
Chalcogen-vacancy group VI transition metal dichalcogenide nanosheets for electrochemical and photoelectrochemical hydrogen evolution†
In Hye
Kwak‡
 ,
Ik Seon
Kwon‡
,
Jong Hyun
Lee
,
Young Rok
Lim
and
Jeunghee
Park
*
,
Ik Seon
Kwon‡
,
Jong Hyun
Lee
,
Young Rok
Lim
and
Jeunghee
Park
*
Department of Advanced Materials Chemistry, Korea University, Sejong 339-700, Republic of Korea. E-mail: parkjh@korea.ac.kr
First published on 16th November 2020
Abstract
Solar photoelectrochemical water splitting using the catalytic hydrogen evolution reaction (HER) is one of the most promising methods for producing hydrogen energy. Herein, we report the facile synthesis of chalcogen vacancy-rich (5–20%) transition metal dichalcogenide (TMD, i.e., MoS2, MoSe2, WS2, and WSe2) nanostructures for the (photo)electrocatalytic HER. Atomically resolved scanning transmission electron microscopy revealed the concentration of chalcogen (S and Se) vacancies, as well as the formation of double chalcogen vacancies at higher concentrations. Photocathodes consisting of a Si nanowire array sheathed in TMD layers exhibited excellent photoelectrocatalytic performance under AM 1.5 irradiation conditions. The photocurrent reached 30 mA cm−2 (at 0 V vs. RHE) with a high faradaic efficiency (90%) and negligible degradation of the HER for 3 h. The free-standing TMD nanosheets exhibit excellent electrocatalytic HER performance (an overpotential of 162–202 mV vs. RHE for 10 mA cm−2 and a Tafel slope of 70–84 mV dec−1 in 0.5 M H2SO4), which can be correlated with the vacancy concentration. A detailed structural analysis suggested that the chalcogen vacancies play a major role in determining the catalytic activity toward the HER, which in turn is responsible for the enhanced PEC performance.
1. Introduction
The drastically increased consumption of fossil fuels has caused serious environmental crises such as global warming and air pollution, prompting scientists to explore renewable and clean energy sources. Hydrogen energy is presently considered the most promising candidate to replace the use of fossil fuels by hydrogen-based energy economy. Solar photoelectrochemical (PEC) water splitting is a carbon neutral technology because it produces hydrogen using practically unlimited resources (i.e., sunlight and water). The first PEC cell was fabricated in 1972 using a TiO2 photocatalyst as the photoanode under UV irradiation.1 Since then, there have been tremendous efforts to develop cost-effective and stable semiconductor photoelectrodes based on earth abundant elements for efficient solar energy harvesting and catalytic water oxidation or reduction reactions.2–10Si has a band gap (Eg) of 1.18 eV, which is narrow enough to efficiently absorb solar energy. Its conduction band position is suitable for the hydrogen evolution reaction (HER). Furthermore, the Si technology is well established, and the material price has decreased significantly in the past few years. Therefore, there is a keen interest in using Si in both the photoanodes and photocathodes of solar PEC cells.2,11–21 A powerful strategy is to apply low-dimensional nanostructures that can increase the surface area for light absorption.11,17,19–21 In particular, nanowires (NWs) can produce a desirable one-dimensional pathway for charge collection along the electrode/electrolyte interface.
Despite the significant progress in employing Si photoelectrodes for solar water splitting PEC cells, their durability and energy conversion efficiency are still far below the goal (>2000 h and 10%, respectively). The Si photoelectrode is intrinsically unstable due to its poor catalytic activity toward the water splitting reaction, which causes surface photo-corrosion. These problems could be avoided by employing efficient catalysts to increase the reaction rate and retard the surface oxidation. Recently, two-dimensional (2D) earth abundant metal dichalcogenide (TMD) nanomaterials with the chemical formula MX2 have been extensively investigated as HER electrocatalysts. Excellent electrocatalytic activities have been reported for Group VI TMDs (i.e., MoS2, MoSe2, WS2, and WSe2).22–24 At the same time, TMDs are often used as catalysts for Si-based photocathodes.25–39 The HER electrocatalytic activity of TMDs can be further improved by introducing S or Se vacancies, because these chalcogen vacancies facilitate the HER by adsorbing proton or water at the metal sites.40–53 However, the enhancement effect of chalcogen vacancies has not been verified for the Si-TMD photocathodes yet.
In this study, we synthesized four TMD nanomaterials (MoS2, MoSe2, WS2, and WSe2) with two distinctive forms: (1) shell layers on the pre-synthesized Si NW array photocathode for water splitting PEC cells (in 0.5 M H2SO4) and (2) freestanding nanosheets for the electrocatalytic HER. Both forms of TMDs are enriched with chalcogen vacancies and synthesized using a common sulfurization/selenization step. Atomically resolved scanning transmission electron microscopy provides evidence for the vacancies at the chalcogen sites. All the Si–MoS2, Si–MoSe2, Si–WS2, and Si–WSe2 photocathodes exhibited excellent PEC performance, with a photocurrent of 20–30 mA cm−2 (at 0 V vs. RHE) in 0.5 M H2SO4 under AM 1.5 irradiation conditions. The electrocatalytic HER performance of freestanding TMD nanosheets was well correlated with the concentration of chalcogen vacancies. A detailed structural analysis shows that S/Se vacancies play a major role in enhancing the HER performance. These results demonstrate that the electrochemical and PEC catalytic efficiencies of the four Group VI TMDs could all be improved by introducing chalcogen vacancies.
