
A new photo switchable azobenzene macrocycle without thermal relaxation at ambient temperature†
Nadi
Eleya‡
 abc,
Souvik
Ghosh‡
ab,
Enno
Lork
d and
Anne
Staubitz
*ab
abc,
Souvik
Ghosh‡
ab,
Enno
Lork
d and
Anne
Staubitz
*ab
aUniversity of Bremen, Institute for Organic and Analytical Chemistry, Leobener Str. 7, 28359 Bremen, Germany. E-mail: staubitz@uni-bremen.de
bUniversity of Bremen, MAPEX Center for Materials and Processes, Bibliothekstr. 1, 28359 Bremen, Germany
cDepartment of Chemistry, Faculty of Science, University of Zakho, KIR, Iraq
dUniversity of Bremen, Institute of Inorganic Chemistry and Crystallography, Leobener Str. 7, 28359 Bremen, Germany
First published on 9th December 2020
Abstract
A new switchable azobenzene macrocyclic derivative has been prepared in 4 steps with an overall yield of 47%. This molecule shows photoisomerization between the E and Z isomers using light of 365 nm and 405 nm respectively. What differentiates this macrocyclic switch from azobenzene is that the λmax for E (ππ*) and Z (nπ*) are well separated by 99 nm. The photostationary state (PSS) after irradiating by 365 nm LED light (measured at 415 nm) had a Z-selectivity of 81% Z and the PSS after irradiating by 405 nm LED light (measured at 316 nm) had an E-selectivity of 88% E. Both were reached after 90 s. The most unusual feature was that there was no thermal relaxation from Z to E observed at 20 °C over a period of 120 days, although photo switching was facile. Even at elevated temperatures of 70 °C and 90 °C, the thermal half-lives of the Z-isomer were as high as 36.4 hours and 2.35 hours respectively. Unusually, the macrocycle does not only photoisomerize in solution but also in the solid state. Therefore, this macrocycle could be useful for data storage and solar fuels applications.
Introduction
Photoactive switchable molecules reversibly isomerize with light of different wavelengths. As both isomers typically have very distinct, different properties, many (potential) applications have emerged in non-linear optics,1 data storage,2–4 biomolecules,5 phase changing materials,6 or making use of the photomechanical effect in liquid crystalline elastomers.7–12 Particularly azobenzenes are important photo switchable compounds. Thermodynamically, the (E)-azobenzene is more stable than the (Z)-azobenzene by 56 kJ mol−1.13 Irradiation of the (E)-azobenzene with light with a wavelength of 365 nm leads to the accumulation of (Z)-azobenzene with a Z-selectivity of 91%.14 The process is reversible both by irradiation with visible light or thermally.15 However, thermal relaxation processes from the Z to the E isomer compromise data storage applications, because a switched molecule will switch back with a certain statistical possibility, and randomly. Ideally, molecules that can be switched only by light are highly desirable. One way to increase life-times is to prepare tetra-ortho-fluoro substituted azobenzenes. The life-time in the longest-lived example was 700 days.16 Another way to extend thermal lifetimes is to incorporate azobenzenes in macrocycles and use geometrical constraints to slow down thermal relaxation. Such molecules might be used in applications in solar cells,17 material science,18 and photo sensing.19 However, even under these geometrical constraints, thermal relaxation takes place for the reported examples. In addition, cyclic azobenzenes can be difficult to prepare. There are reports, in which the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N coupling (reductive20 or oxidative21) is the ring closing step, or C–N-coupling.22 Alternatively, the ring can be closed at a site away from the azobenzene.23 Very few examples of 12 to 16-membered macrocyclic molecules with one azobenzene unit have been reported, and a few with two or more azobenzene units (for a list including photophysical properties in Tables S4 and S5 see ESI†). One reason for the fact that there are only few studies is that the yields for the ring closing step is typically below 40%, which is a severe limitation to the development of applications. Notable exceptions are ring closing reactions giving azobenzenophanes,24,25 or an unusual carbohydrate based macrocyclic azobenzene.26 A further complication for any comparison with the literature is that the switching behavior of many azobenzene macrocycles has often not been reported, mainly because other interesting properties (often complexations with ions) were the focus of the attention.25,27,28 Of some of these systems, the photoisomerization has been reported in detail.29 The thermal half-lives vary greatly: For example, for azobenzenophane-type crown ethers, in which the cyclic linker is highly flexible, the thermal half live is in the range of 3.0 × 10−11 d,30 whereas more rigid systems, with aromatic groups as part of the backbone linker, half-lives of minutes31 to even 8 d27 have been reported. Macrocycles with two azobenzene units are reported with low combined (i.e. Z,Z → Z,E → E,E) half-lives (less than a day32,33); but a remarkable system has been reported with a half live from Z,Z → E,Z with 1600 d.34
