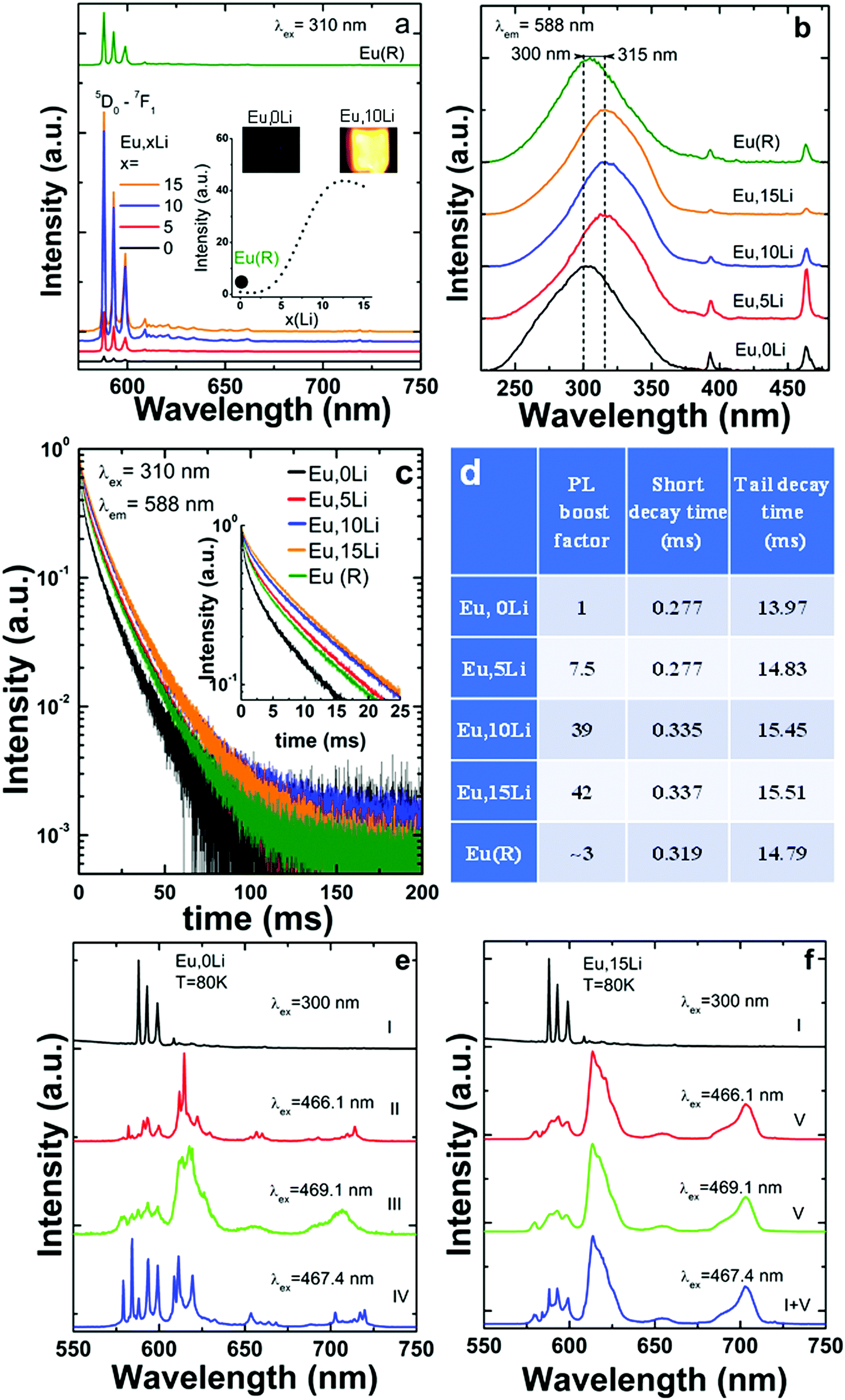 | ||
| Fig. 3 Effects of Li addition on the luminescence properties of Eu–SnO2:(a) luminescence shape and intensity; (b) excitation spectra, (c) emission decays and estimated average lifetimes (d). In (e) and (f) are compared the characteristic emission of Eu centres in Li-free and Li (15%), Eu–SnO2 at low temperature (80 K). The emission/excitation spectra and decays were measured using identical experimental conditions. The digital images in (a) were taken under ambient light. In (d) the short and tail decay times were determined by monoexponential fitting in the 0–5 ms and 50–100 (200) ms, respectively. | ||
Since the comparison of the emission intensity is tricky due to potential variability in the measurement conditions, we took considerable as detailed in the Supplementary Note S2, ESI.† The emission enhancement factors were determined using broad excitation into the maximum of SnO2 absorption (300–315 nm). It is established in the literature that SnO2 absorption represents an efficient sensitizer for the substitutional Ln dopants,50 such as Eu15–21,51 (Fig. 3b) but also for Sm,15,22 Nd12–14 and Er.24–26 Upon excitation into SnO2 absorption, the emission decays of Eu,xLi series measured at 588 nm were compared in Fig. 3c. All decays are strongly nonexponential and display a quasi-persistent behavior with time scales extending up to 200 ms, which are slightly prolonged with Li addition. The long-lived emission was previously assimilated to an atypical persistent emission process suggested to be thermally activated via the co-existence of uniform and exponential distributions in trap depths.16 As shown in Fig. 3d, Li addition increased both short and tail decay times.
