
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA submersible phosphate analyzer for marine environments based on inlaid microfluidics†
Sean
Morgan
 a,
Edward
Luy
b,
Arnold
Furlong
b and
Vincent
Sieben
*a
a,
Edward
Luy
b,
Arnold
Furlong
b and
Vincent
Sieben
*a
aDepartment of Electrical and Computer Engineering, Dalhousie University, 1360 Barrington Street, Halifax, Nova Scotia B3H 4R2, Canada. E-mail: sieben@dal.ca
bDartmouth Ocean Technologies Inc., 25 Parker Street, Suite 21401, Dartmouth, Nova Scotia B2Y 4TS, Canada
First published on 1st December 2021
Abstract
In situ sensors are needed to further our understanding of phosphate flux dynamics in marine environments during short term events such as tidal cycles, algae blooms and runoff periods. Here, we present a fully automated in situ phosphate analyzer based on an inlaid microfluidic absorbance cell technology. The microfluidic device employs colorimetric absorbance spectrophotometry, using the phosphomolybdenum blue (PMB) assay modified by the addition of polyvinylpyrrolidone (PVP), to measure phosphate concentrations in seawater. Bench top calibrations were performed with both copper(II) sulfate dye and the PMB assay, as well as temperature sensitivity studies to characterize the sensor's performance in a range of conditions. It achieves a limit of detection of 15.2 nM, a limit of quantification of 50.8 nM, and a high in situ precision with a relative standard deviation of less than 1.5% across three consecutive measurements. Two consecutive field deployments are conducted as assessments for its intended in situ applications. The sensor is first deployed from a pier at a depth of 6 m, with simultaneous bottle samples taken to perform cross-validation. It is next deployed on the Stella Maris testbed, a multi-sensor seabed platform (MSSP), 100 m offshore and 9 m deep in the inlet to the Bedford Basin in Nova Scotia, Canada. Over 300 successful phosphate measurements were acquired, showing the influence of the tidal cycle, and confirming the sensor's viability in observing nutrient flux dynamics with nanomolar variations.
1. Introduction
Orthophosphate, as well as other forms of phosphorus, is an integral component of the energy storage mechanisms of phytoplankton1 and is therefore considered a key limiting nutrient for phytoplankton growth in many ecosystems.2 Additionally, there are species that have adapted to utilize phosphate to bloom under low nitrate conditions in marine environments.3 Furthermore, nutrient pollution from increased levels of anthropogenic phosphate flux in freshwater or coastal regions can lead to blooms of toxic phytoplankton species such as Karenia brevis.4 To address this, governing bodies have implemented restrictions on allowable levels of phosphate loading from industrial effluent. For example, the Wisconsin Department of Natural Resources in the United States have set a limit of phosphate concentration in effluent to 1 mg L−1 total phosphorus as a monthly average from public and private wastewater sites.5 Furthermore, the Canadian Water Network reports that Canada has implemented provincial restrictions ranging from 0.1 to 1 mg L−1 total phosphorus and less than 0.5 mg L−1 (5.3 μM) ortho-phosphate in effluent.6 Accurate and automatic in situ phosphate analyzers are required to ensure that these policies are effectively realized, and to quantify the effects of anthropogenic phosphate loading on marine environments.There are several well-documented approaches to measuring environmental phosphate in water, including electrochemical techniques,7 biomimetic receptor technologies,8 and fluorescence probes.9 Of the available analytical techniques, the most widely used method for determining orthophosphate concentrations in seawater is flow analysis using reagent-based colorimetry.10,11 The two common reagents used to accomplish this are the yellow vanadomolybdate method and the phosphomolybdenum blue method. There are commercial instruments, such as the Alyza IQ PO4 (YSI Inc., Ohio, United States) that use the yellow vanadomolybdate reagent for phosphate detection. It has the advantage of having a long shelf life, and requires only a single reagent.12 However, it lacks sensitivity compared to the blue method and is considered highly toxic due to the vanadate in the reagent. The more sensitive and less dangerous molybdenum blue phosphate reagent, first introduced by Murphy and Riley in 1962,13 is the more often used alternative. Since 1971, it has been recognized as the Environmental Protection Agency standard (EPA-NERL 365.2) for phosphate determination in water.14 There have been many different variations of the method since its creation, but the basic mechanism occurs in two stages.15 The first stage involves the reaction of the phosphate ion with molybdate under acidic conditions to form a Keggin ion (polyoxometalate species). This molecule is then reduced using an application specific agent in the second stage to form the phosphomolybdenum blue (PMB) assay. Phosphate concentrations are then determined by employing absorbance spectrophotometry on the final PMB complex.
In the laboratory, the spectrophotometric analytical technique for the PMB assay is accomplished using bench top systems, like auto-analyzers, due to their high precision (<1% relative standard deviation), high accuracy (<2% relative error) and low limits of detection (<20 nM for phosphate).16 However, this is an ex situ approach that requires the sample to be collected and removed from the environment. For open ocean sampling, this requires large financial overhead of approximately 10–15 K CAD per day for the ship and crew, and several hours between sample collection and analysis. Furthermore, the samples require on site manual filtering and near immediate freezing to be properly preserved. These considerations severely limit the maximum possible spatial and temporal resolution of the technique, making it difficult to capture short term events such as tidal cycles. In situ sensors provide an effective alternative as they can make more frequent measurements (several samples per hour) directly in the environment.
The HydroCycle PO4 (Sea-Bird Scientific, Washington, United States) is an example of a widely used in situ commercial phosphate analyzer that uses the molybdenum blue method. It is capable of making up to 4 measurements per hour with a detection limit of 24 nM, however it generates 30 mL of waste and consumes from 1.6–2.5 kJ of energy per sample.17 Microfluidic devices on the other hand can perform laboratory analytical techniques such as spectrophotometric analysis on a miniature scale, making them ideal for in situ instruments in environments where power, weight and reagents need to be conserved. Several groups around the world have developed these instruments, with wide reaching applications including nutrient detection. Duffy et al., based out of Dublin, have combined centrifugal microfluidics with absorption spectrophotometry to create an on site phosphate analyzer.18 Donohoe et al. have utilized 3D printing technology to build a phosphate sensing platform based on the yellow vanadomolybdate method,19 and Zhu et al. are looking at using cavity enhancement techniques to perform absorption measurements with limits of detection (LODs) comparable to those of the auto-analyzers.20 In 2010, Sieben et al. explored the use of tinted absorbance cells21–23 to perform in situ absorption measurements applied to nutrient sensing. Since then, the Mowlem group at the National Oceanography Center (NOC) UK, has built on the tinted absorbance cell technology to deploy a nitrite sensor on the seafloor at a depth of 170 m in 2015,24 they have measured nitrate from glacial runoff in Greenland over 14 days in 2017,25 and have incorporated a nitrate sensor onto an autonomous glider to obtain high resolution measurements in the Celtic Sea in 2018.26 Recently, the NOC team have used a phosphate analyzer to identify relationships between nutrients and flow rate in a riverine environment over the course of 9 weeks in 2017,27 and have also deployed a phosphate sensor for 44 days on an autonomous glider to measure the variability of the nutricline in the North Sea in 2021.28 Although the tinted absorbance cell has made it possible to obtain data across a range of deployment environments, it requires the optical electronics of the sensor to be carefully aligned and physically secured to the microfluidic chip via an adhesive, thereby restricting serviceability, easy replacement, and limits reconfigurability. Furthermore, the tinted material inhibits visual inspection for flaws or delamination created during or after the chip manufacturing process. Lastly, the microfluidic devices made from the semi-transparent acrylic cannot be readily adapted to perform other analytical techniques such as fluorescence measurements or cavity ring down spectroscopy (CRDS) that rely on transparent window/optical interfaces.
Here we present a fully automated in situ phosphate analyzer that incorporates our previously reported inlaid microfluidic absorbance cell,29 allowing the electronics to be entirely decoupled from the microfluidic chip through use of embedded beam-steering optical components. We interface our novel chip approach to a custom-made and ruggedized sensor instrument with 3-syringe pumps, 10-valves, and 4-optical measurement channels for enabling a wide range of chemical assays. Additionally, the 3 separate syringe pumps and 10 valves provide increased reconfigurability that can allow techniques such as continuous flow analysis (CFA) and flow injection analysis (FIA)30 to be performed by only replacing the microfluidic chip and modifying the protocol. Furthermore, the transparent windows in the chip enable the ability to perform improved absorption measurements through cavity enhanced absorption spectroscopy (CEAS) and multi-bounce configurations.31 In this report, the sensor uses the well documented PMB assay, but with the addition of polyvinylpyrrolidone (PVP) to inhibit aggregation, decrease temperature sensitivity, and lower the limit of detection.27,32,33 The temperature sensitivity of the absorbance measurements is determined through 4 sequential calibrations done at even temperature intervals between 5 °C and 20 °C. A first standalone deployment from a near-shore pier shows data captured over a period of 36 hours, with bottle samples taken for verification. A second deployment on the Stella Maris multi-sensor seabed platform (MSSP), 100 m offshore in the Bedford Basin, Nova Scotia, Canada (44.66° N, −63.56° W) demonstrates the analyzer's ability to integrate with platforms or vehicles. We anticipate the successful deployment of the fully automated phosphate sensor presented here will permit a readily expandable platform to perform a range of colorimetric chemistries for widespread in situ observations by changing the microfluidic chip, reagents, and automation protocol.