N coupling (reductive20 or oxidative21) is the ring closing step, or C–N-coupling.22 Alternatively, the ring can be closed at a site away from the azobenzene.23 Very few examples of 12 to 16-membered macrocyclic molecules with one azobenzene unit have been reported, and a few with two or more azobenzene units (for a list including photophysical properties in Tables S4 and S5 see ESI†). One reason for the fact that there are only few studies is that the yields for the ring closing step is typically below 40%, which is a severe limitation to the development of applications. Notable exceptions are ring closing reactions giving azobenzenophanes,24,25 or an unusual carbohydrate based macrocyclic azobenzene.26 A further complication for any comparison with the literature is that the switching behavior of many azobenzene macrocycles has often not been reported, mainly because other interesting properties (often complexations with ions) were the focus of the attention.25,27,28 Of some of these systems, the photoisomerization has been reported in detail.29 The thermal half-lives vary greatly: For example, for azobenzenophane-type crown ethers, in which the cyclic linker is highly flexible, the thermal half live is in the range of 3.0 × 10−11 d,30 whereas more rigid systems, with aromatic groups as part of the backbone linker, half-lives of minutes31 to even 8 d27 have been reported. Macrocycles with two azobenzene units are reported with low combined (i.e. Z,Z → Z,E → E,E) half-lives (less than a day32,33); but a remarkable system has been reported with a half live from Z,Z → E,Z with 1600 d.34
It is clear that if one wishes to achieve a high thermal stability of the metastable isomer (Z), combined with facile photoswitchability, a fine balance between flexible and inflexible chain segments in the ring needs to be struck.
This motivated us to design a medium sized azobenzene macrocycle, and to demonstrate its photophysical properties as well as its thermal relaxation properties. It is necessary to point out that our target molecule is structurally related to a diradical that has been postulated as a product of a photochemical enediyne cyclization, but of which was not isolated and of which no analysis exists.31
In this communication, we demonstrate a facile synthetic route to an ether bridged macrocyclic azobenzene molecule in high overall yield (47% over 4 steps from commercial starting materials). The design principle for the ring was to include enough flexibility to allow photo switching, because very rigid systems have been demonstrated to be either photochemically inert or very fast in their thermal relaxation.35–37 Too flexible rings on the other hand are expected to be too similar to unrestrained azobenzenes, which would preclude any benefit from a macrocyclic design.38 The photoisomerization behavior of this compound was very selective with good switching yields in the photostationary equilibria. What renders this compound particularly useful for future applications is its high thermal and photochemical stability, and especially the fact that thermal relaxation of the Z-state is entirely absent at ambient temperature. Moreover, it switches in the solid state.
Results and discussion
The synthesis started with the protection of commercially available amino phenol 1 with Boc anhydride (Boc2O) to obtain 2 in a yield of 71%, followed by a Williamson ether synthesis reaction with 1,2-bis(bromomethyl)benzene 3 (Scheme 1). Deprotection of 4, and an intramolecular cyclization oxidation reaction of compound 5 by MnO2 gave the azobenzene macrocycle 6(E) in a yield of 89%.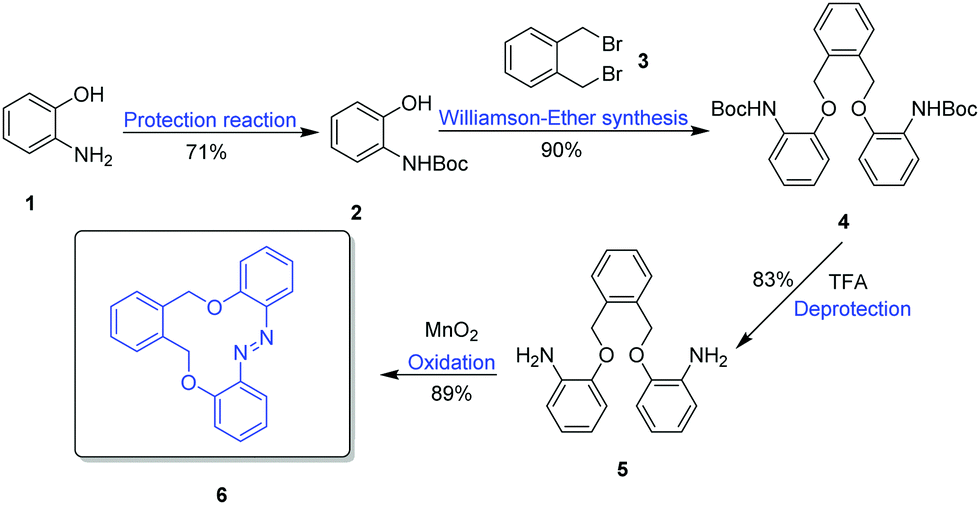 | ||
| Scheme 1 Synthetic route for the azobenzene macrocycle dioxadiazacyclododecine. | ||
The switching behavior was analyzed by UV-vis spectroscopic studies, and NMR studies. It was possible to isolate both the E and the Z isomers in pure form as crystals viewed under a microscope. Thus, the identity and structure of these products could be proved by single crystal X-ray crystallography; the thermal stability was analyzed by thermogravimetric analysis (TGA). A kinetic study on the thermal relaxation was performed by UV-vis and NMR spectroscopy.