As some of us reported recently,15 Eu (also Sm) presents a complex multisite distribution in SnO2, that includes besides the substitutional isolated center (without near-neighbour (NN) local charge - compensation), several Ln-defects associates (with NN local-charge compensation) and a surface center. The characteristic emission of isolated substitutional Eu termed (centre I or C2h) represented in Fig. 3a was identified several decades ago.52,53 Its emission is exclusively excited via SnO2 absorption and characterized by relatively strong three 5D0–7F1 lines around 590 nm consistent with C2h inversion local symmetry. At least three Ln-defect associates, termed as centers II–IV, were identified in ref. 15 with significant shorter average lifetimes and altered emission shapes characteristic of non-inversion local symmetry. A much broader and shorter-lived emission was assigned to Eu surface type center, termed as center V. Since Eu (and Sm distribution) is also confirmed in the present study, we strengthen that such multisite distribution is intrinsic, being independent of the synthesis route (hydrothermal, sol–gel, coprecipitation).
The effects of Li co-doping on the emission properties (intensity and shape) characteristic of each Eu center, were assessed by using similar emission/excitation conditions and illustrated in Fig. 3d and e. We found that Li addition progressively increased the emission of C2h centre I at the expense of the Eu-defects associates (centers II–IV) (Fig. S4, ESI†). At Li concentration of 15%, the characteristic emission of Eu centres II–IV was significantly quenched; as such, the total Eu emission was partitioned between that of C2h (major) and surface center (minor). A similar trend was observed for Sm that shows a luminescence enhancement factor of grossly of up to 40. The emission of Li free, Sm–SnO2 sample was substantially weaker than that of Eu homologue, a trend observed previously for hydrothermal synthesis route.15 Therefore, only selected spectra were included in Fig. S4 (ESI†). Using extensive excitation into SnO2 band gap and Eu/Sm f–f absorptions, we also confirmed that Li addition did not induce additional Eu/Sm centres, others than those identified for Li free samples (Fig. S4 and S5, ESI†).
Er.
As shown in Fig. 3a, following excitation into maximum absorption of SnO2 absorption around 300 nm, only the near-infrared (NIR) emission of Er corresponding to Er 4I13/2–4I15/2 transition (around 1530 nm) was observed, irrespective of Li concentration. Switching excitation from SnO2 based absorption to Er f–f absorptions in the Vis and NIR regions under down-conversion (475 nm) and upconversion (800, 980, and 1530 nm) excitation modes did not induce any observable emission. The observation of only 4I13/2–4I15/2 based emission at 1530 nm, which is of predominant magnetic dipole character (vacuum MD spontaneous emission rate around 10.17 s−154) confirmed that Er substituted Sn exclusively in the centrosymmetric C2h sites without locally charge compensation. It is interesting to note that a similar unique Er center was observed for nanoparticles grown by solvothermal,24 hydrothermal,55 coprecipitation26 and sol–gel56 methods. This suggests that the exclusive location onto C2h Sn sites without defects nearby represents an intrinsic doping property of Er. Li addition enhanced the Er emission, by a maximum factor of 44 for 15% Li. Similar to Eu, the excitation spectra shifted towards greater wavelengths with Li addition57 (Fig. 4b) in opposition to the trend evidenced by the diffuse reflectance spectra (Fig. S2 and Table S1, ESI†). The different trends are explained by the fact that the diffuse reflectance measures the total absorption while the excitation spectra measure only the absorption that contributes to Ln luminescence.