2. Materials and methods
2.1 Chemical preparation
All chemicals and reagents were supplied by Fisher Chemical (New Jersey, United States), unless otherwise stated. The PMB assay was prepared as two reagents: an ammonium molybdate solution and an ascorbic acid solution. To prepare the first reagent, 0.3005 g of potassium antimonyl tartrate (C8H10K2O15Sb2·3H2O, CAS-no. 28300-74-5, Sigma-Aldrich, Missouri, United States) was first dissolved in 100 mL of Milli-Q and set aside. Next, 0.56 g of ammonium molybdate(VI) tetrahydrate (H24Mo7N6O24·4H2O, CAS-no. 12054-85-2) was dissolved in 200 mL Milli-Q in a 1 L volumetric flask. After that, 6.73 mL of concentrated sulfuric acid (H2SO4, CAS-no. 7664-93-9, EMD Millipore, Darmstadt, Germany) was added to the volumetric flask, which was then allowed to cool to room temperature. Lastly, 12.5 mL of the antimonyl potassium tartrate solution was added to the flask, which was then filled to volume with Milli-Q.The second reagent was prepared by dissolving 10 g of L-ascorbic acid (C6H8O6, CAS-no. 50-81-7, Sigma Aldrich, Missouri, United States) in roughly 600 mL of Milli-Q in a 1 L volumetric flask. Next, 0.10 g of polyvinylpyrrolidone (PVP) ((C6H9NO)n, CAS-no 9003-39-8, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, Sigma Aldrich, Missouri, United States) was added to the volumetric flask and mixed in by shaking. Finally, the solution was diluted to the 1 L mark with Milli-Q and shaken again to mix.
000 g mol−1, Sigma Aldrich, Missouri, United States) was added to the volumetric flask and mixed in by shaking. Finally, the solution was diluted to the 1 L mark with Milli-Q and shaken again to mix.
The 1000 μM phosphate standard stock solution was prepared by dissolving 0.1361 g of potassium phosphate monobasic (KH2PO4, CAS-no. 7778-77-0) in 1 L of Milli-Q. The remaining standards were prepared by performing a serial dilution of the appropriate portion of the stock solution with Milli-Q. Both reagents for the PMB assay and the phosphate standard were used for 2–3 months with no observable difference in the color development profiles.
The 50 μM copper(II) sulfate dye stock solution was prepared by dissolving 1.2484 g of copper(II) sulfate pentahydrate (CuSO4·5H2O, CAS-no. 7758-99-8, Sigma Aldrich, Missouri, United States) in 100 mL of Milli-Q and then shaking to mix. The remaining standards were then prepared by again performing a serial dilution of the appropriate portion of stock solution with Milli-Q.
2.2 Microfluidic chip fabrication
The substrate for the microfluidic chip (88.9 mm in diameter and 12 mm thick) was made from extruded opaque black poly(methyl methacrylate) (PMMA) (9M001, Acrylite, Maine, United States) inlaid into extruded clear PMMA (0A000, Acrylite, Maine, United States), according to the method documented by Luy et al.29 A micromill (S103, LPKF Laser and Electronics, Garbsen, Germany) was used to mill the 400 μm deep square channels and other features, and a 50 W laser cutter (Mini Helix, Epilog Laser, Ontario, Canada) was used to cut the chips and inlaid pieces from the larger acrylic sheets. To bond the two halves of the microfluidic chip after the inlaying and milling process was complete, each side was first exposed to chloroform vapour for 45 seconds. They were then slotted into a custom fitting jig before being pressed in a PCB press (MultiPress S, LPKF Laser and Electronics, Garbsen, Germany) at a pressure of 625 N cm−2 and a temperature of 85 °C for 2.25 hours.2.3 Sensor description
The underlying analytical technique for phosphate detection is carried out using the microfluidic chip and its integral absorbance cells. Fig. 1A is a fluid flow diagram highlighting the internal and external processes of the chip. Depending on the stage in the protocol, either the sample or standard syringe is actuated, synchronized with the 2 reagent syringes. The sample is pulled from the environment (seawater) through a 0.45 μm hydrophilic filter membrane to remove larger particulate material and sediment. Filter blockage and clogging is one of the limiting factors for extended deployments, which is dependent on the particulate loading of the environment. However, 1–2 months deployments in marine environments with comparably sized filters has been demonstrated.27,28 The syringes for the sample and standard move fluid at a rate of 2 mL per minute, and the two reagent syringes move fluid at a rate of 1 mL per minute but are always actuated together, resulting in a combined reagent flow of 2 mL per minute. The intake valves (to the left side of the syringes) open as the syringes withdraw, and then close before the output valves (to the right side of the syringes) open for the syringes to dispense. The two reagents mix through diffusion in the first serpentine mixer before mixing with the third fluid in the second serpentine channel to realize 4 mL per minute total flow. The mixed fluid is pumped into the two absorbance cells (short path = SP, long path = LP) and flow is then stopped to allow a colorimetric reaction to take place. Afterwards the fluid is displaced by the next stage and dispensed through one of the two waste ports. One port is connected to the waste bag and is intended to capture the used reagents and chemicals during deployments; however, the other port is meant as an outlet when flushing the system either with seawater or during in-lab testing.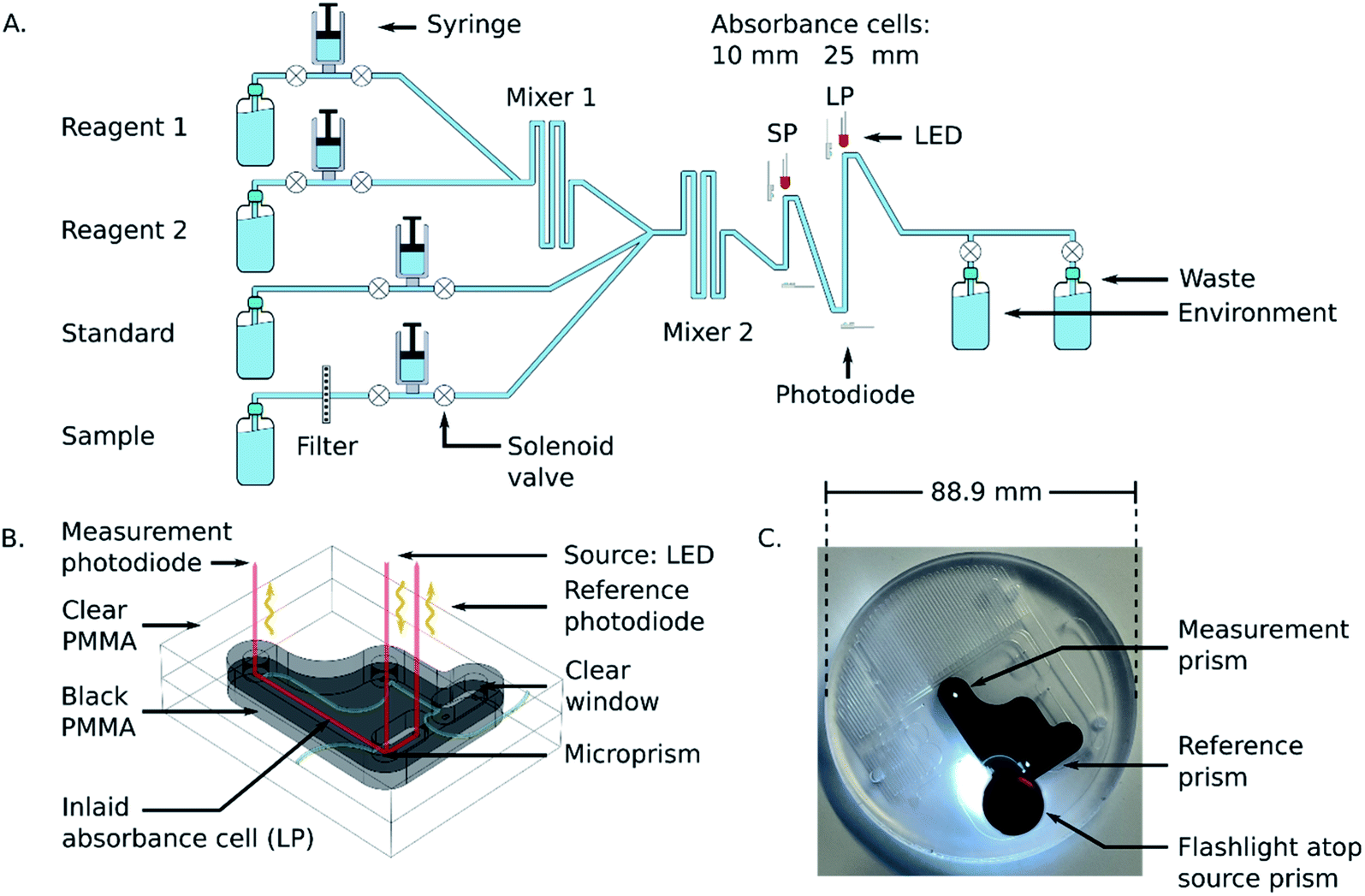 | ||
| Fig. 1 (A) Fluid flow diagram for the microfluidic chip. Fluid from either the standard or sample is mixed with the two reagents, and the resultant product is analyzed in the two absorbance cells. (B) CAD rendering of an inlaid absorbance cell. Microprisms are machined into the clear plastic to direct light from an LED, through the cell, and to a photodiode. Reference photodiodes monitor fluctuations in the light source. (C) Photograph of the bonded microfluidic chip. The mixing chamber and fluid ports are milled into the clear plastic, and the absorbance cell is within the inlaid black plastic. Integral optical windows are realized by routing the microfluidic channel across the clear-black PMMA interface. A flashlight is placed atop the source prism to launch incident light down the optical-fluidic absorbance channel, which then shows light reflecting off the reference and measurement prisms associated with the long absorbance cell. | ||