Single crystal X-ray study
Both the E und Z form of the compound could be crystallized and analyzed by single crystal X-ray diffraction (for selected angles see Table 1). The E and Z form of compound 6 displayed central torsion angles for C1–N1N2–C7 of 172.52(2)° and 1.72(2)° respectively (Fig. 1a). This confirmed that in the E form, there was a severe distortion compared to unsubstituted azobenzene. In the E isomer, the π-systems of the aromatic rings and the NN double bond were almost disjunct. Upon switching, the shape of the cavity changed significantly, which was a direct consequence of the incorporated oxygen atoms that provided the ring with sufficient flexibility to perform this movement, which was observed by us for a similar molecule before.39 The crystal structure of the E isomer was orthorhombic (space group of P212121), whereas the crystal structure of Z-isomer was triclinic with the space group of P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Fig. 1b, see Table S2 in ESI†).
(Fig. 1b, see Table S2 in ESI†).
| 6(E) | 6(Z) | |
|---|---|---|
| Bond angles/° | ||
| N2–N1–C1 | 112.51(2) | 120.37(2) |
| N1–N2–C7 | 114.36(2) | 121.40(2) |
| C2–C1–N1 | 114.90(2) | 119.16(2) |
| O1–C6–C5 | 122.05(2) | 124.10(2) |
| O2–C12–C11 | 112.99(2) | 125.42(2) |
| O2–C12–C7 | 128.37(2) | 114.61(2) |
| Torsion angles/° | ||
| C1–N1–N2–C7 | 172.51(2) | 1.72(2) |
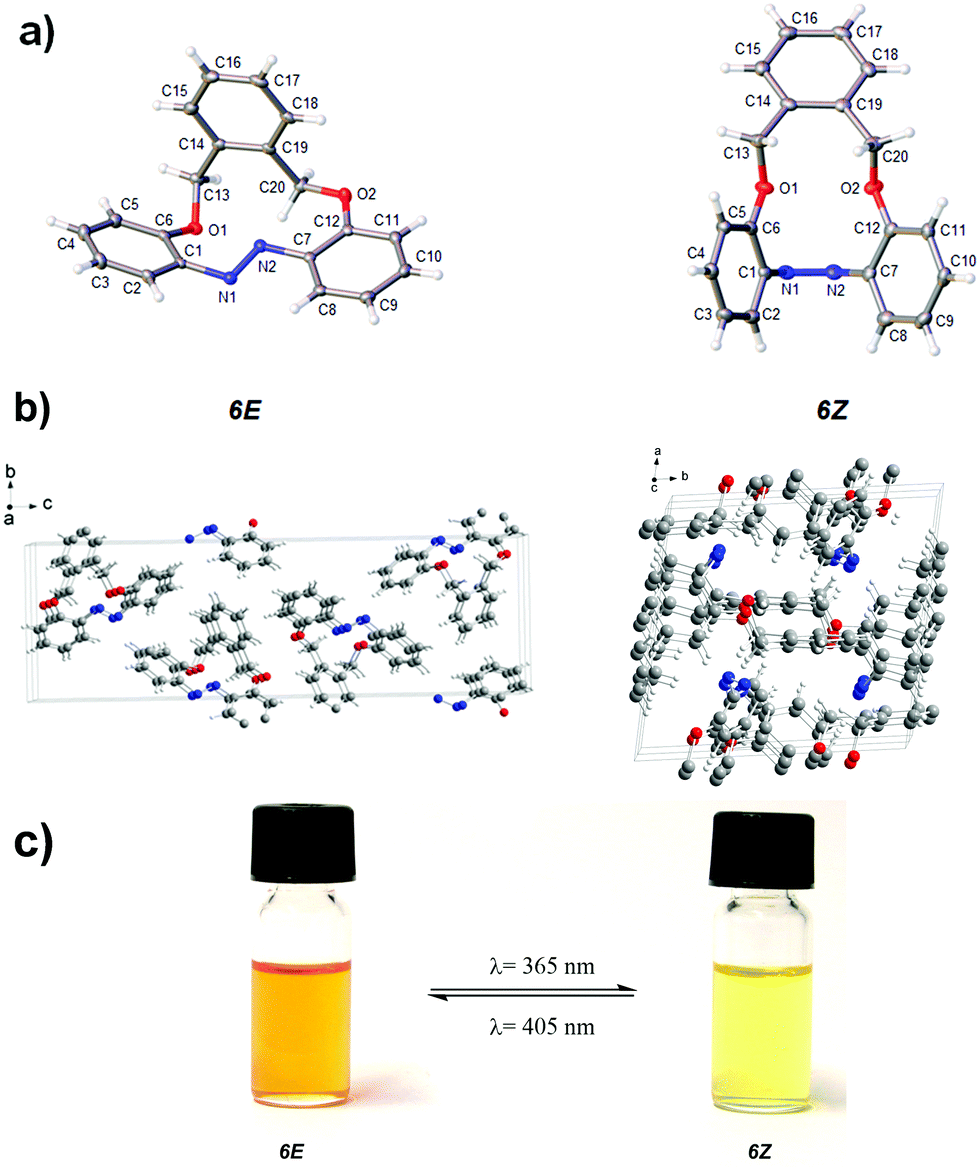 | ||
| Fig. 1 (a) and (b) X-ray structures of 6(E) and 6(Z); (c) Photochromism in acetonitrile. The samples were irradiated with LEDs of the wavelength of 365 nm and 405 nm respectively. | ||
UV-vis spectroscopic study
The photoisomerization between 6(E) and 6(Z) was investigated using dilute sample solutions in acetonitrile. The photoisomerization of 6(E) to 6(Z) was accomplished by irradiating the solution of 6(E) using a LED with wavelength of 365 nm for 90 s. The color change from bright orange to glossy yellow was