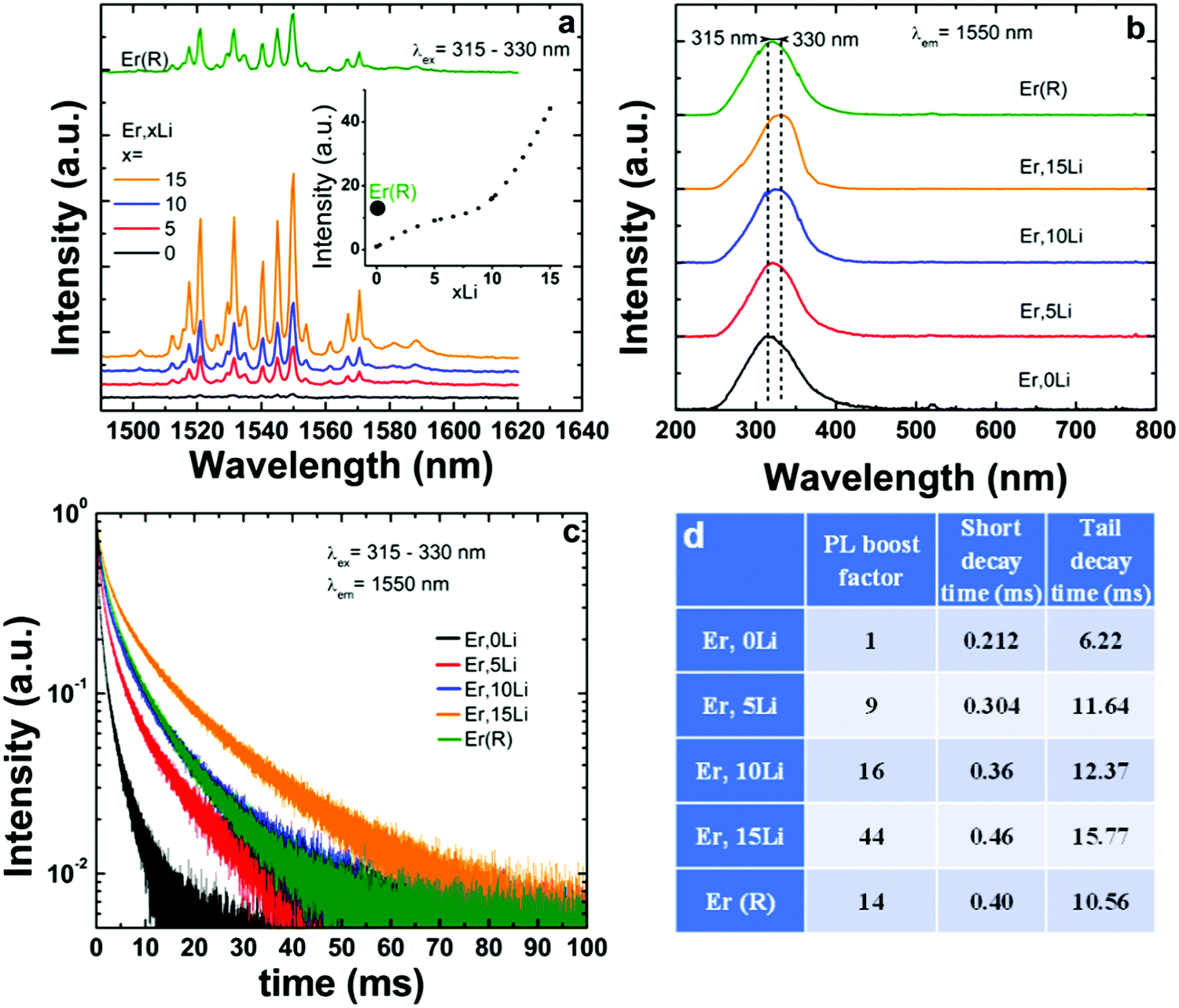 | ||
| Fig. 4 Effects of Li addition on the luminescence properties of Er–SnO2 (a) luminescence shape and intensity; (b) excitation spectra; (c) emission decays with tabulated average lifetimes in (d). In (d) the short and tail decay times were determined by monoexponential fitting in the 0–5 ms and 10/50–100 ms ranges, respectively. | ||
Upon Li co-doping, Er emission decays measured around 1530 nm suffered a more significant lengthening compared to those of Eu: both short and tail decay times were increased by more than 100% from ∼0.21 to 0.46 ms and from 6.2 to 15.8 ms for 0Li and 15Li, respectively (Fig. 4c and Table in Fig. 4d).
Dy and Tb.
In the Li free samples, the luminescence shape and dynamics of Dy and Tb were consistent with surface location, in line with previous report.15 As shown in Fig. 5a, the Dy and Tb emissions are broad (FWHM around 20 nm), are exclusively excited by f–f absorptions, and present short-lived timescales of few hundreds of microseconds. Li addition did not change Dy or Tb emission properties (emission intensity and shape, dynamics) irrespective of Li concentration. By use of time-gated single-photon detection, we could, however, separate for the first time, to our knowledge, the Dy emission characteristic of the substitutional isolated center or C2h center (orange lined spectrum in Fig. 5a). In these measurements, a single photon is counted per pixel of an iCCD camera using a long time delay of up to 2.5 ms. This way, long-lived weak emission of substitutional Dy center was temporally discriminated against the shorter-lived, but much more intense emission of surface Dy. As shown in Fig. 5a, the Dy emission in C2h inversion sites displays a relative intense narrow emission at 770 nm corresponding to 4F9/2–6H9/2, 6F11/2 transition of predominant magnetic dipole character.54 For comparison, also included in Fig. 5a are the characteristic emission spectra of Dy in the inversion symmetry sites of Y2O3 (S6/C3i) and CeO2 (Oh) reported recently by some of us.58,59 Finally, Fig. 5b illustrates the Dy 3d and 4d XPS spectra of Dy,0Li–SnO2, and Dy,15Li–SnO2. In the limit of the instrumental resolution, the 3d spectrum of Dy,15Li sample displays a slightly higher surface/volume ratio of 2.3 compared to 2.2 for Dy,0Li.