2. Experimental section
Synthesis of Si-TMD NW arrays
The Si NW array was synthesized using Ag-assisted chemical etching of a Si(100) substrate, as has been described elsewhere.54 The deposition of TMD nanosheets on the Si NW array was carried out as follows. A substrate of Si NW array was immersed in 0.4 M MoCl5 (or WCl6) ethanol solution for 5 min and then rinsed with ethanol. The metal ions deposited on the Si NW substrate were then sulfurized or selenized as follows. The NW substrate was placed in a quartz tube inside a chemical vapor deposition (CVD) reactor (electrical furnace), and S or Se powder (200 mg) was located in a quartz boat 3 cm away from the Si substrate. Mixed hydrogen (H2)/argon (Ar) gas was continuously flowed at a rate of 20/100 sccm (mL min−1) during the entire process. The temperatures of the S (or Se) powder and the Si NW substrate were, respectively, 200 and 450–550 °C during the sulfurization/selenization, and the reaction time was 30 min.Synthesis of freestanding TMD nanosheets
First, molybdenum oxide (MoO3) and tungsten oxide (WO3) nanocrystals were synthesized using a hydrothermal method. Sodium molybdate dihydrate (Na2MoO4·2H2O, molecular weight (MW) = 241.95 g mol−1, 1 mmol) or sodium tungstate dihydrate (Na2WO4·2H2O, MW = 329.85 g mol−1, 1 mmol) and 30 μL HCl (conc. 37%) were dissolved in deionized (DI) water (20 mL) using a bath sonicator. The pH of the mixture was 1–2. The solution was transferred to a Teflon-lined stainless-steel autoclave reactor. The hydrothermal reaction was performed at 180 °C for 6 h in an electric oven. The product (MoO3 or WO3) was collected by centrifugation, washed thoroughly with DI water and acetone several times, and then vacuum-dried at room temperature. For sulfurization or selenization, 20 mg of MoO3 (or WO3) was placed inside the CVD reactor. The reaction time was 60–120 min. The other conditions are the same as those of the Si NW samples. The characterization method and (photo)electrochemical measurements are described in the ESI.†3. Results and discussion
Fig. 1 shows a schematic of the synthesis procedure. (1) TMD nanosheets were synthesized directly on a pre-grown Si NW array. The NW surface was coated with metal ions by soaking the substrate in a MoCl5 or WCl6 ethanol solution. (2) Subsequent sulfurization/selenization was performed at 450–550 °C via the chemical vapor transport of the S or Se powder under H2/Ar gas flow. (3) Separately, the freestanding TMD nanosheets were synthesized via the sulfurization/selenization of wire-like MoO3 or WO3 nanocrystals under the same conditions as those for the NW sample. The MoO3 and WO3 were synthesized using the hydrothermal reaction of NaMoO4 and Na2WO4 at 180 °C, respectively. Fig. S1 (ESI†) shows the scanning electron microscopy (SEM) and high-resolution transmission electron microscopy (HRTEM) images, X-ray energy-dispersive fluorescence spectroscopy (EDX) data, and X-ray diffraction (XRD) patterns for the wire-like MoO3 and WO3 nanocrystals. The XRD patterns of the two series of TMD materials are shown in Fig. S2 (ESI†). The hexagonal (2H, space group = P63/mmc) phase was identified for MoS2 (JCPDS No. 87-2416, a = 3.160 Å, c = 12.290 Å), MoSe2 (JCPDS No. 29-0914, a = 3.287 Å, c = 12.925 Å), WS2 (JCPDS No. 08-0237, a = 3.154 Å, c = 12.362 Å), and WSe2 (JCPDS No. 38-1388, a = 3.285 Å, c = 12.982 Å).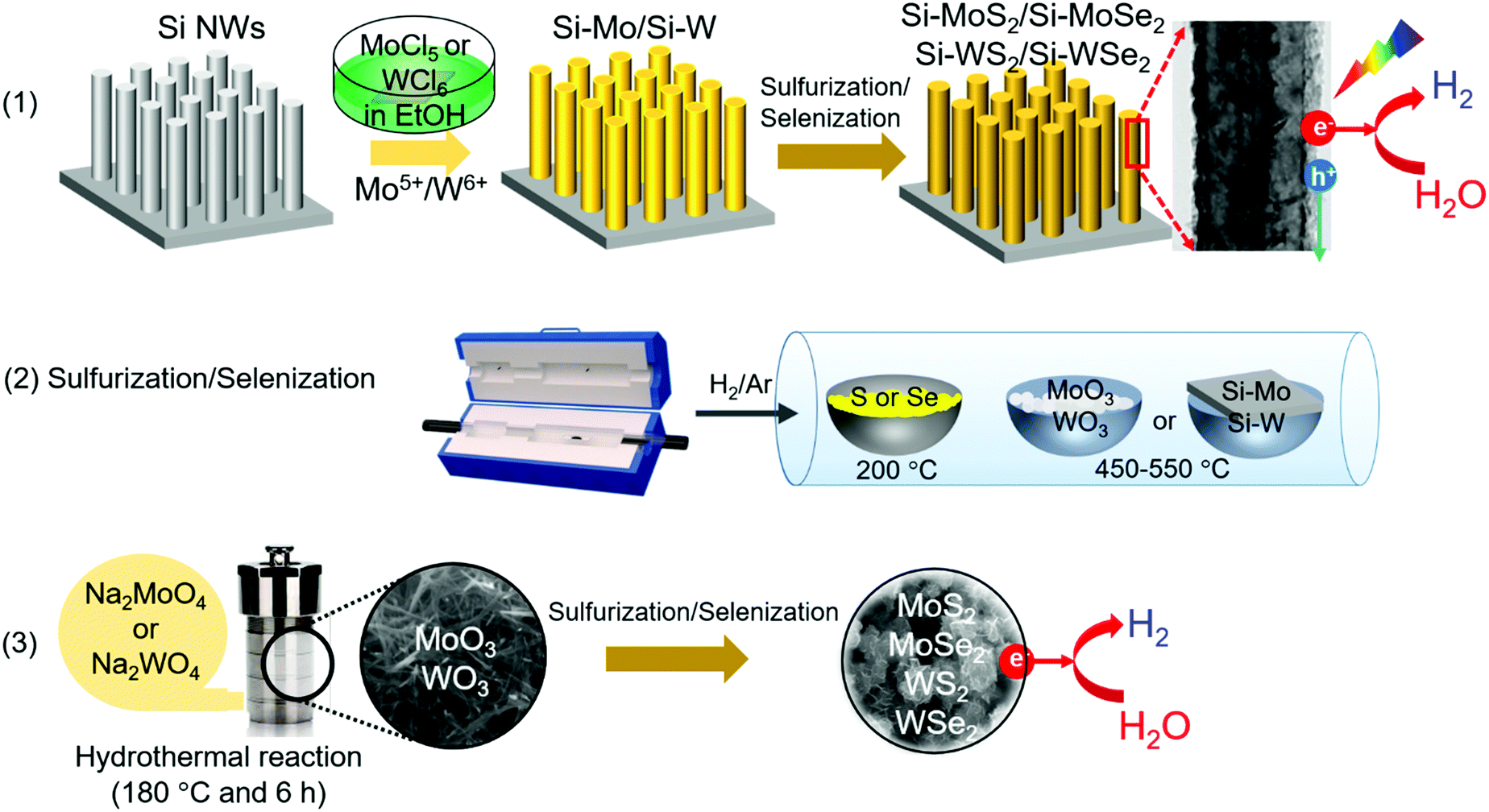 | ||
| Fig. 1 Schematic of the synthesis of two forms of TMD (MoS2, MoSe2, WS2, and WSe2) nanosheets. (1) For the photoelectrochemical HER, the shell layers of TMD on the Si NW array (Si-TMD) were synthesized in two steps: metal ion coating and sulfurization/selenization. (2) Sulfurization/selenization step using the thermal chemical transport of S and Se powders under H2/Ar gas flow. (3) For the electrochemical HER, freestanding TMD nanosheets were synthesized in two steps: hydrothermal synthesis of MoO3/WO3 nanocrystals and sulfurization/selenization. | ||
The TMD nanosheets were synthesized as shell layers on Si NWs, and these samples were referred to as Si–MoS2, Si–MoSe2, Si–WS2, and Si–WSe2. The tilted SEM view in Fig. 2a shows that the Si-TMD NWs were grown on a large area of Si substrates via Ag-assisted etching of Si(100) wafers. The side view shows that the Si NWs had a length of approximately 4 μm (Fig. 2b). The top views confirm that TMDs were grown on the Si NWs (Fig. 2c). Moreover, HRTEM images indicate that the surface of Si NWs was coated homogeneously with MoS2 layers with a thickness of approximately 20 nm (Fig. 2d). Lattice-resolved TEM images revealed that the MoS2 nanoflakes assembled into a polycrystalline shell sheathing the Si NW core. The d-spacing of the (002) planes was identified as 6.1 Å, which is consistent with the reference value of 6.145 Å. Fig. 2e shows the HRTEM images of Si–MoSe2, in which the polycrystalline MoSe2 layers have d002 = 6.5 Å, consistent with the reference value of 6.46 Å. Fig. 2f and g show the HRTEM images of Si–WS2 and Si–WSe2, respectively. These shell layers also consisted of the nanoflake assembly. The WS2 and WSe2 nanoflakes have uniform d002 = 6.2 and 6.5 Å, which are also consistent with the respective reference values of 6.18 and 6.49 Å.
 | ||
| Fig. 2 SEM images of Si-TMD NWs in (a) tilted and (b) side views. (c) Magnified images of the top part, showing the homogeneous deposition of TMD nanosheets on the Si NWs. HRTEM images of (d) Si–MoS2, (e) Si–MoSe2, (f) Si–WS2, and (g) Si–WSe2. The distance between adjacent (002) planes (d002) in the TMD is 6.1, 6.5, 6.2, and 6.5 Å, respectively. | ||
The high-angle annular dark-field scanning TEM (HAADF-STEM) image and EDX elemental mapping spectra (using the Mo L-shell, W M-shell, S K-shell, Se L-shell, and Si K-shell peaks) revealed that only the Mo and X elements were present in the shell part, while Si was localized in the Si NWs (Fig. S3, ESI†). The EDX and X-ray photoelectron spectroscopy (XPS) analyses provided [S]/[Mo] = 1.85, [Se]/[Mo] = 1.9, [S]/[W] = 1.9, and [Se]/[W] = 1.8 (see Fig. S4, ESI†). The corresponding concentrations were 7%, 5%, 5%, and 10%, which are half of those for the freestanding TMD nanosheets (i.e., 15%, 10%, 10%, and 20%).