so strong that it was visible to the naked eye (Fig. 1c). The thermodynamically stable 6(E) exhibited an absorption maximum at 316 nm, which was attributed to a π–π* transition, and a further absorption maximum at 450 nm, which arises from a n–π* transition. On the other hand, 6(Z) exhibited a π–π* absorption maximum at 290 nm and a n–π* absorption maximum at 415 nm (Fig. 2a). Such a difference in absorption has also been observed for very small azobenzene containing cycles, for example diazocine, which also shows very distinct differences between the maxima for the E and Z isomers and for which high selectivities can be achieved in the photostationary state.40,416(Z) was switched back to 6(E) by using irradiation with an LED light at 405 nm for 90 s. There was no evidence of any thermal relaxation to the E-isomer even after more than 120 days (see Fig. S13 in ESI†).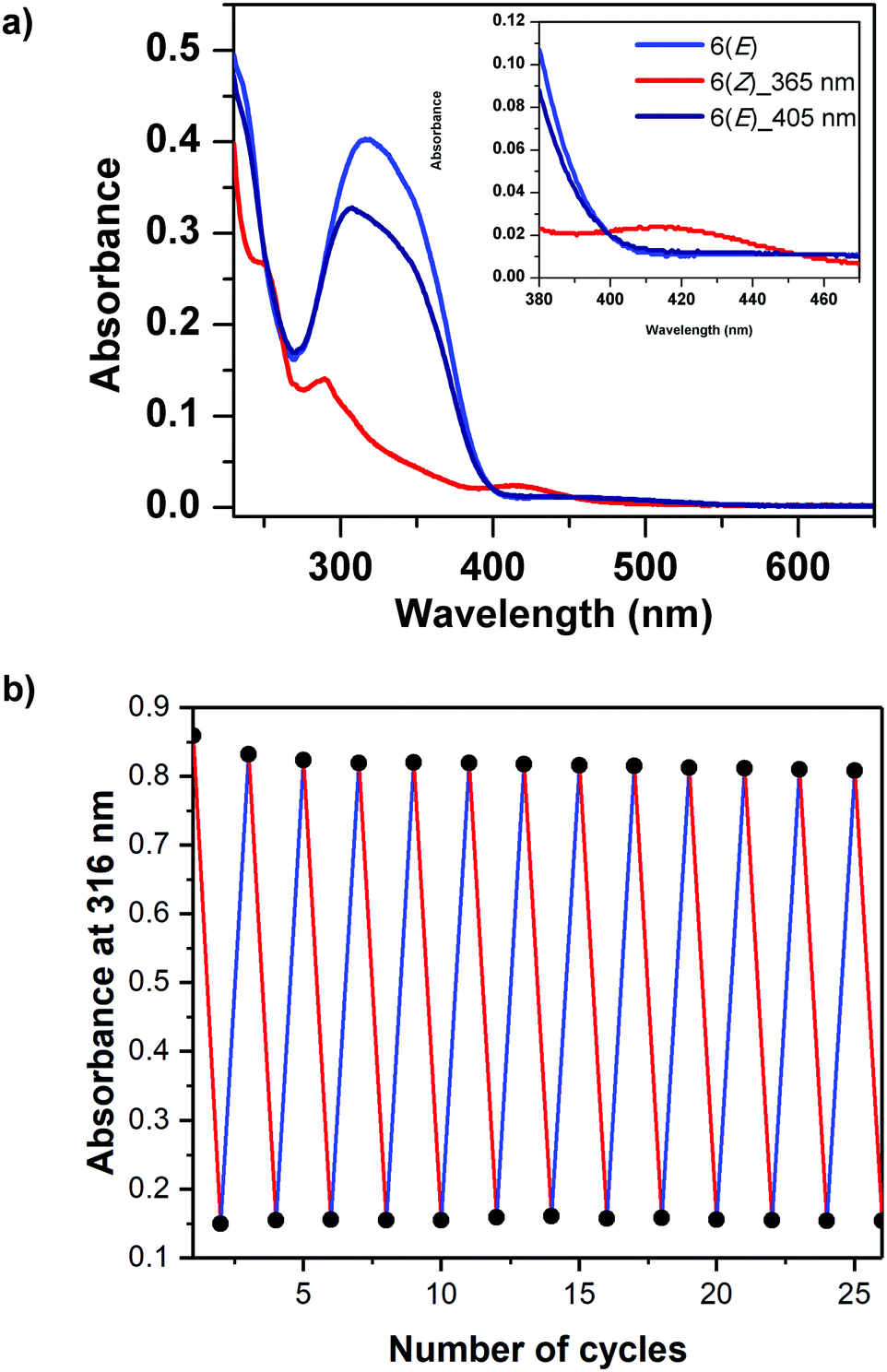 | ||
| Fig. 2 (a) Light blue line: UV/vis spectrum (0.025 mg mL−1 in acetonitrile) of 6(E) as synthesized; red line: after irradiation of this sample using a 365 nm LED light (giving mostly 6(Z)); dark blue line: after irradiation of 6(Z) using a 405 nm LED light. Inset: Enlargement of the visual region of the spectrum. (b) Absorptions at 316 nm with alternating irradiations at 365 and 405 nm demonstrating reversible photoisomerization cycles of compound 6 (0.035 mg mL−1 in acetonitrile). | ||
In addition, the solid form of 6(E) was irradiated under UV light (365 nm LED light). This led to a color change from orange to bright yellow (Fig. S18b in the ESI†). UV-vis spectra measured of the powder in reflection mode revealed that 6(E) had an absorption maximum at 326 nm, which was attributed to a π–π* transition and is slightly red shifted compared to the absorption in solution (Fig. S18a in the ESI†). Furthermore, there was a n–π* absorption maximum of low intensity. The other isomer, 6(Z), exhibited a π–π* absorption maximum at 289 nm and a n–π* absorption maximum at 355 nm, which is substantially hypsochromically shifted compared to the solution state (Fig. S18a in the ESI†).