Fig. 1B shows the working principle of the inlaid optical absorbance cell. Incident light from an LED centered at 880 nm and a 60 nm FWHM (MTE8800NK2, Marktech Optoelectronics, New York, United States) is directed by embedded microprisms using total internal reflection (TIR) into the absorbance cell of length, l, and again up into a photodiode (TSL257, AMS, Premstaetten, Austria) for detection. The black PMMA attenuates all non-directional light, allowing the photodiode to measure only light that has passed through the absorbance cell. The blank voltage, V0, is defined as the photodiode output when only reagents or water have been pumped into the cell. The sample voltage, V, is the photodiode output when a dye has formed in the cell, either from a standard or a sample reacting with the two reagents. The negative logarithm of the ratio of these two voltages determines how much of the initial light has been absorbed, A, which according to the Beer–Lambert law:
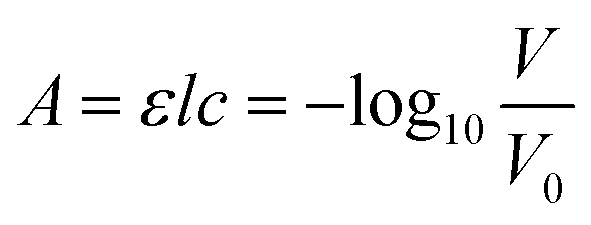 | (1) |
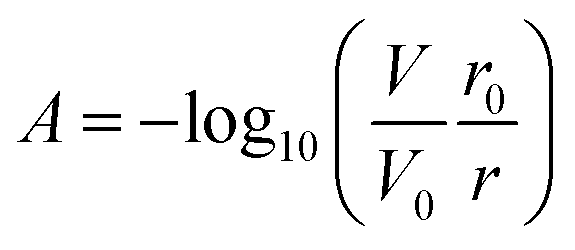 | (2) |
Fig. 1C is a photograph of the completed microfluidic chip, with the inlaid absorbance cells and mixing channels fully integrated. The fluid ports and mixing serpentines are milled into the clear plastic to make it easier to visually inspect the device for collapsed channels or flaws resulting from the manufacturing process. The transparent material also makes it possible to see when and where bubbles have entered the system during laboratory testing. The channels and absorbance cells have square profiles, with 400 μm depth and width. The total fluid volume on the chip, including the 13 μL dead volume in each of the 10 valves, is approximately 450 μL. Of the total volume, roughly 34 μL of sample is required to reach the mixer 2 Y-junction in Fig. 1A. However, since the standard and reagents fluids are not changing between measurements, the only relevant dead-volume on-chip is the sample-to-waste fluid path, which is 253 μL. This value is doubled in the automation protocol to provide a minimum flushing volume whilst avoiding crosstalk between samples.
Fig. 2A shows the Stella Maris multi-sensor seabed platform (MSSP) with the phosphate analyzer integrated as one of the sensors. The MSSP is a submersible tripod testbed that supports over 20 different marine sensing instruments, with a subsea cable that provides power and a communications link with the shore station. The reagents and waste are contained in four separate fluid bags, three 100 mL bags for reagents 1 and 2 and the 2 μM on-board standard, while the waste was a 1 L bag. All reagent bags were housed in a perforated aluminum cannister. Both the sensor and reagent cannister are physically mounted to the frame of the Stella Maris, and communications and power are handled via the platform itself. The data is stored locally on an SD card in the sensor in memory mode, and, also transmitted in real-time to the shore via ethernet. To accomplish this, an RS232-to-ethernet hub is integrated within the tripod MSSP and is accessible through a subsea ethernet connection. This provides constant access to the sensor and data via the MSSP cabin onshore. Fig. 2B is a rendering of the internal components of the phosphate sensor. There are three independently actuatable syringe pumps in total: one for reagent syringes, one for a standard and one for the sample intake. The pumps are comprised of four syringes actuated by three stepper motors threaded into moving plates on guide rails; the two reagent syringes are coupled to the same stepper motor. Limit switches are used to indicate when a syringe is at the end of its stroke. Three pumps provide extensibility and flexibility for a variety of chemical protocols and microfluidic devices compared to prior work with a single actuation mechanism.25 Fluid routing is accomplished with the 10 solenoid valves (LFN series, Lee Company, Connecticut, United States), along with their associated printed circuit board (PCB), housed in a separate compartment at the base of the sensor that can be oil filled for higher pressure deployments. A custom penetrator SubConn cable (MacArtney Canada Ltd) establishes communication and power between the upper valve compartment and the lower main compartment. The microfluidic chip is sandwiched between the two compartments and fluidically interfaces with each cannister via O-ring face seals. The entire sensor is housed in an anodized aluminum pressure casing, and communications and power are handled using a second SubConn cable. An external cannister houses the reagents, standards, and waste bags. Fluid connections are accomplished using 1/16 in inner diameter poly-tetrafluoroethylene (PTFE) tubing and ¼-28 polyether ether ketone (PEEK) connectors (P-359X, IDEX Health and Science, Washington, United States). Power is supplied by either an optional third cannister containing 12 D-cell batteries, not shown, or from an external platform such as the Stella Maris.
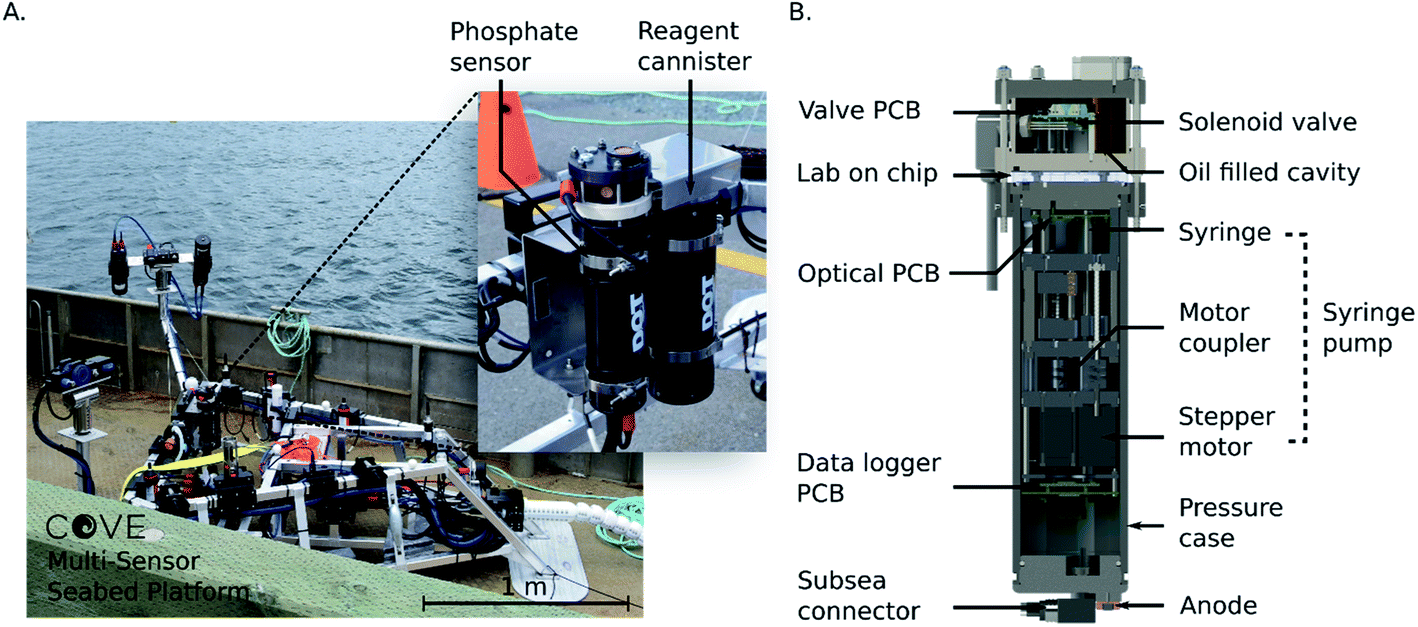 | ||
| Fig. 2 (A) Stella Maris sensor platform on-board the ship prior to deployment in the Bedford Basin. Inset photograph: phosphate analyzer and reagent cannister mounted to the frame of the Stella Maris. (B) Cross section view of the interior of the phosphate sensor. Fluid is pulled and directed into the lab-on-chip by stepper motor-actuated syringes and active solenoid valves. Valve control, data logging and optical control are handled using three separate electronics boards. | ||
The sensor electronics were controlled by three custom made PCBs with custom firmware and software. The automation, data collection and logging was accomplished with firmware written in C++, run on the data logger PCB. The optical PCB handled the stepper motors and optical elements; the LED voltages were controlled using a constant current driver on the optical board, and the measured photodiode voltages were streamed to the data logger PCB and stored on a memory card. The valve PCB was controlled by the data logger board through an RS-232 interface. The data logger also communicated to the outside world via RS-232 and ethernet. The sensor was designed to accept voltages from 7–24 VDC, and nominally consumed 0.1 A during the optical measurements and 1.2 A during pumping. The entire sensor was designed for operation to depths up to 450 m.