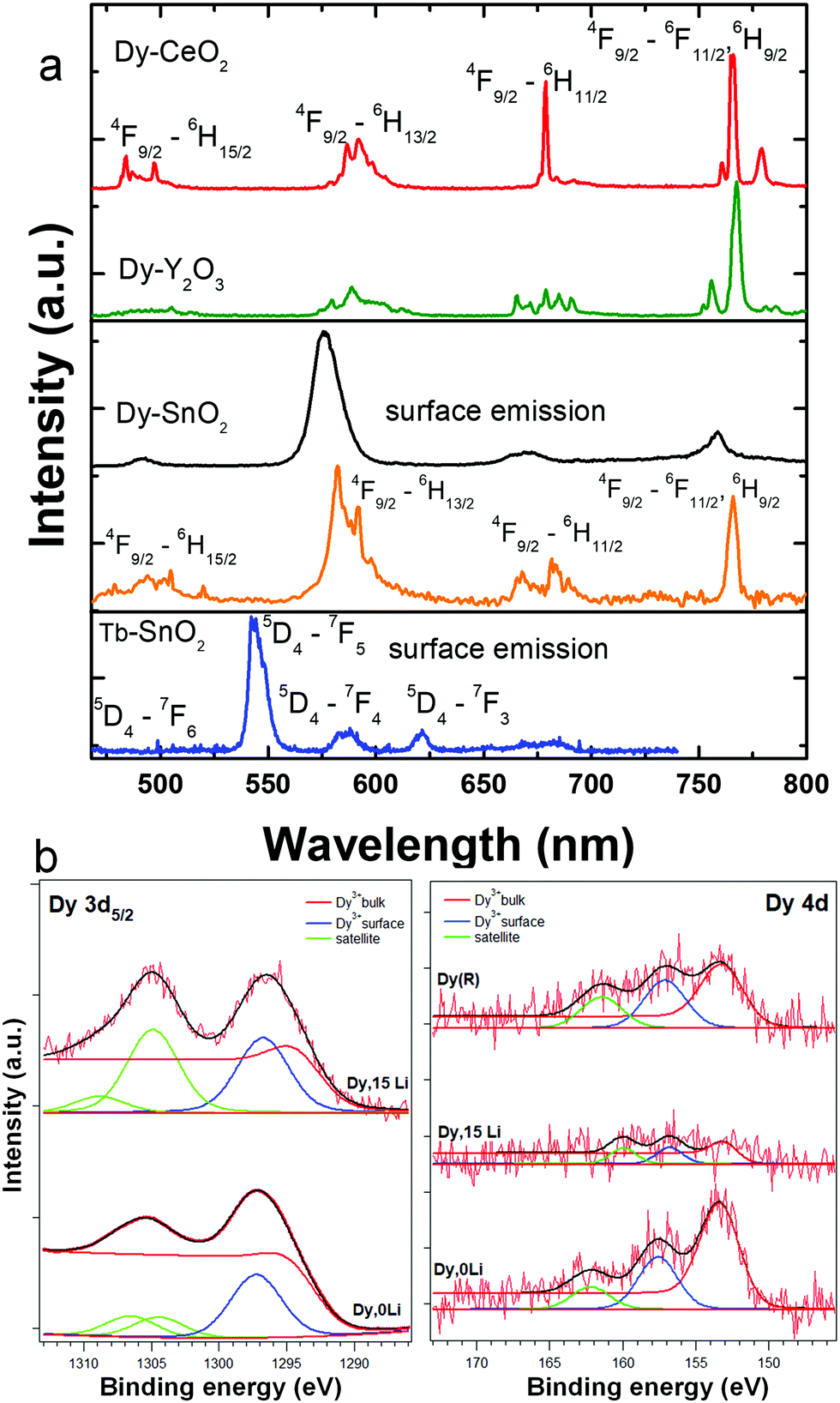 | ||
| Fig. 5 (a) Luminescence spectra of surface Dy–SnO2 (black line) and Tb–SnO2 (blue line). Li co-doping has no effects on Dy/Tb emission intensity and shape. With orange line is illustrated the luminescence spectrum of isolated substitutional (C2h) Dy center. For comparison only, the luminescence spectra of Dy in the inversion symmetry sites (S6, Y2O3) and (Oh, CeO2) are also included. (b) Dy 3d and 4d XPS spectra of Dy,0Li–SnO2 and Dy,15Li–SnO2. | ||
III. Discussion
Li induced improved crystallization or/and local structure distortion around Ln activator are the most frequently invoked mechanisms to explain the luminescence enhancement in Ln based materials34,35,60 We, therefore, assess in more detail to what extent the two mechanisms contribute to the luminescence enhancement observed for Eu, Sm, and Er substitutional dopants.Improved crystallinity
Li co-doping of Ln–SnO2 increases the crystallite/particle size up to 50–60%, from 10–11 nm for Li free to 16–18 nm for Ln, 15Li–SnO2, irrespective of Ln type (Fig. 1 and Table S1, ESI†). It is well-known that a greater particle size enhances the luminescence by reducing the number of surface Ln exposed to efficient non-radiative emission quenching.61 To mask the particle size effects on the Ln luminescence, we have structurally and optically investigated Li free Ln–SnO2 thermally treated at 1000 °C, which was labeled through the text and figures as reference (R) sample. The annealing temperature was selected among three values of 900, 1000, and 1100 °C by considering the closeness in particle size to Ln,10/15Li–SnO2. According to Fig. 1, besides particle size, the reference sample also has a similar morphology, enhancement trend of OH band (Fig. 1), and vibrational properties. The emission enhancement factors measured with these reference samples were estimated at around 4 (Eu), 19 (Sm) and 14 (Er), definitely smaller than 44, 40, or 46 measured with Eu, Sm or Er,10/15Li–SnO2. Therefore, Li induced improved crystallinity cannot stand as a major mechanism for the emission enhancement. In fact, the emission enhancement by simply increase of annealing temperature/(or particle size) of Ln–SnO2 is not a successful strategy due to emission quenching by Ln segregation onto the surface.15,33,62 This is also supported here by the XPS data (Fig. 2) that show a significant increase of surface Eu in R sample (57%) compared to Eu,15Li–SnO2 (44%) and Eu,0Li–SnO2 (46%) samples.21Distortion of local symmetry
Li co-doping can distort the local symmetry at the Ln site, as a result of the additional oxygen vacancies generated by charge-compensation. These defects can locate nearby the Ln, distorting, thus the local crystal-field. While the elucidation of the can complex defects chemistry is outside the scope of this study, the Ln luminescence can give significant information on Ln-defect interactions that are not reachable with other techniques. When Ln substitute for Sn C2h inversion symmetry sites with no defect nearby (isolated substitutional or C2h center), the emission has a predominant magnetic dipolar nature, as the electric dipole transitions are forbidden.52,53,63Scheme 1 includes simplified electronic levels of Eu, Sm, Dy, and Er with the magnetic dipole emission transitions highlighted. Any deviation from the inversion symmetry would alter the f–f radiative and non-radiative transition probabilities,64,65 reflecting in both the alteration of the emission shapes and acceleration of the