The electronic structures of the samples were analyzed using XPS, showing that the binding energies were lower than those in the bulk, also owing to the S/Se vacancies (Fig. S5, ESI†). The magnitude of the redshift is nearly the same for all samples, probably because the vacancy concentration varied in the narrower range compared to that of freestanding nanosheets as described later. The lower concentration of chalcogen vacancies in Si-TMD is ascribed to the shorter time of the sulfurization/selenization step. The optimized reaction time was 30 min for the TMD shell layers. In contrast, the freestanding TMD nanosheets need at least 60 min to obtain a complete conversion of the oxide.
The SEM images of freestanding TMD nanosheets show that the thin nanosheets assembled into flower-like microspheres (Fig. S6, ESI†). Fig. 3a shows the HRTEM images of WS2 nanosheets. The average thickness of the nanosheets is 2 nm based on the statistical distribution. The lattice-resolved image (Fig. 3b) of the basal plane (magnified in the marked area) shows that the distance between adjacent (100) planes (d100) is 2.7 Å, which is close to the value of 2H phase WS2 (2.73 Å). The HAADF-STEM image and EDX elemental mapping of MoS2, MoSe2, WS2, and WSe2 nanosheets show uniform distributions of metal (Mo or W) and chalcogen (S or Se) atoms over the entire samples (Fig. 3c). All four types of nanosheets have significant chalcogen vacancies since [S]/[Mo] = 1.7, [Se]/[Mo] = 1.8, [S]/[W] = 1.8, and [Se]/[W] = 1.6. These ratios were supported by the XPS analysis (Fig. S7, ESI†). Therefore, the chalcogen vacancies were estimated to be 15%, 10%, 10%, and 20%, respectively, for MoS2, MoSe2, WS2, and WSe2.
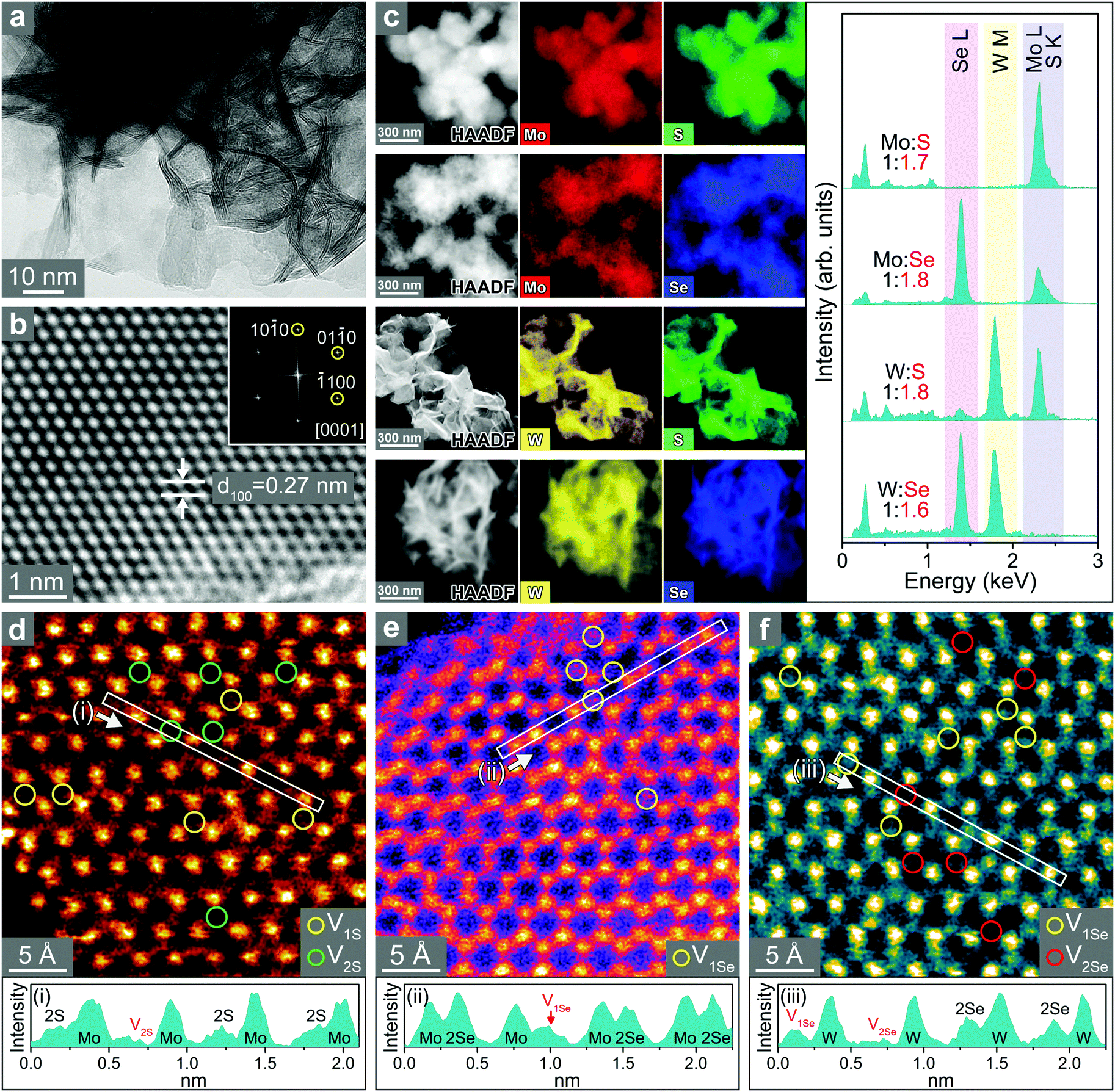 | ||
| Fig. 3 (a) High-resolution transmission electron microscopy (HRTEM) image of WS2 nanosheets and (b) lattice-resolved TEM image for their basal plane. The distance between adjacent (100) planes (d100) is 2.7 Å. (c) HAADF-STEM image; EDX elemental mapping of Mo (L shell), W (M shell), S (K shell), and Se (L shell); and EDX spectra for MoS2, MoSe2, WS2, and WSe2. Atomically resolved HAADF-STEM images of the basal plane for (d) MoS2, (e) MoSe2, and (f) WSe2, and line profiles for the selected areas (i), (ii), and (iii). The single (V1S and V1Se) and double (V2S and V2Se) chalcogen vacancies are marked by circles. | ||
Atomic-resolution HAADF STEM images for the basal planes of MoS2, MoSe2, and WSe2 were obtained, and the corresponding contour maps are shown in Fig. 3d–f, respectively. They all consisted of a well-defined hexagonal pattern of 3(M–X) corresponding to the 2H phase of the TMD. Darker intensities at the atomic sites suggest that these atoms are missing. The contour map clearly distinguishes the single/double vacancies from the bright yellow S or Se atoms.55 In MoS2, S sites missing one and two S atoms are marked by V1S and V2S, respectively, and they are approximately equal in number. In contrast, MoSe2 has only sites missing one Se atom (marked by V1Se). In the case of WSe2, vacancies with one and two missing Se atoms (marked by V1Se and V2Se, respectively) are formed in equal numbers. The production of double vacancies (V2S and V2Se) in MoS2 and WSe2 is ascribed to the higher concentration of vacancies (20%) compared to that in MoSe2 (10%). The line profiles for regions (i)–(iii) clearly show the two types (i.e. single and double) of chalcogen vacancies. The presence of S/Se vacancies was also supported by electron paramagnetic resonance (EPR), as shown in Fig. S8 (ESI†). A stronger signal was found in MoS2 and WSe2 than that in MoS2 and WS2, which is correlated with the higher concentration of vacancies.43,51
The electronic structures of the samples were analyzed using XPS. The binding energy was lower compared to the corresponding bulk crystals, indicating increased metallicity that is probably due to the presence of S or Se vacancies (Fig. S9, ESI†). The higher concentration of vacancies in MoS2 and WSe2 induced a more significant redshift compared to MoS2 and WS2.