2.4 Sensor calibration
The optical system of the sensor, consisting of the LEDs, absorbance cells and photodiodes, was first tested on its own by creating a calibration curve using copper(II) sulfate dye. This first step was necessary to verify that the system works reliably without the added uncertainty of reaction kinetics and mixing ratios that are introduced when using the phosphate assay. Copper(II) sulfate was chosen as a dye because it absorbs in the infrared region and has an absorption peak that overlaps with the emission peak of the 880 nm LED.To create the dye calibration curve, five standards were made ranging in concentrations from 31.25 μM to 500 μM using a serial dilution. Each standard was pumped into the cell for measurement three times, each time followed by a flush cycle. For each concentration, a blank measurement of only Milli-Q water was taken before the triplicate readings. The average voltage from the dye measurements and each preceding blank measurement was used to calculate the absorbance of each concentration.
After the stable dye tests, the phosphate assay was performed using premade standards for calibration. Six phosphate standards ranging from 0.2 μM to 10 μM were prepared by serial dilution, and the two reagents for the PMB assay were prepared in the method previously described. To generate the calibration curve, each standard was pumped through the standard port into the cell for measurement three times, each time followed by a flush cycle. Three blank measurements were taken before each set of triplicate standard readings. In this case, the blanks were entirely Milli-Q water, with no reagent. The standards were pumped into the absorbance cell at a 1:1 ratio with the two-part reagent solution (1:1:2, R1:R2:S) and allowed to diffusively mix and undergo color development for 10 minutes before being flushed out. The absorbance of each standard measurement was calculated using the averaged voltage from the last 5 seconds of the color development and the averaged voltage from the last of the three blank measurements. The measured absorbances of each standard were plotted against their concentrations to determine the attenuation coefficient of the system.
The above sample handling was fully automated using the syringe pumps and solenoid valves in the sensor itself for fluid control, as well as an external 10-port selector valve (Cheminert C65 series, VICI, Texas, United States) for switching between standards and flushing. The selector valve linked the eight standards via PTFE tubing to the standard port on the sensor. The two reagent reservoirs were directly connected to the reagent ports on the sensor using PTFE tubing.
2.5 Temperature sensitivity analysis
To characterize the effects of temperature on the sensor performance, the entire unit, along with the reagents and standards, was submerged in a temperature-controlled water bath. A recirculating cooling unit (NESLAB RTE-7, Thermo Scientific, Massachusetts, United States) was used to fix the bath temperature at 5, 10, 15 and 20 °C. At each of the four temperatures, a three-standard calibration was performed using the same triplicate method as the room temperature calibration. However, for this test, the selected standards were 10 μM, 2 μM, and 0.5 μM PO43−, to cover a wide range. The bath temperatures were measured using both a standard laboratory glass thermometer with 0.5 °C precision and the built-in temperature sensor on the chilling unit itself.2.6 In situ verification and deployments
To test the repeatability and precision of the measurements in an unchanging in situ environment, the unit was fully submersed in a tank of water that had an unknown baseline phosphate concentration (tap water). First a blank measurement, and then three consecutive sample measurements, each preceded by a flushing cycle were taken and analyzed.The sensor was then deployed at a depth of 6 m off a pier in the inlet to the Bedford Basin in Nova Scotia, Canada for 36 hours. Concurrent bottle samples were taken from the water next to the instrument at five different times using a Niskin bottle and were then processed and analyzed in duplicate in a benchtop auto-analyzer (Skalar San++). The sensor was then deployed for a second time on the Stella Maris, 100 m offshore and 9 m deep, again in the inlet to the Bedford Basin.
The sampling protocol and fluid usage volumes for the pier deployment was as follows. First, the system was flushed with 0.5 mL of the standard (2 μM PO43−). Next, 0.25 mL of each reagent was injected into the system. After a 10 minutes wait, the last 5 seconds from the measurement photodiode voltage was recorded to use as the blank voltage of the standard. After that, 0.5 mL of the standard, along with 0.25 mL of each reagent was injected and allowed to mix and react for 10 minutes to allow dye formation to occur. The voltage from the last 5 seconds was recorded and used as the sample voltage of the standard. This step was then repeated once to ensure there was no drift or carry over in the standards. The system was then thoroughly flushed with 0.5 mL of seawater, and monitored for 10 minutes, providing a blank measurement for the seawater sample. This step was then repeated to ensure there was no crosstalk between samples. Next, 0.5 mL of seawater and 0.25 mL of each reagent (1 mL total fluid) was injected into the cell and given a 10 minutes reaction time. The last 5 seconds of the measurement photodiode voltage was recorded to use as the sample voltage. This step was repeated 10 times, generating 10 sample measurements and then the entire cycle was restarted. The calibration curve generated in the lab was used to convert the measured absorbances of the samples and standards into phosphate concentrations from the field deployments.
The sampling protocol for the MSSP deployment was the same as for the pier deployment, but with the fluid volumes increased. The seawater flushes were 3 mL instead of 1 mL to be certain of no crosstalk or sampling smearing, which proved to be un-necessary as shown in the results section. Likewise, the blank for the standard measurements also used 3 mL of fluid. The fluid volumes for both the sample measurements and the standard measurements used 3 mL of sample/standard and 3 mL of mixed reagent (1.5 mL of each reagent). Other than the differences in volumes, the automated protocol remained identical to the pier deployment.
3. Results and discussion
The optical subsystem was verified using a series of stable copper(II) sulfate mixtures with varying concentrations to evaluate the measured absorbances of the complete system. The raw voltage measurements for each standard and blank are displayed in the top panel of Fig. 3. The plot shows one blank measurement, indicated by the grey shaded regions, for every three copper sulfate measurements. The noisy periods preceding each voltage plateau (more clearly displayed in the long path cell) for the blank and sample measurements are the result of the fluid being displaced during pumping. The processed and averaged voltage data used to generate the calibration curve can be found in the ESI, Fig. S1.† The bottom left panel of Fig. 3 shows the calibration curve generated from the voltage data. Both the long path and short path length absorbance cells produced the expected Beer–Lambert linear result, each with an R2 value over 0.99 and root-mean-square-error (RMSE) values of 0.23 and 0.20 mAU for the short and long paths respectively. The bottom right panel is a residual plot for the linear regression of both path lengths, short and long. At the lower concentrations (125 μM and lower), the spread on the residuals begins to increase presumably due to the weighting of the linear fit. However, the distribution of residuals is random and thus indicates no detectable systematic errors.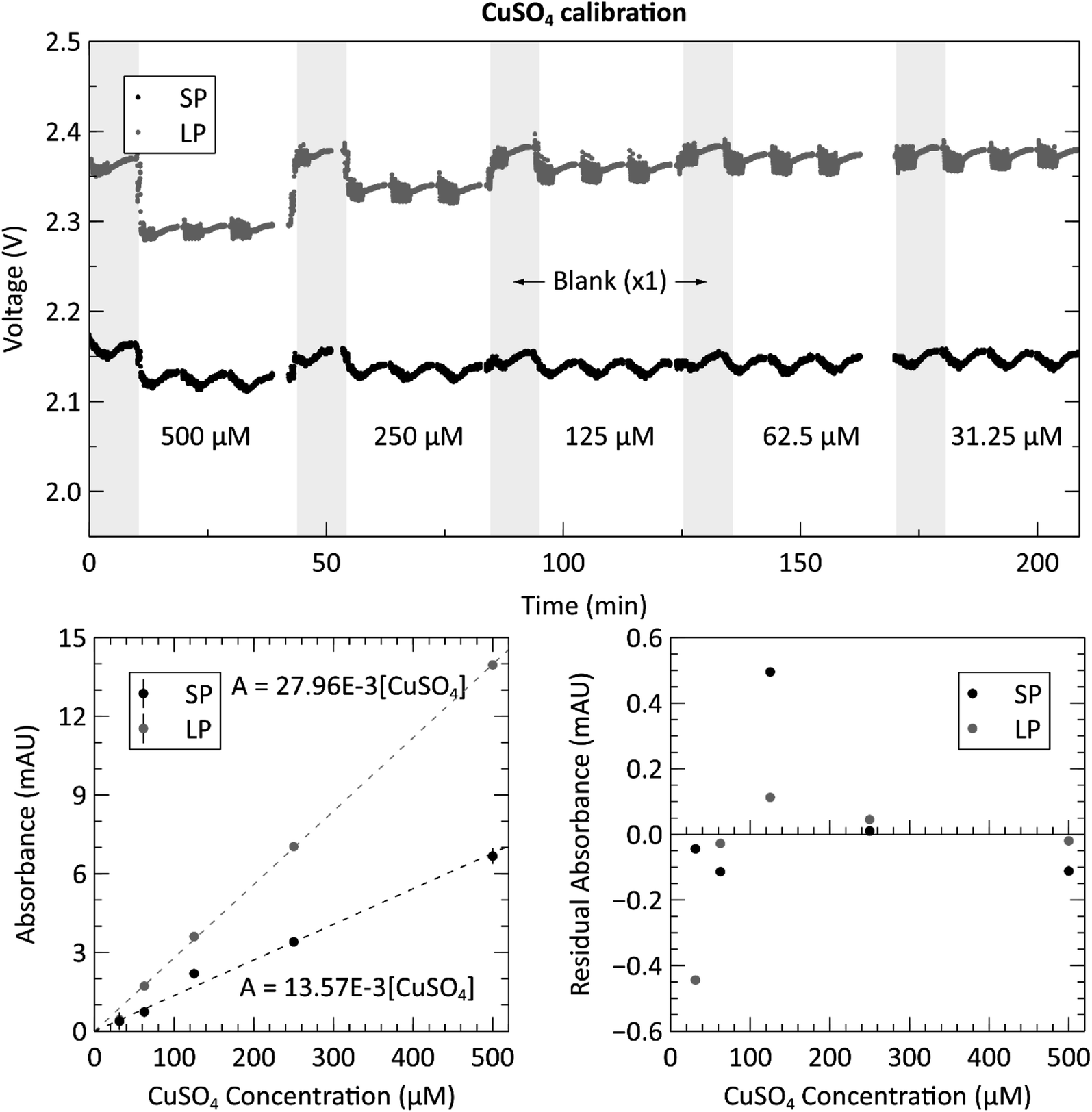 | ||