emission decays. Our extensive site-selective, time-gated luminescence investigations did not reveal additional Ln centeres induced by Li while the emission shapes associated with C2h centeres were fully preserved. In the case of Eu, the peak values (588.24, 593.04, and 599.08 nm), their relative intensities (1/0.76/0.59) of the three 5D0–7F1 Eu Stark emissions and asymmetry ratio (around 0.17) (Fig. 3a) were identical in Li free and Eu, Li samples, irrespective of Li concentration. The emissions decays were slightly (Eu, Fig. 3c and d) or more significantly lengthened (Er, Fig. 4c and d). Preservation of the emission shapes together with lack of decays acceleration exclude Li induced local structure distortion as a possible mechanism for Ln emission enhancement. Up to a certain concentration, Li incorporates into Sn lattice site generating three 3 holes localized on O atoms coordinated to the dopant being also able to trap an extra hole.29 In the detailed center to center comparison made for Li free Eu and Eu,15Li in Fig. 3e and f, Li removes the vacancies from the nearest-neighbor (NN) position of Eu. The luminescence enhancement appears to be mainly the effect of an increased concentration of substitutional isolated centres at the expense of Eu – defect associates. The Li induced oxygen defects and their distribution around the Ln activator play a distinct role in phase changing oxides with tetravalent cations. As such, Li co-doping induced complete tetragonal to monoclinic (ZrO2,66) or anatase to rutile (TiO267) phase transitions. The mechanism of luminescence enhancement cannot be translated from Eu(and likely Sm) to Er. For Er, only a unique substitutional isolated center with no Er-defects associates was observed which is markedly different from the case of Eu or Sm.15 Also, different from Eu, Li induced emission enhancement does not saturate with Li concentration (Fig. 4a). Er is the smallest Ln investigated as dopant, with an ionic radius of 0.89 Å (compared to 0.947 Å for Eu) and thus closest to the ionic radius of Sn4+ (0. 0.69 Å, all in octahedral coordination32). We can only speculate that smaller Ln repels defects from the nearest-neighbor (NN) position, which would be the opposite case established for Ln–CeO2.58,68 The emission enhancement by Li co-doping relates thus to a more homogenous distribution of Er dopants onto Sn sites25,69 which decreases the emission quenching effect70 and lengthens the decays (Fig. 4c and d). We note that Er–SnO2 represents a rare example of broad UV to NIR (300 to 1500 nm) downshifting material. Therefore, Li induced emission enhancement of Er–SnO2 may be attractive for photon down shifter layers in solar cells which were recently demonstrated for Nd–SnO2.12,14,71
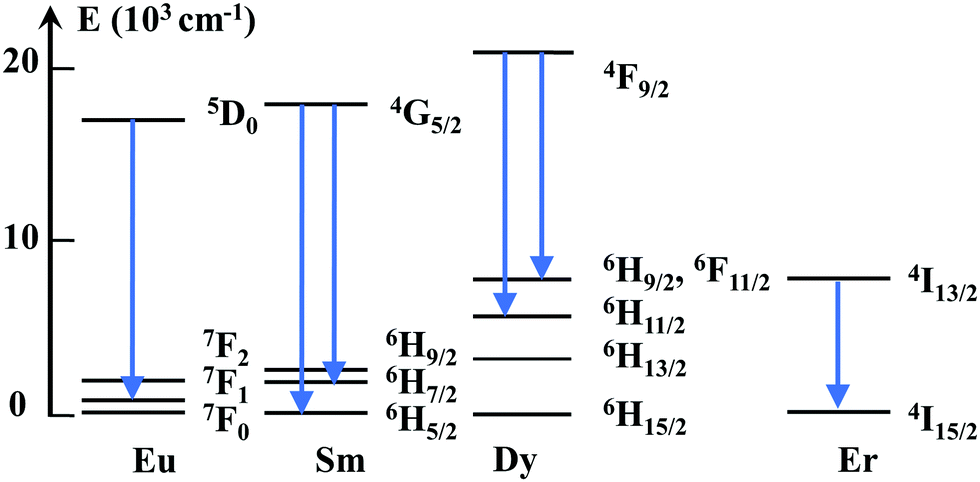 | ||
| Scheme 1 Simplified energy level diagram indicating the (magnetic dipole) emission transitions of Eu, Sm, Dy and Er allowed in the C2h inversion local symmetry. | ||
Li co-doping did not enhance the Dy or Tb solubility into the lattice even though it improves the crystallinity (Fig. S1, ESI†). This was also consistent with rather similar surface fractions of Dy in Li free, Dy–SnO2, and Dy,15Li–SnO2 derived from the XPS data (Fig. 5b). Few Dy succeeded in substituting for Sn as substitutional isolated center as shown by time-gated single-photon counting luminescence in Fig. 5a. Obviously, due to low emission intensity, host sensitization phenomenon, or “quasi persistent” emission decay shared by Eu, Sm, and Er substitutional