When synthesizing the freestanding TMD nanosheets or the TMD shell layers of Si NWs, a H2 gas flow was essential because the sulfurization/selenization was not efficient. However, the H2 gas flow also inevitably depleted the S/Se, owing to the reaction H2 + S (or Se) → H2S (or H2Se) during/after the growth of nanosheets. In fact, H2 annealing is a common method for producing vacancies in TMD nanomaterials.45,50 Guo et al. reported the vacancy formation energy for monolayer MoS2, MoSe2, WS2, and WSe2 to be 2.35, 2.82, 3.38, and 2.81 eV, respectively.56 Lee et al. calculated these values as 2.21, 2.64, 2.37, and 2.70 eV.57 A lower formation energy implies more favored vacancy formation. Thus, these formation energies can explain the higher vacancy concentration of MoS2 nanosheets than that of MoSe2 nanosheets. However, there is a discrepancy in the values of WS2 and WSe2, which needs further studies to explain our results.
Now, we discuss the I–V curves measured for water splitting PEC cells using photocathodes of Si, Si–MoS2, Si–MoSe2, Si–WS2, and Si–WSe2 in 0.5 M H2SO4 (pH 0) under solar irradiation (AM 1.5G, 100 mW cm−2). For the first and second scanned linear sweep voltammetry (LSV) curves, the current density (mA cm−2) vs. applied potential (V vs. RHE) is plotted in Fig. 4a. A quick photocurrent response was observed during light irradiation in 2 s on/off cycles (first scan). The second scan was performed under the light-on condition. The onset potential (=open circuit voltage, VOC) was 0.25, 0.22, 0.24, and 0.15 V, respectively, for Si–MoS2, Si–MoSe2, Si–WS2, and Si–WSe2, which was approximately 0.2 V for all Si-TMD samples. The photocurrent at 0 V vs. RHE (JSC, short circuit current) was 30, 26, 23, and 22 mA cm−2, respectively, for Si–MoS2, Si–MoSe2, Si–WS2, and Si–WSe2, and 0 for pristine (bare) Si NWs. The maximum current reaches 32 mA cm−2. The PEC performance of the four TMDs is much enhanced compared to the bare Si NWs and is summarized in Table 1.
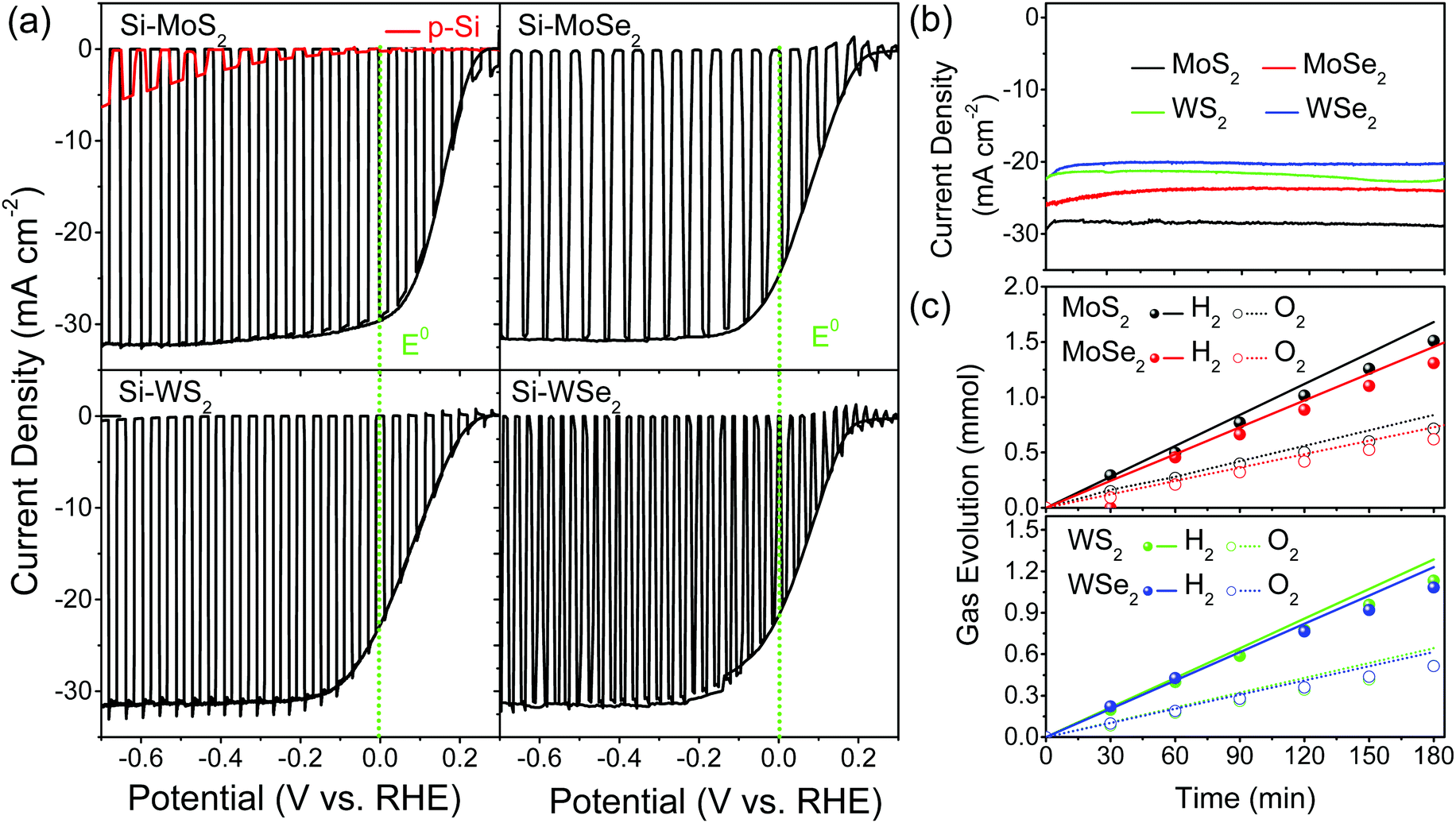 | ||
| Fig. 4 (a) Current density (mA cm−2) vs. potential (V vs. RHE) for Si, Si–MoS2, Si–MoSe2, Si–WS2, and Si–WSe2 photocathodes, measured in 0.5 M H2SO4 (pH 0) under AM 1.5G irradiation (100 mW cm−2), with the light-on/off cycles (first scan) and light-on (second) scan. The scan rate is 20 mV s−1. E0 represents the 0 V vs. RHE. (b) CA response of photocurrent and (c) H2/O2 evolution vs. time (min). Applied potential of 0 V vs. RHE. The line represents the calculated H2/O2 evolution from current generation with FE = 100%, and the circle symbol for the data. | ||
| Samples | S or Se vacancies | PEC | R ct | E fb | |||
|---|---|---|---|---|---|---|---|
| J SC | V OC | Degradation for 3 hc | FEH2d | ||||
| a Short circuit current: current density (mA cm−2) at 0 V vs. RHE. b Open circuit voltage (V vs. RHE). c Degradation of current density at 0 V vs. RHE for the 3 h CA test. d Average faradaic efficiency for 3 h H2 gas generation. e Charge transfer resistance (Ω) determined using Nyquist plots at 0 V (vs. RHE) under light irradiation. f Flat band potential (V vs. RHE) determined using Mott–Schottky plots. | |||||||
| Si–MoS2 | 7% | 30 | 0.25 | 0% | 90% | 172 | 0.17 |
| Si–MoSe2 | 5% | 26 | 0.22 | 3% | 88% | 112 | 0.23 |
| Si–WS2 | 5% | 23 | 0.24 | 0% | 86% | 109 | 0.25 |
| Si–WSe2 | 10% | 22 | 0.18 | 8% | 85% | 203 | 0.20 |
Fig. 4b shows the chronoamperometric (CA) response of the photocurrent (at 0 V vs. RHE) with a degradation of 3% over 3 h. In order to confirm water splitting in the PEC cell at pH 0, we monitored the evolution of hydrogen (H2) and oxygen (O2) gases at 0 V (vs. RHE). Fig. 4c displays the H2 and O2 evolution data (in mmol). The corresponding faradaic efficiencies (FEs) for 2H+ + 2e− → H2(g) and 2H2O → O2(g) + 4H+ + 4e− were calculated using the equations 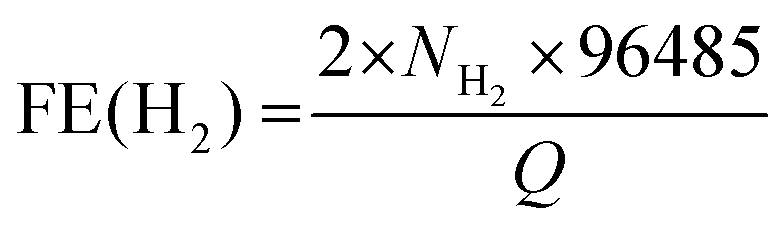 and