| Fig. 3 Top: raw voltage from analyzing three repetitions of each of the five copper(II) sulfate (CuSO4) dye concentrations (ranging from 31.25–500 μM), with a blank measurement (shaded grey regions) between each new concentration. Data from both the short path length (SP = 10.4 mm) and long path length (LP = 25.4 mm) absorbance cells are displayed. Bottom left: calibration curve from the triplicate measurements of the CuSO4 calibration. The absorbance values are determined from the voltage measurements shown in the top panel. Bottom right: residual plot of the linear regressions for the long and short path lengths. | ||
Next, we proceeded to evaluate the sensor by performing the phosphate assay on-chip with known pre-made standards. Fig. 4 shows the calibration data acquired from the automated system when supplying the sensor with six phosphate standards ranging from 0.2 μM to 10 μM. The top panel shows the measured raw voltages for the three blank measurements, indicated by the grey shaded regions, and the three standard measurements for each concentration of PO43− from both measurement cells. See the ESI, Fig. S2† for the processed and averaged voltage data. The concentrations displayed in the top panel are the actual concentrations of the prepared standards, before mixing, and fed directly into the sample intake port. The concentrations shown on the horizontal axes of the two bottom panels are the PO43− concentrations in the absorbance cell, after being mixed at a 1:1 ratio with the reagents. The calibration curves were created by calculating the absorbance of each concentration using the triplicate standard measurements and the last of each blank measurement. The slopes of the curves are 0.0465 AU μM−1 and 0.0193 AU μM−1 for the long and short path lengths, respectively. By dividing out the lengths of each absorbance cell, the attenuation coefficients of the long and short paths are calculated as 0.0183 cm−1 and 0.0186 cm−1. This is higher than the coefficients reported by Clinton-Bailey et al., who reported 0.00584–0.00612 cm−1 (ref. 27) for the PMB assay modified with 0.01% PVP. This large difference is mostly due to the difference in how the concentrations are reported. Clinton-Bailey et al. reports the concentration of the standard before mixing at 1:1:1 ratio with the two reagents, where as we report it after mixing. Therefore, their slope needs to be multiplied by three yielding 0.01752–0.01836 cm−1, showing that the coefficients are very similar. However, there are also other considerations, such as the difference in the selected reaction times. We use a 10 minutes hold time to allow for more color development and increased sensitivity, whereas Clinton-Bailey et al. use a 5 minutes reaction time for increased sampling frequency. The 10 minutes reaction time in the short path accounts for a 25% increase in the measured absorbance of a 2 μM standard when compared to a 5 minutes reaction time. Please see Fig. S3 in the ESI† for a plot of the color development curve. A second important difference is that we use an LED centered at 880 nm, versus 700 nm in the work by Clinton-Bailey. A spectral analysis of a 2 μM standard revealed that the measured absorbance (averaged over a 5 nm window) at 880 nm was 133% of the measured absorbance at 700 nm. Furthermore, the emission peak of our 880 nm LED has a 60 nm FWHM that is wider than the 700 nm LED with a FWHM of 21 nm. These details indicate that our measured coefficients should be higher than those reported by Clinton-Bailey et al. by 166%; however, the photodiode used by both authors is roughly 50% as sensitive at 880 nm as it is at 700 nm, which may explain why the final coefficients are comparable. Despite the discrepancy between our attenuation coefficients and the reported literature values, the calibration curves were linear, with R2 values of 0.99 for both path lengths and RMSE values of 1.56 and 3.95 mAU for the short and long paths respectively. The bottom right panel of Fig. 4 shows the residual plot of the linear regression for the measurements made in the two absorbance channels. The pattern shows that the residual error is very low at the lower concentrations, which is likely due to the addition of the 0.01% PVP to the reagents. In our previous trials that did not include PVP (data not shown), the residuals observed below 1 μM showed that the results were not linear, nor repeatable between successive tests. The residual analysis also indicates that the 10 μM (5 μM in the cell) measurement might be skewing the regression upwards, because most of the middle-concentration measurements (0.25–2.5 μM) are below the linear fit. The 10 μM data set in the long path length also has the largest error of the concentrations. Nevertheless, the calibration produced the expected linear relationship and showed a dynamic range between 0.2 to 10 μM with relative standard deviations (RSDs) ranging from 12.1% to 3.45%, respectively. To further quantify the repeatability of the sensor, an in situ verification test was performed on tap water (please see ESI Fig. S4†). The RSDs of the unknown concentrations from these three measurements were 1.48% in the long channel and 1.06% in the short channel, demonstrating excellent precision on bench and underwater.
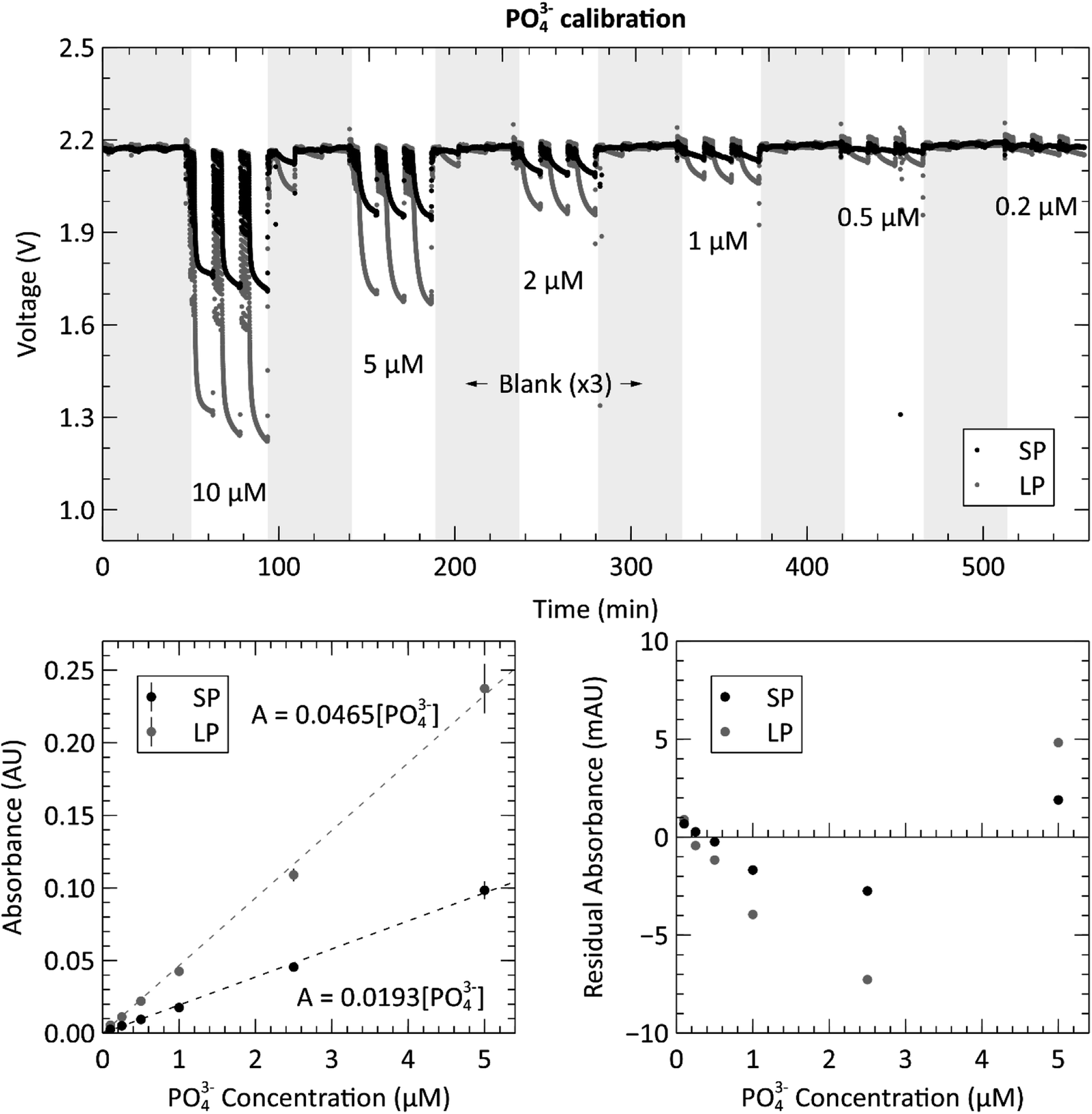 | ||
| Fig. 4 Top: raw voltage from analyzing three repetitions of each of the five phosphate (PO43−) standard concentrations (ranging from 0.2–10 μM), with three blank measurements (shaded grey regions) between each new concentration. Data from both the short path length (SP = 10.4 mm) and long path length (LP = 25.4 mm) absorbance cells are displayed. Bottom left: calibration curve from the triplicate measurements of the PO43− calibration. Bottom right: residual plot of the linear regressions for the long and short path lengths. Measurements conducted at room temperature or 21–23 °C. | ||
To fully characterize the sensor's performance, we calculated the limit of detection (LOD) and limit of quantification (LOQ) from the 18 blank measurements taken during the preceding calibration. For a linear regression, the three-sigma LOD is defined as three times the value of the blank baseline noise.34 The LODs for the short and long paths were therefore calculated to be 0.33 and 0.29 mAU, corresponding to 16.9 and 15.2 nM [PO43−]. However, a more practical metric for the performance of the sensor is the limit of quantification, which is defined as ten times the blank baseline noise for a linear regression, and was calculated as 1.09 and 0.98 mAU, corresponding to 56.5 and 50.8 nM [PO43−] for the short and long paths, respectively.