dopants, could not be confirmed. The weak to absent luminescence related to substitutional Dy and Tb in SnO2 compared to Eu (Sm) and Er may be explained in the framework of the charge-trapping model.50,72 As such, the energy differences between Ln ground and the metastable energy levels relative to the band edges of SnO2 determine significantly the sensitization efficiency of Ln luminescence. In addition, for Tb and Dy, the electron trapping site is susceptible to autoionization which reduces the probability of electron–hole carrier recombination and, consequently, the quantum yield of the luminescence.50
IV. Conclusions
We present a first investigation on the effects induced by Li co-doping on the luminescence performance of lanthanide (LnEu, Sm, Er, Dy, and Tb) doped SnO2 nanoparticles. To this aim, X-ray diffraction, transmission electron microscopy, diffuse reflectance, X-ray photoelectron, and Raman spectroscopies, as well as extensive low temperature, site-selective, time-gated luminescence with excitation above band gap of SnO2 or into Ln f–f absorptions, were performed. The correlation of structural and luminescence responses to Li co-doping reveals a strong selectivity of the luminescence response on the Ln type. The Ln emission is enhanced up to 46 for the substitutional Eu, Sm, and Er dopants but remains unperturbed for the surface Dy and Tb dopants. The luminescence enhancement induced by Li is explained by an interplay of removal of nearby oxygen vacancies (Eu, Sm), improved Ln doping homogeneity (Er) and, improved crystallinity (Eu, Sm, Er).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
DA, BC, CT and CC acknowledge the financial support from CNCS-UEFISCDI, project PN-III-P4-ID-PCE-2016-0305, contract PCE 67/2017. CI acknowledges the Romanian Ministry of Research and Innovation through the Core Program PN19 (contract no. 21/08.02.2019) for financial support. LA is granted by a post-doctoral project PN-III-P1-1.1-PD-2019-0763 funded by the Romanian Ministry of Education and Research. The authors thank Dr Cristi Mihailescu for XPS measurements. The authors also thank Dr Cristian – Mihail Teodorescu for the interpretation of XPS data.References
- L. Xiong, Y. Guo, J. Wen, H. Liu, G. Yang, P. Qin and G. Fang, Adv. Funct. Mater., 2018, 28, 1802757 CrossRef.
- F. Zoller, D. Böhm, T. Bein and D. Fattakhova-Rohlfing, ChemSusChem, 2019, 12, 4140–4159 CrossRef CAS.
- H. Wang and A. Rogach, Chem. Mater., 2014, 26, 123–133 CrossRef CAS.
- H. Wang, F. Sun, Y. Zhang, L. Li, H. Chen, Q. Wu and C. Y. Jimmy, J. Mater. Chem., 2010, 20, 5641–5645 RSC.
- D. Chu, J. Mo, Q. Peng, Y. Zhang, Y. Wei, Z. Zhuang and Y. Li, ChemCatChem, 2011, 3, 371–377 CrossRef CAS.
- F. Gu, S. F. Wang, M. K. Lü, G. J. Zhou, D. Xu and D. R. Yuan, J. Phys. Chem. B, 2004, 108, 8119–8123 CrossRef CAS.
- C. Zhang and J. Lin, Chem. Soc. Rev., 2012, 41, 7938–7961 RSC.
- L. Fang, X. Zu, Z. Li, S. Zhu, C. Liu, L. Wang and F. Gao, J. Mater. Sci.: Mater. Electron., 2008, 19, 868–874 CrossRef CAS.
- S. Roy, A. Joshi, S. Chatterjee and A. Ghosh, Nanoscale, 2018, 10, 10664–10682 RSC.
- A. Ahmed, S. Muhamed, M. Singla, S. Tabassum, A. Naqvi and A. Azam, J. Lumin., 2011, 131, 1–6 CrossRef CAS.
- M. Garcia-Tecedor, D. Maestre, A. Cremades and J. Piqueras, J. Phys. Chem. C, 2016, 120, 22028–22034 CrossRef CAS.
- K. Bouras, G. Schmerber, H. Rinnert, D. Aureau, H. Park, G. Ferblantier, S. Colis, T. Fix, C. Park, W. Kim, A. Dinia and A. Slaoui, Sol. Energy Mater. Sol. Cells, 2016, 145, 134–141 CrossRef CAS.