and 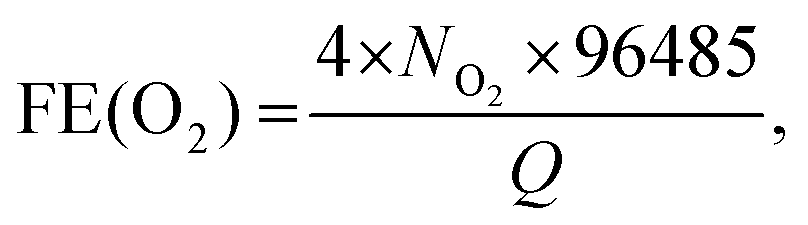 where NH2 and NO2 are the amounts (mol) of H2 and O2, respectively, and Q is the total amount of generated charge in Coulombs (photocurrent × time). The average FE was 90% for H2 and 85% for O2 (the line in the figure represents the calculated H2/O2 evolution from the photocurrents assuming FE = 100%). Table S1 (ESI†) compares the PEC performance with previously reported data for Si-TMD PEC cells, showing that the JSC value of Si–MoS2 is close to that obtained by the Shen group using the MoS2 layer on the pyramid shaped Al2O3/n+p-Si photocathode in 1 M HClO4 (34 mA cm−2).32
where NH2 and NO2 are the amounts (mol) of H2 and O2, respectively, and Q is the total amount of generated charge in Coulombs (photocurrent × time). The average FE was 90% for H2 and 85% for O2 (the line in the figure represents the calculated H2/O2 evolution from the photocurrents assuming FE = 100%). Table S1 (ESI†) compares the PEC performance with previously reported data for Si-TMD PEC cells, showing that the JSC value of Si–MoS2 is close to that obtained by the Shen group using the MoS2 layer on the pyramid shaped Al2O3/n+p-Si photocathode in 1 M HClO4 (34 mA cm−2).32
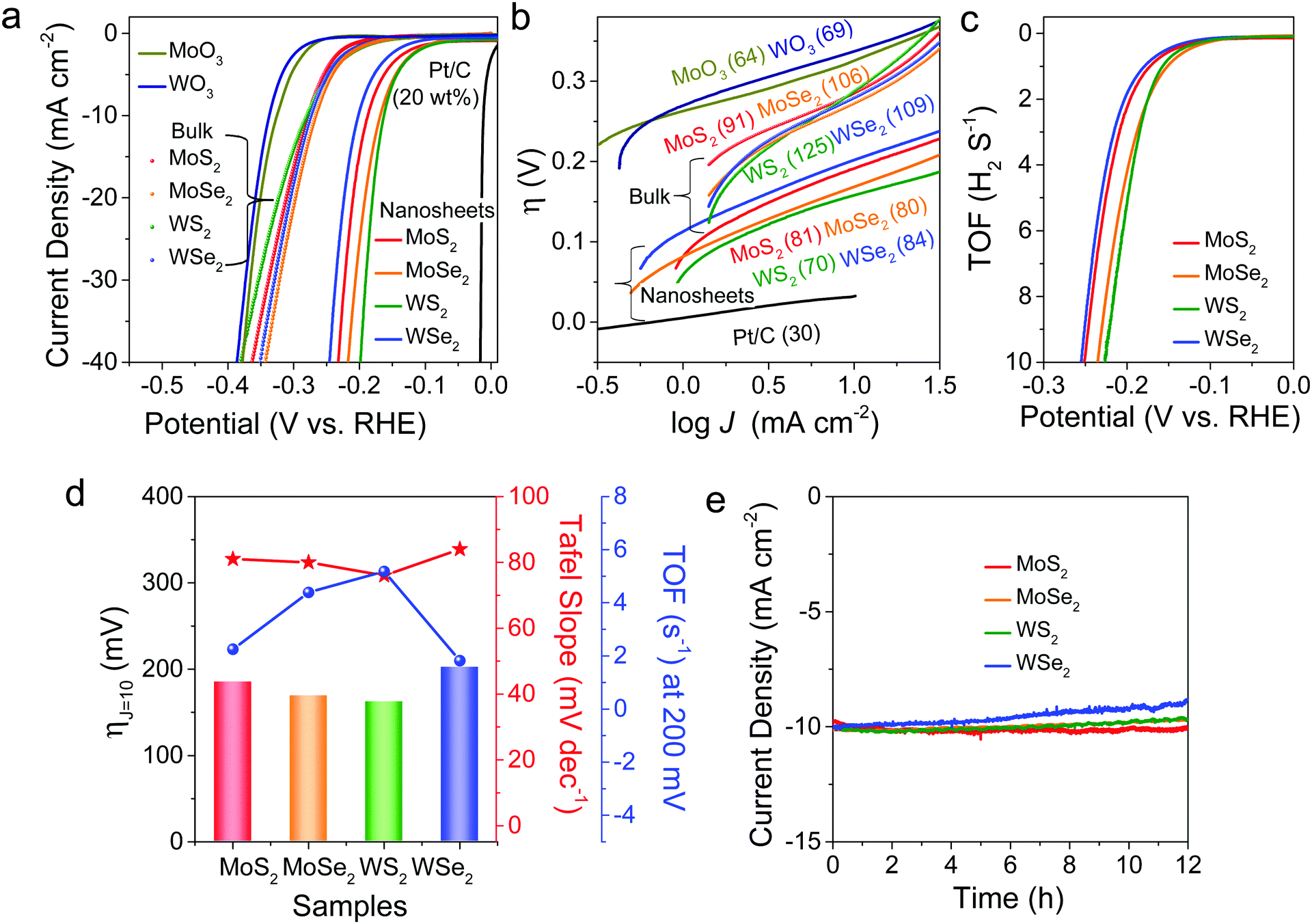 | ||
Fig. 5 HER performance of freestanding MoS2, MoSe2, WS2, and WSe2 nanosheets. (a) LSV curves (scan rate: 2 mV s−1) of the HER in H2-saturated 0.5 M H2SO4 (pH 0) using TMD nanosheets (with bulk and oxide samples) and commercial Pt/C as catalysts. (b) Tafel plots derived from the LSV curves at low potentials that correspond to the activation-controlled region, using the Tafel equation η = b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log(J/J0), where η is the overpotential, b is the Tafel slope (mV dec−1), J is the measured current density (mA cm−2), and J0 is the exchange current density. The b values are given in parentheses. (c) TOF (s−1) curves and (d) comparison of ηJ=10, b, and TOF values at 0.2 V. (e) CA responses at ηJ=10 for 12 h. log(J/J0), where η is the overpotential, b is the Tafel slope (mV dec−1), J is the measured current density (mA cm−2), and J0 is the exchange current density. The b values are given in parentheses. (c) TOF (s−1) curves and (d) comparison of ηJ=10, b, and TOF values at 0.2 V. (e) CA responses at ηJ=10 for 12 h. | ||
We examined the structure and morphology of the samples after PEC H2 evolution for 3 h. The XRD patterns showed that all the crystals retained their crystal phases (Fig. S10, ESI†). The XPS and EDX data indicate that the electronic structures also remained the same afterwards. Electrochemical impedance spectroscopy (EIS) measurements provided the charge transfer resistance (Rct) under light irradiation, as shown in Fig. S11 (ESI†). The Rct values of Si–MoSe2 (112 Ω) and Si–WS2 (109 Ω) are smaller than those of Si–MoS2 (172 Ω) and Si–WSe2 (203 Ω), which is consistent with the freestanding nanosheets. Using Mott–Schottky (MS) plots, the flat-band potential (Efb) was estimated to be approximately 0.2 V (Fig. S12, ESI†). Overall, the PEC performance of Si-TMD is nearly the same because they all have 5–10% chalcogen vacancies, which are optimal according to the HER catalytic activity for freestanding nanosheets with 10–20% chalcogen vacancies.