The submersible lab-on-chip phosphate sensor presented thus far shows that it performs well on the bench, but it is intended to be deployed in marine environments. As such, calibrations like that of Fig. 4, were performed at the expected environmental water temperatures. The left panel of Fig. 5 contains the short path calibration curves resulting from our temperature dependence study. Each of the four curves represent a calibration done at a different set temperature (5, 10, 15 and 20 °C). The average value and standard error of each slope was calculated and plotted in the right panel of Fig. 5 to show the sensitivity change with temperature. The slopes of each sensitivity curve are 2.3 × 10−4 and 5.8 × 10−4 AU (μM °C)−1 for the short and long absorbance cells, respectively. When the path lengths are divided out, the sensitivities for each cell become 2.21 × 10−4 and 2.28 × 10−4 AU (μM °C cm)−1. These sensitivities are slightly higher than the slopes reported by Grand et al., who measured 1.46 × 10−4 AU (μM °C cm)−1 when the path length is divided out.32 The different reaction times and absorbance peaks are the likely source of the discrepancy. All three of the measurements done at 5 °C in the long path were omitted from linear fit as outliers. It is suspected that a bubble was trapped in the long absorbance cell for the duration of the 5 °C triplicate measurements, causing the absorbances to measure higher than they would have otherwise. The data from the short path does not show this outlying behavior, which further supports the idea that a bubble was impeding the light path in the longer cell. This also punctuates the value and robustness provided by having multiple absorbance cells. Nevertheless, the linear regressions from the sensitivity measurements of the two absorbance cells had R2 values of 0.92 and 0.97 and RMSE values of 0.38 and 0.44 mAU μM−1 for the short and long cells, respectively.
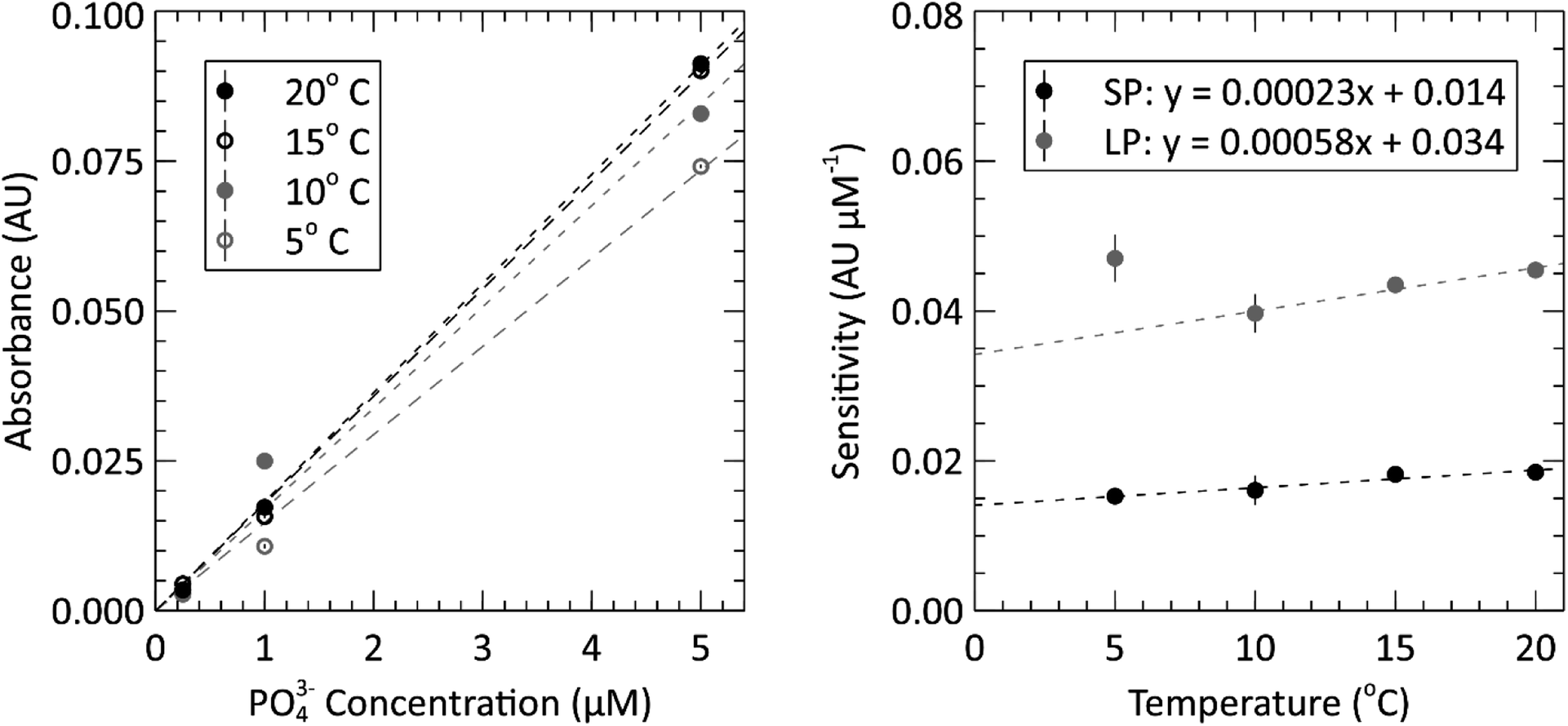 | ||
| Fig. 5 Left: in situ sensor calibration curves for the short path length at four temperatures between 5 °C and 20 °C. Each test was done using a 10 μM standard (5 μM in the cell), a 2 μM standard (1 μM in the cell) and a 0.5 μM standard (0.25 μM in the cell). Right: temperature sensitivity analysis of the sensor; where, linear relationship describes the absorbance measurements of each flow cell changes with temperature. The 5 °C point on the long path length data was omitted from the fit. | ||
Finally, we proceeded to deploy the fully lab-characterized phosphate sensor in marine environments. The two panels of Fig. 6 show the phosphate concentration measurements made during the two in situ deployments, one at a local pier and one on the MSSP subsea platform, described in the methods. The concentrations were calculated from the measured absorbance by using the slope of the calibration curve generated in the laboratory as a conversion factor, i.e. the slopes in Fig. 4 and 5. The gaps in the data displayed on the two plots in Fig. 6 are a result of the previously described sampling protocol. They represent the times when sensor was making blank and standard measurements and therefore could not make sample measurements. The temperature logged on the sensor itself was used to determine the appropriate sensitivity according to the temperature dependence characterization from Fig. 5. During the pier deployment in the top panel, manual bottle samples were taken and analyzed in an independent laboratory, shown with ‘x’ markers in Fig. 6, to validate the sensor readings reported. Except for the first bottle sample, the phosphate concentrations measured by the auto-analyzer for the pier deployment are all within 100 nM of the closest sensor measurements, with the average discrepancy being 42 nM. Overall, the average percentage of difference between the two methods was 27.7%, comparing manual sample and autoanalyzer versus in situ lab-on-chip sensor, for this low concentration range. During the first bottle sample acquisition, the Niskin bottle hit the seabed, likely disturbing the sediment, and releasing phosphate into the surrounding water. This is the probable explanation for the large difference (177 nM) between the auto-analyzer measurement and the sensor measurement at the time of the first bottle sample. All other bottle samples were taken without contacting the ground.