This new macrocyclic azobenzene proved to have a high cycle stability: The photoisomerization between 6(E) and 6(Z) in solution was cycled by alternating irradiation using wavelengths of 365 nm and 405 nm respectively, for 26 cycles without appreciable photo degradation (Fig. 2b).
NMR study
The 1H NMR spectra of 6(E) and 6(Z) were characterized by a change in the chemical shifts of the aromatic protons during photoisomerization. For instance, in a 2.5 mg mL−1 solution of CD3CN, the Hb signal of compound 6(E) shifted from 7.78 ppm (dd, J = 7.9, 1.7 Hz) to 6.61 ppm (dd, J = 7.7, 1.6 Hz) for 6(Z) and signal of He of 6(E) shifted from 7.43 ppm (dd, J = 8.1, 1.1 Hz) to 7.02 ppm (dd, J = 7.9, 1.1 Hz) for 6(Z) (Fig. 3).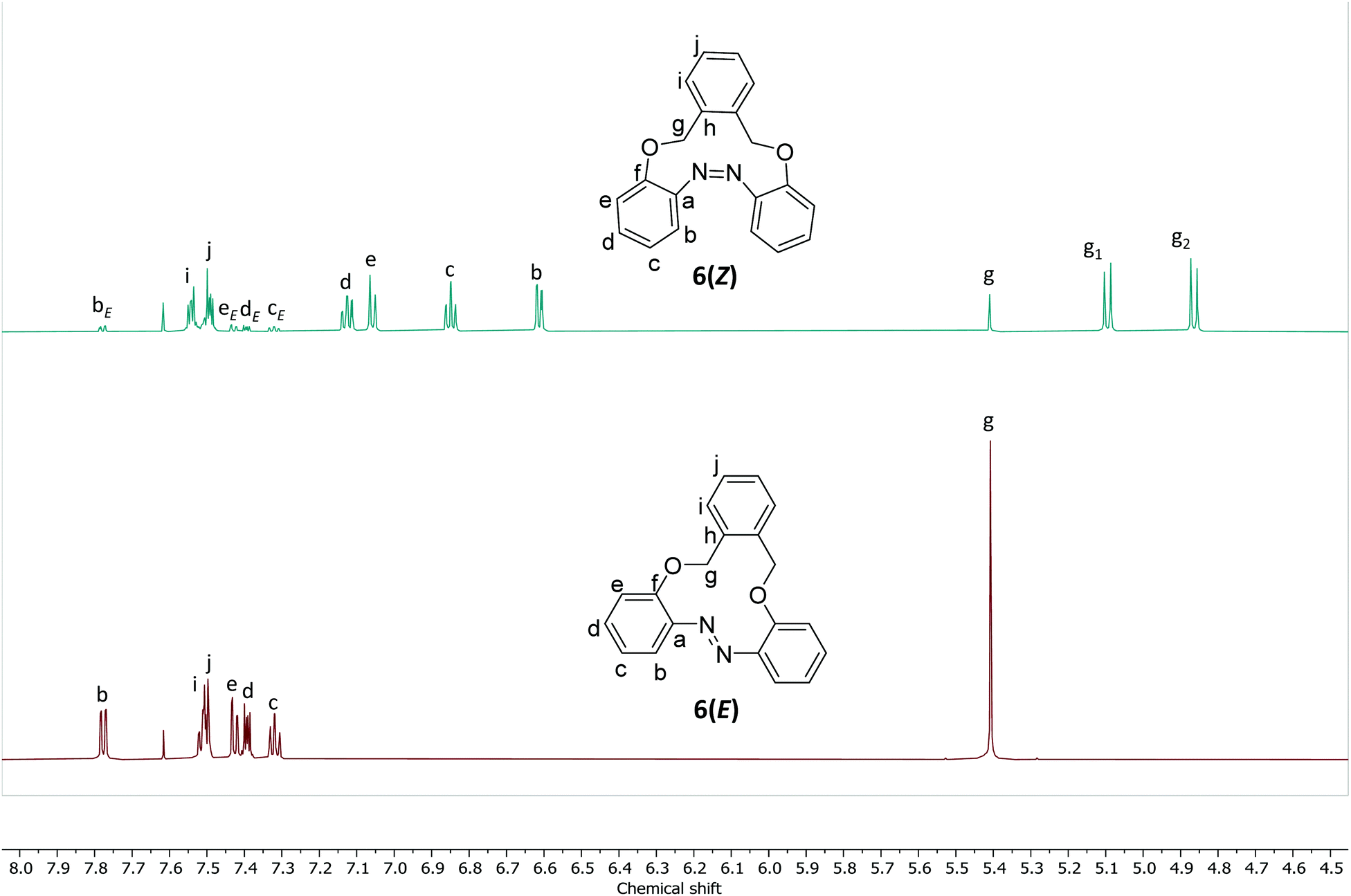 | ||
| Fig. 3 1H NMR spectrum of compound 6 (2.5 mg mL−1 in CD3CN) before and after irradiation with light at λ = 365 nm. | ||
The most interesting finding was the thermal behavior of the Z-isomer: Typically, Z-azobenzenes do not only convert photochemically to E-azobenzenes, but also thermally. After this solution of 6(E) in CD3CN was irradiated by 365 nm LED light for 15 minutes, the ratio of 6(E)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6(Z) was 19:81. This solution was kept in the dark at 20 °C and 1H NMR spectrum was measured once in every 10 days. There was no evidence of any thermal relaxation to the E-isomer even after 120 days. However, if the same solution was then irradiated with light at 405 nm for 30 mins, the 1H NMR spectrum showed that the compound switched back to the E-isomer and the ratio of 6(E):6(Z) was 88:12.