The extent of band bending (Eb) that occurred in the surface-electrolyte interface can be determined by E − Efb, where E is the applied potential. If Efb is more positive, Eb becomes more negative at the cathodic potential E, which provides a driving force for electron transfer to the surface and benefits the catalytic HER at the active sites of TMDs. Because Efb = 0.2 V for all considered Si-TMDs, a similar downward band bending would be expected. The metallicity of the TMDs due to the chalcogen vacancies increases the carrier concentration to induce band bending. The good catalytic activity of TMD nanosheets with chalcogen vacancies resulted in the efficient separation of the photoexcited electron–hole pairs. Therefore, their excellent HER catalytic activity gives rise to excellent PEC performance.
As a next step, the electrocatalytic HER performances of freestanding MoS2, MoSe2, WS2, and WSe2 nanosheets were investigated via LSV, using these samples as working electrodes in a typical three-electrode setup. Fig. 5a shows the LSV curves measured at pH 0, where the potentials are referenced to the reversible hydrogen electrode (RHE). The overpotential required to obtain a current density of 10 mA cm−2 (ηJ=10) is 185, 169, 162, and 202 mV for MoS2, MoSe2, WS2, and WSe2 nanosheets, respectively. For comparison, the LSV curves measured for commercially available bulk powders (purchased from Alfa Aesar) provided the following values: ηJ=10 = 274, 261, 272, and 269 mV, respectively, for MoS2 (purity 99%), MoSe2 (99%), WS2 (99.8%), and WSe2 (99.8%). As shown in the earlier XPS and EPR data (see Fig. S7 and S8, ESI†), these bulk powders have no S/Se vacancies. MoO3 and WO3 also showed much lower catalytic activity (ηJ=10 = 326 and 336 mV, respectively). A commercial 20 wt% Pt/C catalyst exhibited ηJ=10 = 27 mV.
Fig. 5b displays the Tafel plots, η(V) vs. log[J (mA cm−2)], in the low potential region. The slope of the linear fit provides the Tafel slope (b). The b values are 81, 80, 70, and 84 mV dec−1, respectively, for MoS2, MoSe2, WS2, and WSe2 nanosheets. Respective bulk powders show b = 91, 106, 125, and 109 mV dec−1. In comparison, Pt/C, MoO3, and WO3 showed b = 30, 64, and 69 mV dec−1, respectively. The catalytic activity can be compared using the turnover frequency (TOF) per active site. We quantified the active sites using cyclic voltammetry (CV) curves at pH 7 (phosphate buffer solution) in the region of −0.2 to 0.6 V vs. RHE (see “Experimental details” in the ESI†). Fig. 5c shows the TOF curves derived from LSV data by normalizing the active sites in the samples. The TOF values at η = 0.2 V are 2.3, 4.4, 5.2, and 1.8 s−1 for MoS2, MoSe2, WS2, and WSe2, respectively. Fig. 5d compares the values of ηJ=10, b, and TOF for the four samples, showing that MoSe2 and WS2 display better HER performance than MoS2 and WSe2.
Electrochemical impedance spectroscopy (EIS) measurements provided the charge transfer resistance (Rct), as shown in Fig. S13 (ESI†). Its values at −0.2 V (vs. RHE) are 41.0, 20.0, 9.1, and 66.4 Ω for MoS2, MoSe2, WS2, and WSe2, respectively, in the reverse order of their HER performance. Since a smaller Rct value implies more facile electron transfer kinetics that enhances the catalytic activity, the Rct values are well correlated with the relative HER performance. As shown in Fig. S14 (ESI†), the double-layer capacitance (Cdl) was measured using CV scans in the non-faradaic region (0.1–0.2 V vs. RHE) to be 10.7, 11.6, 14.7, and 8.2 mF cm−2 for MoS2, MoSe2, WS2, and WSe2, respectively. Thus, the sample with higher catalytic activity consistently exhibits a larger charge capacitance value. Fig. 5e shows the CA responses of the samples at ηJ=10 for 12 h. There was no current attenuation for MoS2 after 12 h, indicating that it had the best stability among the samples. In contrast, WSe2 showed a 12% current decrease, but it was only 3% until 3 h. In the cases of MoSe2 and WS2, no degradation occurred until 6 h and the decrease was 3% after 12 h.