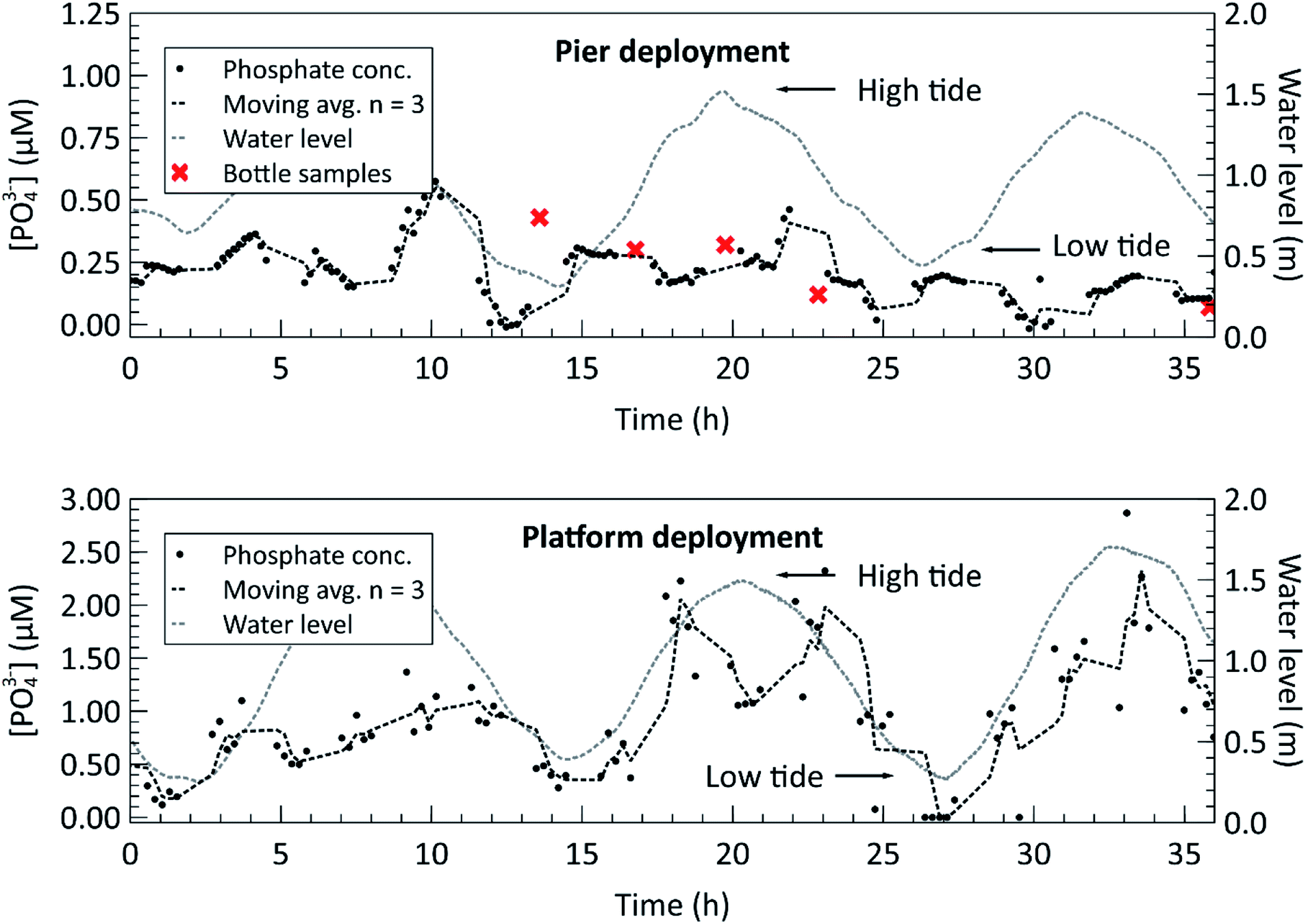 | ||
| Fig. 6 Top: in situ phosphate concentration measurements from a near-shore pier at a depth of 6 m. Concurrent bottle sample measurements are plotted alongside the sensor measurements at five different points in time. The tidal cycle is overlaid to show the mixing effect close to shore. Bottom: in situ phosphate concentration measurements from a multi-sensor seabed platform (MSSP), 100 m offshore and 9 m deep in the inlet to the Bedford Basin. The tidal cycle is overlaid to show the mixing effect in the deployment region. | ||
For both deployments, the water level, measured by the nearby Bedford Institute of Oceanography tide gauge at station BIO 00491, is overlaid on the deployment plots to highlight the correlation between the semi-diurnal tidal cycle and phosphate concentration. A correlation coefficient of 0.59 was found between the measured phosphate concentration and the water level for the platform deployment, indicating a moderate positive correlation. A plot of the phosphate concentration versus water height can be found in the ESI, Fig. S5.† Furthermore, the magnitudes of the measured phosphate concentrations increase during a water level transition. The trend is observable in the pier deployment data in the top panel and in the MSSP deployment data in the bottom panel. The inlet to the Bedford Basin is a shallow, less than 20 m deep region, known as the “narrows” and is subject to strong currents during the ebb and flow phases of the tide.35 This leads to turbulent vertical mixing, in which nutrient rich water from the depths of the Scotian Shelf mixes with the surface waters during the incoming tide, and nutrient rich runoff water from the Basin mixes during the outgoing tide.36 This can explain the phosphate concentration profile observed during both deployments, in which the nutrient concentrations peak when the water current in either direction is strongest. However, further long-term deployments, potentially with multiple sensors positioned around the basin are required to verify these findings. The Bedford Institute of Oceanography has done a thorough analysis of the seawater matrix in the Bedford Basin,37 including weekly phosphate measurements carried out using the blue method on the Skalar SAN++ autoanalyzer. In doing so, they have demonstrated that the phosphomolybdenum blue method is valid for the Bedford Basin, despite possible matrix effects or interferences in other bodies of water. Our future plans are to collect multi-parameter measurements to extract the necessary metadata for contextualizing nutrient concentrations with other biogeochemical measurements such as salinity, irradiance, nitrate, and chlorophyll-a, across a range of marine environments.
4. Conclusions
Robust, submersible, and fluid-efficient nutrient sensors are required if we are to improve our understanding of nutrient dynamics and fluxes in coastal regions. Here, we have presented a microfluidic phosphate analyzer capable of making precise in situ measurements, with less than 1.5% RSD underwater, that cross-correlate within 42 nM [PO43−] to the measurements performed by an auto-analyzer. It achieves a limit of detection of 15.2 nM and a quantification limit of 50.8 nM over a dynamic range of 0.2 to 10 μM. The sensor is based on inlaid microfluidic absorbance cells, that permit decoupling of the lab-on-chip (LOC) from the electronics package, allowing configurability for different LOC designs. It is readily serviceable in the event of clogging or chip failure, and has self-aligned optical components, requiring no physical adhesives to the LOC. We conducted two field deployments to monitor the change in phosphate concentrations during the semi-diurnal tidal cycle and highlight the sensor's ability to capture short term events in a way that discrete ex situ methods cannot. This provides further insight into phosphate dynamics in the Bedford Basin, and other bodies of water, where phosphate concentrations could be sampled at appropriate temporal and spatial resolutions that inform biogeochemical models and ensure local regulatory compliance. Long term deployments with tens of sensors are the next phase in demonstrating suitable field endurance and will enable insight into biogeochemical observations with increased monitoring capacity. We believe that such field portable and in situ lab-on-chip systems will be highly impactful in aquaculture, oceanographic science, waste-water treatment, and industrial runoff monitoring.Conflicts of interest
Arnold Furlong and Vincent Sieben declare share holdings in Dartmouth Ocean Technologies Inc. There are no other conflicts of interest to declare.Acknowledgements
Acknowledgments are extended to Connor Mackie for the acquisition and preparation of bottle samples, Anadiuska Rondon Vivas for processing our samples on the auto-analyzer, and to Heather Daurie for supplying Milli-Q water. Also, the authors are thankful to Bruce MacDougall, Sheila Paterson, Tom Knox and Jason Hopper at the Centre for Ocean Ventures and Entrepreneurship (COVE) for facilities use and access to the MSSP platform. Recognition also goes to Prof. Doug Wallace and Prof. Julie LaRoche for their scientific guidance on nutrient measurements in the Bedford Basin. Finally, acknowledgements go to the staff and contractors from Dartmouth Ocean Technologies Inc. (DOT) for their role in the sensor instrument: Merle Pittman, Lee Miller, Iain Grundke, James Smith, Mark Wright, Kirk Phelan, and Colin Sonnichsen. Finally, gratitude is expressed towards the Marine Environmental Observation, Prediction and Response Network (MEOPAR), the Natural Sciences and Engineering Research Council (NSERC), the Canada First Research Excellence Fund (CFREF) through the Ocean Frontier Institute (OFI), and to the Innovacorp Early Stage Commercialization Fund (ESCF) for funding.References
- J. R. Casey,
et al., Phytoplankton taxon-specific orthophosphate (Pi) and ATP utilization in the western subtropical North Atlantic, Aquat. Microb. Ecol., 2009, 58(1), 31–44, DOI:10.3354/ame01348
.
- A. C. Martiny, Y. Huang and W. Li, Occurrence of phosphate acquisition genes in Prochlorococcus cells from different ocean regions, Environ. Microbiol., 2009, 11(6), 1340–1347, DOI:10.1111/j.1462-2920.2009.01860.x
- E. J. Lessard, A. Merico and T. Tyrrell, Nitrate: Phosphate Ratios and Emiliania huxleyi Blooms, Limnol. Oceanogr., 2005, 50(3), 1020–1024 CrossRef CAS
- M. Garrett, J. Wolny, E. Truby, C. Heil and C. Kovach, Harmful algal bloom species and phosphate-processing effluent: field and laboratory studies, Mar. Pollut. Bull., 2011, 62(3), 596–601, DOI:10.1016/j.marpolbul.2010.11.017
- Wisconsin State Legislature Chapter NR 217.04: Effluent Standards and Limitations for Phophorus, 2011, available: https://docs.legis.wisconsin.gov/code/admin_code/nr/200/217.