:6(Z) was 19:81. This solution was kept in the dark at 20 °C and 1H NMR spectrum was measured once in every 10 days. There was no evidence of any thermal relaxation to the E-isomer even after 120 days. However, if the same solution was then irradiated with light at 405 nm for 30 mins, the 1H NMR spectrum showed that the compound switched back to the E-isomer and the ratio of 6(E):6(Z) was 88:12.
Thermal relaxation at elevated temperatures
The thermal relaxation of compound 6(Z) was measured by NMR and UV-vis spectroscopy at 70 °C and 90 °C. For the NMR study, compound 6(E) was dissolved in CD3CN in a hermetically sealed NMR tube and was irradiated with LED light at a wavelength of 365 nm for 15 min. Subsequently, the NMR tubes were heated to the respective temperatures in water baths shielded from light and a 1H NMR spectrum was measured every 60 min. The thermal backswitching of the Z-isomer to the E-isomer was observed by following the changes of the aromatic proton Hb signals (see Fig. 3 for the assignment) in the 1H NMR spectra over time (see Fig. S15 and S16 in ESI†). The E to Z ratio was calculated by the integration of the chemically shifted proton signals in the 1H NMR spectra. Thermal relaxation of 6(Z) to 6(E) was accomplished at 90 °C within four hours following a first order kinetics with the rate constant (k) 0.393 h−1. At 70 °C, the rate constant (k) was found to be 0.019 h−1. From the kinetics study, half-lives for thermal relaxation of 6(Z) to 6(E) were determined to be 36.4 h and 1.76 h at 70 °C and 90 °C respectively (see Fig. S15 and S16 in ESI†).For the UV-vis study, diluted solutions of compound 6(E) in acetonitrile were prepared to record the UV-vis absorption spectra during thermal relaxation at 70 °C and 90 °C. The samples were irradiated with LED light at 365 nm wavelength for 90 s and then placed in water baths shielded form light at 70 °C and 90 °C. Compound 6(Z) switched back to the E form at 70 °C and 90 °C with rate constants of 0.029 h−1 and 0.294 h−1 respectively (see Fig. S17 in ESI†). The half-lives were 23.8 h at 70 °C and 2.35 h at 90 °C respectively.