Table 2 summarizes the measured HER performance for MoS2, MoSe2, WS2, and WSe2 nanosheets and bulk powders. We highlight the following results: (i) The HER catalytic activity of the bulk powders is much lower than that of the nanosheets. The difference between them is negligible compared to the nanosheets. (ii) MoSe2 and WS2 nanosheets exhibit higher HER performance than MoS2 and WSe2. (iii) MoS2 has the best stability. (iv) WSe2 possesses the worst catalytic capability, especially in terms of stability. These trends in HER performance can be simply explained using the following model. The data of bulk powders suggest that the intrinsic catalytic activity of four TMDs may not be much different. Therefore, the enhanced HER performance of nanosheets can be correlated with their S/Se vacancies. Based on previous studies, the S/Se vacancies enhance the HER performance by producing active sites that favorably form the Mo–H or W–H bond.40–53 When the vacancy concentration exceeds the optimum value, the activity of active sites usually decreases.40,45,50 Therefore, the higher HER performance of MoSe2 and WS2 compared to MoS2 and WSe2 is due to the optimum S/Se vacancies (10% vs. 15% and 20%). The worst performance of WSe2 can be similarly ascribed to the rich Se vacancies. The double chalcogen vacancies at a higher concentration can act as a defective trapping site for the adsorbed protons. Nevertheless, the dependence of the HER performance on the concentration of chalcogen vacancies over the range of 10–20% is not significant. It is also possible that the optimal HER performance occurs at below 10%, which was not feasible to examine in this study. On the other hand, the highest stability of MoS2 may originate from its inherent nature, which needs further investigation.
| Nanosheets | Bulk powders | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| TMD | [X]/[M]a | S or Se vacanciesb | η J=10 | b | TOFe | R ct | C dl | CAh | η J=10 | b |
| a M = Mo or W, X = S, Se. b Determined using EDX and XPS data. c Overpotential (mV vs. RHE) at J = 10 mA cm−2. d Tafel slope (mV dec−1). e TOF (s−1) at η = 0.2 V. f Charge transfer resistance (Ω) obtained using the Nyquist plot of EIS at η = 0.2 V. g Double layer capacitance (mF cm−2). h Degradation of current during the CA test for 12 h. | ||||||||||
| MoS2 | 1.7 | 15% | 185 | 81 | 2.3 | 41.0 | 10.7 | 0% | 274 | 91 |
| MoSe2 | 1.8 | 10% | 169 | 80 | 4.4 | 20.0 | 11.6 | 3% | 261 | 106 |
| WS2 | 1.8 | 10% | 162 | 76 | 5.2 | 9.1 | 14.7 | 3% | 272 | 125 |
| WSe2 | 1.6 | 20% | 202 | 84 | 1.8 | 66.4 | 8.2 | 12% | 269 | 109 |
4. Conclusions
We synthesized MoS2, MoSe2, WS2, and WSe2 nanosheets with chalcogen vacancies as excellent catalysts for solar-driven water splitting PEC cells and the electrocatalytic HER. Using the shared step of gas-phase sulfurization/selenization under H2 flow, these TMDs were prepared in two forms: freestanding nanosheets and shell layers on a pre-grown Si NW array. This process produced 5–20% chalcogen vacancies. High-resolution STEM images revealed that double chalcogen vacancies were formed at higher concentrations while single vacancies formed at lower concentrations. The XPS analysis showed that a higher chalcogen vacancy concentration induces a larger redshift of the binding energy. The Si-TMD NW array with optimal chalcogen vacancies (5–10%) exhibited excellent PEC performance in 0.5 M H2SO4; the photocurrent is 22–30 mA cm−2 (at 0 V vs. RHE) and the onset potential is 0.18–0.25 V. In terms of stability, nearly 100% of the HER performance is retained after 3 h, with a high FE of H2 generation (90%). The enhanced catalytic activity due to the chalcogen vacancies strongly boosts the performance of PEC cells. Meanwhile, we evaluated the electrocatalytic activity of the freestanding nanosheets toward the HER, showing that the catalytic performance in 0.5 M H2SO4 was optimized when there were 10% chalcogen vacancies. For the four TMDs having 10–20% chalcogen vacancies, ηJ=10 = 162–202 mV (vs. RHE) and b = 76–84 mV dec−1. These results demonstrated that the introduction of S/Se vacancies in TMDs can improve the (photo)electrocatalytic activity toward the HER.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by Korea University. The HVEM measurements were supported by the KBSI R&D program (Project No. C030440).Notes and references
- A. A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef
.
- R. N. Dominey, N. S. Lewis, J. M. Bruce, D. C. Bookbinder and M. S. Wrighton, J. Am. Chem. Soc., 1982, 104, 467–482 CrossRef
- A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS
- O. Khaselev and J. A. Turner, Science, 1998, 280, 425–427 CrossRef CAS
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef
- A. Paracchino, V. Laporte, K. Sivula, M. Grätzel and E. Thimsen, Nat. Mater., 2011, 10, 456–461 CrossRef
- D. Bae, B. Seger, P. C. K. Vesborg, O. Hansen and I. Chorkendorff, Chem. Soc. Rev., 2017, 46, 1933–1954 RSC
- Y. Hou, B. L. Abrams, P. C. K. Vesborg, M. E. Björketun, K. Herbst, L. Bech, A. M. Setti, C. D. Damsgaard, T. Pedersen, O. Hansen, J. Rossmeisl, S. Dahl, J. K. Nørskov and I. Chorkendorff, Nat. Mater., 2011, 10, 434–438 CrossRef CAS
- Y. W. Chen, J. D. Prange, S. Dühnen, Y. Park, M. Gunji, C. E. D. Chidsey and P. C. McIntyre, Nat. Mater., 2011, 10, 539–544 CrossRef CAS
- S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers and D. G. Nocera, Science, 2011, 334, 645–648 CrossRef CAS
- M. J. Kenney, M. Gong, Y. Li, J. Z. Wu, J. Feng, M. Lanza and H. Dai, Science, 2013, 342, 836–840 CrossRef CAS
- K. Sun, S. Shen, Y. Liang, P. E. Burrows, S. S. Mao and D. Wang, Chem. Rev., 2014, 114, 8662–8719 CrossRef CAS