- K. Bouras, G. Schmerber, D. Aureau, H. Rinnert, G. Ferblantier, T. Fix, S. Colis, P. Bazylewski, B. Leedahl, A. Etcheberry, G. Chang, A. Dinia and A. Slaoui, RSC Adv., 2016, 6, 67157–67165 RSC.
- K. Bouras, J. L. Rehspringer, G. Schmerber, H. Rinnert, S. Colis, G. Ferblantier, M. Balestrieri, D. Ihiawakrim, A. Dinia and A. Slaoui, J. Mater. Chem. C, 2014, 2, 8235–8243 RSC.
- B. Cojocaru, D. Avram, V. Kessler, V. Parvulescu, G. Seisenbaeva and C. Tiseanu, Sci. Rep., 2017, 7, 1–14 CrossRef.
- J. Kong, W. Zheng, Y. Liu, R. Li, E. Ma, H. Zhu and X. Chen, Nanoscale, 2015, 7, 11048–11054 RSC.
- V. Kiisk, T. Kangur, M. Paalo, T. Tätte, S. Lange, S. Pikker and I. Sildos, Mater. Chem. Phys., 2011, 130, 293–298 CrossRef CAS.
- C. Ma, M. Brik, V. Kiisk, T. Kangur and I. Sildos, J. Alloys Compd., 2011, 509, 3441–3451 CrossRef CAS.
- E. A. Morais, L. V. Scalvi, A. Tabata, J. B. De Oliveira and S. J. Ribeiro, J. Mater. Sci., 2008, 43, 345–349 CrossRef CAS.
- H. Zhang, X. Fu, S. Niu, G. Sun and Q. Xin, J. Lumin., 2005, 115, 7–12 CrossRef CAS.
- T. Moon, S. Hwang, D. Jung, D. Son, C. Kim, J. Kim, M. Kang and B. Park, J. Phys. Chem. C, 2007, 111, 4164–4167 CrossRef CAS.
- L. Singh, M. Luwang and S. Srivastava, New J. Chem., 2014, 38, 115–121 RSC.
- K. Bouras, G. Schmerber, D. Aureau, H. Rinnert, J. L. Rehspringer, D. Ihiawakrim, A. Dinia, A. Slaoui and S. Colis, Phys. Chem. Chem. Phys., 2019, 21, 21407–21417 RSC.
- J. Kong, H. Zhu, R. Li, W. Luo and X. Chen, Opt. Lett., 2009, 34, 1873–1875 CrossRef CAS.
- X. Zhang, S. Lin, T. Lin, P. Zhang, J. Xu, L. Xu and K. Chen, Phys. Chem. Chem. Phys., 2015, 17, 11974–11980 RSC.
- S. Nigam, A. Prasad, V. Sudarsan and R. Vatsa, Solid State Phys., Proc. DAE Solid State Phys. Symp., 58th, 2014, 1591, 524 CAS.
- F. del Prado, A. Cremades, D. Maestre, J. Ramirez-Castellanos, J. Gonzalez-Calbet and J. Piqueras, J. Mater. Chem. A, 2018, 6, 6299–6308 RSC.
- G. Rahman, N. Din, V. Garcia-Suarez and E. Kan, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87(20), 205205, DOI:10.1103/PhysRevB.87.205205.
- D. Scanlon and G. Watson, J. Mater. Chem., 2012, 22, 25236–25245 RSC.
- G. Zhang, C. Xie, S. Zhang, S. Zhang and Y. Xiong, J. Phys. Chem. C, 2014, 118, 18097–18109 CrossRef CAS.
- A. Nag, S. Chakraborty and D. Sarma, J. Am. Chem. Soc., 2008, 130, 10605–10611 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- I. Weber, A. Maciel, P. Lisboa, E. Longo, E. Leite, C. Paiva-Santos, Y. Maniette and W. Schreiner, Nano Lett., 2002, 2, 969–973 CrossRef CAS.
- A. K. Singh, S. K. Singh and S. B. Rai, RSC Adv., 2014, 4, 27039–27061 RSC.
- S. Han, R. Deng, X. Xie and X. Liu, Angew. Chem., Int. Ed., 2014, 53, 11702–11715 CrossRef CAS.
- P. Psuja, W. Strek, S. Jiang, M. Digonnet, J. Glesener and J. Dries, Optical Components and Materials VI, 2009, vol. 7212 Search PubMed.
- G. Chen, H. Liu, H. Liang, G. Somesfalean and Z. Zhang, J. Phys. Chem. C, 2008, 112, 12030–12036 CrossRef CAS.
- S. H. Sun, G. W. Meng, G. X. Zhang, T. Gao, B. Y. Geng, L. D. Zhang and J. Zuo, Chem. Phys. Lett., 2003, 376, 103–107 CrossRef CAS.
- L. Balan, C. Ghimbeu, L. Vidal and C. Vix-Guterl, Green Chem., 2013, 15, 2191–2199 RSC.
- P. Pasierb, S. Komornicki, M. Rokita and M. Rekas, J. Mol. Struct., 2001, 596, 151–156 CrossRef CAS.
- D. Amalric-Popescu and F. Bozon-Verduraz, Catal. Today, 2001, 70, 139–154 CrossRef CAS.
- B. Zhang, Y. Tian, J. Zhang and W. Cai, Mater. Lett., 2011, 65, 1204–1206 CrossRef CAS.
- N. D. Mott, EA, Electronic process in non-crystalline materials, Oxford University Press, 1971 Search PubMed.
- R. Awbery and S. Tsang, J. Nucl. Mater., 2008, 381, 223–230 CrossRef CAS.
- D. Aurbach and I. Weissman, Electrochem. Commun., 1999, 1, 324–331 CrossRef CAS.
- J. Wang, W. Zhou and P. Wu, Appl. Surf. Sci., 2014, 314, 188–192 CrossRef CAS.
- NIST XPS database, https://srdata.nist.gov/xps/main_search_menu.aspx, accessed July 13, 2020.
- N. Apostol, L. Stoflea, G. Lungu, C. Chirila, L. Trupina, R. Negrea, C. Ghica, L. Pintilie and C. Teodorescu, Appl. Surf. Sci., 2013, 273, 415–425 CrossRef CAS.
- L. Tanase, N. Apostol, L. Abramiuc, C. Tache, L. Hrib, L. Trupina, L. Pintilie and C. Teodorescu, Sci. Rep., 2016, 6, 35301 CrossRef CAS.
- P. Manna, G. Debnath, D. Waldeck and P. Mukherjee, J. Phys. Chem. Lett., 2018, 9, 6191–6197 CrossRef CAS.
- Y. Wang, J. Gao, C. Gao, H. Ma, B. Yang, Y. Han, E. Zhou, Q. Cheng, S. Jing and L. Huang, Nanoscale, 2019, 11, 16562–16570 RSC.
- D. F. Crabtree, J. Phys. D: Appl. Phys., 1975, 8, 107 CrossRef CAS.
- T. Matsuoka, Y. Kasahara, M. Tsuchiya, T. Nitta and S. Hayakawa, J. Electrochem. Soc., 1978, 125, 102–106 CrossRef CAS.
- C. M. Dodson and R. Zia, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 125102 CrossRef.
- P. Tuan, L. Hieu, L. Nga, N. Ha, N. Dung and T. Khiem, J. Electron. Mater., 2017, 46, 3341–3344 CrossRef CAS.
- S. Brovelli, N. Chiodini, F. Meinardi, A. Monguzzi, A. Lauria, R. Lorenzi, B. Vodopivec, M. Mozzati and A. Paleari, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 153108 CrossRef.
- A. Kar, S. Kundu and A. Patra, J. Phys. Chem. C, 2011, 115, 118–124 CrossRef CAS.
- D. Avram, M. Sanchez-Dominguez, B. Cojocaru, M. Florea, V. Parvulescu and C. Tiseanu, J. Phys. Chem. C, 2015, 119, 16303–16313 CrossRef CAS.
- D. Avram, B. Cojocaru, M. Florea and C. Tiseanu, Opt. Mater. Express, 2016, 6, 1635–1643 CrossRef CAS.
- S. Gupta, K. Sudarshan, A. Yadav, R. Gupta, D. Bhattacharyya, S. Jha and R. Kadam, Inorg. Chem., 2018, 57, 821–832 CrossRef CAS.
- B. Tissue, Chem. Mater., 1998, 10, 2837–2845 CrossRef CAS.
- W. Luo, R. Li, G. Liu, M. Antonio and X. Chen, J. Phys. Chem. C, 2008, 112, 10370–10377 CrossRef CAS.
- J. Bünzli, in Lanthanide Luminescence, eds. P. Hänninen and H. Härmä, Springer, Berlin, Heidelberg, 2010, vol. 7, ch. Basics of Lanthanide Photophysics, pp. 1–45 Search PubMed.
- B. R. Judd, Phys. Rev., 1962, 127, 750 CrossRef CAS.
- G. S. Ofelt, J. Chem. Phys., 1962, 37, 511 CrossRef CAS.
- D. Avram, C. Colbea, M. Florea, S. Lazar, D. Stroppa and C. Tiseanu, Nanoscale, 2019, 11, 16743–16754 RSC.
- D. Avram, B. Cojocaru and C. Tiseanu, Mater. Res. Bull., 2021, 134, 111091 CrossRef CAS.
- R. Gerhardt-Anderson and A. S. Nowick, Solid State Ionics, 1981, 5, 547–550 CrossRef CAS.
- X. Zhang, T. Lin, X. Jiang, J. Xu, J. Liu, L. Xu and K. Chen, Chin. Opt. Lett., 2012, 10, 091603 CrossRef.
- W. Shi, M. Bass and M. Birnbaum, J. Opt. Soc. Am. B, 1990, 7, 1456–1462 CrossRef CAS.
- R. A. Ferreira, S. F. Correia, A. Monguzzi, X. Liu and F. Meinardi, Mater. Today, 2020, 33, 105–121 CrossRef.
- G. H. Debnath, P. Mukherjee and D. H. Waldeck, J. Phys. Chem. C, 2020 DOI:10.1021/acs.jpcc.0c07548.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tc04582a |
| This journal is © The Royal Society of Chemistry 2021 |