- Canada's Challenges and Opportunities to Address Contaminants in Wastewater, Canadian Water Network, Mar. 2018, available: https://cwn-rce.ca/wp-content/uploads/projects/other-files/Canadas-Challenges-and-Opportunities-to-Address-Contaminants-in-Wastewater/CWN-Report-on-Contaminants-in-WW-Supporting-Doc-2.pdf Search PubMed.
- U. Sivasankaran,
et al., Ultrasensitive electrochemical sensing of phosphate in water mediated by a dipicolylamine-zinc(II) complex, Sens. Actuators, B, 2020, 321, 128474, DOI:10.1016/j.snb.2020.128474
- C. Warwick,
et al., Conductance based sensing and analysis of soluble phosphates in wastewater, Biosens. Bioelectron., 2014, 52, 173–179, DOI:10.1016/j.bios.2013.08.048
- X. Song,
et al., Europium-based infinite coordination polymer nanospheres as an effective fluorescence probe for phosphate sensing, RSC Adv., 2017, 7(14), 8661–8669, 10.1039/c6ra27819a
- M. D. Patey, M. J. A. Rijkenberg, P. J. Statham, M. C. Stinchcombe, E. P. Achterberg and M. Mowlem, Determination of nitrate and phosphate in seawater at nanomolar concentrations, TrAC, Trends Anal. Chem., 2008, 27(2), 169–182, DOI:10.1016/j.trac.2007.12.006
- C. Warwick, A. Guerreiro and A. Soares, Sensing and analysis of soluble phosphates in environmental samples: a review, Biosens. Bioelectron., 2013, 41, 1–11, DOI:10.1016/j.bios.2012.07.012
- M. Bowden and D. Diamond, The determination of phosphorus in a microfluidic manifold demonstrating long-term reagent lifetime and chemical stability utilising a colorimetric method, Sens. Actuators, B, 2003, 90(1), 170–174, DOI:10.1016/S0925-4005(03)00024-8
- J. Murphy and J. P. Riley, A modified single solution method for the determination of phosphate in natural waters, Anal. Chim. Acta, 1962, 27, 31–36, DOI:10.1016/S0003-2670(00)88444-5
- United States. Environmental Protection Agency Method 365.2, Methods for the Chemical Analysis of Water and Wastes (MCAWW) (EPA/600/4-79/020).
- E. A. Nagul, I. D. McKelvie, P. Worsfold and S. D. Kolev, The molybdenum blue reaction for the determination of orthophosphate revisited: opening the black box, Anal. Chim. Acta, 2015, 890, 60–82, DOI:10.1016/j.aca.2015.07.030
- E. V. Dafner, Segmented continuous-flow analyses of nutrient in seawater: intralaboratory comparison of Technicon AutoAnalyzer II and Bran+Luebbe Continuous Flow AutoAnalyzer III, Limnol. Oceanogr.: Methods, 2015, 13(10), 511–520, DOI:10.1002/lom3.10035
- HydroCycle PO4, Sea-Bird Scientific – Downloads, Sea-Bird, https://www.seabird.com/hydrocycle-po/product-downloads?id=54721314201, accessed Apr 06 2021 Search PubMed.
- G. Duffy, I. Maguire, B. Heery, C. Nwankire, J. Ducrée and F. Regan, PhosphaSense, A fully integrated, portable lab-on-a-disc device for phosphate determination in water, Sens. Actuators, B, 2017, 246, 1085–1091, DOI:10.1016/j.snb.2016.12.040
- A. Donohoe, G. Lacour, P. McCluskey, D. Diamond and M. McCaul, Development of a Cost-Effective Sensing Platform for Monitoring Phosphate in Natural Waters, Chemosensors, 2018, 6(4), 57, DOI:10.3390/chemosensors6040057
- J. M. Zhu,
et al., Optofluidic marine phosphate detection with enhanced absorption using a Fabry–Pérot resonator, Lab Chip, 2017, 17(23), 4025–4030, 10.1039/C7LC01016H
- V. J. Sieben, C. F. A. Floquet, I. R. G. Ogilvie, M. C. Mowlem and H. Morgan, Microfluidic colourimetric chemical analysis system: application to nitrite detection, Anal. Methods, 2010, 2(5), 484–491, 10.1039/C002672G
- F.-E. Legiret,
et al., A high performance microfluidic analyser for phosphate measurements in marine waters using the vanadomolybdate method, Talanta, 2013, 116, 382–387, DOI:10.1016/j.talanta.2013.05.004
- A. D. Beaton,
et al., Lab-on-Chip Measurement of Nitrate and Nitrite for In Situ Analysis of Natural Waters, Environ. Sci. Technol., 2012, 46(17), 9548–9556, DOI:10.1021/es300419u
- M. Yücel, A. D. Beaton, M. Dengler, M. C. Mowlem, F. Sohl and S. Sommer, Nitrate and Nitrite Variability at the Seafloor of an Oxygen Minimum Zone Revealed by a Novel Microfluidic In Situ Chemical Sensor, PLoS One, 2015, 10(7) DOI:10.1371/journal.pone.0132785
- A. D. Beaton,
et al., High-Resolution in Situ Measurement of Nitrate in Runoff from the Greenland Ice Sheet, Environ. Sci. Technol., 2017, 51(21), 12518–12527, DOI:10.1021/acs.est.7b03121
- A. G. Vincent,
et al., Nitrate drawdown during a shelf sea spring bloom revealed using a novel microfluidic in situ chemical sensor deployed within an autonomous underwater glider, Mar. Chem., 2018, 205, 29–36, DOI:10.1016/j.marchem.2018.07.005
- G. S. Clinton-Bailey,
et al., A Lab-on-Chip Analyzer for in Situ Measurement of Soluble Reactive Phosphate: Improved Phosphate Blue Assay and Application to Fluvial Monitoring, Environ. Sci. Technol., 2017, 51(17), 9989–9995, DOI:10.1021/acs.est.7b01581
- A. J. Birchill, A. D. Beaton, T. Hull, J. Kaiser, M. Mowlem, R. Pascal, A. Schaap, Y. G. Voyonova, C. Williams and M. Palmer, Exploring Ocean Biogeochemistry Using a Lab-on-Chip Phosphate Analyser on an Underwater Glider, Front. Mar. Sci., 2021, 8, 698102, DOI:10.3389/fmars.2021.698102
- E. A. Luy, S. C. Morgan, J. J. Creelman, B. J. Murphy and V. J. Sieben, Inlaid microfluidic optics: absorbance cells in clear devices applied to nitrite and phosphate detection, J. Micromech. Microeng., 2020, 30(9), 095001, DOI:10.1088/1361-6439/ab9202
- I. R. G. Ogilvie, V. J. Sieben, M. C. Mowlem and H. Morgan, Temporal Optimization of Microfluidic Colorimetric Sensors by Use of Multiplexed Stop-Flow Architecture, Anal. Chem., 2011, 83(12), 4814–4821, DOI:10.1021/ac200463y
- C. M. Rushworth, J. Davies, J. T. Cabral, P. R. Dolan, J. M. Smith and C. Vallance, Cavity-enhanced optical methods for online microfluidic analysis, Chem. Phys. Lett., 2012, 554, 1–14, DOI:10.1016/j.cplett.2012.10.009
- M. M. Grand, G. S. Clinton-Bailey, A. D. Beaton, A. M. Schaap, T. H. Johengen, M. N. Tamburri, D. P. Connelly, M. C. Mowlem and E. P. Achterberg, A Lab-On-Chip Phosphate Analyzer for Long-term In Situ Monitoring at Fixed Observatories: Optimization and Performance Evaluation in Estuarine and Oligotrophic Coastal Waters, Front. Mar. Sci., 2017, 4, 255, DOI:10.3389/fmars.2017.00255
- P. C. Bartels and A. F. Roijers, A kinetic study on the influence of the parameters in the determination of inorganic phosphate by the molybdenum blue reaction, Clin. Chim. Acta, 1975, 61(2), 135–144, DOI:10.1016/0009-8981(75)90307-1
- A. Shrivastava and V. Gupta, Methods for the determination of limit of detection and limit of quantitation of the analytical methods, Chron. Young Sci., 2011, 2(1), 21, DOI:10.4103/2229-5186.79345
- S. Shan and J. Sheng, Examination of circulation, flushing time and dispersion in Halifax Harbour of Nova Scotia, Water Qual. Res. J., 2012, 47(3–4), 353–374, DOI:10.2166/wqrjc.2012.041
- W. J. Burt, H. Thomas, K. Fennel and E. Horne, Sediment-water column fluxes of carbon, oxygen and nutrients in Bedford Basin, Nova Scotia, inferred from 224Ra measurements, Biogeosciences, 2013, 10(1), 53–66, DOI:10.5194/bg-10-53-2013
-
W. K. W. Li, Bedford Institute of Oceanography, Canada, Department of Fisheries and Oceans and Ocean and Ecosystem Sciences Division, The State of Phytoplankton and Bacterioplankton at the Compass Buoy Station: Bedford Basin Monitoring Program 1992–2013, Dartmouth: Fisheries and Oceans Canada, Maritimes Region, Ocean and Ecosystem Sciences Division, Bedford Institute of Oceanography, 2014 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ay01876k |
| This journal is © The Royal Society of Chemistry 2022 |