- L. Ji, M. D. McDaniel, S. Wang, A. B. Posadas, X. Li, H. Huang, J. C. Lee, A. A. Demkov, A. J. Bard, J. G. Ekerdt and E. T. Yu, Nat. Nanotechnol., 2015, 10, 84–90 CrossRef CAS
- Q. Ding, J. Zhai, M. Cabán-Acevedo, M. J. Shearer, L. Li, H.-C. Chang, M.-L. Tsai, D. Ma, X. Zhang, R. J. Hamers, J.-H. He and S. Jin, Adv. Mater., 2015, 27, 6511–6518 CrossRef CAS
- J. C. Hill, A. T. Landers and J. A. Switzer, Nat. Nanotechnol., 2015, 14, 1150–1155 CAS
- H. Zhang, Q. Ding, D. He, H. Liu, W. Liu, Z. Li, B. Yang, X. Zhang, L. Lei and S. Jin, Energy Environ. Sci., 2016, 9, 3113–3119 RSC
- D. Liu, J. Ma, R. Long, C. Gao and Y. Xiong, Nano Today, 2017, 17, 96–116 CrossRef CAS
- Z. Luo, T. Wang and J. Gong, Chem. Soc. Rev., 2019, 48, 2158–2181 RSC
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963–969 CrossRef CAS
- D. Voiry, H. Yamaguchi, J. Li, R. Silva, D. C. B. Alves, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nat. Mater., 2013, 12, 850–855 CrossRef CAS
- P. D. Tran, S. S. Pramana, V. S. Kale, M. Nguyen, S. Y. Chiam, S. K. Batabyal, L. H. Wong, J. Barber and J. Loo, Chem. – Eur. J., 2012, 18, 13994–13999 CrossRef CAS
- Q. Ding, F. Meng, C. R. English, M. C. Acevedo, M. J. Shearer, D. Liang, A. S. Daniel, R. J. Hamers and S. Jin, J. Am. Chem. Soc., 2014, 136, 8504–8507 CrossRef CAS
- L. Zhang, C. Liu, A. B. Wong, J. Resasco and P. Yang, Nano Res., 2015, 8, 281–287 CrossRef CAS
- K. C. Kwon, S. Choi, K. Hong, C. W. Moon, Y.-S. Shim, D. H. Kim, T. Kim, W. Sohn, J.-M. Jeon, C.-H. Lee, K. T. Nam, S. Han, S. Y. Kim and H. W. Jang, Energy Environ. Sci., 2016, 9, 2240–2248 RSC
- C.-J. Chen, K.-C. Yang, C.-W. Liu, Y.-R. Lu, C.-L. Dong, D.-H. Wei, S.-F. Hu and R.-S. Liu, Nano Energy, 2017, 32, 422–432 CrossRef CAS
- Y. Hou, Z. Zhu, Y. Xu, F. Guo, J. Zhang and X. Wang, J. Hydrogen Energy, 2017, 42, 2832–2838 CrossRef CAS
- S. Oh, J. B. Kim, J. T. Song, J. Oh and S.-H. Kim, J. Mater. Chem. A, 2017, 5, 3304–3310 RSC
- R. Fan, J. Mao, Z. Yin, J. Jie, W. Dong, L. Fang, F. Zheng and M. Shen, ACS Appl. Mater. Interfaces, 2017, 9, 6123–6129 CrossRef CAS
- L. A. King, T. R. Hellstern, J. Park, R. Sinclair and T. F. Jaramillo, ACS Appl. Mater. Interfaces, 2017, 9, 36792–36798 CrossRef CAS
- G. Huang, J. Mao, R. Fan, Z. Yin, X. Wu, J. Jie, Z. Kang and M. Shen, Appl. Phys. Lett., 2018, 112, 013902 CrossRef
- R. Fan, G. Huang, Y. Wang, Z. Mi and M. Shen, Appl. Catal., B, 2018, 237, 158–165 CrossRef
- M. Alqahtani, S. Sathasivam, F. Cui, L. Steier, X. Xia, C. Blackman, E. Kim, H. Shin, M. Benamara, Y. I. Mazur, G. J. Salamo, I. P. Parkin, H. Liua and J. Wu, J. Mater. Chem. A, 2019, 7, 8550–8558 RSC
- S. Seo, S. Kim, H. Choi, J. Lee, H. Yoon, G. Piao, J. C. Park, Y. Jung, J. Song, S. Y. Jeong, H. Park and S. Lee, Adv. Sci., 2019, 6, 1900301 CrossRef
- A. Hasani, Q. V. Le, M. Tekalgne, M.-J. Choi, T. H. Lee, S. H. Ahn, H. W. Jang and S. Y. Kim, ACS Appl. Mater. Interfaces, 2019, 11, 29910–29916 CrossRef
- R. Fan, J. Zhou, W. Xun, S. Cheng, S. Vanka, T. Cai, S. Ju, Z. Mi and M. Shen, Nano Energy, 2020, 71, 104631 CrossRef
- H. Li, C. Tsai, A. L. Koh, L. Cai, A. W. Contryman, A. H. Fragapane, J. Zhao, H. S. Han, H. C. Manoharan, F. Abild-Pedersen, J. K. Nørskov and X. Zheng, Nat. Mater., 2016, 15, 48–54 CrossRef CAS
- G. Ye, Y. Gong, J. Lin, B. Li, Y. He, S. T. Pantelides, W. Zhou, R. Vajtai and P. M. Ajayan, Nano Lett., 2016, 16, 1097–1103 CrossRef CAS
- H. Li, M. Du, M. J. Mleczko, A. L. Koh, Y. Nishi, E. Pop, A. J. Bard and X. Zheng, J. Am. Chem. Soc., 2016, 138, 5123–5129 CrossRef CAS
- Y. Yin, J. Han, Y. Zhang, X. Zhang, P. Xu, Q. Yuan, L. Samad, X. Wang, Y. Wang, Z. Zhang, P. Zhang, X. Cao, B. Song and S. Jin, J. Am. Chem. Soc., 2016, 138, 7965–7972 CrossRef CAS
- L. Lin, N. Miao, Y. Wen, S. Zhang, P. Ghosez, Z. Sun and D. A. Allwood, ACS Nano, 2016, 10, 8929–8937 CrossRef CAS
- Y. Sun, X. Zhang, B. Mao and M. Cao, Chem. Commun., 2016, 52, 14266–14269 RSC
- G. Li, D. Zhang, Q. Qiao, Y. Yu, D. Peterson, A. Zafar, R. Kumar, S. Curtarolo, F. Hunte, S. Shannon, Y. Zhu, W. Yang and L. Cao, J. Am. Chem. Soc., 2016, 138, 16632–16638 CrossRef CAS
- D. Gao, B. Xia, Y. Wang, W. Xiao, P. Xi, D. Xue and J. Ding, Small, 2018, 14, 1704150 CrossRef
- L.-B. Huang, L. Zhao, Y. Zhang, Y.-Y. Chen, Q.-H. Zhang, H. Luo, X. Zhang, T. Tang, L. Gu and J.-S. Hu, Adv. Energy Mater., 2018, 8, 1800734 CrossRef
- S. Park, J. Park, H. Abroshan, L. Zhang, J. K. Kim, J. Zhang, J. Guo, S. Siahrostami and X. Zheng, ACS Energy Lett., 2018, 3, 2685–2693 CrossRef CAS
- Q. Zhu, W. Chen, H. Cheng, Z. Lu and H. Pan, ChemCatChem, 2019, 11, 2667–2675 CrossRef CAS
- L. Li, Z. Qin, L. Ries, S. Hong, T. Michel, J. Yang, C. Salameh, M. Bechelany, P. Miele, D. Kaplan, M. Chhowalla and D. Voiry, ACS Nano, 2019, 13, 6824–6834 CrossRef CAS
- J. Lee, C. Kim, K. S. Choi, J. Seo, Y. Choi, W. Choi, Y.-M. Kim, H. Y. Jeong, J. H. Lee, G. Kim and H. Park, Nano Energy, 2019, 63, 103846 CrossRef CAS
- C. Hu, Z. Jiang, W. Zhou, M. Guo, T. Yu, X. Luo and C. Yuan, J. Phys. Chem. Lett., 2019, 10, 4763–4768 CrossRef CAS
- S. Lee, S. Cha, Y. Myung, K. Park, I. H. Kwak, I. S. Kwon, J. Seo, S. A. Lim, E. H. Cha and J. Park, ACS Appl. Mater. Interfaces, 2018, 10, 33198–33204 CrossRef CAS
- J. Hong, Z. Hu, M. Probert, K. Li, D. Lv, X. Yang, L. Gu, N. Mao, Q. Feng, L. Xie, J. Zhang, D. Wu, Z. Zhang, C. Jin, W. Ji, X. Zhang, J. Yuan and Z. Zhang, Nat. Commun., 2015, 6, 6293 CrossRef CAS
- Y. Guo, D. Liu and J. Robertson, Appl. Phys. Lett., 2015, 106, 173106 CrossRef
- J. Lee, S. Kang, K. Yim, K. Y. Kim, H. W. Jang, Y. Kang and S. Han, J. Phys. Chem. Lett., 2018, 9, 2049–2055 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available: Additional data. See DOI: 10.1039/d0tc04715e |
| ‡ I. H. K. and I. S. K equally contributed. |
| This journal is © The Royal Society of Chemistry 2021 |