The kinetics observed in the NMR study and the UV-vis study show slight differences in the absolute numbers, but they are of the same order of magnitude. This is not unusual for azobenzenes.42
Thermogravimetric analysis
For many materials applications, the overall thermal stability is important. The decomposition temperature of this compound was investigated by thermogravimetric analysis (TGA), which showed an onset temperature of decomposition of 210 °C (see Fig. S9 in ESI†).Conclusions
In conclusion, we report the synthesis of a new azobenzene macrocycle (dioxadiazacyclododecine) in high yield. It shows reversible photo switching between the E and the Z form for more than 26 cycles. It also exhibits solid state photoisomerization. This compound is thermally highly stable in its E form. The new strategy of incorporating azobenzenes in semi-flexible small macrocycles to prevent thermal relaxation has been shown to be successful: No thermal relaxation from Z to the E form was observed at 20 °C over a period of 120 days. Even for elevated temperatures, the half-lives were comparatively long with 36.4 hours, 1.76 hours (by NMR spectroscopy) and 23.8 hours, 2.35 hours (by UV-vis spectroscopy) at 70 °C at 90 °C respectively. This has important implications for real life applications in all applications, in which the E and the Z form need to be addressed precisely, with no loss of either information (such as in data storage) or in energy (for example solar fuels).Conflicts of interest
There are no conflicts to declare.Acknowledgements
Nadi Eleya would like to thank Philipp Schwartz Initiative of the Alexander von Humboldt foundation and the international office of Bremen University for their support.Notes and references
- S. K. Yesodha, C. K. Sadasivan Pillai and N. Tsutsumi, Prog. Polym. Sci., 2004, 29, 45–74 CrossRef CAS.
- R. H. Berg, S. Hvilsted and P. S. Ramanujam, Nature, 1996, 383, 505–508 CrossRef CAS.
- S. Hvilsted, C. Sanchez and R. Alcala, J. Mater. Chem., 2009, 19, 6641–6648 RSC.
- S. J. Zilker, T. Bieringer, D. Haarer, R. S. Stein, J. W. Van Egmond and S. G. Kostromine, Adv. Mater., 1998, 10, 855–859 CrossRef CAS.
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- W.-C. Xu, S. Sun and S. Wu, Angew. Chem., Int. Ed., 2019, 58, 9712–9740 CrossRef CAS PubMed.
- C. J. Barrett, J.-I. Mamiya, K. G. Yager and T. Ikeda, Soft Matter, 2007, 3, 1249–1261 RSC.
- J. Cviklinski, A. R. Tajbakhsh and E. M. Terentjev, Eur. Phys. J. E, 2002, 9, 427–434 CrossRef CAS PubMed.
- E. Kizilkan, C. F. Schaber, S. N. Gorb, J. Strueben, X. Jin, R. Adelung and A. Staubitz, R. Soc. Open Sci., 2016, 3, 150700 CrossRef PubMed.
- E. Kizilkan, J. Strueben, A. Staubitz and S. N. Gorb, Sci. Robot., 2017, 2, 9454 CrossRef PubMed.
- T. J. White, S. V. Serak, N. V. Tabiryan, R. A. Vaia and T. J. Bunning, J. Mater. Chem., 2009, 19, 1080–1085 RSC.
- M. Yamada, M. Kondo, J.-I. Mamiya, Y. Yu, M. Kinoshita, C. J. Barrett and T. Ikeda, Angew. Chem., Int. Ed., 2008, 47, 4986–4988 CrossRef CAS PubMed.
- E. Wei-Guang Diau, J. Phys. Chem. A, 2004, 108, 950–956 CrossRef.
- E. Fischer, M. Frankel and R. Wolovsky, J. Chem. Phys., 1955, 23, 1367 CrossRef CAS.
- T. Schultz, J. Quenneville, B. Levine, A. Toniolo, T. J. Martínez, S. Lochbrunner, M. Schmitt, J. P. Shaffer, M. Z. Zgierski and A. Stolow, J. Am. Chem. Soc., 2003, 125, 8098–8099 CrossRef CAS PubMed.
- D. Bléger, J. Schwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597–20600 CrossRef PubMed.
- Y. Jiang, J. Huang, W. Feng, X. Zhao, T. Wang, C. Li and W. Luo, Sol. Energy Mater. Sol. Cells, 2019, 193, 198–205 CrossRef CAS.
- T. Ikeda and O. Tsutsumi, Science, 1995, 268, 1873 CrossRef CAS PubMed.
- J. P. Van der Berg, W. A. Velema, W. Szymanski, A. J. M. Driessen and B. L. Feringa, Chem. Sci., 2015, 6, 3593–3598 RSC.
- S. Li, G. Han and W. Zhang, Macromolecules, 2018, 51, 4290–4297 CrossRef CAS.
- G. Cabre, A. Garrido-Charles, A. Gonzalez-Lafont, W. Moormann, D. Langbehn, D. Egea, J. M. Lluch, R. Herges, R. Alibes, F. Busque, P. Gorostiza and J. Hernando, Org. Lett., 2019, 21, 3780–3784 CrossRef CAS PubMed.
- S. Li, N. Eleya and A. Staubitz, Org. Lett., 2020, 22, 1624–1627 CrossRef CAS PubMed.
- A. Lyapunov, T. Kirichenko, C. Kulygina, R. Zubatyuk, M. Fonari, A. Kyrychenko and A. Doroshenko, J. Incl. Phenom. Macro., 2015, 81, 499–508 CrossRef CAS.
- H.-M. Kang, J.-W. Jung and C.-G. Cho, J. Org. Chem., 2007, 72, 679–682 CrossRef CAS PubMed.
- K. Takaishi, M. Kawamoto, A. Muranaka and M. Uchiyama, Org. Lett., 2012, 14, 3252–3255 CrossRef CAS PubMed.
- C. Lin, S. Maisonneuve, R. Métivier and J. Xie, Chem. – Eur. J., 2017, 23, 14996–15001 CrossRef CAS PubMed.
- E. Luboch, E. Wagner-Wysiecka, Z. Poleska-Muchlado and V. C. Kravtsov, Tetrahedron, 2005, 61, 10738–10747 CrossRef CAS.
- J. F. Luboch, Y. A. Simonov and A. A. Dvorkin, Tetrahedron, 1998, 54, 4977–4990 CrossRef.
- T. Yamamoto, D. Nakamura, G. Liu, K. Nishinaka and A. Tsuda, J. Photochem. Photobiol., A, 2016, 331, 66–75 CrossRef CAS.
- S. Shinkai, T. Minami, Y. Kusano and O. Manabe, J. Am. Chem. Soc., 1983, 105, 1851–1856 CrossRef CAS.
- M. Kar, A. Basak and M. Bhattacharjee, Bioorg. Med. Chem. Lett., 2005, 15, 5392–5396 CrossRef CAS PubMed.
- Y. Norikane, R. Katoh and N. Tamaoki, Chem. Commun., 2008, 1898–1900, 10.1039/B718813G.
- E. Uchida, K. Sakaki, Y. Nakamura, R. Azumi, Y. Hirai, H. Akiyama, M. Yoshida and Y. Norikane, Chem. – Eur. J., 2013, 19, 17391–17397 CrossRef CAS PubMed.
- S. A. Nagamani, Y. Norikane and N. Tamaoki, J. Org. Chem., 2005, 70, 9304–9313 CrossRef CAS PubMed.
- A. H. Heindl, J. Becker and H. A. Wegner, Chem. Sci., 2019, 10, 7418–7425 RSC.
- A. H. Heindl, L. Schweighauser, C. Logemann and H. A. Wegner, Synthesis, 2017, 2632–2639 CAS.
- C. Slavov, C. Yang, A. H. Heindl, T. Stauch, H. A. Wegner, A. Dreuw and J. Wachtveitl, J. Phys. Chem. Lett., 2018, 9, 4776–4781 CrossRef CAS PubMed.
- M. Dowds, D. Bank, J. Strueben, D. P. Soto, F. D. Soennichsen, F. Renth, F. Temps and A. Staubitz, J. Mater. Chem. C, 2020, 8, 1835–1845 RSC.
- N. Eleya, C. Appiah, E. Lork, M. Gogolin, T. M. Gesing, T. Stauch and A. Staubitz, Molecules, 2020, 25, 283 CrossRef CAS PubMed.
- M. Hammerich, C. Schütt, C. Stähler, P. Lentes, F. Röhricht, R. Höppner and R. Herges, J. Am. Chem. Soc., 2016, 138, 13111–13114 CrossRef CAS PubMed.
- R. Siewertsen, H. Neumann, B. Buchheim-Stehn, R. Herges, C. Näther, F. Renth and F. Temps, J. Am. Chem. Soc., 2009, 131, 15594–15595 CrossRef CAS PubMed.
- D. Samanta, J. Gemen, Z. Chu, Y. Diskin-Posner, L. J. W. Shimon and R. Klajn, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9379–9384 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2021638 and 2021639. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0tc05211f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |